

Abstract
DEAD-box proteins are the most common RNA helicases, and they are associated with virtually all processes involving RNA. They have nine conserved motifs that are required for ATP and RNA binding, and for linking phosphoanhydride cleavage of ATP with helicase activity. The Q motif is the most recently identified conserved element, and it occurs ∼17 amino acids upstream of motif I. There is a highly conserved, but isolated, aromatic group ∼17 amino acids upstream of the Q motif. These two elements are involved in adenine recognition and in ATPase activity of DEAD-box proteins. We made extensive analyses of the Q motif and upstream aromatic residue in the yeast translation-initiation factor Ded1. We made site-specific mutations and tested them for viability in yeast. Moreover, we purified various mutant proteins and obtained the Michaelis–Menten parameters for the ATPase activities. We also measured RNA affinities and strand-displacement activities. We find that the Q motif not only regulates ATP binding and hydrolysis but also regulates the affinity of the protein for RNA substrates and ultimately the helicase activity.
Keywords: adenine recognition, ATPase, molecular motor, Saccharomyces cerevisiae, unwind
Introduction
RNA and DNA helicases of superfamilies 1 and 2 (SF1 and SF2) are ubiquitous proteins that are involved in virtually all aspects of gene expression. The ‘helicase core' of the proteins contains seven to nine conserved motifs, and it is the characteristics of these motifs that are used to classify the proteins into different subfamilies (Gorbalenya and Koonin, 1993). The solved crystal structures of proteins from various families show structurally conserved cores consisting of two domains, where the conserved motifs mostly face a cleft formed between the domains (recently reviewed by Caruthers and McKay, 2002). Remarkably, the different structures can be largely superimposed on each other, which suggests that they all use similar molecular mechanisms (Caruthers and McKay, 2002). The amino-terminal motifs I and II, also known as the Walker A and B motifs, are common to NTPases in general (Walker et al, 1982; Smith and Rayment, 1996; Leipe et al, 2003). These two motifs coordinate the phosphates of a bound NTP, and they are thought to link somehow hydrolysis of the β–γ phosphoanhydride bond with enzymatic activity (Walker et al, 1982; Smith and Rayment, 1996; Leipe et al, 2003). With only a few exceptions, most SF1 and SF2 helicase families are specific for ATP or dATP (reviewed by Tanner, 2003). Some helicases have processive unwinding activity of long double-stranded RNA or DNA substrates, but most have feeble activity when tested in vitro (Lohman and Bjornson, 1996; Soultanas and Wigley, 2000; Tanner and Linder, 2001; Silverman et al, 2003).
The DEAD-box family of SF2 is the largest family of RNA helicases (Rocak and Linder, 2004). They are so named by the characteristic Asp-Glu-Ala-Asp (D-E-A-D) sequence of motif II (Linder et al, 1989). We were interested in studying the structural–functional relationships of the different domains in these proteins when we discovered a previously unidentified conserved element that occurs ∼17 amino acids (aa) upstream of motif I (Tanner et al, 2003). It contains the sequence G-F-c-c-P-T-P-I-Q, where c is a charged sidechain residue (Figure 1A). The glutamine is invariant, and consequently we call this new element the Q motif. There is an additional highly conserved aromatic group (F) ∼17 aa upstream of the Q motif. Comparisons with published crystal structures of DEAD-box proteins (Benz et al, 1999; Caruthers et al, 2000; Story et al, 2001) and secondary structure predictions indicate that this new motif is part of a highly conserved structure consisting of a loop–helix–loop, where the isolated conserved aromatic group interacts with the conserved central proline of the Q motif. This loop–helix–loop structure forms a cap on top of motif I, and is capable of forming extensive interactions with motif I and a bound ATP. This new motif is both unique to and characteristic of DEAD-box proteins (Tanner et al, 2003). We made site-specific mutations of the Q motif and upstream aromatic group in the translation-initiation factor eIF4A in the yeast Saccharomyces cerevisiae, and showed that the conserved residues were important for in vivo viability and for ATPase activity of the purified proteins (Tanner et al, 2003).
Figure 1.
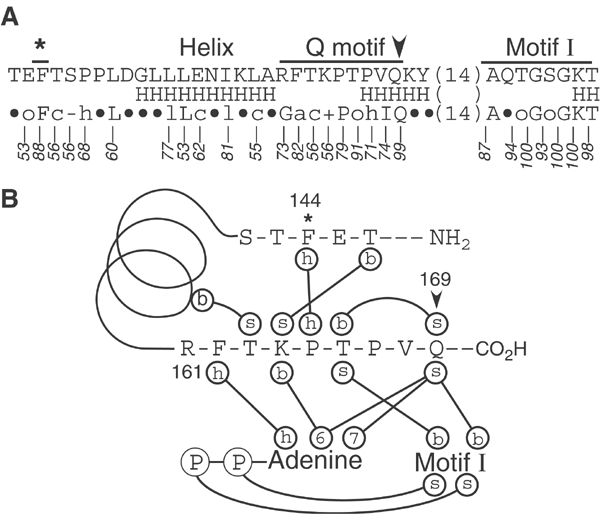
The Q motif and flanking sequences. (A) The amino-acid (aa) sequence of Ded1 around the Q motif (residues 142–193). The predicted helical (H) structure is shown below (PredictProtein; Rost and Sander, 1994), as well as the consensus from 277 DEAD-box sequences in the database (a=F,Y,W; c=D,E,H,K,R; −=D,E; +=H,K,R; l=I,L,V; ○=S, T; h=A,I,L,M,P,V; •=any amino acid; Bork Consensus; http://www.bork.embl-heidelberg.de). The numbers indicate the percentage of occurrence of each residue or functional group. (B) Predicted network of interactions of Ded1 of the Q motif and flanking sequences based on the solved crystal structure of eIF4A with a bound ADP (Benz et al, 1999). The sidechain (s) of the highly conserved Q169 interacts with the N6 and N7 positions of adenine and with the peptide backbone (b) of motif I. The conserved aromatic group (F162) forms hydrophobic stacking interactions (h) with the base. *, A highly conserved, but isolated, aromatic group (F and less often W) that typically appears 17 aa upstream of the Q motif;  , the essentially invariant Gln that typically appears 17 aa upstream of motif I. Note that a short helix is invariably predicted to occur between the isolated upstream aromatic residue and the Q motif within randomly selected sequences in the database.
, the essentially invariant Gln that typically appears 17 aa upstream of motif I. Note that a short helix is invariably predicted to occur between the isolated upstream aromatic residue and the Q motif within randomly selected sequences in the database.
Our previous analyses were limited because of the poor ATPase and unwinding activities of eIF4A in vitro. Consequently, we expanded our analysis with Ded1, which is another yeast translation-initiation factor (Chuang et al, 1997; de la Cruz et al, 1997). It was previously shown that purified Ded1 has good ATPase and strand-displacement activities (Iost et al, 1999). In this work, we show that mutations of the Q motif and upstream aromatic group give in vivo phenotypes and in vitro ATPase activities that are consistent with those obtained for eIF4A. However, we also find a close relationship between ATPase, RNA-binding and strand-displacement activities, which also are correlated strongly with the in vivo phenotypes. In particular, we detect at least three different protein conformations that are associated with free, ADP-bound and ATP-bound forms of the proteins, and these different conformations have different affinities for RNA substrates. Thus, the Q motif and upstream aromatic residue regulate ATP binding, and consequently they regulate substrate affinity and helicase activity.
