

Abstract
Tumor multidrug resistance (MDR) is a serious clinical challenge that significantly limits the effectiveness of cytotoxic chemotherapy. As such, complementary therapeutic strategies are being explored to prevent relapse. The altered metabolic state of cancer cells, which perform aerobic glycolysis, represents an interesting target that can enable discrimination between healthy cells and cancer cells. We hypothesized that co-silencing of genes responsible for aerobic glycolysis and for MDR would have synergistic antitumor effect. In the current study, siRNA duplexes against pyruvate kinase M2 (siPKM-2) and multidrug resistance gene-1 (siMDR-1) were encapsulated in hyaluronic acid (HA)-based self-assembling nanoparticles. The particles were characterized for morphology, size, charge, encapsulation efficiency and transfection efficiency. In vivo studies included biodistribution assessment, gene knockdown confirmation, therapeutic efficacy, and safety analysis. The benefit of active targeting of cancer cells was confirmed by modifying the particles’ surface with a peptide targeted to epidermal growth factor receptor (EGFR), which is overexpressed on the membranes of the SKOV-3 cancer cells. To augment the studies involving transplantation of a PTX-resistant cell line, an in vivo paclitaxel (PTX) resistance model was developed by injecting repeated doses of PTX following tumor inoculation. The nanoparticles accumulated significantly in the tumors, hindering tumor volume doubling time (p<0.05) upon combination therapy in both the wild type (2-fold) and resistant (8-fold) xenograft models. Whereas previous studies indicated that silencing of MDR-1 alone sensitized MDR ovarian cancer to PTX only modestly, these data suggest that concurrent silencing of PKM-2 improves the efficacy of PTX against MDR ovarian cancer.
Keywords: Multidrug resistance, ovarian cancer, PKM-2, MDR-1, hyaluronic acid-based nanoparticles, siRNA, paclitaxel
1. Introduction
Ovarian cancer is one of the most common female gynecological malignancies and has an average 5-year survival rate of 44% (1). Because the majority of patients present with advanced stages of the disease, monotherapies typically do not confer meaningful efficacy. As a result, innovative strategies to prolong disease remission are being explored. Paclitaxel (PTX) is a standard treatment for ovarian cancer, though its effectiveness often diminishes over the course of treatment owing to the emergence of multi-drug resistance (MDR). MDR develops as a consequence of several factors, including poor systemic drug delivery due to vascular abnormalities, insufficient intracellular availability, and micro-environmental selection pressures(2). Amongst these, one of the most studied resistance mechanisms is the reduction in intracellular drug concentration by efflux transporter proteins that pump drugs out of cells. Many of these transporters are members of the ATP-binding cassette transmembrane protein super-family, including P-glycoprotein (P-gp) and MDR protein-1 (MRP-1). Although increasing the dosing of anticancer agents can transiently increase intracellular drug concentrations, dose-limiting toxicities can necessitate drug-free recovery periods that lead to reversion to MDR.
Along with drug resistance, cancer cells also undergo adaptions in cellular respiration to enhance their tumorigenic and metastatic potential (3). Cellular respiration is a series of catabolic reactions that yield ATP. In healthy cells, this occurs by the energy-efficient oxidative phosphorylation (OXPHOS) system (yielding between 30 to 36 ATP's for every glucose molecule) whereas cancer cells show a propensity to rely on a less energy efficient process termed aerobic glycolysis (which yields 2 ATPs for every glucose molecule) for energy. Although this dependence of cancer cells on aerobic glycolysis would seem detrimental energetically, cancer cells are not limited for ATP and prioritize conversion of glucose to biomass for dividing cells. From a therapeutic perspective, these metabolic differences between cancer cells and healthy cells can be used to discriminate these cell types, thereby avoiding the harsh side effects of cytotoxic therapies.
Pyruvate kinase (PK) is a critical enzyme in the glycolysis pathway that has been avidly explored as a potential target for cancer therapy. It catalyzes the final irreversible rate-limiting step of glycolysis by dephosphorylating phosphoenolpyruvate (PEP) into pyruvate and produces one molecule of ATP in the process. PK has four isoforms (L, R, M1 and M2) with a specific tissue distribution pattern(4). PKM-1 and PKM-2 are different splicing products of the same mRNA transcribed from the PKM gene. The PKM-1 isoform is expressed in energy-intensive organs such as muscles, brain, heart and kidney, while PKM-2 is expressed in lungs, fat, pancreatic islets and in cells with a high rate of nucleic acid synthesis, such as embryonic cells, adult stem cells, and tumor cells. Due to a high expression in tumor cells, novel inhibitors (5, 6) and nucleic acid therapies (7-9) have been designed to target PKM-2 to achieve anticancer activity.
RNA interference (RNAi) is a biological process in which RNA molecules (miRNA or siRNA) inhibit gene expression, typically by causing the destruction of complementary mRNA molecules. It has emerged as a powerful strategy for cancer therapy, especially MDR cancer. Frustratingly, delivery of these therapies to specific tumor tissues and cells followed by intracellular release from endosomal/lysosomal compartments into the cytoplasm still remains a major hurdle. Naked RNAi-based therapy is unfavorable due to the charge, size, and instability of short interfering RNAs (siRNAs). While viral vectors are efficient tools for gene delivery, concerns regarding their toxicity have limited their clinical applications, necessitating development of safer alternatives. Non-viral/polymeric nanoparticle based delivery platforms are becoming viable alternatives for delivery of RNAi-based therapies. Indeed, siRNA against PKM-2 has been successfully delivered by nanoparticles to induce cell apoptosis and tumor regression (7, 10). Goldberg et al., had previously reported the delivery of M2 isoform specific siPKM2 in lipidoid formulations in HepG2 and SKOV-3 tumor bearing mice. The siRNA nanoparticles showed a 76-85% decrease in tumor volume relative to the controls (7). Similarly, we have previously used RNAi-based therapeutic approaches to silence MDR-1 expression, enhancing the uptake and therapeutic efficacy of anticancer drugs (11, 12). In our experience, encapsulation of siMDR-1 and PTX in polymeric nanoparticles has provided MDR-1 gene silencing and subsequent enhancement in cytotoxicity attributed to an increase in intracellular drug concentration in SKOV-3TR cells (11). Similarly use of lipid functionalized dextran doxorubicin nanoparticles has shown five to ten fold higher anti-proliferative activity in drug sensitive and drug resistant osteosarcoma (KHOS) and ovarian cancer (SKOV-3) cells lines (12). Recently we have also assessed the delivery of MDR-1 siRNA to ovarian cancer cells followed by paclitaxel treatment to induce inhibition of tumor growth, decreased Pgp expression and increased apoptosis (13, 14).
