

Background: Leber congenital amaurosis 9 (LCA9) is a severe retinal degeneration condition caused by mutations in the NAD+ biosynthetic enzyme NMNAT1.
Results: Many of LCA9-associated NMNAT1 mutants have relatively normal enzymatic and neuroprotective activities, but manifest stress-induced instability.
Conclusion: Stress-induced instability of NMNAT1 could be responsible for retinal abnormalities in LCA9.
Significance: Our results identify potential mechanisms that promote retinal degeneration observed in LCA9.
Keywords: metabolic disease, neuroprotection, nicotinamide adenine dinucleotide (NAD), retinal degeneration, structural biology, Leber congenital amaurosis, NMNAT
Abstract
Leber congenital amaurosis 9 (LCA9) is an autosomal recessive retinal degeneration condition caused by mutations in the NAD+ biosynthetic enzyme NMNAT1. This condition leads to early blindness but no other consistent deficits have been reported in patients with NMNAT1 mutations despite its central role in metabolism and ubiquitous expression. To study how these mutations affect NMNAT1 function and ultimately lead to the retinal degeneration phenotype, we performed detailed analysis of LCA-associated NMNAT1 mutants, including the expression, nuclear localization, enzymatic activity, secondary structure, oligomerization, and promotion of axonal and cellular integrity in response to injury. In many assays, most mutants produced results similar to wild type NMNAT1. Indeed, NAD+ synthetic activity is unlikely to be a primary mechanism underlying retinal degeneration as most LCA-associated NMNAT1 mutants had normal enzymatic activity. In contrast, the secondary structure of many NMNAT1 mutants was relatively less stable as they lost enzymatic activity after heat shock, whereas wild type NMNAT1 retains significant activity after this stress. These results suggest that LCA-associated NMNAT1 mutants are more vulnerable to stressful conditions that lead to protein unfolding, a potential contributor to the retinal degeneration observed in this syndrome.
Introduction
Leber congenital amaurosis (LCA)2 is a severe retinal degenerative condition that results in blindness at birth or within the first few years of life. Causative mutations in at least 17 different genes have been identified in LCA. These genes are those with largely retina-specific expression (e.g. RPE65, AIPL1, LCA5, CRX, CEP290, and IMPDH) as well as those that are expressed broadly in many tissues (e.g. GUCY2D, NMNAT1, RDH12, RPGRIP1, and CRB1) (1). A 10-centimorgan region on 1p36 was identified as LCA9 locus in a Pakistani family (2) and further analysis identified NMNAT1 mutations in families with LCA, most of which had associated macular atrophy (3–6). Almost all of these patients harbored compound heterozygote mutations, with the most common being a missense E257K mutation (3–6). A new study reports that individuals with a homozygous E257K mutation in one family did not have ocular abnormalities, suggesting this is a hypomorphic mutation with incomplete penetrance (7).
The retinal dystrophy in LCA is characterized by photoreceptor cell dysfunction and death with almost undetectable electroretinogram responses in most cases by the end of the first year of life (1). Many genes whose mutation results in photoreceptor degeneration in LCA or retinitis pigmentosa are ubiquitously expressed, yet the pathology is localized to the retina. The susceptibility of retinal photoreceptor cells to these mutations could be associated with their unique physiology and environment. For example, the visual cycle, which regenerates the light-sensitive 11-cis-retinal from the light-stimulated all-trans-retinal form, produces oxidative stress and requires continual supplies of NAD+ for the necessary dehydrogenase reactions (9). The accumulation of all-trans-retinal due to dysfunction of retinal pigment epithelium cells, which engulf photoreceptor cells to maintain homeostasis, also results in photoreceptor abnormalities (10). In addition, the high levels of unsaturated lipids like docosahexaenoic acid in photoreceptor outer segments make them highly vulnerable to oxidative stress (11).
NMNAT1 is an enzyme that synthesizes NAD+ from NMN and ATP. NMNAT1 is localized to the nucleus, whereas related enzymes NMNAT2 and NMNAT3 are located in the Golgi/cytoplasm and mitochondria, respectively. NMNAT1 mutations in LCA are located throughout the entire molecule; no single domain or motif appears to be targeted. The mutations are thought to reduce NMNAT enzymatic activity based on analysis of patient cells in two cases and limited analysis of other mutants in vitro (5, 6). NMNAT1 appears to be an essential gene as Nmnat1-deficient mice die during early embryogenesis, whereas Nmnat1 heterozygotes are reported to have no overt deficits (8). Thus, it would appear unlikely that the LCA-associated NMNAT1 mutations are total loss-of-function, because the patients have no other overt phenotypes. Much recent excitement has been generated by the discovery that overexpression of NMNAT1 (or other Nmnat enzymes) prevents degeneration of injured axons (12, 13). Moreover, NMNAT enzymatic activity is essential and sufficient for axonal protection (14). In contrast, studies in Drosophila show that dNMNAT chaperone activity, but not enzymatic activity, is required to prevent degeneration of photoreceptors (15) or axons of mushroom body neurons after JNK inactivation (16). In this study, we used multiple assays to characterize a series of NMNAT1 mutants associated with LCA to gain insights into how these mutations lead to retinal degeneration.
Experimental Procedures
Plasmids
NMNAT1 mutants were constructed with either N-terminal HA or FLAG epitope tag by gene synthesis and inserted into the BamHI site of pUC57 (GeneScript). The clones constructed included: FLAG-wild type, FLAG-V9M, FLAG-A13T, FLAG-D33G, FLAG-R66W, FLAG-I217N, FLAG-E257K, FLAG-STP280Gln, HA-wt, HA-M69V, HA-V98G, HA-V151F, HA-D173G, HA-R207W, HA-R237C, and HA-L239S. The fragments were transferred into the BamHI site of the lentivirus vector FCIV that includes a downstream IRES Venus for monitoring expression (12). The E257Q and E257D NMNAT1 mutants were generated using mega primer PCR and cloned into the BamHI site of FCIV by InFusion (Clontech). For bacterial expression of wild type and mutant NMNAT1 proteins, the respective cDNAs were subcloned into the BamHI site of pET30a.
Cell Culture
Mouse dorsal root ganglion (DRG) were dissected from E13.5 CD1 embryo and cultured in 96-well plates coated with poly-d-lysine (0.1 mg/ml; Sigma) and laminin (3 μg/ml; Invitrogen) as previously described (14). After cell attachment, 100 μl of complete medium (Neurobasal E (Gibco) containing B27 (2%; Invitrogen), nerve growth factor (100 ng/ml; 2.5S Harlan Bioproducts), uridine (1 μm; Sigma), 5-fluoro-2′-deoxyuridine (1 μm; Sigma), and penicillin/streptomycin) was added. Lentiviruses were added (1–10 × 103 pfu) after 3 days in vitro (DIV) where indicated.