Results
In vivo analysis of the Q motif
From comparisons of the solved crystal structures of domain 1 of eIF4A, with and without a bound ligand, and from additional comparisons with the solved crystal structure of the nearly full-length MjDEAD, it was clear that residues of the Q motif were making a network of interactions between the adenine base, motif I and the isolated upstream aromatic group (Benz et al, 1999; Caruthers et al, 2000; Story et al, 2001). Moreover, the isolated aromatic group (generally F) appeared to be in the center of a cluster of charged or polar residues, and it was possible that these residues were making important interactions that we had not detected in the solved crystal structures. We decided to change systematically the equivalent amino acids in Ded1 to alanine or leucine to see what affect they would have on yeast cell growth. We also randomly mutagenized the residues at positions F144, F162, T166 and Q169 with oligonucleotides that were completely degenerated for the encoding codon. These were the most highly conserved residues (Figure 1A) and they were making the most significant interactions (Figure 1B). For the random mutagenesis, we screened 200 clones at F144, 380 clones at F162, 200 clones at T166, 600 clones for the invariant Q169 and a number of clones for each of the residue-specific mutants (A or L) that were selected on 5-fluoroorotic acid (5-FOA) plates.
Representative results are presented in Figure 2A and the entire screen is summarized in Figure 2B. As expected from our previous experiments with eIF4A (Tanner et al, 2003), F144, F162 and Q169 in Ded1 were very sensitive to changes that altered the character of the sidechain. Only large hydrophobic residues supported wild-type growth at positions F144 and F162; this was consistent with these moieties forming the hydrophobic stacking interactions shown in Figure 1B. Phenylalanine was not recovered at position F144. However, thymidine was rarely found in the second codon position (one out of 21 clones sequenced), which indicated that the oligonucleotide we used was not completely randomized at this position. Nevertheless, aromatic residues were recovered from four out of five wild-type clones. Aromatic groups were recovered four out of eight times at position F162, and there was a clear preference for large hydrophobic residues. To verify this, we site-specifically changed the residue to alanine and found that this clone did not support growth. To our surprise, we found that a wide variety of changes were tolerated at position T166, despite its high level of conservation (91% S or T; Figure 1A). The threonine sidechain was predicted to form hydrogen-bond interactions with motif I (Figure 1B), and it was recovered five times out of 13 wild-type clones. Three out of the four possible codons were used. The other substitutions were mostly hydrophobic residues (54%). The T166S mutation had a strong cold-sensitive phenotype (Figure 2A), and it was recovered in eight out of 18 conditional clones with three different codons. However, this phenotype was specific to Ded1, as the same (T to S) or converse (S to T) mutations within other DEAD-box proteins in yeast did not show any growth defect (Tanner et al (2003); O Cordin and J Banroques, unpublished results). Arginine was recovered in about 22% of the conditional-growth clones. We found multiple different codons for the various amino acids recovered, which indicated that our screens were extensive.
Figure 2.
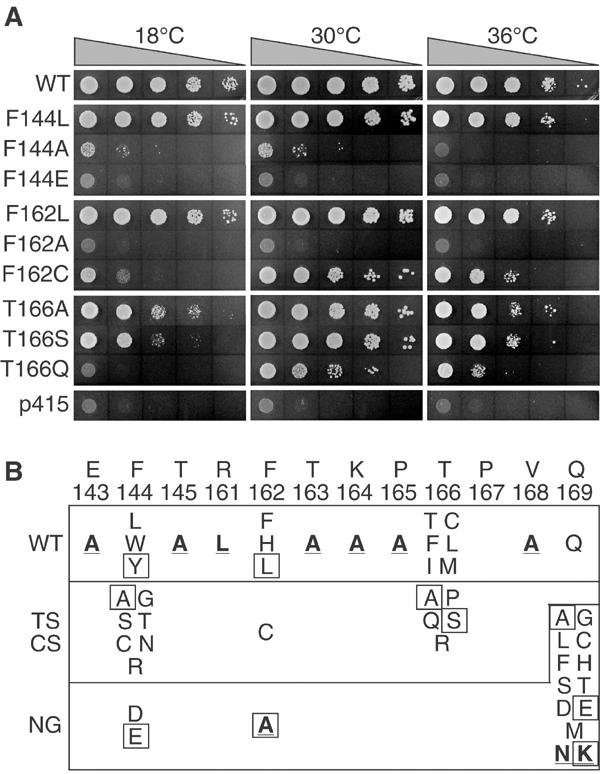
Complementation of the Q motif mutants in yeast. (A) Representative growth of yeast transformed with various mutants at different temperatures. Cells were grown in liquid medium lacking leucine to approximately the same density, serially diluted, and each dilution was spotted onto 5-FOA-containing plates. Plates were incubated at the indicated temperatures for 3 days (30 and 36°C) or 4 days (18°C). WT, wild-type Ded1; p415, plasmid alone control. (B) Resume of the in vivo analysis. Residues in bold and underlined were made by site-specific mutagenesis; all others were made with oligonucleotides that were completely randomized at the three nucleotide positions of the codon. Proteins of the boxed residues were purified. WT, wild-type or nearly wild-type growth; TS/CS, temperature-sensitive or cold-sensitive phenotypes; NG, no detectable growth.
The most pronounced results were obtained at position Q169. We made two independent screens using two independent sets of degenerated oligonucleotides. Of the 600 clones screened, we recovered glutamine 16 times (both codons) from viable clones. No other residue supported growth. To verify this, we site-specifically changed the residue to lysine and to asparagine, which have sidechains similar to glutamine; both mutants had nongrowth phenotypes. Consequently, as predicted from our sequence alignments, the glutamine was the only residue tolerated at this position, and this clearly demonstrated its essential role in Ded1 for cell viability.
Despite the predicted network of interactions, changing the other conserved residues of the Q motif, and the residues flanking the isolated upstream phenylalanine (F144), to alanine or leucine had no strong effect on yeast growth. This was perhaps because the interactions they were making were not important, or more likely, because the interactions were redundant with those at other positions. Additional, multiple mutations will be needed to address this question. In all cases, protein expression was assayed; no differences were seen between any of the proteins, which indicated that they were all stably expressed, although it is possible that some mutations affected the protein conformation.
In vitro ATPase activity
We subcloned representative mutant DED1 genes into a pET expression vector containing a carboxyl-terminal His6 tag. This permitted us to express the proteins in Escherichia coli and to purify them using a nickel resin column. We were able to purify successfully the wild type and the mutants F162A, F162L, T166A, T166S, Q169A and Q169E at high yields and purity. We also purified a mutant in motif I (K192A) as a control; the equivalent mutation in the P loop of eIF4A eliminated ATPase activity (Tanner et al, 2003). All of the F144 mutants we attempted to purify (F144A, F144E and F144Y) were insoluble. We assayed the overall folding of the purified proteins by subjecting them to limited trypsin proteolysis and then analyzing the fragment pattern (data not shown). In all cases, the patterns were identical, which indicated that no gross structural alterations were present.