To our knowledge, this is the first report of the combination of an siRNA that directly induces apoptosis and an siRNA that mitigates against chemotherapy efflux prior to cytotoxic chemotherapy. Herein, we describe the dramatically improved efficacy of this combination relative to either siRNA monotherapy prior to cytotoxic chemotherapy in a two MDR models of ovarian cancer. In addition to the combination therapeutic approach, we explored the targeting capability of our hyaluronic acid (HA)-based self-assembling systems by adding targeting moieties to epidermal growth factor receptors, which are highly expressed on ovarian cancer cells (15). To explore this combination therapy in a model that would maximally recapitulate the biology of MDR ovarian cancer, we also developed and assessed our therapies in an in vivo-induced PTX resistant model. Our data again confirmed the synergistic potential of combination RNAi and cytotoxic therapy for MDR ovarian cancer.
2. Materials and Methods
2.1 Materials
Sodium hyaluronate (HA) with an average molecular weight of 20 kDa was obtained from Lifecore Biomedical Co. (Chaska, MN). Poly(ethylene imine) (PEI MW 20,000 Da) was obtained from Polysciences Inc, (Warrington, PA). Mono-functional poly(ethylene glycol)-amine (PEG2K-NH2, MW=2000 Da) was purchased from Creative PEG Works, Inc. (Winston Salem, NC). Paclitaxel (PTX) was purchased from Fisher Scientific.
2.2 Cell lines
Human ovarian adenocarcinoma cell line SKOV-3 was obtained from ATCC (Manassas, VA). A PTX-resistant version of this cell line (SKOV-3TR) was obtained from Massachusetts General Hospital, (Boston, MA). The cells were cultured in RPMI-1640 medium supplemented with 10% FBS and grown at 37°C, 5% CO2. SKOV-3TR cells were cultured in 20 nM PTX to maintain their drug-resistant phenotype. The resistant phenotype of these cells was regularly assessed by cytotoxicity analysis to determine IC50 values.
2.2 Formation and characterization of siRNA-loaded HA nanoparticles: determination of size, charge, morphology, siRNA encapsulation, and siRNA release
We have previously published information regarding preparing combinatorial designed HA formulations (16). The HA-PEI and HA-PEG conjugates were prepared using this combinatorial approach (16). For the synthesis of HA-PEG-EGF, 50 mg of maleimide-PEG-amine was added to EDC/NHS activated HA. Following synthesis of HA-PEG-maleimide, an EGFR specific peptide, YHWYGYTPQNVI designated as GE11 was used for conjugation with maleimide. The GE11 peptide was originally synthesized and screened as an EGFR specific peptide by Li and colleagues (17). For this study, the GE11 peptide with a spacer sequence of GGGGC was synthesized at Tufts University Core Facility, Boston, MA. We have previously successfully prepared EGFR targeted polymeric nanoparticles using this peptide sequence (18). The carboxyl group of terminal cysteine of the peptide was reacted with the maleimide of maleimide-PEG-HA in HEPES buffer (pH 7.4) at 1:1 molar ratio while mixing under N2 at 4°C for 24 h. The peptide conjugate was then purified by dialysis (3.5 kDa), lyophilized and characterized by NMR spectroscopy. Similarly for the preparation of HA-rhodamine 123, 50 mg of rhodamine 123 was added to the EDC/NHS activated HA, the solution stirred overnight, dialyzed and lyophilized.
The HA-PEI, HA-PEG solutions (3 mg/ml) were prepared by dissolving the polymer in PBS. Nanoparticle size and charge were determined on a Malvern Nano ZS (Malvern Instruments, UK). Transmission electron microscopy (JEOL, JEM-1000, Tokyo, Japan) was performed to assess the formation of siRNA-loaded nanoparticles (NP's). Uranyl acetate ribonucleic acid stain was used to demarcate siRNA from the polymer. The dark staining of siRNA by heavy metals such as uranyl acetate provides a high contrast compared to hyaluronic acid polymer that can help to confirm loading of siRNA in the polymeric NP's. The ability of these complexes to release siRNA was determined by treating them with poly(acrylic acid) (PAA), followed by gel electrophoresis.
2.3 Assessment of HA-siRNA uptake by confocal microscopy
For the purpose of uptake studies, fluorescein-labeled scrambled siRNA was formulated with HA-PEI conjugated with rhodamine dye. Particles containing 10 nM siRNA were incubated with SKOV-3WT and SKOV-3TR cells at 37 °C and cell uptake was assessed after 5 min, 15 min or 30 min.
2.4 Transfection of siRNA against MDR-1 and PKM-2 to assess silencing of target mRNA expression
SKOV-3 cells were transfected with siRNA-loaded NP's (100 nM siRNA) incubated for 72 h or 96 h. The cells were then harvested, and RNA was extracted and used to run quantitative PCR to assess mRNA levels. mRNA knockdown was determined by normalizing the expression of MDR-1 or PKM-2 to β-actin levels.
2.5 Cytotoxicity assay for single and combination siRNA therapy followed by PTX
SKOV-3WT and SKOV-3TR cells were initially transfected with siMDR-1 and/or siPKM-2. After 72 h, PTX was added to assess the effect of co-sensitization therapy. Cellular cytotoxicity was assessed by MTT assay 72 h after PTX treatment and expressed as the percentage of viable cells.
2.6 Establishment of subcutaneous SKOV-3WT tumors
Animal procedures were performed according to a protocol approved by Northeastern University, Institutional Animal Care and Use Committee (NU-IACUC). The tumor models for this study were developed in nude mice obtained from Charles River Laboratories (Wilmington, MA). 5-6 week old nude mice were injected subcutaneously (s.c.) with SKOV-3WT tumor cells (5 × 106 cells + Matrigel) on the right flank. Tumor sizes were measured at least once or twice a week to monitor tumor growth.
2.7 Qualitative biodistribution studies of HA nanoparticles using a NIR-encapsulated dye
The biodistribution of HA-PEI/PEG and dual-targeted HA NP's was assessed by encapsulating indocyanine green (ICG) into the NP's. Tumor-bearing mice were inoculated as described above, and the study commenced when the tumors reached an average size of ~200 mm3. The particles were then injected i.v. into mice bearing SKOV-3WT or SKOV-3TR tumors. Mice were imaged at 1 h, 2 h, 6 h, and 24 h after the injection to monitor the distribution of the particles using an Xenogen IVIS Imaging System (Xenogen Corporation, Alameda, CA) (Ex: 785nm, Em: 820nm).
