

Abstract
The hormone/cytokine prolactin (PRL) is implicated in breast cancer cell invasion and metastasis. PRL-induced pathways are mediated by two non-receptor tyrosine kinases, JAK2 and Src. We previously demonstrated that prolactin stimulates invasion of breast cancer cells TMX2-28 through JAK2 and its target serine/threonine kinase PAK1. We hypothesize herein that the actin-binding protein cortactin, a protein involved in invadopodia formation and cell invasion, is activated by PRL. We demonstrate that TMX2-28 cells are more invasive than T47D breast cancer cells in response to PRL. We determine that cortactin is tyrosyl phosphorylated in response to PRL in a time and dose-dependent manner in TMX2-28 cells, but not in T47D cells. Furthermore, we show that PRL mediates cortactin tyrosyl phosphorylation via Src, but not JAK2. Finally, we demonstrate that maximal PRL-mediated TMX2-28 cell invasion requires both Src and JAK2 kinase activity, while T47D cell invasion is JAK2- but not Src-dependent. Thus PRL may induce cell invasion via two pathways: through a JAK2/PAK1 mediated pathway that we have previously demonstrated, and Src-dependent activation and tyrosyl phosphorylation of cortactin.
Keywords: cortactin, prolactin, Src kinase, JAK2 kinase, tyrosine kinase, cell invasion
Graphical Abstract
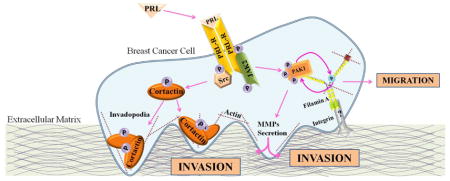
Introduction
Breast cancer is the most common malignancy among women. Prolactin (PRL), the hormone which regulates lactation and mammary gland development, is also implicated in promoting breast cancer cell proliferation, survival, motility and angiogenesis, and the prolactin receptor (PRL-R) is expressed in nearly 98% of human breast cancers (reviewed in [1]). Importantly, PRL mediates breast cancer cell invasion and metastasis. Administration of PRL in animal models increases metastasis, while loss of the PRL-R prevents the progression of neoplasia into invasive carcinoma [2,3]. Overexpression of degradation-resistant PRL-R in MCF10A cells increases cell proliferation and invasion, while PRL-R knockdown in T47D cells significantly reduces cell invasion and matrix-degrading matrix metalloproteinase (MMP)-9 expression [4]. Additionally, PRL, along with IGF-1, increases MMP-2 expression and cell invasion in a MAPK-dependent manner [5]. We have previously provided evidence of a novel PRL-mediated pathway during cell invasion. We demonstrated that PRL increases MMPs secretion and invasion of TMX2-28 cells, a highly invasive variant of the MCF-7 breast cancer cell line, in a JAK2/p21-activated kinase (PAK1)-dependent manner [6,7]. Cell migration is an important step in metastasis and PRL acts as a chemoattractant to breast cancer cells, promoting their motility [8,9]. Cell migration relies on actin cytoskeletal reorganization mediated by small Rho-GTPases Rac1 and CDC42. PRL activates Rac1 through Tec tyrosine kinase/Vav1 and serine/threonine kinase Nek3 [9,10,11,12,13]. Additionally, PRL activates CDC42 [11]. We have demonstrated two novel mechanisms that regulate PRL-mediated breast cancer cell motility; (1) through PAK1 and filamin A and (2) through regulation of adhesion turnover [14,15].
Prolactin-mediated pathways are regulated by non-receptor tyrosine kinases (nRTKs), as the PRL-R has no intrinsic kinase activity. In response to PRL, receptors dimerize and activate downstream nRTKs. One nRTK activated by PRL is JAK2, a protein that participates in cell cycle progression, apoptosis, genetic instability and histone modification (reviewed in [16]). PRL-R dimerization leads to activation and autophosphorylation of JAK2 at tyrosines 1007/1008 [17,18,19]. JAK2 activation stimulates several important signaling cascades, including signal transducers and activators of transcription, mitogen activated protein kinases (MAPKs), and phosphoinositol-3 kinase pathways (reviewed in [1]). PRL-mediated JAK2 activation is implicated in breast cancer progression, as eliminating JAK2 activity suppresses PRL-mediated tumorigenesis [20]. In addition to JAK2, PRL can also signal through another nRTK, the Src family kinases [21,22]. Src activation in response to PRL has also been implicated in breast cancer invasion. In the presence of a stiff collagen matrix, PRL signals through Src/FAK to promote secretion of MMP-2 and works synergistically with estrogen to promote breast cancer cell proliferation [23,24].