Lentiviruses were produced in HEK293T cells as previously described (12). Cells were seeded at a density of 1 × 106 cells/35-mm well the day before transfection. FCIV lentivirus constructs harboring NMNAT1 cDNAs (400 ng) were cotransfected with vesicular stomatitis virus-G (400 ng) and pSPAX2 (1.2 μg; Dr. Didier Trono, EPFL) using X-tremeGene (Roche Applied Science). The virus-laden culture medium was collected 2 days after transfection. The lentivirus particles were purified and concentrated from the cleared culture supernatant with Lenti-X concentrator (Clontech).
In Silico Protein Oligomerization Analysis
We mapped the LCA-associated NMNAT1 mutations onto the experimentally determined structure for NMNAT1 (PDB code 1KQN) (17). We visually classified mutations as candidates for affecting oligomerization based on the participation of the involved residues in contacts at the monomer-monomer interface. The mutations were modeled in silico using the RosettaDesign program (18) to predict conformational changes and alterations in protein stability. This analysis predicted that NMNAT1 R66W, V151G, M69V, and V98G mutants would form unstable oligomers.
NMNAT Enzymatic Assay
HEK293T cells were transfected with NMNAT1 FCIV expression vector that includes an IRES-Venus cassette to co-express Venus fluorescent protein. After 4 days, the cells were lysed by sonication in PBS containing protease inhibitor mixture (Roche) and centrifuged to obtain a clear supernatant. NMNAT enzymatic activity in the cell supernatant was measured by quantifying NAD+ by fluorescence. The fluorescence generated by conversion of non-fluorescent resazurin to resorufin was monitored using a coupled reaction with diaphorase and NADH that is converted from NAD+ by alcohol dehydrogenase.
NMNAT enzymatic assays were performed in buffer containing HEPES (pH 7.4, 10 mm), MgCl2 (4 mm), EtOH (1 mm), resazurin (0.04 mm), alcohol dehydrogenase (0.5 units), diaphorase (0.02 units), ATP (100 μm), and NMN (100 μm) (all chemicals and enzymes were obtained from Sigma). Resorufin fluorescence (excitation = 544 nm, emission = 590 nm) is proportional to the NAD+ concentration and was monitored at 37 °C from 0 to 20 min using a POLARStar Optima plate reader. NAD+ concentrations were calculated from standard curves obtained from reactions containing 0.025 to 20 μm NAD+. To quantify the amount of NMNAT1 in each reaction, the fluorescence intensity (excitation = 485 nm, emission = 520 nm) from the co-expressed Venus was measured. We then used Western blotting to establish a NMNAT1/Venus ratio for each mutant (Fig. 1e) so that we could use fluorescent intensity to calculate NMNAT1 levels in all assays. Finally, the NAD+ concentration in each reaction was normalized by the relative NMNAT1 amount determined by the NMNAT1/Venus ratio. Data were averaged from 3 independent wells for each mutant and wild type NMNAT1 protein.
FIGURE 1.

NMNAT enzymatic activity of LCA-associated NMNAT1 mutants. a, schematic displaying locations of LCA-associated mutations in NMNAT1 (arrows). Asterisks indicate mutations analyzed in this study. b, lysates from HEK293T cells (left column) or DRG neurons (right column) expressing Venus or FLAG-tagged wild type NMNAT1 (left column) were analyzed by Western blotting using antibodies to NMNAT1, FLAG, GFP, or α-tubulin. Note that endogenous NMNAT1 levels are undetectable with this antibody in both HEK293T cells and DRG neurons. c, lysates from HEK293T cells expressing FLAG- or HA-tagged wild type or mutant NMNAT1 from the FCIV expression construct containing an IRES-Venus cassette were analyzed by Western blotting using antibodies to FLAG, HA, or GFP. Note that expression of R66W and M69V mutants was significantly lower than other NMNAT1 proteins. Venus expression serves as transfection control. d, longer exposure of Western blot of R66W and I217N NMNAT1 mutants shows proteins of normal size but very low expression in case of R66W. e, quantification of protein expression of NMNAT1 (FLAG- or HA-tagged), Venus, and α-tubulin (endogenous marker) in HEK293T cells. The ratio of NMNAT1 to Venus, NMNAT1 to α-tubulin, or Venus to α-tubulin are shown for each NMNAT1 mutant. The values were normalized to the corresponding either FLAG- or HA-tagged wild type NMNAT1. Asterisks denote significant reduction of NMNAT1 expression compared with wild type NMNAT1 (p < 0.005 t test).
NMNAT1 Kinetic Assays
Recombinant NMNAT1 proteins were purified as described below. Enzymatic activity was assayed at varying ATP levels (15 to 500 μm) with 16, 63, 250, or 500 μm NMN. Reactions were carried out for 2 min at 37 °C in solution containing 10 mm HEPES (pH 7.4), 4 mm MgCl2, and 0.2 μg/ml of NMNAT1 protein. Reactions were stopped by adding an equal volume of 0.5 m HClO4 and then neutralized with 3 m K2CO3. NAD+ was quantified by HPLC (LC20AT, Shimadzu) using a C18 column (LC18T, Supelco). Samples were loaded in 50 mm phosphate buffer (pH 7.0) and eluted with increasing MeOH concentration (0% for 5 min, 0–5% for 1 min, 5% for 5 min, 5–15% 2 min, 15% for 10 min). Initial reaction velocities (nmol of NAD+/min μg of protein) were fitted to Lineweaver-Burk plots to obtain apparent Vmax. A secondary plot of apparent Vmax against substrate concentration was used to obtain Km and kcat.
Axonal Degeneration and CCCP Neuronal Death Assay
DRG neurons were infected with NMNAT1 or control lentiviruses at DIV3. Four days later the axons were severed and distal axon segments were imaged with ×20 objective at 0, 9, 24, and 48 h after transection using a high content imager (Operetta, PerkinElmer). Axon degeneration (degeneration index) was quantified using 24 images from 4 wells per condition as previously described (14, 19). For dose-dependent axon degeneration studies, neurons were infected with varying amounts of virus (1–10 × 103). Venus fluorescent intensity of the cell bodies (as a measure of NMNAT1 expression level) was monitored immediately after axotomy (4 fields per well). Average Venus fluorescence intensity was calculated after background subtraction using ImageJ (NIH). Briefly, total fluorescent intensity was divided by the area occupied by the cell bodies. Axon degeneration indices were plotted against the averaged Venus intensity arbitrary units (a.u.) (expression level) to determine dose dependence.
DRG neurons expressing wild type or mutant NMNAT1 proteins were incubated with the mitochondria uncoupling reagent CCCP (100 μm; Sigma) at DIV7. Cell death was monitored using ethidium homodimer-1 (1 μm; Invitrogen) and nuclei were visualized with bis-benzamide (12.5 μg/ml; Sigma). NMNAT1 expression was verified by monitoring Venus fluorescence as above. Images of 6 fields per well were taken immediately after addition of CCCP (control, non-degenerated condition) and the same fields were imaged again 24 h later (CCCP-induced cell death) using a high content imager. The percentage of dead cells (ethidium homodimer-1 positive cell) versus total cells (bis-benzamide positive cells) was calculated using background subtracted binarized images of each field with built-in ImageJ macros (Find Maxima with segmentation option and Particle Analyzer). A total of 24 images from 4 independent wells per condition (wild type and mutant NMNAT1) were analyzed.
