

Abstract
BACKGROUND
Pin1 is an intracellular signaling molecule which plays a critical but opposite role in the pathogenesis of Alzheimer’s disease (AD) and many human cancers.
SCOPE OF REVIEW
We review the structure and function of the Pin1 enzyme, the diverse roles it plays in cycling cells and neurons, the epidemiologic evidence for the inverse association between cancer and AD, and the potential therapeutic implications of Pin1-based therapies.
MAJOR CONCLUSIONS
Pin1 is a unique enzyme that has effects the function of target proteins by “twisting” them into different shapes. Cycling cells use Pin1 to help coordinate cell division. It is over-expressed and/or activated by multiple mechanisms in many common human cancers, and acts on multiple signal pathways to promote tumorigenesis. Inhibition of Pin1 in animal models has profound anti-tumor effects. In contrast, Pin1 is down-regulated or inactivated by multiple mechanisms in AD brains. The absence of Pin1 impairs tau function and amyloid precursor protein processing, leading to tangle- and amyloid-related pathologies and neurodegeneration in an age-dependent manner, resembling human AD. We have developed cis and trans conformation-specific antibodies to provide the first direct evidence that tau exists in distinct cis and trans conformations and that Pin1 accelerates its cis to trans conversion, thereby protecting against tangle formation in AD.
GENERAL SIGNIFICANCE
Available studies on Pin1 suggest that cancer and AD may share biological pathways that are deregulated in different directions. Pin1 biology opens exciting preventive and therapeutic horizons for both cancer and neurodegeneration.
1. Introduction
The curious relationship between cancer and neurodegenerative diseases has drawn increasing attention as converging evidence suggests that one family of diseases provides protection against the other. This “inverse comorbidity” is unusual, and suggests that these conditions may share biological pathways which are deregulated in different directions [1]. We hypothesized over a decade ago that a predisposition to cancer might decrease the risk of AD based on our work with the protein Pin1, which plays a critical but opposite role in both diseases [2]. In this article we will show how the enzyme Pin1 is intimately involved in the pathogenesis of both cancer and AD, and serves as one molecular explanation of the inverse association between them.
We (KPL) originally identified Pin1 during a screen for anti-neoplastic agents as a human protein that can not only physically interact with the mitotic kinase NIMA, but also functionally suppresses its ability to induce mitotic catastrophe in yeast [3]. Pin1 is now known to play an important role in many cellular processes, including the cell cycle and cell signaling, regulation of transcription and splicing, and maintenance of neuronal proteins including beta-amyloid and tau [4]. The Pin1 enzyme “twists” proteins into different shapes after proteins are phosphorylated on specific Ser or Thr residues preceding a Pro residue (pSer/Thr-Pro), so called Pro-directed phosphorylation [5, 6].
Pro-directed phosphorylation is a major signaling mechanism in the cell [7–9]. The enzymes that are responsible for such phosphorylation are called proline-directed protein kinases, whose well-known members include mitogen-activated protein kinases (MAP kinases), cyclin-dependent kinases (CDKs) and glycogen synthase kinase-3 (GSK-3). Proline has an interesting stereochemistry due to the presence of a 5-membered ring on its peptide backbone. This allows it to flip between a cis or trans orientation, thereby changing the 3-D structure of the molecule. The recent identification of Pin1 as a peptidyl-prolyl cis-trans isomerase (PPIases) that specifically catalyzes cis-trans isomerization of certain pSer/Thr-Pro motifs led to the hypothesis of a new signaling mechanism, whereby Pin1 catalytically regulates the conformation of substrates after their phosphorylation to further control protein function [3, 10–12]. Subsequent studies have shown that Pin1-catalyzed conformational regulation, which can now be detected by cis and trans conformation-specific antibodies [13], can have a profound impact on many key proteins involved in diverse cellular processes [2, 14–17]. Pin1 has emerged as a novel molecular timer that modulates its multiple targets at various steps of a given cellular process to synergistically control the amplitude and duration of a cellular response or process [18]. Importantly deregulation of Pin1 has a major impact on the development of disease and offers attractive new therapeutic strategies, notably for treating cancer and Alzheimer’s disease [14, 19, 20], the focus of this review.