As PRL promotes breast cancer cell invasion and modulation of the actin cytoskeleton, we decided to study the actin-binding protein cortactin as a possible target of PRL signaling in breast cancer cells. Cortactin is localized to the cortical actin cytoskeleton where it mediates actin nucleation, endocytosis, and actin polymerization during cell motility and adhesion (reviewed in [25]). Notably, the cortactin gene CTTN lies on chromosome locus 11q13, which is often amplified in metastatic breast cancers, and cortactin overexpression has been implicated in tumor aggressiveness and poor prognoses in breast cancer([26], reviewed in [25]). Furthermore, tyrosyl phosphorylation of cortactin is required for its full activation, and increased levels of tyrosyl phosphorylated cortactin correspond with increased cell motility and invasion (reviewed in [25], [27,28,29,30]). Cortactin can be tyrosyl phosphorylated by several kinases, including the Src family kinases, Fer kinase and c-Met [31,32,33]. Interestingly, ablation of cortactin phosphorylation by stable expression of phospho-tyrosine-deficient mutant cortactin in highly invasive MDA-MB-231 breast cancer cells reduces metastasis in nude mice [34]. Furthermore, depletion of cortactin in breast cancer cells overexpressing active Src diminishes invadopodia formation [35].
In this study we provide evidence that cortactin is a novel target in PRL signaling and demonstrate that PRL promotes tyrosyl phosphorylation of cortactin in highly invasive TMX2-28 breast cancer cells, but not in less invasive T47D cells. Our data suggest that tyrosyl phosphorylation of cortactin in response to PRL is mediated through Src but not JAK2 tyrosine kinase. Finally, both JAK2 and Src activity are required for PRL-mediated TMX2-28 cell invasion, thus, introducing a novel PRL-dependent mechanism that regulates breast cancer cell invasion.
Material and Methods
Antibodies
Monoclonal αphospho-tyrosine (αPY; clone 4G10; EMD Millipore), monoclonal αJAK2 (Invitrogen), polyclonal αPY416 Src Family Kinases (Cell Signaling), and polyclonal αPY1007/1008 JAK2 (Invitrogen) were used for immunoblotting. Monoclonal αcortactin (clone 4F11; Millipore) and polyclonal αSrc (Cell Signaling) were used for immunoprecipitation and immunoblotting. JAK2 was immunoprecipitated using αJAK2 antiserum provided by Dr. Carter-Su (The University of Michigan). Prolactin was purchased from Dr. Parlow (National Hormone and Peptide Program, NIDDK).
Cell Cultures
T47D and TMX2-28 (sub-line of MCF-7 cells [6]) cells were maintained in RPMI 1640 medium or DMEM (Corning Inc), respectively, with all supplements. Deprivation media for T47D and TMX2-28 cells consisted of RPMI 1640 or DMEM, respectively, supplemented with 1% bovine serum albumin (Sigma-Aldrich).
Cell Invasion Assay
Equal numbers of T47D and TMX2-28 cells (5x105) were placed in deprivation media with or without inhibitors (0.1μM PP1, 50μM AG490) in the upper chamber of a Boyden chamber (Corning Inc.) coated with Matrigel (BD Biosciences). Deprivation media with or without 500ng/ml PRL was placed in the lower chamber. After 48 hours, cells from five separate fields that had invaded the Matrigel were counted after fixation with 4% formalin (Sigma) and staining with Differential Quik Stain (Polysciences, Inc).
Immunoprecipitation
Cells were deprived of serum for 72hr before treatment with or without PRL. Proteins were immunoprecipitated from the cell lysates using the indicated antibodies and protein A-agarose. Proteins were resolved by SDS-PAGE followed by immunoblotting. Fold phosphorylation of cortactin was determined by densitometric analysis of αPY bands normalized to αcortactin bands using Image J software.
Assessing Src and JAK2 Kinase Inhibition
To assess inhibition by PP1, endogenous Src and JAK2 were immunoprecipitated with αSrc or αJAK2 from cells deprived from serum for 72 hours and treated with PP1 (Calbiochem, 0.1μM, 2 hours) and PRL (200ng/ml, 20 minutes) and subjected to an in vitro kinase assay in the presence of 10μCi of [γ-32P] ATP (MP Biomedicals). Relative levels of incorporated 32P into Src and JAK2 were assessed by autoradiography and estimated by a phosphoimager. The same membrane was blotted with αSrc and αJAK2 antibodies.