Circular Dichroism (CD) Analysis
pET30a plasmids encoding His6-tagged wild type or mutant NMNAT1 proteins were transformed into Rosetta(DE3)pLysS (EMD Millipore) competent cells. Expression of NMNAT1 recombinant protein was induced by isopropyl 1-thio-β-d-galactopyranoside for 5 h at 37 °C. Bacterial pellets were harvested by centrifugation and homogenized by sonication in buffer containing 150 mm NaCl, 50 mm phosphate buffer (pH 8.0), and protease inhibitor mixture (Complete EDTA; Roche). Recombinant proteins were purified with HIS-Select® HF nickel affinity gel (Sigma). Purified NMNAT1 recombinant proteins were dialyzed against PBS and examined by electrophoresis on SDS-PAGE. Protein concentrations were determined by BCA protein assay kit (Thermo Scientific). CD spectra were collected on a Chirascan CD spectrometer (Applied Photophysics) using 20 μg of NMNAT1 protein in 200 μl of 50 mm phosphate buffer (pH 7.5). Wavelength scan measurements were performed from 200 to 280 nm. Thermal denaturation was followed at 220–225 nm as the temperature was ramped from 5 to 80 °C in 5 °C increments. Thermal denaturation curves were fit to a two-state model by the nonlinear least squares method (20).
NMNAT1 Oligomer Analysis by HPLC Gel Filtration
Cell lysates were prepared without detergents from HEK293T cells expressing wild type or mutant NMNAT1 proteins along with Venus fluorescent protein. The lysate from ∼5 × 105 cells was applied to an HPLC column (BIO GFC-500, 30 cm × 7.8 mm, 5-μm particle; Supelco) under isocratic conditions (137 mm NaCl, 1 mm MgCl2, 0.2 m phosphate buffer, pH 7.0) with flow rate of 1 ml/min (Waters 600). Protein elution profiles were monitored using 241 nm absorbance and fractions were collected every 5 s. The retention time (i.e. size) of wild type and mutant NMNAT1 protein fractions was determined by analyzing the individual fractions using Western blotting with FLAG or HA antibody. The column was calibrated using molecular mass standards (200-kDa β amylase, 150-kDa alcohol dehydrogenase, 66-kDa bovine serum albumin, 29-kDa carbonic anhydrase; Sigma). The standards were mixed with Venus-expressing HEK293T lysate at a protein ratio of about 5:1 and retention time of each standard was determined by absorbance at 241 nm. The retention time for Venus protein was measured with 485 nm excitation and 520 nm emission in each HPLC experiment as internal control. The standard curve was calculated by plotting the molecular weight against the relative retention time of each protein standard and Venus. The average molecular weight of each NMNAT1 species (i.e. hexamer, dimer, or monomer) was determined using relative retention time of each NMNAT1 against internal Venus retention time. Results from 3 independent HPLC experiments were averaged.
Western Blot and Immmunohistochemistry
Lysates prepared from HEK293T cells or DRG neurons expressing NMNAT1 proteins were analyzed by Western blotting or immunohistochemistry using anti-HA (9110 rabbit; Abcam), anti-FLAG (M2 mouse; Sigma), anti-Nmnat1 (mouse; Abcam), anti-α-tubulin (mouse; Sigma), or anti-GFP (rabbit; Invitrogen) antibodies using previously described conditions (21). ImageJ (NIH) gel analysis tool was used to quantify the amount of each band.
Results
LCA9 is an autosomal recessive retinal degenerative condition that is caused by mutations in NMNAT1 (1–4). Presently, 34 different mutations, mostly missense mutations, spread across the entire protein-coding region have been reported (Fig. 1a). Most cases harbor compound heterozygous mutations (48 out of 54 LCA9 cases; 3–6), with very few reports of homozygous mutations. A mutation of E257K is the most common and occurs in more than 70% of LCA9 cases. To gain insight into how the NMNAT1 mutation results in retinal degeneration, we investigated the biochemical properties of a number of NMNAT1 mutants. Fourteen LCA-associated NMNAT1 mutants including E257K were chosen for study based on a number of criteria including their position in the protein, the type of mutation, the potential effects on protein oligomerization, and their coincidence with other mutations in LCA cases (Fig. 1a, asterisks). LCA-associated NMNAT1 mutants were tagged with FLAG or HA on the N terminus and cloned into a vector containing a downstream IRES Venus cassette. The level of NMNAT1 expression from this vector in HEK293T and DRG cells was determined by Western blotting using anti-NMNAT1 antibody (Fig. 1b). In both HEK293T cells and DRG neurons, the exogenous wild type NMNAT1 was easily detected using this antibody. However, endogenous NMNAT1 was below the level of detection, suggesting that the contribution of endogenous NMNAT1 is minimal in our enzymatic assays performed with cell lysates.
Next, NMNAT1 mutant proteins were expressed in HEK293T cells. All NMNAT1 mutants were detected at the same molecular weight as wild type NMNAT1 by Western blotting (Fig. 1, c and d). The expression level of the LCA-associated mutants was comparable with wild type NMNAT1 with the exception of the significantly lower expression of the R66W, M69V, V98G, and D173G NMNAT1 mutants (Fig. 1c). For each mutant, we used Western blotting to quantify the level of NMNAT1 protein, the level of the co-expressed Venus, and endogenous α-tubulin (Fig. 1e). The transfection efficiency defined by the Venus/α-tubulin ratio was comparable for each experiment. Mutant NMNAT1 expression, normalized by either α-tubulin or Venus, was comparable with wild type NMNAT1 in most instances. However, this ratio was much less for R66W, M69V, V98G, and D173G mutants (p < 0.005), indicating that these mutant proteins were less stable than wild type NMNAT1. These ratios, established by Western blotting, were used in combination with Venus fluorescent intensity measurements to quantify NMNAT1 in subsequent assays.
Protein localization was analyzed by immunohistochemistry using HEK293T cells expressing wild type versus mutant NMNAT1. All LCA-associated NMNAT1 mutants co-localized with a nuclear marker, indicating that the mutations did not disrupt the normal nuclear localization of the protein (Fig. 2, a and b). Overall, we found no abnormalities in the nuclear localization of LCA-associated NMNAT1 mutants.
FIGURE 2.

Subcellular localization of wild type and LCA-associated NMNAT1 mutants. Subcellular localization of wild type or mutant NMNAT1 with antibodies to (a) FLAG or (b) HA epitope tags was analyzed by immunohistochemistry. All NMNAT1 mutant proteins were localized exclusively to the nucleus (stained with bis-benzamide (bis-Bz)). The specificity of the staining was confirmed by correlation between FLAG or HA signal and the co-expressed Venus protein florescence. c, NMNAT enzymatic activity of the mutants was measured by monitoring NAD+ production (see ”Experimental Procedures“). Activity was normalized to Venus expression level and expressed as NAD+ production rate (μm/min). Asterisks denote significant alteration of NMNAT activity compared with wild type NMNAT1 (p < 0.01, t test).