The optimal reaction conditions were determined for the wild-type protein. Peak activity was obtained with 5 mM Mg2+; there was 40–50% less activity at 1 or 10 mM concentrations. The pH optimum was around 7.5, and there was little activity at pH 6 or below. No difference was seen between using acetate and chloride ions. The reaction profile was linear between 15 and 150 nM protein, so the protein was functioning probably as a monomer. Our protein preparations had less than 1–2% background ATPase, which was not altered by preincubation with RNaseA. Nevertheless, it is possible that the background ATPase activity was due to a small amount of RNaseA-resistant (potentially short) RNA. We used whole yeast RNA for the reactions, because the RNA substrate is unknown. However, we noticed significant differences between lots of RNA as we previously noted for eIF4A (Tanner et al, 2003). Interestingly, the lot optimal for Ded1 was not necessarily optimal for eIF4A or vice versa, which indicated that different substrates were preferred. As a consequence, comparisons were always carried out with the same lot of RNA. The reaction was essentially saturated at 25 ng/μl of RNA with 15 nM of protein. However, if we assumed an ∼10-nucleotide-long binding site that resulted in the hydrolysis of a single ATP and that all sites were equivalent, then each potential site was reused at least 10 times during the course of a 40 min reaction. Little or no loss of activity was observed after a 90 min incubation. Ded1 had 100- to 150-fold higher ATPase activity than eIF4A (data not shown). Both ATP and dATP were efficiently hydrolyzed in the presence of RNA, whereas other nucleotide triphosphates gave less than 2% background.
Figure 3 shows representative results from our ATPase assays, and the derived Michaelis–Menten parameters are shown in Table I. Note that we cannot be sure of the rate-limiting step in the reaction, and moreover some of the mutants could not be fully saturated for ATP in our assays; therefore, these results should be considered as kinetic parameters and not necessarily as true thermodynamic values. Nevertheless, it was clear that mutations that were lethal in vivo (F162A, Q169A and Q169E; Figure 2A) had 100- to 300-fold reduced kcat/Km. This was reflected both in reduced affinities for the ATP and lower catalytic rates. The K192A mutation, which did not support growth in yeast, gave only background levels of hydrolysis (Figure 3A and B). In contrast, mutations that resulted in conditional (T166A, T166S) or nearly wild-type (F162L) growth had only slightly reduced affinities for ATP and slightly slower reaction rates. Oddly, T166A and T166S mutants had essentially the same apparent affinity for ADP as the wild type, despite their lower affinities for ATP. This result was consistent with what we later observed in our electrophoretic mobility shift assays (EMSA; see RNA-binding activity). The F162L mutant was an exception; it showed a higher loss of affinity for ADP than for ATP. This might explain why this Ded1 mutant supported growth nearly as well as the wild type despite its four-fold lower enzymatic efficiency in vitro (Figure 2A). As for the wild type, the mutated proteins hydrolyzed both ATP and dATP, and they showed no clear reaction with other NTPs. The kinetic parameters we obtained are essentially the same as those previously obtained for the wild-type Ded1 (Iost et al, 1999).
Figure 3.
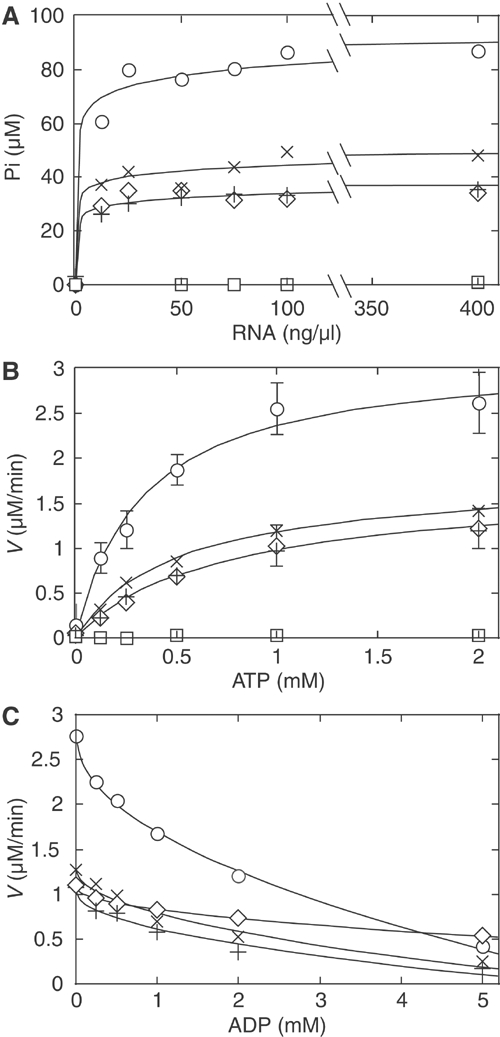
ATPase activities of the wild type and selected mutants. (A) ATPase activity using increasing concentrations of whole yeast (type III) RNA. All comparisons were made with the same lot of RNA. Reactions were carried out for 40 min at 30°C with 1 mM ATP and 15 nM proteins. (B) Michaelis–Menten kinetics of the ATPase reaction using 15 nM of protein and 400 ng/μl of whole yeast RNA. The standard deviations, from three independent sets of experiments, were deleted from some points because they were not significant. Reaction velocities were determined from the initial linear range of the reaction, which corresponded to less than 7% reaction. The reaction profiles of F162A, Q169A and Q169E are not shown; they were slightly above K192A at this plot scale. (C) Competitive inhibition of the ATPase reaction by ADP. ○, wild type; ×, T166A; +, T166S; ⋄, F162L; □, K192A.
Table 1.
Kinetic parameters for RNA-dependent ATPase activitya
| Km (mM) | Vmax (%wt) | kcat b (min−1) | kcat/Km c (M−1 min−1) | Ki d (mM) | |
|---|---|---|---|---|---|
| WT | 0.34±0.05 | 100±4 | 210±20 | 620 000±90 000 | 0.36±0.07 |
| F162A | 3.5±0.4 | 9.3±6.9 | 20±2 | 5600±700 | ND |
| F162L | 0.78±0.11 | 55±6 | 120±10 | 150 000±20 000 | 3.0±0.1 |
| T166A | 0.51±0.04 | 56±3 | 120±10 | 230 000±20 000 | 0.36±0.08 |
| T166S | 0.66±0.05 | 51±3 | 110±10 | 160 000±20 000 | 0.34±0.07 |
| Q169A | 33±12 | 29±33 | 61±20 | 1900±700 | ND |
| Q169E |
1.1±0.2 |
1.2±7 |
2.6±0.2 |
2300±400 |
ND |
| a Values derived from nonlinear fits to the Michaelis–Menten equation using the mean values of three independent experiments. The standard errors of estimation were derived from the curve fits. | |||||
| b A minimum of 10% error was assumed in the protein concentration. Larger errors were derived from the standard errors of estimation. | |||||
| c The largest errors of kcat or Km were carried forward. | |||||
| d Competitive inhibition of ADP. | |||||
| ND: not determined. | |||||
RNA-binding activity
It was shown previously in DEAD-box helicases, including Ded1, that ATP binding enhances the binding of RNA and vice versa (e.g., Lorsch and Herschlag, 1998a; Iost et al, 1999; Polach and Uhlenbeck, 2002). Consequently, we wondered if the mutations in the Q motif would similarly affect the affinity for the substrate. We used a 44-nucleotide-long RNA, which was also used later for the duplex displacement assay (see below). It was 5′-32P endlabeled and incubated on ice with various concentrations of protein in the presence or absence of ADP or adenosine 5′-(β,γ-imido)triphosphate (AMP-PNP; a nonhydrolyzable analog of ATP) for various times. The products were separated by nondenaturing PAGE at 4°C. These assays showed significant variability in the intensity of the shifted bands between experiments (more or less smearing), which prevents us from reliably quantifying the results. Nevertheless, the amount of RNA displaced by the protein was similar and a clear qualitative pattern was observed.