2. Pin1 structure and function
The conformational significance of the pSer/Thr-Pro motif was not appreciated before the discovery of Pin1, which specifically catalyzes the cis/trans isomerization of specific pSer/Thr-Pro motifs (Figure 1) [10]. It takes substantial energy to flip from cis to trans after phosphorylation, making it a naturally slow process. Pin1 accelerates this conformation change by over 1,000-fold, and thus serves as a regulator of proline-directed phosphorylation [10, 11, 21]. Although there are a number of peptidyl-prolyl cis-trans isomerases (PPIases), Pin1 is the only one known so far that specifically targets the pSer/Thr-Pro sequence [22]. Pin1’s specificity derives from its two-domain structure. The WW domain binds only to specific pSer/Thr-Pro motifs, while the PPIase domain catalyzes the conformational change [4]. The role of Pin1 in regulating pro-directed phosphorylation in illustrated in (Figure 2.)
Figure 1. Structure of Pin1.
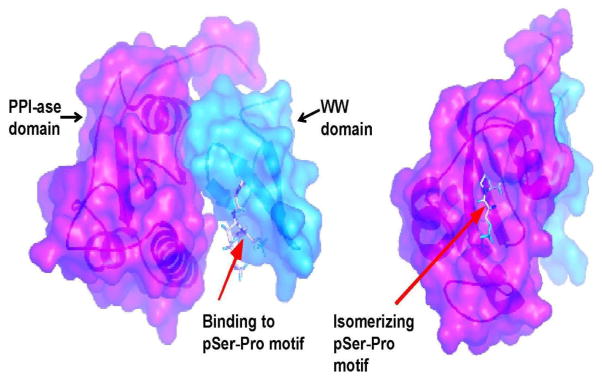
Pin1 has a unique substrate specificity that derives from its two domain structure. The WW domain specifically binds to the phosphorylated serine/threonine residue followed by a proline, and the PPI-ase domain flips the protein’s orientation around the proline bond.
Figure 2. Action of Pin1 on target proteins.
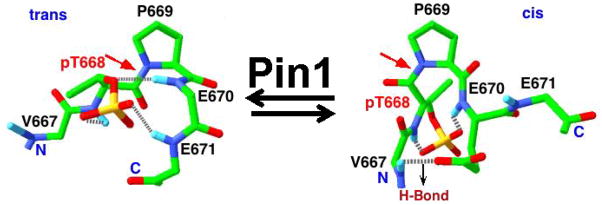
Pin1 is the only known enzyme thus far that can efficiently catalyze conformational change of the targets of proline-directed phosphorylation. H-bond = hydrogen bond. PT688 is the target of proline-directed phosphorylation, and other landmark amino acids are listed in black.
The changes in conformation catalyzed by Pin1 can affect a spectrum of substrate activities. The change in shape may serve as an “on-off” switch for target proteins- for example, by activating or deactivating an enzyme’s catalytic site. Pin1 can also serve a “maintenance” role by returning proteins from a dysfunctional “cis” conformation back into functional “trans.” In addition to affecting the shape and function of individual proteins, Pin1 has also been shown to act as a “molecular timer” that can act on many targets within a complex cellular process such as mitosis at different times and by multiple mechanisms [4]. Pin1’s dual role in the regulation of cell signaling and maintenance of protein folding helps explain why its expression levels vary widely in different tissues. Pin1 usually has very low expression in cells that are not proliferating. Expression increases with cell proliferative capacity and Pin1 over-expression is seen in most human cancers [23–25]. Pin1 is also activated in cancer by posttranslational modifications including dephosphorylation [26], phosphorylation [27, 28] and desumoylation [29]. Pin1 activity is dramatically suppressed by the tumor suppressor gene BRCA-1 [30]. Pin1 catalytic activity and oncogenic function are also effectively suppressed by the tumor suppressor DAPK1 [15]. It is thus easy to see why Pin1 is tightly regulated in cells with mitotic potential. In stark contrast, Pin1 is highly expressed in neurons from the beginning of neuronal differentiation, suggesting that it serves a completely different purpose in these post-mitotic cells [31, 32].