2.8 Quantitative biodistribution and pharmacokinetic studies using siPKM-2 encapsulated in HA nanoparticles
siRNA duplexes silencing PKM-2 was encapsulated in HA-PEI/HA-PEG NP's. These particles were injected into mice (n=6/group) bearing ovarian tumors (SKOV-3WT) at 0.5 mg/kg twice a day on days 1, 3, and 5. Blood samples, major organs (liver, spleen, lung, heart, kidney), and tumors were collected 1 h, 6 h, or 24h following the last injection. The organs were thenhomogenized using Qiagen Tissue Lyser (Qiagen, Germantown, MD) to prepare tissue lysates. The homogenized tissue lysates were subsequently diluted 1:1000, cDNA was generated, and qRT-PCR was performed. The PKM-2 siRNA sequence, reverse primer, forward primer, and an anti-primer sequence are listed below.
PKM-2 siRNA
Sense: 5′ CCAUAAUCGUCCUCACCAA3′Antisense: 5′UUGGUGAGGACGAUUAUGG3′
Primers
Reverse: GGAAGCCGACCCATAATCG
Forward: /56-FAM/ACTCCCTCCCTCGATTTTTGGTGAGGACGA
Anti-primer: AAATCGAGGGAGGGAG/3BHQ_1/
siRNA was denatured and annealed to the RT primer (6 μl diluted siRNA and 18 μl reverse primer, 100 nM). siRNA was denatured by incubating at 95 °C for 5 min. Primers were then annealed by 2 min incubation at 80, 70, 60 and 45°C with 4°C hold. Then the reverse transcription reaction was carried out as follows. A master mix was made by mixing the following components: RT-PCR buffer (6.25 μl), forward primer (10 μM, 0.12 μl), reverse primer (10 μM, 0.12 μl), anti-primer (100 μM, 0.12 μl), 25 U RT-PCR enzyme (0.5 μl), and water (1.5 μl).
A total of 8.5μl of this master mix was then mixed with 3.5 μl of sample and run on the PCR at the following listed conditions: 50 °C (10 min), 9 °C (10 min); 40 cycles of 95 °C (15 s), 45 °C (60 s). The amplified siRNAs were finally detected and quantitated by running a standard curve using lysate from untreated mouse tissue spiked with known concentrations of siRNA.
2.9 Evaluating target gene knockdown in established subcutaneous SKOV-3 tumors
When subcutaneous tumors were ~ 150-200 mm3 in size, the animals were randomized into groups such that each group had similar tumor size. The animals were given an siRNA dose of 0.5 mg/kg twice a day on days 1, 3, and 5. 24 h after the last dose, the tumors were excised, RNA was extracted, and qRT-PCR was performed to determine the extent of MDR-1 and PKM-2 silencing.
2.10 Efficacy studies and toxicity analysis to determine safety of siRNA-encapsulated nanoparticles in SKOV-3WT-tumor bearing mice
Animals bearing SKOV-3WT tumors were initially given an siRNA dose of 0.5 mg/kg twice a day every alternate days 1, 3, and 5. PTX was administered at a dose of 20 mg/kg once per week for two weeks. Tumor volumes were measured using the following formula to monitor the tumor growth inhibition: Tumor volume = ((length × width2)/2). The mice were weighed the day the treatments commenced and every day during the dosing period. To measure liver enzyme levels, blood was collected from all groups, and ALT/AST levels were assessed. Liver, kidney, and spleen samples from mice were also collected for histopathological analysis at the end of the study.
2.11 In vivo-induced PTX resistance development, efficacy studies, and safety analysis for combination therapy of siRNA with PTX
For development of the in vivo-induced PTX-resistant mouse model, the mice were inoculated subcutaneously with SKOV-3WT cells. When subcutaneous tumors were ~ 200 mm3 in size, the animals were treated with PTX solution at a dose of 20 mg/kg every alternate day for 10 doses. One week following completion of PTX therapy, the animals were randomized to different treatment groups, and tumor volume was measured to monitor for tumor growth inhibition. Body weight measurements were conducted throughout the entire period of efficacy studies. Liver, kidney, and spleen toxicity studies were conducted as described in section 2.10.
3. Results
3.1 Formation of siRNA loaded HA nanoparticles determination of size, charge, morphology, siRNA encapsulation and release by polyanion competition by gel retardation assay
Figure 1A, provides a schematic representation of negatively charged siMDR-1/siPKM-2 encapsulation in HA-PEI/PEG particles. The self-assembling NP's showed a spherical morphology in TEM (Figure 1B) with particles in the size range of 106-125 nm, polydispersity index (PDI) of 0.1 to 0.3 and surface charge in the range of (-) 25 to (-) 28 mV (Table 1). Electrophoretic retardation analysis of siRNA binding by HA-PEI with the release of intact siRNA by PAA showed 92-95% siRNA encapsulation efficiency.
Figure 1.
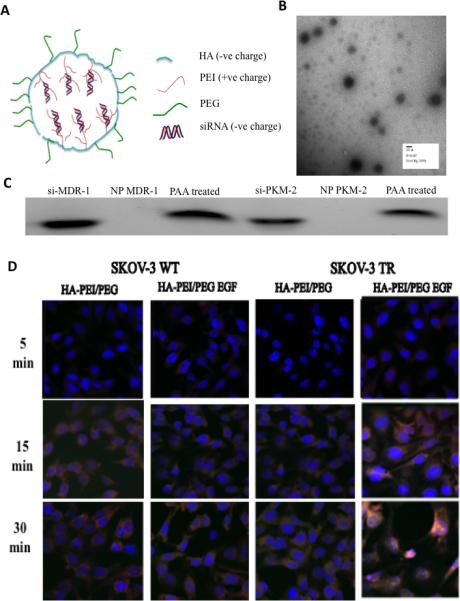
Characteristics of HA derivative/siRNA particles. (A) Schematic representation of HA-PEI/PEG/EGF nano-assemblies with negatively charged siRNA. (B) The self-assembling nanoparticles showed a spherical morphology in TEM. (C) Electrophoretic retardation analysis of siRNA binding by HA-PEI with the release of intact siRNA by poly(acrylic acid). (D) Cell uptake studies in SKOV-3 WT and TR model. HA-rhodamine-123 and scramble siRNA labeled with fluorescein were incubated with the cells for 5, 15 and 30 min. The cells showed uptake within 15 min of incubation with the dual targeted system showing higher fluorescence signal compared to the CD44 targeted system. The cell nuclei were stained with DAPI (blue).