Figure 4 shows some typical results from these experiments. The RNA migrated as a single band in the absence of protein. In the absence of a nucleotide, all the proteins had the same moderate affinity for the substrate, which resulted in a smear on the gel, and occasionally one or more distinct bands were evident near the top of the gel. The bandshifting assay was strongly dependent on the protein concentration, and typically a 40- to 80-fold excess of protein to RNA was needed. The bandshifting could be eliminated by adding a large excess of unlabeled RNA, and no difference was seen when unlabeled RNA was added before or after the labeled RNA; thus, equilibrium was rapidly established in solution under our assay conditions and the complexes were soluble in the reaction mix. All the proteins showed significantly weaker affinity for the substrate in the presence of ADP, including the motif I mutant K192A. This latter result was somewhat surprising because the P loop is known to form specific interactions with the α and β phosphates of the nucleotide, and these interactions should have been perturbed by the mutation (Caruthers and McKay, 2002). However, the fact that most of the mutants had essentially the same reduced affinity for the substrate in the presence of ADP was consistent with the ADP inhibition results in the ATPase assays, which showed that the affinity for ADP was often unchanged even when the affinity for ATP was reduced (Table I).
Figure 4.
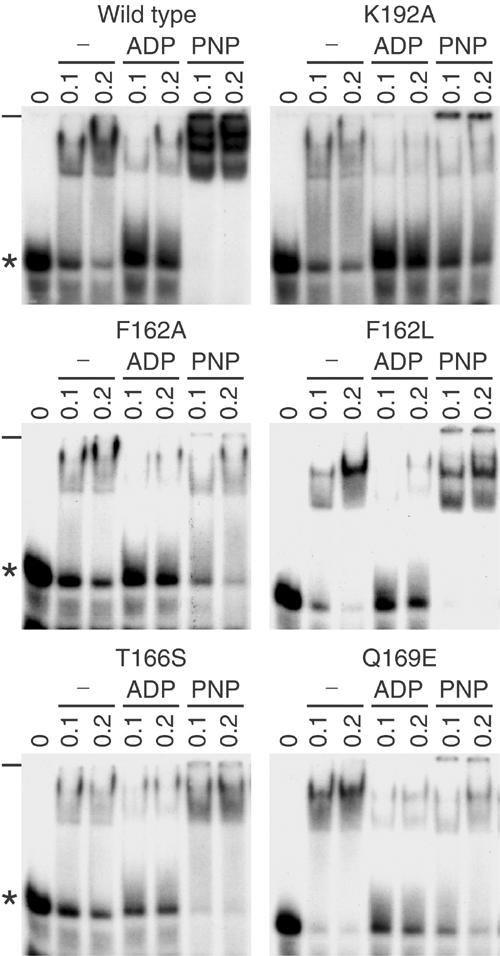
Electrophoretic mobility shift of Ded1-bound 44-mer RNA. In all, 2.5 nM of 32P-labeled RNA (shown in Figure 5) was incubated with the indicated concentrations of Ded1 (in μM) and electrophoretically separated on a 7.5% nondenaturing polyacrylamide gel that was run at 4°C. The ADP and AMP-PNP (PNP) concentrations were 5 mM. We could account for most of the added radioactivity by quantifying the gels with a phosphoimager. There was little or no RNA degradation, and the apparent loss of radioactivity in some lanes is caused by smearing, which is more obvious on longer exposures. *, free RNA; −, origin of gel.
The main difference between the proteins in the EMSA assay was in the presence of AMP-PNP. Proteins that supported nearly wild-type growth in vivo, and that had good ATPase activity in vitro, showed strong bandshifting; often three bands could be clearly resolved (Figure 4). In contrast, mutants that had little ATPase activity in vitro and no viability in vivo showed weaker bandshifting that was similar to, or even less than, that obtained in the absence of nucleotide. This was particularly evident in the K192A mutant, indicating that the protein had different, although perhaps overlapping, determinants for binding ADP and ATP. However, we do not take this as evidence for two different binding sites, but rather as a potential alteration of the same site. Consistent with this, in competition experiments, ADP reduced the AMP-PMP-specific bands in a concentration-dependent fashion (data not shown). As expected, the F162L mutant was much less sensitive to the addition of ADP than other constructs. The multiple bands observed in the presence of AMP-PNP could be the protein binding at different positions on the RNA substrate, different protein conformations, or due to additional interactions between proteins.
Duplex-displacement (helicase) activity
The substrate for the unwinding assay was a variant of that used by Rogers et al (2001) in their analysis of eIF4A (Figure 5A). The substrate consisted of a 44-nucleotide-long RNA with a 16-nucleotide-long DNA hybridized at the 3′ end (Figure 5). The duplex contained a 25-nucleotide-long 5′ single-stranded region, which was needed for initial binding of the protein (Iost et al, 1999), and a 16 base pair duplex (ΔG°=−19.8 kcal/mol under standard conditions). The wild-type Ded1 had no strong directionality; that is, it unwound duplexes in the 5′–3′ and 3′–5′ directions (NK Tanner, unpublished). The RNA substrate contained a short 3′ single-stranded region because T7 polymerase is known to leave heterogeneous ends (Pleiss et al, 1998). This design assured that the duplex population would have the same stability.
Figure 5.
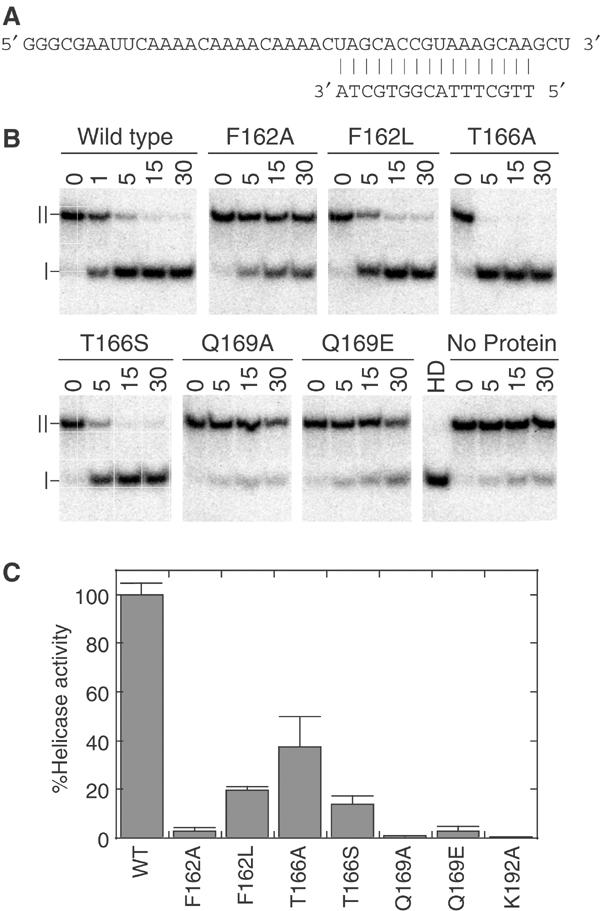
Strand-displacement activity of Q motif mutants. (A) RNA–DNA duplex used for the unwinding assay. The DNA oligonucleotide was 5′-32P-labeled and hybridized to unlabeled RNA. The calculated ΔG° of the duplex was −19.8 kcal/mol, which gave a Tm of ∼52°C under our salt and duplex concentrations. (B) Time course for ATP-dependent unwinding of the duplex. In all, 200 nM of duplex was incubated with 30 nM (wild type) or 120 nM (mutant) protein with 1 mM ATP, for the indicated times in minutes at 37°C. To prevent reannealing of the displaced 32P-labeled oligonucleotide, 5 μM of cold DNA oligonucleotide was added as a competitor. Products were separated on a 15% polyacrylamide gel, which was then subjected to autoradiography. ∣, free DNA oligonucleotide; ∥, RNA–DNA duplex; HD, heat-denatured at 95°C and then quick-cooled on ice. (C) Relative helicase activity of mutants compared to the wild type, based on the extent of reaction after 20 min at various protein concentrations. All values were normalized to the same protein concentration. Standard errors are as shown.