3. Pin1 and aging
Studies of Pin1-deficient mice suggest that it works to preserve cellular integrity in the face of aging. Pin1- knockout mice appear normal until about half-way through their lifespan, when they develop diffuse signs of premature aging, including neurodegeneration, osteoporosis, atrophy of skin and retina, loss of body mass, and accelerated telomere shortening (Figure 3) [32–34]. There are a number of mechanisms by which Pin1 may help promote healthy aging through maintaining genomic integrity and regulating the cellular response to stress. The p53 gene is generally considered the “guardian of the genome” and can trigger senescence or apoptosis in response to DNA damage [35]. p53 is therefore a tumor suppressor and is commonly deleted or mutated in cancer cells. Pin1 preserves the function of p53 in the setting of response to DNA damage by preventing its degradation by the ubiquitin proteasome system [36, 37]. It also enhances the DNA-binding activity of p53 to its targets, and is actually required to maintain the DNA damage checkpoints which allow cells to repair critical DNA damage [37].
Figure 3. Pin1 and regulation of aging.

Compared to wild-type mice (WT), Pin1 knockout mice (Pin1−/−) develop widespread signs of premature aging but are very resistant to cancer.
Pin1 is also involved in the maintenance of telomeres- the critically important protective caps on the ends of linear chromosomes. Telomere shortening is related to many age-related diseases including some cancers, cardiovascular disease and neurodegeneration. Pin1 regulates the stability of the telomeric DNA-binding protein TRF1 [38]. When in its cis-conformation, TRF1 protein is stable and inhibits telomere elongation by binding to telomeres. Pin1 flips TRF1 into trans, TRF1 is susceptible to proteasome-mediated degradation, thereby allowing telomere elongation to occur via the enzyme telomerase. Pin1 also helps to limit oxidative damage by its negative regulation of the CDK inhibitor p27kip1 through binding to FOXO4, a protein involved in the response to mitochondrial and oxidative stress [39]. The fact that Pin1 is highly expressed in neurons and is oxidized and inactivated in the hippocampus of patients with MCI and AD [40, 41] suggest that it may take part in the early response to oxidative stress. Together, these data point to Pin1 as a key regulator of healthy aging. As we will now see, these and other anti-aging properties of Pin1 have strong neuroprotective effects.
4. Pin1 and neuronal health
The physiological function of Pin1 in neurons is not fully understood, but it is expressed at very high levels. Pin1 is known to regulate important neuronal proteins such as tau, amyloid precursor protein (APP), myeloid cell leukemia sequence-1 (MCL-1) and gephyrin [42–45]. The function of APP remains poorly understood, but tau is known to be necessary for the stabilization of the microtubules that form the neuronal scaffolding and serve as transport channels between cell body and synapse.
Although familial forms of AD are uniformly associated with mutations in genes involved in the processing of amyloid precursor protein (APP), the strongest risk factor for sporadic AD, which accounts for 95% of all cases, is ageing. Pathologically, AD is characterized by the aggregation of abnormal beta-amyloid (Aβ) peptides into oligomers and plaques, and the accumulation of hyperphosphorylated tau protein into neurofibrillary tangles (NFTs). While the pathology of early and late onset disease is indistinguishable, sporadic AD is generally not associated with mutations in tau or APP. There is evidence that age-related processes such as oxidative damage and disrupted energy metabolism may precede abnormalities in these proteins [46, 47], and the link between Aβ and tau pathology remains obscure.