Table 1.
Nanoparticle size range, polydispersity index (PDI), surface charge and encapsulation efficiency.
| Formulation | Size (d.nm) | PDI | Zeta potential (mV) | Encapsulation efficiency |
|---|---|---|---|---|
| HA-PEI/PEG blank | 106±16 | 0.124 | −27±1.7 | - |
| HA-PEI-PEG/MDR-1 | 117±9 | 0.327 | −28±1.6 | 95±5 |
| HA-PEI-PEG/PKM-2 | 125±26 | 0.286 | −25±0.8 | 92±8 |
3.2 Assessment of siRNA encapsulated HA nanoparticle uptake by confocal microscopy
Figure 1D, shows cell uptake studies in SKOV-3WT and SKOV-3TR model following administration of nano-assemblies formed with rhodamine 123 tagged HA and fluorescein tagged scrambled siRNA for 5, 15 and 30 min. The cells showed uptake within 15 min of incubation in the wild and resistant SKOV-3 cell lines. The dual targeted system showed higher fluorescence signal compared to the HA-PEI/PEG system in both cell lines.
3.3 Transfection of MDR-1 and PKM-2 targeted siRNA to assess silencing of target gene expression
Transfection studies with siMDR-1 and siPKM-2 were conducted in SKOV-3WT (Figure 2A) and SKOV-3TR cells (Figure 2B) for 72h and 96h. Earlier and later time points were also assessed in preliminary studies however as these time points did not indicate a significant down-regulation in gene expression these were not assessed further. Likewise concentrations ranging from 50 nM – 200 nM were assessed for activity. A concentration dependent down-regulation of gene expression was observed and a concentration of 100 nM was selected (lowest concentration with effective gene down-regulation). Also in our previous work with HA-PEI based delivery systems we had observed, 100 nM siRNA encapsulated in the HA-based systems provided effective down-regulation with minimal cellular toxicity from PEI. Figure 2A, measures the transfection efficiency of siMDR-1 and siPKM-2 in SKOV-3WT cells. A significant down-regulation of MDR-1 was not observed even with Lipofectamine as the basal levels of MDR-1 expression in SKOV-3 WT cells is fairly low siPKM-2 on the other hand showed ~80% down-regulation with Lipofectamine and both the NP's. Figure 2B, reports the transfection efficiency of the delivery systems in SKOV-3TR cells. Both Lipofectamine® and the NP's showed 80-85% gene expression down-regulation with the NP's showing greater efficiency after longer periods of incubation compared to transfection achieved following use of Lipofectamine®.
Figure 2.
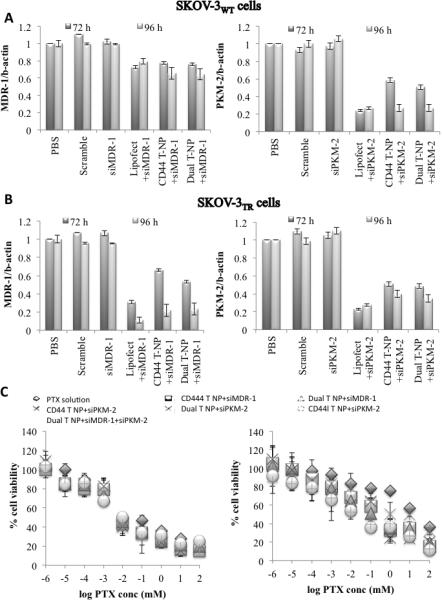
HA-PEI/siRNA mediated MDR-1 and PKM-2 gene silencing in SKOV-3WT (A) and SKOV-3TR (B). Cells were treated with scramble, naked siRNA, siRNA formulated in Lipofectamine and siRNA formulated in HA-PEI/PEG nanoparticles. MDR-1 and PKM-2 expression was measured by qPCR. Data represented as a mean SD (n=3). (C) Cytotoxicity assay following single and combination therapy in SKOV-3WT (left) and SKOV-3TR cells (right).
3.4 Cytotoxicity assay for single and combination siRNA therapy in combination with paclitaxel
Figure 2, shows the effect of single and combination therapy on PTX IC50 in SKOV-3WT and SKOV-3TR cell lines. In SKOV-3WT cells, both the single and combination therapy did not show a significant change in PTX IC50 values (p>0.05). In SKOV-3TR cells, single therapy with siPKM-2 showed a significant decrease in PTX IC50 values (p<0.05). A 1.6 fold difference in the IC50 value was observed between siPKM2 encapsulated in dual targeted NP's compared to HAPEI/PEG NP's. Between single and combination therapy, combination therapy showed a 1.3 fold decrease in IC50 value (p<0.05) compared to siMDR-1 therapy.
3.5 Qualitative biodistribution studies using NIR-encapsulated dye in the HA nanoassemblies
The whole body NP distribution in live animals was evaluated using a NIR dye, ICG encapsulated in HA NP's. ICG was loaded into HA-PEI/PEG NP's using a similar technique as used to encapsulate siRNA. These ICG loaded NP's had similar characteristics as siRNA encapsulated HA-PEI/PEG NP's (size range of ~ 200 nm, surface charge -15mV). The ICG loaded NP's were i.v. injected in SKOV-3WT (Figure 3A) and SKOV-3TR (Figure 3B) tumor bearing mice at a dose equivalent to the dose used in efficacy studies. The NP's were stable during circulation following iv injection and the NIR signal was measured at different time points to capture the distribution pattern in live mice. Strong NIR signal was observed throughout the whole body 1h post administration with both the HA-PEI/PEG and dual targeted NP's. A gradual decrease in signal intensity was observed over 6h with majority of the signal coming from liver and tumor regions 24h after ICG NP administration. Figure 3C shows ex vivo images of liver, kidney, spleen, heart, lungs and tumors 24h post ICG administration. Amongst these tissues, liver showed the highest level of signal intensity, followed by the tumor, kidney and spleen. We have previously observed that i.v. injection of free ICG in healthy mice shows a signal which is detectable in the liver as early as 3 min post-injection, reaching a peak level between 5 and 10min, indicative of a rapid hepatic clearance from the systemic circulation. The relatively short circulation time of free ICG in vivo could be attributed to fluorescence quenching of free ICG in physiological environments.