To assess inhibition by AG490, deprived cells were treated with 0, 25, 50, 100, and 125μM AG490 (Calbiochem) overnight. Before harvesting, cells were treated with PRL (200ng/mL) for 20 minutes. Proteins were resolved using SDS-PAGE and immunoblotted using αpY1007/1008 JAK2 antibody to determine JAK2 autophosphorylation and αPY416 Src Family Kinase antibody to determine Src autophosphorylation. The same membrane was probed with αJAK2 and αSrc antibodies.
Statistical Analysis
Data from at least 3 separate experiments were pooled and analyzed using 1-way ANOVA plus Tukey’s honest significant difference test. Differences were considered to be statistically significant at P < 0.05. Results are expressed as the mean ± SE.
Results and Discussion
TMX2-28 cells are more invasive than T47D cells
We have previously demonstrated that PRL stimulates the invasion of TMX2-28 cells via a JAK2/PAK1 pathway [7]. In an attempt to identify additional mechanisms that regulate PRL-dependent cell invasion, we decided to compare the invasiveness of TMX2-28 and the poorly invasive T47D breast cancer cells. 100ng/ml of PRL did not stimulate invasion in neither T47D nor TMX2-28 cells after 48 hours (data not shown). However, treatment of both cell lines with a higher concentration of PRL (500 ng/ml) for 48h led to greater invasion of TMX2-28 cells than T47D cells through Matrigel (Fig. 1, black bars). Basal invasion in serum-free medium without treatment was also attenuated in T47D cells as compared to TMX2-28 cells (Fig. 1, white bars). Thus, PRL stimulates invasion in both T47D and TMX2-28 cells and to a greater extent in TMX2-28 cells.
Figure 1. TMX2-28 cells are more invasive than T47D cells.
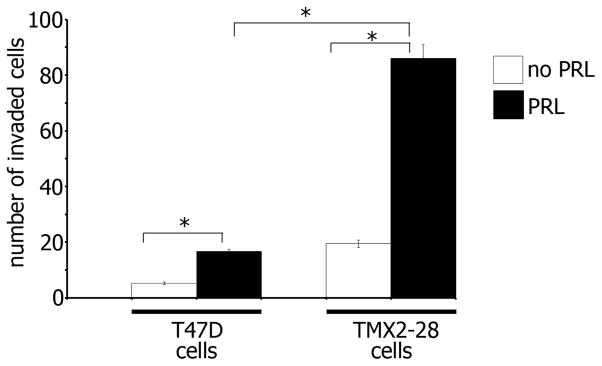
T47D and TMX2-28 cells were serum deprived and equal amounts of cells were loaded into the upper part of the Boyden chamber coated with Matrigel. The number of cells that migrated to the lower part of the chamber towards PRL (500ng/ml) (black bar) or buffer control (white bar) after 48 hours were counted in 5 randomly chosen fields and plotted. Bars represent mean ± SE. *P<0.05 for three independent experiments.
Prolactin stimulates tyrosyl phosphorylation of cortactin in TMX2-28 but not T47D cells
To define a mechanism that regulates cell invasion differently in TMX2-28 and T47D cells, we focused on cortactin since it plays a significant role in invasion [35,36,37]. Since tyrosyl phosphorylation of cortactin is important for cortactin activation [25], we tested whether PRL causes tyrosyl phosphorylation of cortactin. We treated T47D cells with PRL over a time-course and analyzed the immunoprecipitated endogenous cortactin for tyrosyl phosphorylation. Tyrosyl phosphorylation of endogenous cortactin over basal levels in response to PRL was not observed in T47D cells (Fig. 2A). On the contrary, when TMX2-28 cells were treated with PRL over the same time course, maximal tyrosyl phosphorylation of cortactin appeared at 20 minutes of PRL treatment and was transient (Fig. 2B). Furthermore, we treated TMX2-28 cells with increasing concentrations of PRL and showed that a minimum of 200ng/ml of PRL was required for cortactin tyrosyl phosphorylation (Fig. 2C). Increasing PRL concentration above 200ng/ml did not further increase cortactin phosphorylation. Tyrosyl phosphorylation of cortactin upon PRL stimulation observed in TMX2-28 cells which was lacking in T47D cells may explain why TMX2-28 cells are more invasive than T47D cells. Bowden et. al demonstrated that cortactin colocalizes with phospho-tyrosine in complexes termed “invadopodia complexes” [38]. Increasing the amount of phospho-tyrosine at these cortactin-rich invadopodia increased proteolytic activity in these areas, suggesting that increased tyrosyl phosphorylation of cortactin in invadopodia contributes to cell invasion. Importantly, PRL does not stimulate tyrosyl phosphorylation of cortactin in T47D in our study. T47D cells are not known to form invadopodia and basal level T47D invasion is potentiated only after cortactin overexpression [35,39]. It is also important to note that the lack of cortactin phosphorylation in T47D was not due to low levels of expressed endogenous cortactin protein, as the amount of immunoprecipitated cortactin in T47D cells was comparable to TMX2-28 cells (Fig. 2A–C, αcortactin blots). To determine the mechanism in which PRL induces cortactin phosphorylation, we focused on the two major nRTKs activated in response to PRL.