The major function of NMNAT1 is to convert NMN to NAD+, a key molecule in cellular bioenergetics. To test whether LCA-associated NMNAT1 mutants have deficits in enzymatic activity, HEK293T lysates expressing wild type or mutant NMNAT1 proteins were analyzed for NMNAT function (see “Experimental Procedures”). The NAD+ produced using lysates prepared from HEK293T cells expressing wild type NMNAT1 after a 60-min incubation was 121 ± 23.5 μm, whereas that of non-transfected cells (endogenous NMNAT activity) was negligible (4.2 ± 0.3 μm). Under these conditions, lysates prepared from cells expressing the less stable R66W, V98G, and D173G NMNAT1 mutants had significantly lower enzymatic activity, the V151F and R237C mutants showed reduced NMNAT1 activity, whereas all others showed relatively normal NAD+ synthetic activity (Fig. 2c). To summarize, most of the LCA-associated NMNAT1 mutations did not show severe decrements in basal enzymatic activity using this transfected cell lysate-based NMNAT enzymatic assay.
To further characterize the effects of selected mutations on NMNAT enzymatic activity, we compared the kinetic parameters of wild type versus several NMNAT1 mutants using purified recombinant protein. The V98G NMNAT1 mutant was inactive in the lysate assay (Fig. 2c). Consistent with this result, its substrate affinity for both ATP and NMN was greatly reduced, with the Km for NMN more than 100 times greater than wild type (Table 1). The most common LCA9 mutant, E257K, showed a 3-fold increase in Km for ATP and NMN compared with wild type. However, the measured catalytic activity for ATP was comparable, whereas that for NMN was only modestly reduced. The L239S mutant showed reduced affinity for NMN but normal affinity for ATP, and its catalytic activity was higher than wild type. These results suggest that some LCA9 NMNAT1 mutants have reduced enzymatic activity when substrate availability is limited.
TABLE 1.
Kinetic parameters of LCA9 mutant NMNAT1
Kinetic parameters of NMNAT1 wild type and mutants. Recombinant NMNAT1 was mixed with various amount of ATP and NMN and determined NAD+ produced by HPLC. Data are average of three independent reactions and values for wild type NMNAT1 are comparable to a previous report (28).
| ATP |
NMN |
|||
|---|---|---|---|---|
| Km | kcat | Km | kcat | |
| μm | s−1 | μm | s−1 | |
| WT | 57.1 ± 32.2 | 68.4 ± 11.4 | 26.7 ± 17.1 | 73.8 ± 10.3 |
| E257K | 187.8 ± 66.4 | 54.4 ± 14.6 | 91.3 ± 47.8 | 45.6 ± 9.9 |
| V98G | >300 | 456 ± 123 | >2800 | 713 ± 120 |
| L239S | 57.8 ± 11.0 | 153.1 ± 81.1 | 57.9 ± 31.3 | 153.0 ± 81.0 |
It is well known that cell soma degeneration is a leading cause of various neurodegenerative diseases including retinal degeneration (22). We next explored whether the disease-associated mutants were equivalent to wild type NMNAT1 in protecting against cell death. For these experiments, we used an assay in which DRG neurons are treated with the mitochondrial uncoupling agent CCCP, which causes energetic failure, ROS accumulation, and cell death (23). CCCP-induced ROS accumulation in DRG neurons was confirmed using the ROS-sensitive dye DCFDA (Fig. 3, a and b). DRG neurons were infected with lentivirus expressing wild type or mutant NMNAT1 or Venus alone. Neurons expressing Venus only were dead 24 h after addition of CCCP (100 μm), whereas those expressing wild type NMNAT1 largely survived CCCP treatment (Fig. 3, c and d). Most of the LCA-associated NMNAT1 mutants delayed CCCP-induced neuronal death similar to wild type; however, several mutants including those with reduced protein stability and enzymatic activity (R66W, V98G, and D173G) as well as L239S were significantly less effective (Fig. 3, c and d). Interestingly, whereas analysis of enzyme kinetics showed reduced substrate affinity for both E257K and L239S mutants, only L239S fails to prevent CCCP-induced cell death.
FIGURE 3.
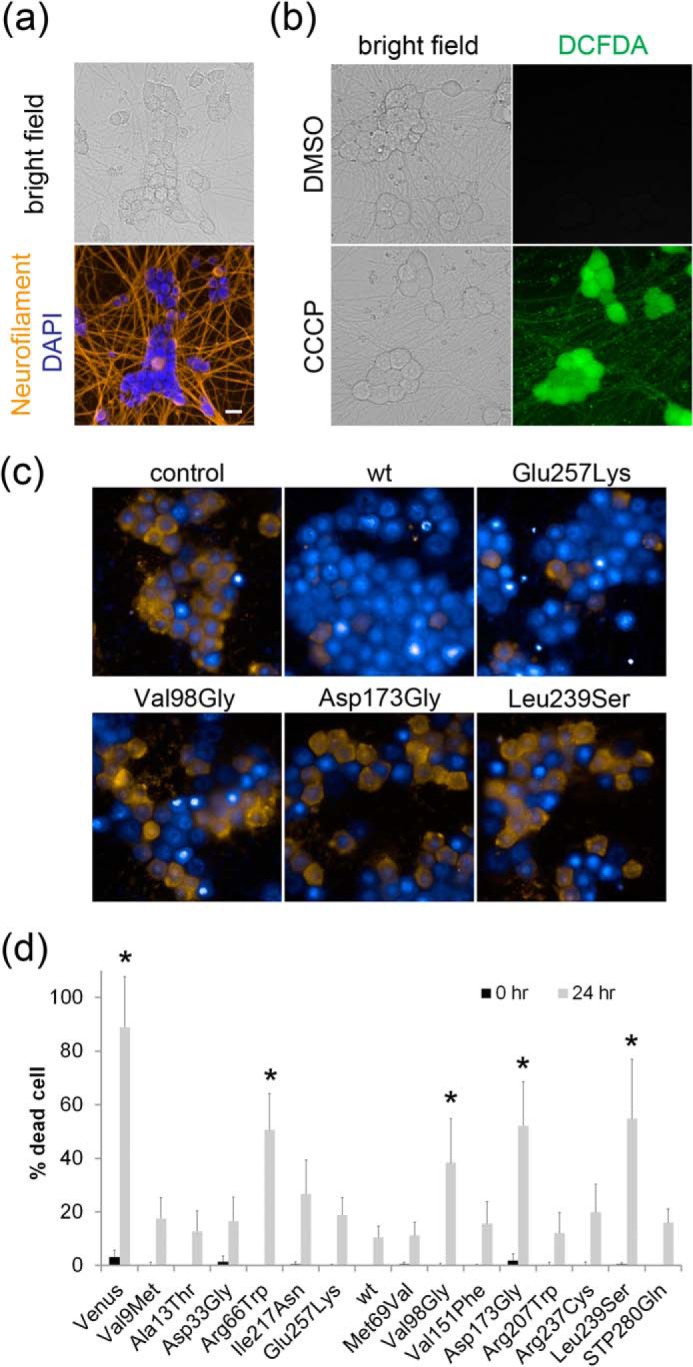
Most LCA-associated NMNAT1 mutants protect neurons from CCCP-induced cell death. a, DRG neurons (7 DIV) were fixed and stained with neurofilament middle chain antibody (DSHB) and nuclear marker DAPI. b, fluorescent and bright field images of DRG neurons (7 DIV) treated with DMSO or 100 μm CCCP for 4 h and incubated with 0.5 mm CM-H2DCFDA ROS sensor (Invitrogen). ROS accumulation in CCCP-treated cells is evident. c, DRG neurons were infected with lentivirus expressing wild type or LCA-associated NMNAT1 mutants. The neurons were treated with CCCP (100 μm) and cell viability was assessed by staining with ethidium homodimers. Representative images of neurons treated with CCCP for 24 h are shown. All nuclei were visualized with bis-benzamide (blue) and dead cells were identified by ethidium homodimer (yellow) incorporation. d, percentage of dead cells at baseline and at 24 h after CCCP treatment. Asterisks denote significant reduction in cell survival when compared with wild type NMNAT1 (p < 0.01, t test). Scale bar = 20 μm.