It was shown previously that an RNA–DNA duplex is efficiently unwound by Ded1 and that DNA is not a substrate for ATPase activity (Iost et al, 1999). In accordance with this, we could not detect any binding of the DNA oligonucleotide in our EMSA experiments. This reduced the potential problem of the DNA oligonucleotide acting as a competitive inhibitor of the binding of the RNA–DNA duplex. A 25-fold excess of the unlabeled DNA oligonucleotide was added to quench re-formation of the duplex with the displaced 32P-labeled 16-mer, because it would efficiently reanneal under our assay conditions. This was important evidence that the duplex was thermodynamically stable under our assay conditions, because it is known that the melting temperature (Tm) is strongly dependent on both the salt and the duplex concentrations (SantaLucia, 1998).
Representative results of our ATP-dependent, strand-displacement reactions are shown in Figure 5B and quantified comparisons of the unwinding efficiencies are shown in Figure 5C. A small amount of strand displacement was apparent in the absence of protein, but it was never more than ∼10% of the initial duplex. Nevertheless, strand exchange due to thermal denaturation could clearly occur. There was a good correlation between ATP-dependent unwinding activity and the protein concentration, which provided further evidence that the protein was functional as a monomer. As the duplex re-formed during the course of the reaction, it was difficult to obtain reliable kinetic parameters for the reaction, and our results may have underestimated the differences between the wild-type and mutant proteins. Nevertheless, the results were consistent with our expectations from the ATPase and EMSA assays. Experiments where the unlabeled competitor was complementary to the displaced 16-mer, and hence formed a blunt-ended DNA–DNA duplex, gave similar results (NK Tanner, unpublished data). This indicated that the RNA–DNA duplex was not a substrate per se in the reaction, but rather that the protein bound the single-strand RNA and then displaced the 16-nucleotide-long DNA, when present, during the reaction cycle. The wild-type Ded1 protein had nearly 1200-fold higher displacement activity than eIF4A with the same substrate (NKT, unpublished results). It had a minimum turnover of 6.7 duplexes per protein in a 30 min reaction, but because the duplex re-formed the actual turnover was much higher. This was much higher activity than that previously described, but in that work the authors used a more stable RNA–RNA duplex (calcd. ΔG°=−25.6 kcal/mol; Iost et al, 1999).
Discussion
Our in vivo and in vitro results for Ded1 are largely consistent with those expected from our in silico analysis of DEAD-box proteins, and from our in vivo and in vitro experiments with yeast eIF4A (Tanner et al, 2003). Aromatic groups occur 96 and 82% of the time at positions equivalent to F144 and F162, respectively, among 277 DEAD-box sequences. In the solved crystal structure of eIF4A with a bound ADP (Benz et al, 1999), these phenylalanines form hydrophobic stacking interactions with a conserved proline and with the adenine base (Figure 1B). Indeed, large hydrophobic groups occur at these two positions in over 97% of the sequences and in all of the mutants of Ded1 with wild-type activity. Similarly, P165, which is the putative partner of F144, is a hydrophobic residue 93% of the time. However, the P165A mutation has a wild-type phenotype, whereas the F144A mutation barely grows (Figure 2A). Thus, the isolated aromatic group is playing a more important role than simply stacking on P165, and it may be involved in other interactions—perhaps conformational changes—not apparent in the crystal structures. The wide variety of mutations at T166 was initially an enigma: normally either a threonine (56%) or a serine (35%) is found at this position. However, phenylalanine (5%) and methionine (1.4%) are also seen, while all other polar groups, besides serine or threonine, are rarely seen (1% of the time). Cysteines are rare in proteins and are not seen in the sequence alignments at this position; nevertheless, they are capable of forming hydrogen bonds with carboxyl groups in a manner similar to serine or threonine (Pal and Chakrabarti, 1998). This indicates that the alcohol (or thio) residues are making unique interactions that cannot be readily replaced by other residues, and that the absence of these interactions is less critical than potentially forming the wrong ones. This is consistent with the residue interacting with motif I because part of the interactions is predicted to be duplicated by the highly conserved glutamine. Notably, serine and threonine are highly conserved in other DEAD-box motifs as well (motifs I, Ia, Ib, III, V and VI; Tanner and Linder, 2001; Silverman et al, 2003). The equivalent serine to threonine mutation in eIF4A has wild-type growth (Tanner et al, 2003).
The V168 position is aliphatic in 99% of the sequences, and its primary role probably is to facilitate formation of the helix seen in the crystal structures and in the predicted secondary structures (Figure 1A). Likewise, a glycine appears at the position equivalent to R161 in 73% of the sequences, and it may be needed to facilitate a sharp turn that proceeds the predicted conserved helix that is seen also in all the solved crystal structures. In this respect, Ded1 is unusual because an arginine occurs only 4% of the time. Most of the other tested positions are generally polar residues: E143 (83%), T145 (81%), T163 (88%) and K164 (86%). They form sidechain–backbone interactions in the solved crystal structures, but none of them are essential in our in vivo assay. This is most likely because the interactions are redundant, and multiple changes may be needed to see a phenotype. The most critical position remains the Q169, which is essentially invariant in the sequence alignments. The few exceptions are undoubtedly sequencing errors in the database (Tanner et al, 2003). All of the amino-acid substitutions were nonviable in yeast. Asparagine has the potential of forming the equivalent hydrogen bonds, but it is never found in the alignments nor does it support growth (Figure 2B). Moreover, in the alignments of the other SF1 and SF2 helicases, there are conserved glutamines upstream from motif I, that are never substituted for asparagine (Tanner, 2003; Tanner et al, 2003).
The in vitro activities of the various purified proteins closely parallel the in vivo results. Mutations that are lethal in vivo (F162A, Q169A, Q169E) have at least 100-fold lower kcat/Km for ATPase activities (Table I), reduced affinities for RNA in the presence of AMP-PNP (Figure 4) and more than 30-fold reduced helicase activities. Mutations that are wild type or have conditional phenotypes have no more than four-fold lower kcat/Km, slightly lower affinities for RNA and no more than seven-fold reduced helicase activities. Notably, both F162 and Q169, which are predicted to make direct contacts with adenine, show reduced binding affinities when substituted with other residues. This is consistent with the Q motif regulating ATP binding, and subsequently RNA affinity and unwinding activities. Oddly, the F162L mutant supports wild-type growth in yeast even though its kinetic parameters are the same as T166S, which has strong conditional growth. However, it has a lower affinity (higher Ki) for ADP, which might stimulate a higher turnover in vivo at lower temperatures.