Figure 2. Cortactin is tyrosyl phosphorylated in TMX2-28 cells, but not T47D cells, in response to PRL.

A–B) Endogenous cortactin was immunoprecipitated from T47D (A) or TMX2-28 (B) cell lysates after PRL (150ng/ml) treatment for the indicated time points and probed for tyrosyl phosphorylation with αphospho-tyrosine antibody (αpY). C) Endogenous cortactin was immunoprecipitated from TMX2-28 cell lysates after 20 minutes of PRL treatment at the indicated concentrations and probed for tyrosyl phosphorylation with αpY. All graphs (A–C) represent the densitometric analysis of the band obtained for phosphorylated cortactin normalized to total cortactin for each lane of at least 3 independent experiments. Bars represent mean ± SE. *P<0.05 compared with the same cells untreated with PRL.
Src kinase, but not JAK2 kinase, tyrosyl phosphorylates cortactin in response to PRL
PRL signaling is mediated by two main non-receptor tyrosine kinases; Src and JAK2. In order to determine which pathway mediates cortactin tyrosyl phosphorylation, we utilized two selective inhibitors, PP1 [40] and AG490 [41], to inhibit either Src or JAK2 respectively. To assess the effectiveness of PP1 on Src kinase inhibition, TMX2-28 cells were treated with PP1 before treatment with PRL. Endogenous Src was immunoprecipitated and the kinase activity of Src was analyzed in an in vitro kinase assay. Src kinase activity was reduced in cells pre-treated with PP1 when compared to those without inhibitor (Fig. 3A). To confirm that PP1 treatment would not also reduce the kinase activity of JAK2, TMX2-28 cells were pre-treated with PP1, and JAK2 was immunoprecipitated after PRL treatment. JAK2 kinase activity in response to PRL was unaffected by PP1, as demonstrated by a JAK2 in vitro kinase assay (Fig. 3A). This is consistent with previously published data indicating that PP1 does not inhibit JAK2-dependent PRL-R phosphorylation and activation of the JAK2/STAT5 pathway in W53 cells and JAK2 autophosphorylation is unaffected by PP1 in T47D cells [42,43]. Next, we assessed the inhibition of JAK2 kinase activity by AG490. TMX2-28 cells were pre-treated with increasing concentrations of AG490 followed by treatment with PRL. Cell lysates were blotted using a phospho-JAK2-specific antibody that recognizes pY1007/1008, the sites of JAK2 autophosphorylation. We showed that AG490 inhibited JAK2 autophosphorylation in a dose dependent manner and a minimum of 50 μM of AG490 was needed to efficiently inhibit JAK2 autophosphorylation (Fig. 3B). To test the effect of AG490 on Src kinase activity, we probed the same membranes with αPY416-Src antibody and demonstrated that Src kinase activity in response to PRL was not reduced with AG490 treatment (Fig.3B). These data are in agreement with previously published data demonstrating that PRL stimulates Src activation in mammary epithelial cells isolated from mid-pregnant JAK2-null mice and suggests that JAK2 activity is not necessary for PRL-mediated Src activation [44].
Figure 3. Src kinase, but not JAK2 kinase, tyrosyl phosphorylates cortactin in response to PRL.