Studies of the wlds mutant mouse led to the discovery that overexpression of NMNAT1 delays the degeneration of axons in response to a wide variety of injuries including mechanical severing, mitochondrial inhibitors, and chemotherapeutic agents (12, 24, 25). This axonal phenotype is exerted locally via extra-nuclear localization of overexpressed NMNAT1 and requires NMNAT enzymatic activity (14). To determine whether defects in NMNAT-mediated axonal protection contributed to the retinal degeneration in LCA, we expressed wild type or mutant NMNAT1 in DRG neurons via lentivirus. After DIV7, the axons were severed and the extent of axonal degeneration was quantified at multiple time points. Axons from neurons overexpressing wild type NMNAT1 remained intact for 48 h post-axotomy (Fig. 4, a and b), whereas those from control neurons (expressing Venus only) completely degenerated in 9 h (Fig. 4, a and b). Most of the LCA-associated NMNAT1 mutants promoted axonal protection that was essentially equivalent to that mediated by wild type NMNAT1. The mutants that showed significantly reduced expression or enzymatic activity in previous assays (R66W, M69V, V98G, V151F, D173G, R237C) were all deficient in their ability to maintain axonal stability after injury (Fig. 4, a and b). Surprisingly, the L239S and the common E257K NMNAT1 mutants failed to prevent axon degeneration, yet their enzymatic activity is relatively normal. In these two mutants, enzymatic kinetic assays showed a reduced substrate affinity (Table 1), suggesting that they could have reduced activity if substrate availability is limited after axonal injury.
FIGURE 4.

Many LCA-associated NMNAT1 mutants prevent axon degeneration after axotomy. Axons from DRG neurons infected with lentivirus expressing wild type or LCA-associated NMNAT1 mutants were severed at DIV7 and axon segments distal to the cut site were imaged. a, representative images of distal axon segments 24 h after severing are shown. b, axon degeneration was quantified at 0, 9, 24, and 48 h after axotomy. The LCA-associated mutants showed varying degrees of axon protective activity. Some mutants protected axons for up to 48 h post-transection, similar to wild type NMNAT1. Other mutants showed essentially no protection with axons degenerating completely within 9 h, similar to Venus (control) expressing neurons. Asterisks denote significant a increase in degeneration index (lack of axon protection) compared with wild type NMNAT1 (p < 0.01, t test).
Previously, we demonstrated that NMNAT1 overexpression gave rise to limited amounts of NMNAT1 in the axon and that axonal protection is correlated with the amount of NMNAT in the axon compartment (13). To further explore the inability of the enzymatically active E257K and L239S NMNAT1 mutants to prevent axonal degeneration, we compared the dose dependence of wild type versus mutant protein using IRES-derived Venus fluorescence intensity as a measure of NMNAT1 expression level (Fig. 1e). Axons from DRG neurons expressing Venus alone degenerated completely by 24 h after axotomy at all Venus fluorescence intensity levels. In contrast, axons from wild type NMNAT1-expressing neurons were protected at Venus fluorescence intensity as low as 700 a.u. (Fig. 5a). The E257K NMNAT1 mutant did not protect axons at low expression levels (i.e. Venus fluorescence intensity between 700 and 1000 a.u.). However, axon degeneration indices decreased with increasing Venus fluorescence intensity such that axons remained intact (degeneration index <0.2) at >1300 a.u. Similarly, other mutants defective in preventing axon degeneration (i.e. M69V, V98G, D173G) also showed dose-dependent increases in axon protection activity (data not shown), suggesting that retinal deficits associated with the NMNAT1 mutation could be dependent on the level of expression. In contrast, the L239S mutant failed to block axon degeneration at all doses tested (Fig. 5a), suggesting that this mutant protein has additional unidentified defects.
FIGURE 5.

Dose-dependent axonal protection by LCA-associated NMNAT1 mutants. Axons from DRG neurons were infected with different amounts of lentivirus expressing wild type, E257K, or L239S NMNAT1 mutants. Axons were transected at DIV7 and axonal degeneration was quantified at 24 h after axotomy. Degeneration indices were plotted against relative Venus fluorescent intensity derived from co-expressed IRES-Venus. a, axonal degeneration of Venus, wild type, or E257K expressing neurons was plotted against expression level (Venus intensity of a.u. Note that E257K mutant protects axons when expressed at higher levels. b, axonal degeneration of E257K, E257Q, or E257D-expressing neurons plotted against relative expression level (Venus intensity). Axons from E257Q or E257D were protected at much lower expression level than E257K. Degeneration indices of E257K from (a) are plotted for reference purpose.
These results suggest that LCA-associated NMNAT1 mutations could affect protein stability or trafficking to the axon compartment. To examine this further, we concentrated on the most common mutation, E257K, which is an acidic to basic substitution. We asked whether the character of the replacement amino acid was important, and tested NMNAT1 molecules with neutral (E257Q) and acidic (E257D) substitutions in the axonal degeneration assay. We found that both E257D and E257Q mutants protected axons similarly to wild type NMNAT1 (Fig. 5b). The enzymatic activities of all three mutants at position 257 were comparable with each other (E257K, 3.3 ± 0.5 μm/min; E257Q, 4.4 ± 0.6 μm/min; E257D, 3.4 ± 0.8 μm/min) and wild type NMNAT1, thus the negative charge at position 257 appears to influence NMNAT1 functions other than its enzymatic activity (for example, see Fig. 6c). Interestingly, a recent report suggested that the E257K mutation could be hypomorphic and could be a genetic modifier of other LCA subtypes (7).
FIGURE 6.