In many respects, our results with the Q-motif mutants are similar to the K192A mutation of the P loop of motif I, which affects binding of the nucleotide phosphates (Caruthers and McKay, 2002). Nevertheless, there are subtle differences between the mutants that reveal much about the properties of DEAD-box proteins. For example, Q169E and Q169A both have essentially the same values for kcat/Km, but Q169E has nearly three times more strand-displacement activity than Q169A, despite a 20-fold lower kcat for ATP hydrolysis. However, Q169E has a higher affinity for ATP, which is consistent with glutamate forming at least some of the same hydrogen bonds to adenine as those to glutamine. These interactions may be essential for linking nucleotide binding with helicase activity, which may be more important than the ATPase activity (kcat) per se. This is more pronounced for eIF4A, where the equivalent mutation (Q to E) has 48% of the wild-type ATPase activity and three-fold higher UV crosslinking of the nucleotide to the protein in vitro (Tanner et al, 2003). However, eIF4A is active at a lower pH, which is closer to the pKa of glutamate. Glutamate is known to interact with the N6 position of purines, although it more commonly forms hydrogen bonds with guanine than with adenine (Moodie et al, 1996; Nobeli et al, 2001). Another revealing property is that T166A and T166S both have the same apparent affinity for ADP as the wild type, even though their affinities for ATP are significantly reduced (Table I). Moreover, the affinity for ADP in the F162L mutant is much more reduced than its affinity for ATP. We obtained similar results with a Q motif mutant of eIF4A (Tanner et al, 2003), which suggests that this is a general feature of DEAD-box proteins. These results indicate that the determinants for binding ADP are not identical to those used to bind ATP, and they imply that the adenine-binding site undergoes conformational changes that are related to the presence or absence of the gamma phosphate. These results are consistent with those obtained for DbpA and eIF4A using limited proteolysis digestion (Lorsch and Herschlag, 1998b; Henn et al, 2002).
It has been shown for DEAD-box helicases that there is cooperative binding between ATP and RNA; that is, the bound nucleotide enhances the affinity of the protein for the substrate (Lorsch and Herschlag, 1998a; Iost et al, 1999; Polach and Uhlenbeck, 2002). We obtain a similar result in our EMSA experiments. In the absence of a nucleotide, Ded1 has a moderate affinity for a 44-nucleotide-long single-stranded RNA, which is strongly enhanced by adding AMP-PNP. In contrast, ADP-bound Ded1 has a weaker affinity. This indicates that there are at least three different protein conformations that have different affinities for the substrate, and that these conformations are controlled by the properties of the bound nucleotide and ultimately by the properties of the Q motif. Similarly, DbpA has an enhanced affinity for RNA in the presence of AMP-PNP (Polach and Uhlenbeck, 2002). AMP-PNP is known to be an imperfect mimic of ATP in RNA helicases (Lorsch and Herschlag, 1998a; Polach and Uhlenbeck, 2002), so the effects may be even more pronounced with ATP. This implies that the energy of hydrolysis of ATP is not used to drive the strand-displacement activity per se, but rather is used to recycle the protein from a high RNA affinity to a low RNA affinity conformation. This also implies that the DEAD-box proteins do not have processive unwinding activity, and that they only function as stoichiometric motors. This would explain why the DEAD-box proteins are often used in vast excess of the substrate in helicase assays and why all the assayed proteins have limited strand-displacement activity in vitro (Tanner and Linder, 2001; Silverman et al, 2003; Rocak and Linder, 2004). In our case, we could show a good turnover of Ded1 with a short duplex, while previous work needed a large excess of protein for a duplex that was only about 30% more stable (Iost et al, 1999). This makes good biological sense as well because the identified substrates in vivo are often very small, and only a single reaction cycle would be needed (e.g., Staley and Guthrie, 1998).
There is precedence for this idea because most ‘classical' NTP-dependent molecular motors are thought to use primarily the energy of nucleotide binding to make work rather than the energy of hydrolysis, although that can also be a contributing factor (Vale, 1996; Vale and Milligan, 2000). This does not preclude other SF1 or SF2 helicases from directly using the energy of ATP hydrolysis to unwind duplexes, but it would not be necessary either since proteins like kinesins have evolved to take advantage of conformational changes to processively move down polymers (Vale, 1996; Vale and Milligan, 2000). Further support for this idea comes from recent work on ATP-dependent protein chaperones, where it was shown that ATP hydrolysis is not needed for productive folding (Chaudhry et al, 2003). Nevertheless, distinguishing between these models in helicases is difficult, particularly because the bound ATP is rapidly hydrolyzed during the reaction cycle. Moreover, there is no evidence that the free gamma phosphate remains associated with the protein. Further work will be needed with other nonhydrolyzable ATP analogs or with fast spectroscopic measurements that are sensitive to protein conformational changes.
This work is the second example that shows that the Q motif and upstream aromatic group are involved in adenine recognition. Since this motif is highly conserved and the same structures are seen in all the solved crystal structures (six structures; three proteins), it seems likely that this is a characteristic and predictive feature of all DEAD-box proteins. The highly conserved glutamine of DEAD-box proteins makes hydrogen bonds to the N6 and N7 positions of adenine, and the base is stacked on an aromatic group. These features would be necessary and sufficient to distinguish unambiguously adenine from the other three bases. Indeed, most ATP-dependent SF1 and SF2 helicases have conserved aromatic groups and glutamines that could make similar interactions, and these are seen in various solved crystal structures with a bound nucleotide (Tanner and Linder, 2001; Tanner et al, 2003). This is a highly unusual property because base recognition by most adenine-binding proteins involves a general network of hydrophobic and electrostatic interactions that vary widely between proteins even within the same family; this has resulted in the concept of a ‘fuzzy'recognition site (Moodie et al, 1996; Nobeli et al, 2001; Cappello et al, 2002). Moreover, hydrogen bonds to adenine—particularly those from glutamine—are surprisingly scarce, and they are often through water molecules (Moodie et al, 1996; Nobeli et al, 2001; Cappello et al, 2002).
The roles of the other residues of the Q motif and of the isolated upstream aromatic group are still a mystery. Most of the changes that we made are neutral in vivo. Most likely, some of the residues are structural elements. For example, the residues at positions R161 and V168 are needed to promote the helix–loop–helix transitions. The alcohol at position T166 may be needed to stabilize the interactions with motif I, which are also predicted to be made by the glutamine. The other residues may be involved in the network of interactions shown in Figure 1B, but additional work is needed to show this. However, it is clear that the distinctive structure created by the Q motif, conserved helix and isolated upstream aromatic group is much more extensive than would be required simply to function as an adenine-binding motif. Indeed, there are now two solved crystal structures of two different DEAD-box proteins that show the P loop of motif I in an unusual ‘closed' position in the absence of a ligand that would not be available for binding the nucleotide phosphates (Caruthers et al, 2000; Carmel and Matthews, 2004). This is consistent with the Q motif being a molecular on–off switch that controls ATP binding, and subsequently helicase activity. Regardless, it is clear that the Q motif can directly regulate the affinity of the protein for the substrate through conformational changes associated with nucleotide binding, and that this ultimately regulates helicase activity.
Materials and methods
Cloning, vectors and strains
All vectors, yeast strains, amino-acid-specific mutations and cloning were as previously described (Tanner et al, 2003). All specific mutations were verified by sequencing. Amino acids at positions 144, 162, 166 and 169 were randomly mutagenized using oligonucleotides that had all the four nucleotides inserted in equal amounts at the three positions of the corresponding codon. All plasmids that supported growth on 5-FOA plates were recovered by plasmid rescue, retransformed into yeast and reselected by 5-FOA to verify the phenotype. In the case of random mutagenesis, all the plasmids that supported growth were subsequently sequenced to identify the mutation. We also sequenced a selection of plasmids that did not support growth in yeast. Some of the constructs were subcloned into a pET-22b vector (Novagen).