Figure 3.
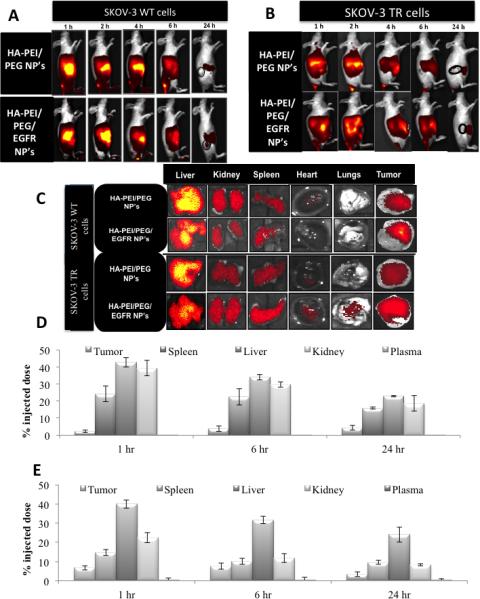
In vivo (A-B) and ex vivo (C) biodistribution of ICG/HA-PEI/PEG and ICG/HAPEI/PEG-EGF nanoparticles in SKOV-3WT and SKOV-3TR tumor bearing mice over 1, 2, 4, 6 and 24 h. Quantitative biodistribution studies to assess distribution of siPKM-2 following intravenous dosing of HA-PEI-siPKM-2 as CD44-targeted and dual targeted delivery systems. Quantitative biodistribution studies to assess distribution of siPKM-2 following intravenous dosing of HA-PEI-siPKM-2 as HA-PEI/PEG and dual targeted delivery systems in SKOV-3WT (D) and SKOV-3TR (E) tumor models.
3.6 Quantitative biodistribution and pharmacokinetic studies using siPKM-2 encapsulated in HA nanoassemblies
After six i.v. injections of 0.5 mg/kg siRNA doses (in HA NP) into tumor bearing mice, blood and tissue samples were collected for siRNA quantitation at 1 h, 6 h and 24 h post last administered dose. Accurate siRNA quantitation was studied using the anti-primer quenching based real time PCR method. A fluorescently labeled PCR primer was designed to anneal to the template RNA and to a universal anti-primer. Following initial PCR, the temperature was lowered to allow the anti-primer to bind to unincorporated primer to quench the fluorescence. As double stranded PCR product would not bind with the anti-primer an increase in fluorescent signal would enable siRNA quantitation. The siRNA was quantitated in each tissue and the % input dose per whole organ was calculated based on the starting siRNA dose. Figure 3D shows the biodistribution profile of siPKM-2 in HA-PEI/PEG NP's whereas Figure 3E indicates the distribution of the dual targeted particles. With both the delivery systems 30-40% of the siRNA is detected in the liver, kidney and spleen within 1h of administration. A very small proportion of the siRNA was detected in the plasma whereas 3-9% of the siPKM-2 was detected in the tumor tissues.
3.7 Evaluating target gene knockdown in SKOV-3 tumor bearing mice
Figure 4A depicts in vivo gene down-regulation following administration of siMDR1 and siPKM2 in HA-PEI/PEG and the dual targeted NP's. siMDR-1 delivery in SKOV-3WT tumor bearing mice showed a 20% down-regulation of MDR-1 expression. A low level of endogenous MDR-1 expression in SKOV-3WT mice would be attributable to the lower degree of down-regulation observed with siMDR1. With siPKM2 on the other hand, a 70% PKM2 down-regulation was observed. Figure 4B, shows in vivo gene down-regulation following administration of the nano-assemblies in SKOV-3TR tumor bearing mice. The HA-PEI/PEG particles provided 40% MDR-1 down-regulation and the dual targeted NP's provided 65% MDR-1 down-regulation. On the other hand siPKM-2 HA-PEI/PEG and the dual targeted NP's showed 60-70% down-regulation of gene expression.
Figure 4.
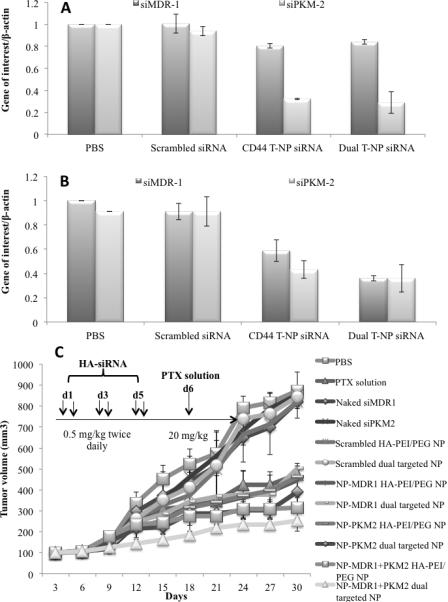
In vivo gene silencing studies of MDR-1 and PKM-2 in (A) SKOV-3WT and (B) SKOV-3TR tumor bearing mice n=6. (C) Effect of the combination of paclitaxel treatment and MDR1 and PKM2 silencing on growth of SKOV3WT tumors.
3.8 Efficacy studies and toxicity analysis to determine safety of HA-encapsulated nanoparticles
Figure 4C shows the effect of single and combination therapy of siRNA and PTX in SKOV3WT tumor bearing mice. Tumor bearing mice were initially administered siMDR-1 and siPKM-2 as single or combination therapy twice a day every alternate day for three days. 24h after the last dose the animals were injected with i.v. dose of PTX at 20 mg/kg. This treatment regimen was repeated over two weeks and tumor volume measurements were measured every alternate day for 4 weeks. siRNA therapy in combination with PTX showed significant improvement in efficacy. The siMDR-1 NP's showed a tumor growth profile similar to PTX solution indicating that combination of siMDR-1 and PTX has minimal effect on tumor growth. This effect could be correlated to low level of MDR1 expression in SKOV-3WT tumor bearing mice. Treatment with siPKM2 in both the nanoparticle systems showed slightly delayed tumor growth compared to PTX solution (p>0.05). Dual siRNA therapy showed a significant decrease in tumor volume compared to control animals. Table 2 shows the tumor growth inhibition (TGI) and tumor volume doubling (TVD) time for all the treatment groups. Single siRNA therapy with PTX showed 50-60% tumor inhibition (p<0.05) compared to saline group. In comparison, combination therapy of dual siRNA and PTX showed 72-84% tumor growth inhibition (p<0.05). Similarly, this combination therapy group showed almost a two-fold increase in tumor volume doubling time. Compared to PTX solution a significant increase in TGI and TVD time was not observed. Safety and toxicity analysis showed minimal effect on body weight, ALT and AST levels (Supplementary Figure 1).