Most LCA-associated NMNAT1 mutants have unstable secondary structures. Analysis of CD spectra of wild type and selected NMNAT1 mutants using bacterially expressed recombinant proteins. a, representative CD spectra of wild type, E257K, V98G, and L239S NMNAT1 proteins. b, CD spectra at 222 nm at a range of temperatures is shown. The midpoint of the unfolding transition (Tm) was calculated as described under “Experimental Procedures.” All three mutants exhibited decreased secondary structure stability against thermal unfolding. c, NMNAT1 proteins were monitored for enzymatic activity after heat shock (10 min at 68 °C). NAD+ production was measured for each mutant in the native state and after heat shock. The ratio of NMNAT activity (heat shocked versus native) at 25 min after substrate addition was plotted for each mutant. Most mutants lost all or most of their NAD+ synthetic activity after heat shock compared with wild type NMNAT1. d, E257K, E257Q, and E257D NMNAT enzymatic activities were monitored with or without heat shock as described in c. Both E257Q and E257D but not E257K retained NMNAT enzymatic activity after heat shock.
Structural studies show that human NMNAT1 exists as a hexamer. To test whether the LCA-associated NMNAT1 mutations alter oligomer formation, we performed a number of studies (see “Experimental Procedures”) on selected mutants. The mutants tested included those predicted by in silico analysis to affect oligomer formation as well as those deficient in the CCCP-induced death and axotomy assays. The mutant proteins were expressed in HEK293T cells and the size of the NMNAT1 oligomers was determined by size exclusion chromatography. We found a major peak of reactivity for wild type and all mutant NMNAT1 proteins at 218 ± 43 kDa (n = 21), which is close to the predicted size of the epitope-tagged NMNAT1 hexamer (198 kDa) (calculated NMNAT1 monomer = 32 kDa). Importantly, there were no significant differences in the peaks observed between wild type and LCA-associated NMNAT1 mutants, suggesting that the mutants do not adversely affect hexamer formation (Table 2).
TABLE 2.
Oligomer formation of LCA9 mutant NMNAT1
Lysates of HEK293T cells expressing wild type or mutant NMNAT1 were fractionated by a size exclusion chromatogram column under non-denaturing conditions. The molecular weights of the NMNAT1 oligomers were determined using a standard curve. Average molecular weights and standard deviations were calculated from 3 independent experiments.
| NMNAT1 | Mr (average) | S.D. |
|---|---|---|
| WT | 219 | 41 |
| V9M | 241 | 45 |
| E257K | 256 | 48 |
| M69V | 201 | 48 |
| V98G | 214 | 56 |
| D173G | 205 | 29 |
| L239S | 188 | 38 |
The native protein conformation is the most thermodynamically stable form of the molecule with the given amino acid sequence. This structure can be disrupted by a single amino acid substitution, reducing the effective protein concentration or resulting in a toxic gain-of-function change. To investigate the effect of LCA-associated mutations on the NMNAT1 secondary structure, we expressed and purified wild type and mutant NMNAT1 proteins from Escherichia coli, and examined them using circular dichroism (CD) spectroscopy. Wild type NMNAT1 showed a CD spectrum typical of an α-helical-rich protein. The NMNAT1 mutants we examined displayed CD spectra similar to wild type NMNAT1 (Fig. 6a). To determine whether LCA-associated mutations rendered NMNAT1 susceptible to thermal stress, we followed thermal unfolding by CD spectroscopy at 222 nm. α-Helical structures of all LCA-associated mutants begin unfolding at a lower temperature than wild type NMNAT1 (Fig. 6b). The midpoint of the unfolding transition (Tm) was calculated using theoretical curve fitting (26) for wild type NMNAT1 = 69.2 ± 0.38 °C, E257K = 66.6 ± 0.59 °C, V98G = 65.8 ± 0.55 °C, and L239S = 61.4 ± 0.86 °C. These results suggest that the secondary structures of E257K, V98G, and L239S NMNAT1 mutants are less stable than that of wild type NMNAT1.
To further investigate the effect of thermal stress on wild type and LCA-associated mutant function, NMNAT enzymatic activity was measured after a heat shock, which causes formation of protein aggregates and has been used previously to examine enzyme stability (27). Wild type and mutant NMNAT1 proteins were either incubated on ice (native) or incubated at 68 °C for 10 min (heat shock) prior to measuring NMNAT enzymatic activity 25 min later. Wild type NMNAT1 retained ∼30% of native activity after the 68 °C incubation; however, most of the LCA-associated mutants including E257K and L239S completely lost their enzymatic activity after heat shock (Fig. 6c). To explore the relationship of these findings to other NMNAT1 functions, we focused on mutations at Glu257. In examining a series of mutations at this residue to the LCA-associated Lys (basic), Gln (neutral), or Asp (acidic), we found that change from an acidic to basic residue did not disrupt baseline enzymatic function but did inhibit NMNAT1 axon protective function (Fig. 5b). We therefore tested each of these three mutants (E257K, E257Q, and E257D) in the heat shock assay and found that NMNAT enzymatic activity of the E257Q and E257D mutants was retained after heat shock, whereas no activity was detectable after heat shock of the LCA-associated E257K mutant (Fig. 6C). The retention of activity was particularly robust for the E257D mutation, indicating that a negative charge at this position contributes to the stability of the enzyme. These results suggest that the secondary structure of many LCA-associated NMNAT1 mutants is less stable than wild type, particularly under stressful conditions such as those that may exist in the metabolically hyperactive photoreceptor. This may result in aberrant functions that are not apparent when basal NMNAT activity is examined and could contribute to the retinal deficits observed in LCA.
Discussion
We have analyzed the biochemical properties of LCA-associated NMNAT1 mutants to gain insight into how these mutations lead to retinal degeneration and blindness. We examined multiple facets of these enzymes including their expression level, cellular localization, NAD+ synthetic activity, prevention of injured axon degeneration, protection from CCCP-induced neuronal cell death, protein secondary and tertiary structure, and protein stability under stressful conditions. To summarize the data, we scored the mutants based on a compilation of results from all the assays we performed (0 = equivalent to wild type, −1 = lower than wild type and higher than negative control, −2 = equivalent to negative control; see Table 3). Although, no consistent pattern emerged for all the mutations from this analysis, several appear to be overall more disruptive including R66W, M69V, V98G, D173G, L239S, and E257K.
TABLE 3.
Biochemical scores of LCA9 mutant NMNAT1
Summary of scores from indicated assays (blank or 0 = equivalent to wild type; −1 = greater than wild type and less than negative control; −2 = equivalent to negative control).
| Expression | Nuclear localization | NMNAT activity | Cell body protection | Oligomerization | Axon protection | Refolding | Total score | |
|---|---|---|---|---|---|---|---|---|
| V9M | −1 | −1 | ||||||
| A13T | 0 | |||||||
| D33G | 0 | |||||||
| R66W | −2 | −2 | −1 | −2 | NDa | −7 | ||
| M69V | −1 | −2 | −2 | −5 | ||||
| V98G | −1 | −1 | −1 | −2 | −2 | −7 | ||
| V151F | −1 | −1 | −2 | |||||
| D173G | −1 | −1 | −1 | −2 | −2 | −7 | ||
| R207W | −2 | −2 | ||||||
| I217N | −2 | −2 | ||||||
| R237C | −1 | −2 | −3 | |||||
| L239S | −1 | −2 | −2 | −5 | ||||
| E257K | −2 | −2 | −4 | |||||
| STP280Gln | −1 | −1 |
a ND, not able to determine because of low protein expression.