In contrast to cancer, Pin1 function is inhibited in human AD by multiple mechanisms, including Pin1 downregulation [48], Pin1 oxidation [41, 49], Pin1 phosphorylation [50] or Pin1 sequestration [51] or even possible Pin1 promoter polymorphisms [52], Pin1 knockout mice develop a syndrome similar to AD characterized by hyper-phosphorylated tau and neurodegeneration [32]. They also display pathogenic processing of APP, leading to Aβ accumulation [43, 53]. Currently, Pin1 is the only gene known so far that can produce both tangle and plaque pathology when deleted. When in their trans-conformations, tau and APP are healthy and functional. After phosphorylation, the proteins tend to switch to cis- and literally become “out of shape.”
We have developed conformation specific antibodies that can detect whether tau phosphorylated at Threonine 231 (pT231) is in cis or trans and have confirmed that cis is the early pathogenic species in MCI and AD [54]. Pin1 isomerization thus prevents the accumulation of cis and thus the buildup of pathogenic tau and Aβ, as illustrated in Figure 4. If not reversed to trans by Pin1, cis phosphorylated tau not only loses its normal function to bind and promote microtubule assembly, but also gain toxic function, becoming resistant to protein dephosphorylation and degradation and prone to protein aggregation and tangle formation. This process eventually leads to destabilization of cell structure and eventually to cell death [55, 56]. Protein phosphorylation occurs to some extent as part of the normal wear-and tear of the cell, and thus Pin1 may play the important housekeeping function of maintaining these and other proteins in their healthy conformations. During neurodegeneration, the activity of the kinases that phosphorylate tau, including GSK3 and CDK5, is upregulated [57, 58], and Pin1 seems to co-localize with insoluble tau aggregates. In brain specimens of patients with MCI and AD, levels of functional Pin1 were very low, further suggesting that Pin1 is an important source of protection against the misfolding that occurs when tau is hyperphosphorylated [59]. Neurons with NFTs in affected hippocampal neurons were substantially lower Pin1 than those with little or no NFTs.
Figure 4. Pin1 and Alzheimer’s disease.

Pin1 is highly expressed in adult neurons where it helps to keep tau and APP in healthy shape. Tau plays a key role in stabilizing microtubular structures. While the role of APP is poorly understood, it may have important neurotrophic properties. Once phosphorylated by protein kinases on specific sites, tau and APP can exist in –cis- and trans- conformations. The trans- isomer is still retains its physiological function and can be dephosphorylated and degraded. The cis- isomer is non-functional and is resistant to both dephosphorylation and degradation. Cis-APP is processed by the amyloidogenic pathway into pathogenic Aβ42. Pin1 protects ptau and pAPP by catalyzing their conversion from cis to trans as well as promoting their dephosphorylation. If Pin1 is absent or deficient, the GSK3 hyperphosphorylates tau and the cis conformation accumulates, eventually leading to plaque and tangle formation, neurodegeneration and cell death.
As with tau, the processing of APP can be regulated by phosphorylation under physiological conditions and hyperphosphorylation in the setting of neurodegeneration increases pathogenic Aβ production [7, 60, 61]. Pin1 binds specifically to Threonine668-Pro on APP and accelerates its transition from cis to trans, which not only keeps APP in the cell plasma membrane, favoring non-amyloidogenic processing that releases neurotrophic αAPPs [53, 62] but also increases APP turnover, preventing APP accumulation [63]. If not reversed to trans by Pin1, cis phosphorylated APP is not only resistant to proteasome-mediated degradation, leading to APP accumulation [63], but also internalizes to the endosomes and undergoes amyloidogenic processing, releasing intact Aβ peptide [53, 62] In addition, Pin1 is critical for inhibition of the kinase activity of GSK3. When Pin1 function is inhibited, GSK3 is hyperactivated, leading to hyperphosphorylation of tau and APP [63]. This and other evidence suggests that regulation of isomerization is a strategy to protect cellular proteins from unfavorable post-phosphorylation structural changes.