Protein expression and purification
The Origami (DE3) E. coli strain (Novagen) was used for the expression of the recombinant Ded1 proteins. Protein expression was as previously described, except that it was carried out for 2.5 h with gentle shaking at 20°C (Tanner et al, 2003). Cells were collected by centrifugation and stored at –20°C until needed. Pellets were resuspended in 5 ml of lysis buffer containing 50 mM Tris base, pH 7.5, 500 mM NaCl and 10 mM imidazole. Lysozyme was added at 10 μg/ml and the mixtures were kept on ice for 30 min. Cells were then sonicated until the lysate became clear with a Sonifier Cell Disruptor B-30 (Branson). The material was centrifuged for 45 min at 15 000 rpm in a SS34 rotor (Sorvall) at 4°C, and the supernatant was loaded onto a 1 ml nickel nitrilotriacetic acid–agarose column (Ni-NTA, Qiagen) equilibrated with the lysis buffer. The column was washed with 20 ml lysis buffer containing 30 mM imidazole and the protein was eluted with lysis buffer containing 100 mM imidazole. The protein was 90–95% pure after this step. The protein could be dialyzed, but it often precipitated upon further manipulation. The small amount of salts and imidazole had no detectable effect on the activity. We typically obtain 1–4 mg of protein from 500 ml of culture. Purified proteins were stored in 50% glycerol at –80°C with no loss of activity after several months. Proteins stored at –20°C were stable for several weeks. Protein concentrations were determined by the Bio-Rad Protein Assay and verified by Laemmli SDS–PAGE. Typically it was 0.45–1.0 μg/μl. The trypsin cleavage pattern, used to assay overall protein folding, was assayed by silver staining and by Western blot analysis using anti-Ded1 antibodies.
ATPase assays
We used a colorimetric assay based on molybdate-Malachite green as previously described (Pugh et al, 1999; Tanner et al, 2003) and a radioactive assay based on separating the released [γ-32P]phosphate of ATP by thin-layer chromatography (TLC) plates. The reaction buffer contained 50 mM potassium acetate, 20 mM Tris base, pH 7.5, 5 mM magnesium acetate, 2 mM dithiothreitol (DTT), 0.1 mg/ml bovine serum albumin (BSA) and various concentrations of whole yeast RNA (Type III, Sigma). Reactions were incubated at 30°C for various times and stopped by adding a 10% of volume 0.5 M ethylenediaminetetraacetic acid (EDTA; ∼46 mM final). For the radioactive assay, 2 μl aliquots were spotted onto polyethylenimine-cellulose plates (PEI-cellulose; Merck) and eluted in 0.15 M formic acid, pH 3.0, and 0.15 M LiCl. Radioactivity was quantified with a Cyclone phosphoimager (Packard) and the included OptiQuant software. Data were analyzed with KaleidaGraph 3.5 software (Synergy).
RNA-binding assays
Approximately 3 nmol of a 44-nucleotide T7 RNA transcript (see Strand-displacement assays) was dephosphorylated with 5 U of calf intestinal phosphatase (CIP; Promega) for 1.5–2 h at 37°C, extracted with phenol/chlorophorm/isoamyl alcohol (Biosolve), ethanol precipitated, dried and resuspended in 20 mM Tris base, pH 8.0, with 0.1 mM EDTA. The RNA concentration was measured by the absorption at 260 nm, and 125 pmol of the RNA was kinased with 20 μCi of [γ-32P]ATP and 13 U of T4 polynucleotide kinase (Q-Bio) for 1.5 h at 37°C. The efficiency of labeling was estimated to be 85%. The bandshift assay was performed with 2.5 nM of labeled RNA and variable concentrations of the proteins, which were incubated together for 25 min on ice in the absence or presence (5 mM) of either ADP or AMP-PNP (Roche) in a volume of 20 μl. After incubation, 3 μl of 50% glycerol was added to the samples and they were loaded onto a 0.75 mm thick, 7.5% polyacrylamide (29:1) gel containing 150 mM Tris base, pH 8.8. The gel was subjected to electrophoresis using a Mini-PROTEAN (Bio-Rad) apparatus and a running buffer containing 25 mM Tris base and 250 mM glycine at 4°C for 2 h at 70 V. The gel was dried and then subjected to autoradiography.
Strand-displacement assays
The substrate template was constructed by digesting the double-stranded version of the sequence 5′ GCGCGAATTCAAAACAAAACAAAACTAGCACCGTA AAGCAAGCTTGCGC 3′ with EcoRI–HindIII and then cloning the fragment into the equivalent sites of pGEM-3Z. A 44-nucleotide-long RNA was transcribed off the Hind-III-cut plasmid using the T7 RNA polymerase MEGAshortscript kit (Ambion). The transcribed RNA was purified by PAGE and the concentration was determined at 260 nm with a calculated absorption of 31 μg/ml/cm/OD. The second strand was a 16-nucleotide-long DNA containing the sequence 5′ ATCGTGGCATTTCGTT 3′, which was obtained PAGE-purified from Microsynth (Switzerland). The oligonucleotide (3.3 μM) was 5′ endlabeled with 10 U of T4 polynucleotide kinase and 20 μCi of [γ-32P]ATP (4500 Ci/mmol; Amersham) for 60 min at 37°C in a 20 μl volume. The duplex was formed by incubating 10 μM of RNA with 10 μM unlabeled oligonucleotide, 0.33 μM 32P-labeled oligonucleotide, 20 mM Tris base, pH 8.0, 200 mM potassium acetate and 0.1 mM EDTA in a 25 μl volume. The mixture was heated to 95°C for 30 s, 80°C for 1 min, slowly cooled to 45°C at 0.1°C/s, kept at 45°C for 10 min and then slowly cooled to 4°C at 0.1°C/s in a PCRSprint (Hybaid) PCR machine. This protocol gave 80% or more of the labeled DNA as a duplex, which was stably stored at –20°C. The duplex stability was calculated according to Sugimoto et al (1995) and Turner et al (1988). The Tm was calculated according to SantaLucia (1998), where the effects of the magnesium were determined according to Peyret (2000).
Unwinding assays were carried out in the presence of an excess of unlabeled DNA oligonucleotide because the duplex would efficiently reanneal under our reaction conditions. Reactions were in 10 μl volumes consisting of 200 nM duplex, 5 μM unlabeled oligonucleotide, 20 mM Tris base, pH 7.5, 50 mM potassium acetate, 5 mM magnesium acetate, 10 mM DTT, 0.1 mg/ml BSA, 1 U RNasin (Promega), various concentrations of proteins and 1 mM ATP. Reactions were incubated at 37°C for various times and then quenched by placing them on ice. A 5 μl solution of 40% glycerol, 10 mM EDTA, 0.025% bromophenol blue and 0.025% xylene cyanole blue was added and the samples were loaded onto a 0.75 mm thick 15% polyacrylamide gel (29:1). The gel was subjected to electrophoresis in a MiniPROTEAN apparatus at 4°C for 1 h at 16 W, with 100 mM Tris base, 90 mM boric acid and 1 mM EDTA running buffer. The radioactive bands within the gel were detected with a Storm 820 phosphoimager (Molecular Dynamics) and quantified using the ImageQuant 5.2 software (Molecular Dynamics).