E257K was significantly less effective at preventing axonal degeneration than wild type or most other LCA-associated NMNAT1 mutants even though it had near normal enzymatic activity. Similarly, the L239S has normal enzymatic activity, yet fails in the axon and cell death protection assays. It is possible that these dichotomies occur due to a failure to properly form the hexameric NMNAT complex. However, our predictive modeling studies to estimate the impact of these mutations on oligomerization as well as the size exclusion chromatography data, revealed no evidence for LCA-associated NMNAT1 mutant defects in hexamer assembly. Analysis of the enzyme kinetics revealed a reduced affinity for substrate in the case of the E257K and L239S mutants. The potential limited substrate availability in an injured or stressed retinal cell may lower the activity of these NMNAT1 mutants.
In addition CD spectroscopy revealed reduced stability of E257K and L239S secondary structure under thermal stress. These observations support the idea that LCA-associated NMNAT1 mutants are less stable than wild type enzyme. Similarly, many mutants including E257K and L239S completely lost enzymatic activity after heat shock, suggesting that LCA-associated NMNAT1 mutations render the enzyme susceptible to cellular stress-induced changes in protein folding and, ultimately, activity.
We analyzed a total of 14 different LCA-associated NMNAT1 mutants in this study, including those that occur as compound heterozygous or homozygous mutations. Among these, there are three with no measurable deficits in NMNAT enzymatic activity that appear as homozygous mutations, V9M, D33G, and STP280Gln, suggesting that other NMNAT1 functions are important for retinal degeneration. There are several currently unexplained discrepancies between our results and those previously reported concerning mutant E257K, which has been reported to be enzymatically crippled (5). The relatively high frequency of the E257K variant allele yet low number of LCA patients homozygous for the mutation coupled with the absence of a retinal phenotype in an individual homozygous for the mutation suggest that E257K is a hypomorphic allele (4–7). Although this mutant had relatively normal activity at baseline in our analyses, it was extremely sensitive to stressful conditions, as heat shock destroyed its activity. The V9M protein was enzymatically normal in our assays but has been reported to be severely compromised when analyzed in vitro (6). In support of our studies, NAD+ levels in fibroblasts from patients homozygous for this mutation were essentially normal (6). Given the mutation profile of the currently available genotypes and the enzymatic data of the mutants we examined, we found only one case (R66W; R237Cys compound heterozygote) in which basal NMNAT enzymatic activity would be predicted to be less than 50% of that in control subjects. All other cases are predicted to have more than half of the NMNAT activity observed in control subjects. This level of NMNAT activity should be sufficient to maintain retinal function; in support, an unaffected family member heterozygous for the R66W NMNAT1 allele (a severe loss-of-function mutation) has normal retinal function (family 007, subject I-1 (6)). These results suggest that a reduction in basal NMNAT enzymatic activity may contribute to retinal degeneration in LCA9 patients, but is unlikely to be the primary mechanism.
In our studies, we found that NMNAT1 secondary structure stability is reduced in many LCA-associated mutants. These results suggest that the combination of reduced NMNAT enzymatic activity and increased susceptibility to protein unfolding under stressful conditions should be considered as a mechanism for the NMNAT1 mutation resulting in retinal degeneration. In support, we summed the total NMNAT1 biochemical scores (Table 3) for each LCA9 case in Table 4 and found that all cases had negative scores except for one case (homozygous N33G). The NMNAT1 N33G mutant, which is homozygous in one LCA9 case, did not show deficits in any of the assays we performed. This result indicates that additional assays are needed to identify the molecular defect in this enzyme or, alternatively, that mutations in other loci may also be present in some LCA9 cases.
TABLE 4.
Summary of biochemical scores for LCA9 cases
Summary of mutations found in LCA9 patients combined with biochemical scores of NMNAT1 from Table 2. The mutations present in individual subjects are listed in columns. The biochemical score for each mutant tested is summed to provide a total mutant score. The left-most column lists the positions and identities of the mutated residues.

NMNAT1 is required for proper embryonic development as Nmnat1-deficient embryos die at embryonic day 10 (8); however, there is no obvious pathology outside of the retina in patients with NMNAT1 mutations (3–6). Why is the retina so sensitive to NMNAT1 mutation? Indeed, photoreceptor cells in the Drosophila eye are also extremely sensitive to dNmnat loss; however, in this organism the deficit can be rescued by enzymatically inactive dNmnat (15). The cell type(s) that are primarily affected in the retina of patients with LCA-associated NMNAT1 mutations is a key question that has not been fully addressed. In many retinal degeneration diseases, including retinitis pigmentosa and LCA, the photoreceptors and retinal pigment epithelium (RPE) cells are often the major affected cell types. The visual cycle is a chain of reactions that regenerate photosensitive 11-cis-retinal from the light stimulated form, all-trans-retinal, whose accumulation results in retinal degeneration. RPE cells are constantly engulfing photoreceptor outer segments and their renewal is essential for photoreceptor cell homeostasis and avoiding accumulation of oxidized unsaturated fatty acids. These retina-specific environments may conspire to make retinal cells more susceptible to altered NMNAT1 function (9, 10). Our finding that NMNAT1 overexpression protects neurons from CCCP-induced cell death may be an important clue for the role of NMNAT1 in retina homeostasis.
Results from the axonal protection assays revealed that most LCA-associated NMNAT1 mutants that failed to protect degenerating axons at low expression levels could prevent axon degeneration when expression was increased. The reduced penetrance of these alleles in some cohorts may be due to different expression levels caused by other genetic or epigenetic factors. This observation suggests that increasing expression of these NMNAT1 mutant proteins in the retina could potentially prevent the deficits and, perhaps, the blindness caused by these mutations. It will be important to study the role of other components in the NAD+ biosynthetic pathway in the retina to fully understand the importance of NAD+ metabolism in retina homeostasis.
Author Contributions
Y. S. and J. M. designed the study and wrote the paper. Y. S., Z. M., B. B., and J. J. H. designed and performed the experiments and analyzed the data. J. J. H. performed in silico protein oligomerization analysis. All authors reviewed results and approved the final version of the manuscript.
Acknowledgment
We thank Kelli Simburger for experimental assistance.
This work was supported by the National Institutes of Health Grants R01AG013730 (to J. M.), R01NS065053 (to J. M. and A. D.), RO1NS078007 (to J. M. and A. D.), R01NS087632 (to J. M.), and R01GM101602 (to J. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- LCA
- Leber congenital amaurosis
- DIV
- days in vitro
- RPE
- retinal pigment epithelium
- DRG
- dorsal root ganglion
- CCCP
- carbonyl cyanide p-chlorophenylhydrazone
- IRES
- internal ribosome entry site
- H2DCFDA
- 2′,7′-dichlorodihydrofluorescein diacetate
- a.u.
- arbitrary unit.