Table 2.
Percentage tumor growth inhibition and tumor volume doubling time following treatment with single or combination therapy of siRNA and PTX in SKOV-3WT tumor bearing mice. PBS – phosphate buffered saline, PTX – paclitaxel, NP – nanoparticle.
| Therapeutic groups | Tumor growth inhibition (%) | Tumor volume doubling time (days) |
|---|---|---|
| PBS | 7.4±4.8 | |
| PTX solution | 56 | 10.9±13.7 |
| Naked siMDRl | 9 | 7.7±6.6 |
| Naked siPKM2 | 5 | 7.6±3.4 |
| Scrambled HA-PEI/PEG NP | 6 | 7.6±6.5 |
| Scrambled dual targeted NP | 12 | 7.8±3.2 |
| NP-MDR1 HA-PEI/PEG NP | 59 | 11.1±5.6 |
| NP-MDR1 dual targeted NP | 49 | 10.0±3.9 |
| NP-PKM2 HA-PEI/PEG NP | 54 | 10.5±6.5 |
| NP-PKM2 dual targeted NP | 65 | 12.2±5.4 |
| NP-MDR1+PKM2 HA-PEI/PEG NP | 74 | 14.2±4.6 |
| NP-MDR1+PKM2 dual targeted NP | 82 | 17.9±8.8 |
3.9 In vivo paclitaxel resistance development, efficacy studies and safety analysis for combination therapy of siRNA with PTX
The in vivo PTX resistance model was developed in SKOV-3WT tumor bearing mice as described earlier. MDR-1/Pg-P development was assessed using PCR and IHC. Figure 5A depicts the MDR-1 expression 1 week and 3 weeks post PTX administration. A two to three fold increase in MDR-1 expression was observed compared to control. PKM-2 expression was unaltered in this model for the time period, which was assessed. IHC studies (Figure 5B) confirmed this analysis where an increase in P-gp expression was observed compared to the PBS control group. Once development of resistance was confirmed, further SKOV-3WT tumor bearing mice were recruited to a similar PTX regimen to develop the in vivo resistance model. Following model development the animals were randomized to the treatment groups and efficacy studies were conducted with the following treatment groups: PBS, NP with scramble siRNA and dual targeted NP's with siRNA as single therapy and combination therapy. Figure 5C, shows the treatment efficacy following single and dual siRNA therapy with PTX. All the treatment groups showed similar tumor volume measurements two weeks into therapy. However two weeks post therapy a difference in tumor volume was observed with combination of the two siRNA and PTX showing the highest anti-tumor efficacy. Although therapy with a single siRNA did show reduced tumor volume growth compared to the control groups there was a steady increase in tumor volume over the period of tumor assessment. In contrast with combination therapy, tumor volume sizes were maintained in the size range of 100 to 220 mm3. Table 3, indicates the TGI and TVD time. Single siRNA therapy showed 57-75% TGI (p<0.05). However only siPKM2 based therapy showed a significant increase in TVD when the siRNA were administered as single therapy. Combination therapy showed a significant increase in percentage TGI and a 8-fold (p<0.05) increase in TVD time compared to the control group. Safety and toxicity analysis showed minimal effect on body weight, ALT and AST levels.
Figure 5.
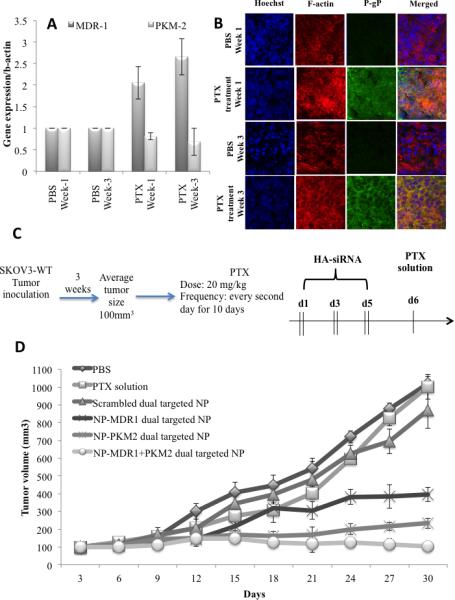
In vivo PTX resistance model development: (A) qPCR studies to assess MDR-1 and PKM-2 expression (B) Immunofluorescence of tumor tissues from Pg-P and PKM-2 expression in PBS treated and PTX treated animals. (C) Schematic representation of the dosing schedule adopted to develop the SKOV-3TR mouse model followed by nanoparticle treatment. (D) Effect of the combination of paclitaxel treatment and MDR1 and PKM2 silencing on growth of SKOV3TR tumors.
Table 3.