A) PP1 inhibits Src, but not JAK2. TMX2-28 cells were treated with PP1 (0.1μM, 2hrs) before treatment with PRL (200ng/ml, 20min). Endogenous Src or JAK2 was immunoprecipitated and subjected to an in vitro kinase (IVK) assay and probed with αSrc or αJAK2. B) AG490 inhibits JAK2, but not Src. TMX2-28 cells were treated with indicated concentrations of AG490 overnight then with PRL (200ng/ml, 20min). Whole cell lysates were immunoblotted with the indicated antibodies. αpY-JAK2 antibody recognizes pY1007/1008 on JAK2 and αpY-Src antibody recognizes pY416 on Src family kinases. C) Src tyrosyl phosphorylates cortactin in response to PRL. TMX2-28 cells were treated with indicated inhibitors before treatment with PRL. Endogenous cortactin was immunoprecipitated from TMX2-28 cells and probed for tyrosyl phosphorylation with αpY. Each figure represents the same blot reblotted with indicated antibodies. All blots are representative of at least 3 experiments.
To determine which kinase is responsible for PRL-mediated cortactin tyrosyl phosphorylation, TMX2-28 cells were pre-treated with PP1, AG490, or both inhibitors in combination before treatment with PRL. The cortactin immunoprecipitates were assessed for tyrosyl phosphorylation. PRL induced cortactin phosphorylation (Fig. 3C, lanes 1 and 2), however, pre-treatment with the Src inhibitor PP1 completely abolished tyrosyl phosphorylation of cortactin (Fig. 3C, lane 3). Conversely, treatment with the JAK2 inhibitor AG490 had no effect on cortactin tyrosyl phosphorylation (Fig. 3C, lane 4). The addition of AG490 to PP1 treatment had no significant impact on PP1’s ability to inhibit PRL-mediated cortactin tyrosyl phosphorylation (Fig. 3C, lane 5). These data suggest that PRL induces tyrosyl phosphorylation of cortactin through Src, and not JAK2. In support, it has been previously demonstrated that reducing Src kinase activity by expressing kinase-inactive Src into MDA-MB-231 cells reduced phospho-tyrosine signal in cortactin-rich invadopodia [38]. Additionally, cortactin was discovered as a target of oncogenic v-Src in transformed chicken fibroblasts and expression of active forms of pp60-Src leads to tyrosyl phosphorylation of cortactin [31].
Both JAK2 and Src kinase activities are required for maximal PRL-mediated TMX2-28 cell invasion
To study whether both JAK2- and Src-dependent pathways regulate PRL-induced cell invasion, we assessed invasion of TMX2-28 and T47D cells in the presence of JAK2 and Src inhibitors, AG490 and PP1 respectively. Treatment with one of inhibitors without PRL did not reduce basal cell invasion of either T47D or TMX2-28 cells (Fig. 4, white bars). Src inhibition significantly attenuated PRL-induced invasion of TMX2-28 but not T47D cells while JAK2 inhibition significantly reduced PRL-mediated invasion of both TMX2-28 and T47D cells (Fig. 4, black bars). The combination of PP1 and AG490 further reduced both PRL-mediated (black bar) and basal (white bar) invasion of TMX2-28, but not T47D cells, suggesting that both JAK2 and Src kinases are required for TMX2-28 cell invasion. It is important to note that Src inhibition did not significantly reduce T47D cell invasion and the combination of both Src and JAK2 inhibitors did not further attenuate either basal or PRL-induced T47D cell invasion. This data suggest that the more invasive TMX2-28 cells utilize two PRL-dependent pathways, JAK2- and Src-dependent, to promote cell invasion while in less invasive T47D cells only JAK2 pathway is implicated in invasion in response to PRL. It may explain why TMX2-28 cells are more invasive than T47D cells.
Figure 4. Both JAK2 and Src kinase activity are required for maximal PRL-mediated TMX2-28 cell invasion.

T47D and TMX2-28 cells were serum deprived and equal amounts of cells were loaded into the upper part of the Boyden chamber coated with Matrigel with or without PP1 (0.1μM), AG490 (50μM), or both inhibitors together. The number of cells that migrated to the lower part of the chamber towards PRL (500ng/ml) (black bar) or buffer control (white bar) after 48 hours were counted in 5 randomly chosen fields and plotted. Bars represent mean ± SE. *P<0.05 for three independent experiments.
Here we propose a novel PRL-mediated pathway during breast cancer cell invasion. PRL promotes cell invasion to a greater extent in TMX2-28 cells when compared to less invasive T47D cells. This increase in cell invasion in TMX2-28 cells may depend on the tyrosyl phosphorylation of cortactin which does not occur in T47D cells upon PRL stimulation. Through this study, we provide evidence that PRL-mediated tyrosyl phosphorylation of cortactin is through the nRTK Src, and not through the nRTK JAK2. Inhibition of either JAK2 or Src reduces PRL-mediated TMX2-28 cell invasion, while only inhibition of JAK2 reduces T47D cell invasion. Thus, PRL can stimulate breast cancer cell invasion via two different pathways: (1) through JAK2/PAK1 and secretion of MMPs which we have previously demonstrated and (2) through Src/cortactin, a novel PRL-mediated pathway.