As one might expect, we have shown that transgenic overexpression of Pin1 in postnatal neurons in mice is able to protect against age-dependent neurodegeneration [64]. Moreover, in a Chinese population, we identified a functional polymorphism in the Pin1 promoter that leads to increased Pin1 expression as a result of a failure of AP4 to repress the promoter activity of the C variant. This polymorphism correlates with a 3-year delay in the age of onset of sporadic AD, further supporting the neuroprotective effect of Pin1 [65]. These results suggest that modulation of Pin1 might be a novel therapeutic target in AD.
5. Pin1 and cancer
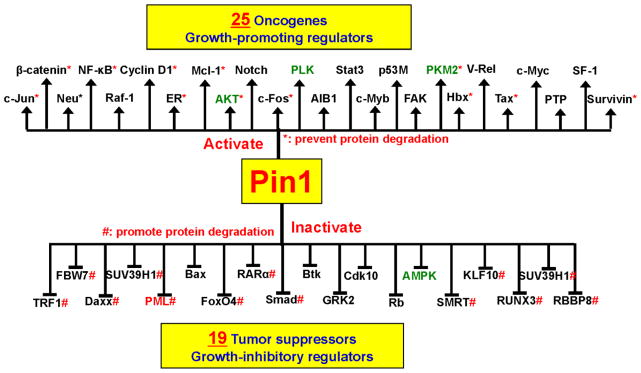
The cell uses proline-directed phosphorylation to help coordinate signaling pathways in cell proliferation, and so Pin1 overexpression can lead to the simultaneous upregulation of many oncogenic pathways [4]. (Figure 5) lists the oncogenes that can be activated by Pin1 and the tumor suppressor genes that it can deactivate. Under physiologic conditions, Pin1 expression and activity are highly regulated, playing a key role in the progression of the normal cell cycle. Progression is regulated by cyclins and cyclin dependent kinases (CDKs), and transition from the resting state (G0) to the first phase of the cell cycle (G1) is controlled by the cyclin D-cdk4,6 complex. Pin1 increases the expression and stability of Cyclin D1 and so fosters progression through G1 phase to S phase. During S phase, Pin1 is necessary for the coordination of DNA synthesis and centrosome duplication. The centrosome organizes the formation of the mitotic spindle poles needed to separate duplicated chromosomes [66, 67]. Pin1 also regulates many other mitotic phosphoproteins by switching them “on or off” through conformational change.
Figure 5. Pin1 helps coordinate mitosis in cycling cells.
Entry into and progression through the cell cycle requires the cell to release its tight control on the expression of mitotic proteins. Pin1 helps to coordinate the required suppression of the many proteins that suppress growth and mitosis with the expression of mitogens. In this sense, Pin1 acts as a molecular “switch,” allowing the cell to transition from maintenance to reproductive mode.
Overexpression of Pin1 promotes oncogenesis by acting on at least three major signaling pathways, all of which increase expression of Cyclin D1, as illustrated in Figure 6 [25, 34, 68, 69]. Pin1 overexpression also leads to the production of excess centrosomes, leading to genomic instability. Various oncogenes may promote cancer by increasing Pin1, which in turn activates and amplifies other oncogenic pathways. Thus, Pin1 is not an oncogene in itself, but can profoundly potentiate the effect of oncogenes. Pin1 overexpression is found in most human cancers, including the most common typeslung, prostate, breast and colon [24]. It portends a poor prognosis and can help predict risk of cancer recurrence [70, 71]. Inhibition of Pin1 in cancer cells leads either to apoptosis or reversal of transformation [3, 72], and Pin1-deficient mice are almost completely resistant to breast cancers induced by over-expression of the oncogene Ras or Neu [73], or by deletion of tumor suppressor p53 [74]. Moreover, a functional polymorphism in the Pin1 promoter that reduces Pin1 expression is associated with reduced risk for multiple cancers, further supporting the tumor-promoting activity of Pin1 [75]. Overexpression of Pin1 may also promote oncogenesis by increasing the levels of functional tau and APP. The normal function of APP is not known, but there is evidence it may stimulate growth in developing neurons [76]. Aberrant expression of APP has been associated with lung, breast, colon, pancreas and prostate cancer [77]. Tau protein stabilizes microtubules, which are necessary for mitosis. Overexpression of tau predicts less sensitivity to taxane therapy and a worse overall prognosis in breast, ovarian and a number of other cancers [78, 79].