Acknowledgments
We thank Ulrich Baumann for providing us with the PDB coordinates for eIF4A and Michael Zuker for his advice on determining the salt effects on the calculated Tm. We thank Pinak Chakrabarti for his insight into the properties of cysteine residues, and we are grateful to Thierry Lacombe, Joëlle Marie and Sanda Rocak for fruitful discussions. We are very much indebted to Costa Georgopoulos for his undying support. This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Swiss National Science Foundation and by the University of Geneva. JB was also supported by a Short Term European Molecular Biology Organization Fellowship. We dedicate this work to the memory of Françoise Cordin.
References
- Benz J, Trachsel H, Baumann U (1999) Crystal structure of the ATPase domain of translation initiation factor 4A from Saccharomyces cerevisiae—the prototype of the DEAD box protein family. Struct Fold Des 7: 671–679 [DOI] [PubMed] [Google Scholar]
- Cappello V, Tramontano A, Koch U (2002) Classification of proteins based on the properties of the ligand-binding site: the case of adenine-binding proteins. Proteins 47: 106–115 [DOI] [PubMed] [Google Scholar]
- Carmel AB, Matthews BW (2004) Crystal structure of the BstDEAD N-terminal domain: a novel DEAD protein from Bacillus stearothermophilus. RNA 10: 66–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruthers JM, Johnson ER, McKay DB (2000) Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc Natl Acad Sci USA 97: 13080–13085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruthers JM, McKay DB (2002) Helicase structure and mechanism. Curr Opin Struct Biol 12: 123–133 [DOI] [PubMed] [Google Scholar]
- Chaudhry C, Farr GW, Todd MJ, Rye HS, Brunger AT, Adams PD, Horwich AL, Sigler PB (2003) Role of the gamma-phosphate of ATP in triggering protein folding by GroEL-GroES: function, structure and energetics. EMBO J 22: 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang RY, Weaver PL, Liu Z, Chang TH (1997) Requirement of the DEAD-box protein Ded1p for messenger RNA translation. Science 275: 1468–1471 [DOI] [PubMed] [Google Scholar]
- de la Cruz J, Iost I, Kressler D, Linder P (1997) The p20 and Ded1 proteins have antagonistic roles in eIF4E-dependent translation in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 94: 5201–5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya AE, Koonin EV (1993) Helicases: amino acid sequence comparisons and structure–function relationships. Curr Opin Struct Biol 3: 419–429 [Google Scholar]
- Henn A, Shi SP, Zarivach R, Ben-Zeev E, Sagi I (2002) The RNA helicase DbpA exhibits a markedly different conformation in the ADP-bound state when compared with the ATP- or RNA-bound states. J Biol Chem 277: 46559–46565 [DOI] [PubMed] [Google Scholar]
- Iost I, Dreyfus M, Linder P (1999) Ded1p, a DEAD-box protein required for translation initiation in Saccharomyces cerevisiae, is an RNA helicase. J Biol Chem 274: 17677–17683 [DOI] [PubMed] [Google Scholar]
- Leipe DD, Koonin EV, Aravind L (2003) Evolution and classification of P-loop kinases and related proteins. J Mol Biol 333: 781–815 [DOI] [PubMed] [Google Scholar]
- Linder P, Lasko PF, Ashburner M, Leroy P, Nielsen PJ, Nishi K, Schnier J, Slonimski PP (1989) Birth of the D-E-A-D box. Nature 337: 121–122 [DOI] [PubMed] [Google Scholar]
- Lohman TM, Bjornson KP (1996) Mechanisms of helicase-catalyzed DNA unwinding. Annu Rev Biochem 65: 169–214 [DOI] [PubMed] [Google Scholar]
- Lorsch JR, Herschlag D (1998a) The DEAD box protein eIF4A. 1. A minimal kinetic and thermodynamic framework reveals coupled binding of RNA and nucleotide. Biochemistry 37: 2180–2193 [DOI] [PubMed] [Google Scholar]
- Lorsch JR, Herschlag D (1998b) The DEAD box protein eIF4A. 2. A cycle of nucleotide and RNA-dependent conformational changes. Biochemistry 37: 2194–2206 [DOI] [PubMed] [Google Scholar]
- Moodie SL, Mitchell JB, Thornton JM (1996) Protein recognition of adenylate: an example of a fuzzy recognition template. J Mol Biol 263: 486–500 [DOI] [PubMed] [Google Scholar]
- Nobeli I, Laskowski RA, Valdar WS, Thornton JM (2001) On the molecular discrimination between adenine and guanine by proteins. Nucleic Acids Res 29: 4294–4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Chakrabarti P (1998) Different types of interactions involving cysteine sulfhydryl group in proteins. J Biomol Struct Dyn 15: 1059–1072 [DOI] [PubMed] [Google Scholar]
- Peyret N (2000) Prediction of Nucleic Acid Hybridization: Parameters and Algorithms. Chemistry. Detroit: Wayne State University [Google Scholar]
- Pleiss JA, Derrick ML, Uhlenbeck OC (1998) T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA 4: 1313–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, Uhlenbeck OC (2002) Cooperative binding of ATP and RNA substrates to the DEAD/H protein DbpA. Biochemistry 41: 3693–3702 [DOI] [PubMed] [Google Scholar]
- Pugh GE, Nicol SM, Fuller-Pace FV (1999) Interaction of the Escherichia coli DEAD box protein DbpA with 23 S ribosomal RNA. J Mol Biol 292: 771–778 [DOI] [PubMed] [Google Scholar]
- Rocak S, Linder P (2004) DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5: 232–241 [DOI] [PubMed] [Google Scholar]
- Rogers GW Jr, Lima WF, Merrick WC (2001) Further characterization of the helicase activity of eIF4A. Substrate specificity. J Biol Chem 276: 12598–12608 [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C (1994) Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19: 55–72 [DOI] [PubMed] [Google Scholar]
- SantaLucia J Jr (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA 95: 1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman E, Edwalds-Gilbert G, Lin RJ (2003) DExD/H-box proteins and their partners: helping RNA helicases unwind. Gene 312: 1–16 [DOI] [PubMed] [Google Scholar]
- Smith CA, Rayment I (1996) Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys J 70: 1590–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas P, Wigley DB (2000) DNA helicases: ‘inching forward'. Curr Opin Struct Biol 10: 124–128 [DOI] [PubMed] [Google Scholar]
- Staley JP, Guthrie C (1998) Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell 92: 315–326 [DOI] [PubMed] [Google Scholar]
- Story RM, Li H, Abelson JN (2001) Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii. Proc Natl Acad Sci USA 98: 1465–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto N, Nakano S, Katoh M, Matsumura A, Nakamuta H, Ohmichi T, Yoneyama M, Sasaki M (1995) Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry 34: 11211–11216 [DOI] [PubMed] [Google Scholar]
- Tanner NK (2003) The newly identified Q motif of DEAD box helicases is involved in adenine recognition. Cell Cycle 2: 18–19 [DOI] [PubMed] [Google Scholar]
- Tanner NK, Cordin O, Banroques J, Doere M, Linder P (2003) The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol Cell 11: 127–138 [DOI] [PubMed] [Google Scholar]
- Tanner NK, Linder P (2001) DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell 8: 251–262 [DOI] [PubMed] [Google Scholar]
- Turner DH, Sugimoto N, Freier SM (1988) RNA structure prediction. Annu Rev Biophys Biophys Chem 17: 167–192 [DOI] [PubMed] [Google Scholar]
- Vale RD (1996) Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J Cell Biol 135: 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Milligan RA (2000) The way things move: looking under the hood of molecular motor proteins. Science 288: 88–95 [DOI] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, Gay NJ (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1: 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]