Highlights.
Prolactin induces cortactin tyrosyl phosphorylation in invasive breast cancer cells
Src but not JAK2 phosphorylates cortactin in response to prolactin
As shown before prolactin induces cell motility/invasion via PAK1/FilaminA and MMPs
We propose here a novel prolactin-mediated mechanism for cell invasion via Src/cortactin
Acknowledgments
The authors are grateful to Dr. Carter-Su (The University of Michigan, Ann Arbor, MI, USA) for providing αJAK2 serum. This work was supported by a grant from the National Institutes of Health (R01DK88127 to MD).
Abbreviations
- PRL
prolactin
- JAK2
Janus kinase 2
- PRL-R
prolactin receptor
- nRTKs
non-receptor tyrosine kinases
- MMP
matrix metalloproteinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Alan Hammer, Email: alan.hammer@utoledo.edu.
Sneha Laghate, Email: sdlaghate@gmail.com.
Maria Diakonova, Email: mdiakon@utnet.utoledo.edu.
References
- 1.Clevenger CV, Furth PA, Hankinson SE, Schuler LA. The role of prolactin in mammary carcinoma. Endocr Rev. 2003;24:1–27. doi: 10.1210/er.2001-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liby K, Neltner B, Mohamet L, Menchen L, Ben-Jonathan N. Prolactin overexpression by MDA-MB-435 human breast cancer cells accelerates tumor growth. Breast Cancer Res Treat. 2003;79:241–252. doi: 10.1023/a:1023956223037. [DOI] [PubMed] [Google Scholar]
- 3.Oakes SR, Robertson FG, Kench JG, Gardiner-Garden M, Wand MP, Green JE, Ormandy CJ. Loss of mammary epithelial prolactin receptor delays tumor formation by reducing cell proliferation in low-grade preinvasive lesions. Oncogene. 2007;26:543–553. doi: 10.1038/sj.onc.1209838. [DOI] [PubMed] [Google Scholar]
- 4.Plotnikov A, Varghese B, Tran TH, Liu C, Rui H, Fuchs SY. Impaired turnover of prolactin receptor contributes to transformation of human breast cells. Cancer Res. 2009;69:3165–3172. doi: 10.1158/0008-5472.CAN-08-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carver KC, Schuler LA. Prolactin does not require insulin-like growth factor intermediates but synergizes with insulin-like growth factor I in human breast cancer cells. Mol Cancer Res. 2008;6:634–643. doi: 10.1158/1541-7786.MCR-07-2069. [DOI] [PubMed] [Google Scholar]
- 6.Fasco MJ, Amin A, Pentecost BT, Yang Y, Gierthy JF. Phenotypic changes in MCF-7 cells during prolonged exposure to tamoxifen. Mol Cell Endocrinol. 2003;206:33–47. doi: 10.1016/s0303-7207(03)00256-9. [DOI] [PubMed] [Google Scholar]
- 7.Rider L, Oladimeji P, Diakonova M. PAK1 regulates breast cancer cell invasion through secretion of matrix metalloproteinases in response to prolactin and three-dimensional collagen IV. Mol Endocrinol. 2013;27:1048–1064. doi: 10.1210/me.2012-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maus MV, Reilly SC, Clevenger CV. Prolactin as a chemoattractant for human breast carcinoma. Endocrinology. 1999;140:5447–5450. doi: 10.1210/endo.140.11.7245. [DOI] [PubMed] [Google Scholar]
- 9.Miller SL, Antico G, Raghunath PN, Tomaszewski JE, Clevenger CV. Nek3 kinase regulates prolactin-mediated cytoskeletal reorganization and motility of breast cancer cells. Oncogene. 2007;26:4668–4678. doi: 10.1038/sj.onc.1210264. [DOI] [PubMed] [Google Scholar]
- 10.Kline JB, Moore DJ, Clevenger CV. Activation and association of the Tec tyrosine kinase with the human prolactin receptor: mapping of a Tec/Vav1-receptor binding site. Mol Endocrinol. 2001;15:832–841. doi: 10.1210/mend.15.5.0631. [DOI] [PubMed] [Google Scholar]
- 11.Akhtar N, Streuli CH. Rac1 links integrin-mediated adhesion to the control of lactational differentiation in mammary epithelia. J Cell Biol. 2006;173:781–793. doi: 10.1083/jcb.200601059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aksamitiene E, Achanta S, Kolch W, Kholodenko BN, Hoek JB, Kiyatkin A. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cell Signal. 2011;23:1794–1805. doi: 10.1016/j.cellsig.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller SL, DeMaria JE, Freier DO, Riegel AM, Clevenger CV. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol Endocrinol. 2005;19:939–949. doi: 10.1210/me.2004-0443. [DOI] [PubMed] [Google Scholar]