Percentage tumor growth inhibition and tumor volume doubling time following treatment with single or combination therapy of siRNA and PTX in SKOV-3TR tumor bearing mice. PBS – phosphate buffered saline, PTX – paclitaxel, NP – nanoparticle
| Therapeutic groups | Tumor growth inhibition (%) | Tumor volume doubling time (days) |
|---|---|---|
| PBS | 10.7±8.6 | |
| PTX solution | 2.1 | 12.3±7.9 |
| Scrambled dual targeted NP | 16.3 | 11.0±11.6 |
| NP-MDR1 dual targeted NP | 57.9 | 14.7±12.8 |
| NP-PKM2 dual targeted NP | 75.6 | 32.0±10.7 |
| NP-MDR1+PKM2 dual targeted NP | 89.7 | 79.4±10.2 |
4. Discussion
The aim of our current investigation was to study the effectiveness of combination therapy with siMDR-1 and siPKM-2 in SKOV-3WT and SKOV-3TR human ovarian adenocarcinoma cell lines and xenograft models. Previously, combination of MDR-1 gene silencing and chemotherapy administration has been assessed using polymeric NP's (11, 12, 19). These delivery systems have provided improved cytotoxicity attributed to an increased intracellular accumulation of anti-cancer agents upon MDR-1 gene silencing. Similarly for PKM-2 based therapy several nanocarrier based delivery approaches have been reported in literature for anticancer activity. Goldberg, et al. screened a library of siPKM-2 and identified a specific PKM-2 siRNA (si156), which when encapsulated in lipidoid NP's induced cell apoptosis showing tumor regression in HepG2 and SKOV3 xenograft tumor models (7). Another report showed that short hairpin RNA (shRNA) loaded in liposomal formulation showed effective PKM-2 silencing in human A549 lung cancer xenograft model and an increased efficacy in combination with cisplatin as an anticancer drug (10). PKM-2 silencing by shRNA loaded in Lipofectamine® has also shown increased efficacy of docetaxel in a similar model. Thus use of drug delivery systems to deliver nucleic acid to silence PKM-2 provides novel prospects of treating cancer. We have previously reported the development of CD44 targeting HA based NP's for the delivery of siRNA to A549 non-small lung cancer cells and tumors (20-22). In this study we wanted to extend the capability of our delivery system to assess the synergistic activity following the administration of combination of siRNA (targeting MDR-1 and PKM-2 expression) with PTX in the treatment of ovarian cancer. As SKOV-3 cells have a high surface EGFR expression we also examined the effect of actively targeted NP's in the delivery of these therapeutic agents (15). While testing these delivery systems in vivo we explored the possibility of developing an in vivo PTX resistance model, which would provide a realistic recapitulation of PTX resistance development. Traditionally in xenograft models PTX resistance is developed by subcutaneous injection of cancer cells which have been established by the reverse transformation technique, where the sublines are established in vitro by repeated or prolonged exposure to increasing doses of antineoplastic agents (23). However these cells usually show strongly suppressed metastatic and/or tumorigenic properties in vivo which makes them incompatible comparators to recurrent cancers which show increased metastatic and/or tumor growth potential. Several groups have reported development of xenograft resistant models by administration of PTX in a subcutaneous model followed by excision and re-transplantation over 6 months and the final tumor-derived cells being established and maintained as in vivo resistance sublines (24). In the current study we aimed to develop in vivo PTX resistance by repeated administration of PTX to recapitulate clinical resistance development and assess the effectiveness of our therapies in these models.
The siRNA nano-assemblies were prepared by charged based encapsulation of siRNA into HA-PEI particles. HA is a naturally occurring mucopolysaccharide composed of alternating disaccharide units of D-glucuronic acid and N-acetyl-D-glucosamine (NAG) with β(1-4) interglycosidic linkage. It is a highly anionic biopolymer present in the extracellular matrix and synovial fluids. It is biodegradable, non-toxic, non-immunogenic and non-inflammatory, which makes it an ideal carrier polymer for systemic drug delivery applications. The HA backbone specifically recognizes CD44, an integral membrane glycoprotein overexpressed on several tumor cell surfaces including tumor initiating stem cells. Similarly, PEI is widely used for nucleic acid delivery, including siRNA delivery where nucleic acids are condensed by electrostatic interactions to form dense particles (complexes), which protect the genetic material from enzymatic degradation and promote their cellular uptake by absorptive endocytosis. PEI also has an intrinsic endosomal escape mechanism known as the ‘proton sponge’ effect, which causes osmotic swelling and rupture of the endosome membrane, triggering the release of PEI complexes into the cytosol. In this study, for the preparation of the complexes, a 54:1, polymer:siRNA ratio was adapted from our previous studies where optimal encapsulation and release was observed with this ratio (21). At these ratios we obtained ~ 92-95% encapsulation efficiency of siRNA. Similarly for in vitro studies we selected a siRNA concentration of 100 nM due to effect of higher concentrations on cell viability (Supplementary Figure 2). For the incorporation of EGFR targeted HA, in our preliminary investigation we altered the ratio of HAEGF and determined the transfection efficiency (Supplementary Figure 3). Earlier transfection periods showed increased down-regulation of gene expression however at later times points altering ratios did not have a significant effect on down-regulation of MDR-1 expression.
Following the preparation of these particles uptake and transfection studies showed efficient uptake and transfection with both the nanoparticle systems showing similar decrease in gene expression in both SKOV-3WT and SKOV-3TR cell lines. Synergistic activity of dual siRNA therapy showed minimal effect on SKOV-3WT but had a significant effect on IC50 of PTX in SKOV-3TR cell lines. In SKOV-3WT cells as there is a low level of basal MDR-1, the cells would be sensitive to PTX thus HA-siMDR-1 expression would have minimal effect on cytotoxicity. In contrast, SKOV-3TR cells have an increased MDR-1 expression thus single therapy with siMDR-1 would affect cytotoxicity. Additionally combination therapy with dual siRNA showed a further improvement in cytotoxicity, which could be attributed to synergistic effect of siMDR-1 on P-gP expression and PKM-2 on the glycolytic pathway. A minimal effect on cytotoxicity was observed with both the nanoparticle delivery systems which could be attributed to similar uptake profile of these delivery systems in vitro.
In vivo biodistribution studies indicated a high degree of accumulation of the NP's in the liver, kidney and spleen due to preferential uptake of these particles by macrophages residing in these tissues. Although a small proportion of the total siRNA dose was detected at the tumor site, in vivo knockdown studies showed a significant down-regulation of gene expression. This down-regulation translated to a small improvement in anti-tumor efficacy following combination therapy of dual siRNA and PTX with minimal toxicity.
Post in vivo studies in SKOV-3WT tumor bearing models the efficacy of the delivery systems was assessed in the in vivo PTX resistant model. This model was developed over 6 weeks of therapy and MDR-1 expression was determined. As a significant difference in an increase in MDR-1 expression was not observed between 4 and 6 weeks post PTX treatment we initiated in vivo studies in TR model within 4 weeks of therapy. Post model development and dosing with NP with a similar regimen as was adopted in the WT model there was an improvement in anti-tumor efficacy with siRNA-based therapy. Dual siRNA based approach further augmented the anti-tumor effect possibly due to synergistic effect of siMDR-1 on PgP expression leading to increased cellular PTX concentration and the effect of siPKM-2 on the glycolysis pathway affecting cancer cell metabolism. Thus combination of siRNA with cytotoxic agents provided synergistic effect and enhanced anti-tumor efficacy due to its actions on different resistance mechanism in cancer.