References
- 1. den Hollander A. I., Roepman R., Koenekoop R. K., Cremers F. P. (2008) Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye. Res. 27, 391–419 [DOI] [PubMed] [Google Scholar]
- 2. Keen T. J., Mohamed M. D., McKibbin M., Rashid Y., Jafri H., Maumenee I. H., Inglehearn C. F. (2003) Identification of a locus (LCA9) for Leber's congenital amaurosis on chromosome 1p36. Eur. J. Hum. Genet. 11, 420–423 [DOI] [PubMed] [Google Scholar]
- 3. Perrault I., Hanein S., Zanlonghi X., Serre V., Nicouleau M., Defoort-Delhemmes S., Delphin N., Fares-Taie L., Gerber S., Xerri O., Edelson C., Goldenberg A., Duncombe A., Le Meur G., Hamel C., Silva E., Nitschke P., Calvas P., Munnich A., Roche O., Dollfus H., Kaplan J., Rozet J. M. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet. 44, 975–977 [DOI] [PubMed] [Google Scholar]
- 4. Chiang P. W., Wang J., Chen Y., Fu Q., Zhong J., Chen Y., Yi X., Wu R., Gan H., Shi Y., Chen Y., Barnett C., Wheaton D., Day M., Sutherland J., Heon E., Weleber R. G., Gabriel L. A., Cong P., Chuang K., Ye S., Sallum J. M., Qi M. (2012) Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat. Genet. 44, 972–974 [DOI] [PubMed] [Google Scholar]
- 5. Koenekoop R. K., Wang H., Majewski J., Wang X., Lopez I., Ren H., Chen Y., Li Y., Fishman G. A., Genead M., Schwartzentruber J., Solanki N., Traboulsi E. I., Cheng J., Logan C. V., McKibbin M., Hayward B. E., Parry D. A., Johnson C. A., Nageeb M., Finding of Rare Disease Genes (FORGE) Canada Consortium, Poulter J. A., Mohamed M. D., Jafri H., Rashid Y., Taylor G. R., Keser V., Mardon G., Xu H., Inglehearn C. F., Fu Q., Toomes C., Chen R. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 44, 1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Falk M. J., Zhang Q., Nakamaru-Ogiso E., Kannabiran C., Fonseca-Kelly Z., Chakarova C., Audo I., Mackay D. S., Zeitz C., Borman A. D., Staniszewska M., Shukla R., Palavalli L., Mohand-Said S., Waseem N. H., Jalali S., Perin J. C., Place E., Ostrovsky J., Xiao R., Bhattacharya S. S., Consugar M., Webster A. R., Sahel J. A., Moore A. T., Berson E. L., Liu Q., Gai X., Pierce E. A. (2012) NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 44, 1040–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siemiatkowska A. M., Schuurs-Hoeijmakers J. H., Bosch D. G., Boonstra F. N., Riemslag F. C., Ruiter M., de Vries B. B., den Hollander A. I., Collin R. W., Cremers F. P. (2014) Nonpenetrance of the most frequent autosomal recessive leber congenital amaurosis mutation in NMNAT1. JAMA Ophthalmol. 132, 1002–1004 [DOI] [PubMed] [Google Scholar]
- 8. Conforti L., Janeckova L., Wagner D., Mazzola F., Cialabrini L., Di Stefano M., Orsomando G., Magni G., Bendotti C., Smyth N., Coleman M. (2011) Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J. 278, 2666–2679 [DOI] [PubMed] [Google Scholar]
- 9. Rózanowska M., Sarna T. (2005) Light-induced damage to the retina: role of rhodopsin chromophore revisited. Photochem. Photobiol. 81, 1305–1330 [DOI] [PubMed] [Google Scholar]
- 10. Sparrow J. R., Boulton M. (2005) RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 80, 595–606 [DOI] [PubMed] [Google Scholar]
- 11. Beatty S., Koh H., Phil M., Henson D., Boulton M. (2000) The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 45, 115–134 [DOI] [PubMed] [Google Scholar]
- 12. Araki T., Sasaki Y., Milbrandt J. (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 305, 1010–1013 [DOI] [PubMed] [Google Scholar]
- 13. Sasaki Y., Vohra B. P., Baloh R. H., Milbrandt J. (2009) Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 29, 6526–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sasaki Y., Vohra B. P., Lund F. E., Milbrandt J. (2009) Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 29, 5525–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhai R. G., Cao Y., Hiesinger P. R., Zhou Y., Mehta S. Q., Schulze K. L., Verstreken P., Bellen H. J. (2006) Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 4, e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rallis A., Lu B., Ng J. (2013) Molecular chaperones protect against JNK- and Nmnat-regulated axon degeneration in Drosophila. J. Cell Sci. 126, 838–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou T., Kurnasov O., Tomchick D. R., Binns D. D., Grishin N. V., Marquez V. E., Osterman A. L., Zhang H. (2002) Structure of human nicotinamide/nicotinic acid mononucleotide adenylyltransferase: basis for the dual substrate specificity and activation of the oncolytic agent tiazofurin. J. Biol. Chem. 277, 13148–13154 [DOI] [PubMed] [Google Scholar]
- 18. Kuhlman B., Baker D. (2000) Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U.S.A. 97, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gerdts J., Sasaki Y., Vohra B., Marasa J., Milbrandt J. (2011) Image-based screening identifies novel roles for IκB kinase and glycogen synthase kinase 3 in axonal degeneration. J. Biol. Chem. 286, 28011–28018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenfield N. J. (2004) Analysis of circular dichroism data. Methods Enzymol. 383, 282–317 [DOI] [PubMed] [Google Scholar]
- 21. Sasaki Y., Araki T., Milbrandt J. (2006) Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J. Neurosci. 26, 8484–8491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Athanasiou D., Aguilà M., Bevilacqua D., Novoselov S. S., Parfitt D. A., Cheetham M. E. (2013) The cell stress machinery and retinal degeneration. FEBS Lett. 587, 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Summers D. W., DiAntonio A., Milbrandt J. (2014) Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J. Neurosci. 34, 9338–9350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Press C., Milbrandt J. (2008) Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J. Neurosci. 28, 4861–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bhattacharya M. R., Gerdts J., Naylor S. A., Royse E. X., Ebstein S. Y., Sasaki Y., Milbrandt J., DiAntonio A. (2012) A model of toxic neuropathy in Drosophila reveals a role for MORN4 in promoting axonal degeneration. J. Neurosci. 32, 5054–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Borgo B., Havranek J. J. (2012) Automated selection of stabilizing mutations in designed and natural proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 1494–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nguyen V. T., Morange M., Bensaude O. (1989) Protein denaturation during heat shock and related stress: Escherichia coli β-galactosidase and Photinus pyralis luciferase inactivation in mouse cells. J. Biol. Chem. 264, 10487–10492 [PubMed] [Google Scholar]
- 28. Sorci L., Cimadamore F., Scotti S., Petrelli R., Cappellacci L., Franchetti P., Orsomando G., Magni G. (2007) Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry 46, 4912–4922 [DOI] [PubMed] [Google Scholar]