- 14.Hammer A, Oladimeji P, Casas LE, Diakonova M. Phosphorylation of tyrosine 285 of PAK1 facilitates betaPIX/GIT1 binding and adhesion turnover. FASEB J. 2014 doi: 10.1096/fj.14-259366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammer A, Rider L, Oladimeji P, Cook L, Li Q, Mattingly RR, Diakonova M. Tyrosyl phosphorylated PAK1 regulates breast cancer cell motility in response to prolactin through filamin A. Mol Endocrinol. 2013;27:455–465. doi: 10.1210/me.2012-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 17.Rui H, Kirken RA, Farrar WL. Activation of receptor-associated tyrosine kinase JAK2 by prolactin. J Biol Chem. 1994;269:5364–5368. [PubMed] [Google Scholar]
- 18.Rui H, Lebrun JJ, Kirken RA, Kelly PA, Farrar WL. JAK2 activation and cell proliferation induced by antibody-mediated prolactin receptor dimerization. Endocrinology. 1994;135:1299–1306. doi: 10.1210/endo.135.4.7925093. [DOI] [PubMed] [Google Scholar]
- 19.Feng J, Witthuhn BA, Matsuda T, Kohlhuber F, Kerr IM, Ihle JN. Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol. 1997;17:2497–2501. doi: 10.1128/mcb.17.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakamoto K, Triplett AA, Schuler LA, Wagner KU. Janus kinase 2 is required for the initiation but not maintenance of prolactin-induced mammary cancer. Oncogene. 2010;29:5359–5369. doi: 10.1038/onc.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clevenger CV. Role of prolactin/prolactin receptor signaling in human breast cancer. Breast Dis. 2003;18:75–86. doi: 10.3233/bd-2003-18108. [DOI] [PubMed] [Google Scholar]
- 22.Berlanga JJ, Fresno Vara JA, Martin-Perez J, Garcia-Ruiz JP. Prolactin receptor is associated with c-src kinase in rat liver. Mol Endocrinol. 1995;9:1461–1467. doi: 10.1210/mend.9.11.8584023. [DOI] [PubMed] [Google Scholar]
- 23.Barcus CE, Keely PJ, Eliceiri KW, Schuler LA. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. J Biol Chem. 2013;288:12722–12732. doi: 10.1074/jbc.M112.447631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barcus CE, Holt EC, Keely PJ, Eliceiri KW, Schuler LA. Dense Collagen-I Matrices Enhance Pro-Tumorigenic Estrogen-Prolactin Crosstalk in MCF-7 and T47D Breast Cancer Cells. PLoS One. 2015;10:e0116891. doi: 10.1371/journal.pone.0116891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weaver AM. Cortactin in tumor invasiveness. Cancer Lett. 2008;265:157–166. doi: 10.1016/j.canlet.2008.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bekri S, Adelaide J, Merscher S, Grosgeorge J, Caroli-Bosc F, Perucca-Lostanlen D, Kelley PM, Pebusque MJ, Theillet C, Birnbaum D, Gaudray P. Detailed map of a region commonly amplified at 11q13-->q14 in human breast carcinoma. Cytogenet Cell Genet. 1997;79:125–131. doi: 10.1159/000134699. [DOI] [PubMed] [Google Scholar]
- 27.Huang C, Liu J, Haudenschild CC, Zhan X. The role of tyrosine phosphorylation of cortactin in the locomotion of endothelial cells. J Biol Chem. 1998;273:25770–25776. doi: 10.1074/jbc.273.40.25770. [DOI] [PubMed] [Google Scholar]
- 28.Huang J, Asawa T, Takato T, Sakai R. Cooperative roles of Fyn and cortactin in cell migration of metastatic murine melanoma. J Biol Chem. 2003;278:48367–48376. doi: 10.1074/jbc.M308213200. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Huang C, Zhan X. Src is required for cell migration and shape changes induced by fibroblast growth factor 1. Oncogene. 1999;18:6700–6706. doi: 10.1038/sj.onc.1203050. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Tondravi M, Liu J, Smith E, Haudenschild CC, Kaczmarek M, Zhan X. Cortactin potentiates bone metastasis of breast cancer cells. Cancer Res. 2001;61:6906–6911. [PubMed] [Google Scholar]