Conclusions
In summary, anti-cancer therapeutics employing RNAi mechanism holds promise for sequence-specific silencing of target genes. However targeted delivery to tumor sites remains a major hurdle for cancer therapy. Thus in this study we evaluated encapsulation and delivery of siMDR-1 and siPKM-2 in HA-PEI based self-assembling nanosystems for the treatment of wild-type and resistant ovarian cancer. The nano-assemblies showed effective transfection and improved cytotoxic effect of PTX in cancer cells. Although the effect was not pronounced in wild-type animal models, dual therapy provided action of these therapeutic agents on multiple pathways in cancer cells providing synergistic activity for cancer therapy. Although an in vivo PTX resistance model was developed to assess the effectiveness of our therapies, future studies in PTX resistance model development in orthotopic or genetically modified ovarian cancer mice would indicate further clinical relevance of these therapies.
Supplementary Material
Acknowledgments
This study was partially supported by the National Cancer Institute's (NCI) Cancer Nanotechnology Platform Partnership (CNPP) grant U01- CA151452, the Dana-Farber Cancer Institute-Northeastern University Collaborative Grant on Development of Cancer Therapeutics and the Ovarian Cancer Research Fund. The authors would also like to acknowledge Dr. Arun Iyer, Dr. Amit Singh, and Dr. Shanthi Ganesh for their expertise on hyaluronic acid nanoparticle synthesis and formulation development.
Footnotes
Conflict of Interest Statement: The authors declare no conflict of interest.
References
- 1.Society AC. Survival rates for ovarian cancer, by stage. 2014 [updated 2015; cited 2015! April 23]. Available from: http://www.cancer.org/cancer/ovariancancer/detailedguide/ovarian-cancer-survival-rates.
- 2.Jabr-Milane LS, van Vlerken LE, Yadav S, Amiji MM. Multi-functional nanocarriers to overcome tumor drug resistance. Cancer treatment reviews. 2008;34:592–602. doi: 10.1016/j.ctrv.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert opinion on biological therapy. 2014;14:1145–59. doi: 10.1517/14712598.2014.912270. [DOI] [PubMed] [Google Scholar]
- 4.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–3. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 5.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Frontiers in pharmacology. 2011;2:49. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, et al. Identification of small molecule inhibitors of pyruvate kinase M2. Biochemical pharmacology. 2010;79:1118–24. doi: 10.1016/j.bcp.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. The Journal of experimental medicine. 2012;209:217–24. doi: 10.1084/jem.20111487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo W, Zhang Y, Chen T, Wang Y, Xue J, Zhang Y, et al. Efficacy of RNAi targeting of pyruvate kinase M2 combined with cisplatin in a lung cancer model. J Cancer Res Clin Oncol. 2011;137:65–72. doi: 10.1007/s00432-010-0860-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi HS, Li D, Zhang J, Wang YS, Yang L, Zhang HL, Wang, et al. Silencing of PKM-2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci. 2010;101:1447–53. doi: 10.1111/j.1349-7006.2010.01562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–80. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Yadav S, van Vlerken LE, Little SR, Amiji MM. Evaluations of combination MDR-1 gene silencing and paclitaxel administration in biodegradable polymeric nanoparticle formulations to overcome multidrug resistance in cancer cells. Cancer Chemother Pharmacol. 2009;63:711–22. doi: 10.1007/s00280-008-0790-y. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi E, Iyer AK, Hornicek FJ, Amiji MM, Duan Z. Lipid-functionalized dextran nanosystems to overcome multidrug resistance in cancer: a pilot study. Clinical Orthopaedics and Related Research®. 2013;471:915–25. doi: 10.1007/s11999-012-2610-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang X, Iyer AK, Singh A, Milane L, Choy E, Hornicek FJ, et al. Cluster of Differentiation 44 Targeted Hyaluronic Acid Based Nanoparticles for MDR1 siRNA Delivery to Overcome Drug Resistance in Ovarian Cancer. Pharm Res. 2014:1–13. doi: 10.1007/s11095-014-1602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, Singh A, Choy E, Hornicek FJ, Amiji MM, Duan Z. MDR1 siRNA loaded hyaluronic acid-based CD44 targeted nanoparticle systems circumvent paclitaxel resistance in ovarian cancer. Scientific reports. 2015;5:1–9. doi: 10.1038/srep08509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Milane L, Duan Z, Amiji M. Development of EGFR-Targeted Polymer Blend Nanocarriers for Combination Paclitaxel/Lonidamine Delivery To Treat Multi-Drug Resistance in Human Breast and Ovarian Tumor Cells. Molecular Pharmaceutics. 2010;8:185–203. doi: 10.1021/mp1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganesh S, Iyer AK, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials. 2013;34:3489–502. doi: 10.1016/j.biomaterials.2013.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Z, Zhao R, Wu X, Sun Y, Yao M, Li J, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. The FASEB journal. 2005;19:1978–85. doi: 10.1096/fj.05-4058com. [DOI] [PubMed] [Google Scholar]
- 18.Xu J SA, Amiji M. Redox-responsive targeted gelatin nanoparticles for delivery of combination wt-p53 expressing plasmid DNA and gemcitabine in the treatment of pancreatic cancer. BMC Cancer. 2014;14:75. doi: 10.1186/1471-2407-14-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro G, Sawant RR, Biswas S, Essex S, Tros de Ilarduya C, Torchilin VP. P- glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine : nanotechnology, biology, and medicine. 2012;7:65–78. doi: 10.2217/nnm.11.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganesh S, Iyer AK, Weiler J, Morrissey DV, Amiji MM. Combination of siRNA- directed gene silencing with cisplatin reverses drug resistance in human non-small cell lung cancer. Molecular Therapy - Nucleic Acids. 2013:2. doi: 10.1038/mtna.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganesh S, Iyer AK, Weiler J, Morrissey DV, Amiji MM. Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials. 2013;34:3489–502. doi: 10.1016/j.biomaterials.2013.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganesh S, Iyer AK, Gattacceca F, Morrissey DV, Amiji MM. In vivo biodistribution of siRNA and cisplatin administered using CD44-targeted hyaluronic acid nanoparticles. Journal of Controlled Release. 2013;172:699–706. doi: 10.1016/j.jconrel.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamendola DE, Duan Z, Yusuf RZ, Seiden MV. Molecular description of evolving paclitaxel resistance in the SKOV-3 human ovarian carcinoma cell line. Cancer research. 2003;63:2200–5. [PubMed] [Google Scholar]
- 24.Okugawa K, Kobayashi H, Hirakawa T, Sonoda T, Ogura T, Nakano H. In vivo establishment and characterization of a paclitaxel-resistant human ovarian cancer cell line showing enhanced growth properties and drug-resistance only in vivo. J Cancer Res Clin Oncol. 2004;130:178–86. doi: 10.1007/s00432-003-0516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.