- 31.Schuuring E, Verhoeven E, Litvinov S, Michalides RJ. The product of the EMS1 gene, amplified and overexpressed in human carcinomas, is homologous to a v-src substrate and is located in cell-substratum contact sites. Mol Cell Biol. 1993;13:2891–2898. doi: 10.1128/mcb.13.5.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapus A, Di Ciano C, Sun J, Zhan X, Kim L, Wong TW, Rotstein OD. Cell volume-dependent phosphorylation of proteins of the cortical cytoskeleton and cell-cell contact sites. The role of Fyn and FER kinases. J Biol Chem. 2000;275:32289–32298. doi: 10.1074/jbc.M003172200. [DOI] [PubMed] [Google Scholar]
- 33.Crostella L, Lidder S, Williams R, Skouteris GG. Hepatocyte Growth Factor/scatter factor-induces phosphorylation of cortactin in A431 cells in a Src kinase-independent manner. Oncogene. 2001;20:3735–3745. doi: 10.1038/sj.onc.1204474. [DOI] [PubMed] [Google Scholar]
- 34.Webb BA, Jia L, Eves R, Mak AS. Dissecting the functional domain requirements of cortactin in invadopodia formation. Eur J Cell Biol. 2007;86:189–206. doi: 10.1016/j.ejcb.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Hill A, McFarlane S, Mulligan K, Gillespie H, Draffin JE, Trimble A, Ouhtit A, Johnston PG, Harkin DP, McCormick D, Waugh DJ. Cortactin underpins CD44-promoted invasion and adhesion of breast cancer cells to bone marrow endothelial cells. Oncogene. 2006;25:6079–6091. doi: 10.1038/sj.onc.1209628. [DOI] [PubMed] [Google Scholar]
- 36.Rothschild BL, Shim AH, Ammer AG, Kelley LC, Irby KB, Head JA, Chen L, Varella-Garcia M, Sacks PG, Frederick B, Raben D, Weed SA. Cortactin overexpression regulates actin-related protein 2/3 complex activity, motility, and invasion in carcinomas with chromosome 11q13 amplification. Cancer Res. 2006;66:8017–8025. doi: 10.1158/0008-5472.CAN-05-4490. [DOI] [PubMed] [Google Scholar]
- 37.van Rossum AG, Moolenaar WH, Schuuring E. Cortactin affects cell migration by regulating intercellular adhesion and cell spreading. Exp Cell Res. 2006;312:1658–1670. doi: 10.1016/j.yexcr.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 38.Bowden ET, Onikoyi E, Slack R, Myoui A, Yoneda T, Yamada KM, Mueller SC. Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells. Exp Cell Res. 2006;312:1240–1253. doi: 10.1016/j.yexcr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z, Nakamura Y, Fukami K. Lipid rafts and caveolin-1 are required for invadopodia formation and extracellular matrix degradation by human breast cancer cells. Cancer Res. 2009;69:8594–8602. doi: 10.1158/0008-5472.CAN-09-2305. [DOI] [PubMed] [Google Scholar]
- 40.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 41.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 42.Fresno Vara JA, Caceres MA, Silva A, Martin-Perez J. Src family kinases are required for prolactin induction of cell proliferation. Mol Biol Cell. 2001;12:2171–2183. doi: 10.1091/mbc.12.7.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Acosta JJ, Munoz RM, Gonzalez L, Subtil-Rodriguez A, Dominguez-Caceres MA, Garcia-Martinez JM, Calcabrini A, Lazaro-Trueba I, Martin-Perez J. Src mediates prolactin-dependent proliferation of T47D and MCF7 cells via the activation of focal adhesion kinase/Erk1/2 and phosphatidylinositol 3-kinase pathways. Mol Endocrinol. 2003;17:2268–2282. doi: 10.1210/me.2002-0422. [DOI] [PubMed] [Google Scholar]
- 44.Sakamoto K, Creamer BA, Triplett AA, Wagner KU. The Janus kinase 2 is required for expression and nuclear accumulation of cyclin D1 in proliferating mammary epithelial cells. Mol Endocrinol. 2007;21:1877–1892. doi: 10.1210/me.2006-0316. [DOI] [PubMed] [Google Scholar]