Figure 6. Pin1 and upregulation of Cyclin D1.

One way in which Pin1 promotes oncogenesis is by increasing the expression of Cyclin D1, a key promoter of progression through the G1-S phase of the cell cycle. Pin1 accomplishes this by modulating the shapes of C-Jun Fos, β-catenin and NF-κβ proteins.
Together, hese results suggest that inhibition of Pin1 can suppress oncogenesis. However, it should be emphasized that cancer is not one disease but many, and even within a cancer type, each tumor will have its own genetic and metabolic characteristics. While it clearly fosters oncognensis in many cancer types, in one study of renal cell cancer, Pin1 actually suppressed tumor growth [80], and another study suggests its effect may differ in mice of different genetic background [81]. Thus, the relationship between Pin1 and cancer may be complex and remains an area for additional investigation.
6. Epidemiologic evidence for the inverse relationship between cancer and AD
The first evidence of an inverse association between AD and cancer came from autopsy [82, 83] and case control studies [84, 85]. Over the past decade, a number of cohort studies have found convincing evidence that cancer survivors develop less AD than expected, and that patients with AD develop less cancer [86–90].. Based on the observation that a history of cancer was less common in nursing home residents with AD, Roe and Behrens analyzed data from a longitudinal memory cohort [89]. People with AD had a hazard ratio (HR) of 0.39 (95% CI 0.21–0.74) for the development of cancer compared to those without AD, after adjustment for age, gender and level of education. Those with a history cancer at study baseline appeared to develop AD at a slower rate (HR=0.40; 95% CI 0.12–1.13). In a follow-up study using the Cardiovascular Health Study (CHS; n=3,020), the same group found that people with prevalent cancer at baseline had a lower risk of probable AD (HR=0.57; 95%CI=0.36–0.90), and those with prevalent AD had fewer cancer hospitalizations (HR=0.31; 95%CI 0.12–0.86) [90]. In the Framingham Heart Study cohort we showed that diagnosis with cancer decreased the risk of diagnosis with AD (HR=0.67; 95%CI 0.47–0.97), and that incident AD decreased the risk of subsequent diagnosis with cancer (HR=0.29; 95%CI 0.26–0.58)[91]. Multiple well-designed studies have now shown that this “protective” association is not simply due to decreased survival in people with cancer, as the inverse association is the same in those who survive as in those who die during follow-up [87, 91]. It is also not an artifact of decreased cancer detection in those with dementia, as people with vascular dementia and stroke actually have an increased risk of cancer diagnosis [90, 91]. Finally, the association is not likely due to treatment for cancer or AD, since the inverse relation is seen both before and after the diagnosis of each condition [87]. More work in very large cohorts is needed to determine which specific cancer types are inversely related to AD.
Pin1 represents the most extended studied biological mechanism that could help explain this unusual epidemiologic association. However a number of other biological mechanisms have now emerged with similar and opposite impacts on these two major age-related diseases. The propensity of dividing cells to become malignant and that of neurons to undergo apoptosis is based on their very different teleology [92]. Dividing cells rely on mitosis, while neurons must maintain their connections with networked cells indefinitely and do not retain the ability to complete mitosis once mature. A neuron does have adequate machinery for programmed cell death, however, and will undergo apoptosis in the face of overwhelming damage. Thus, differential regulation of genes controlling the cell cycle, cell survival and apoptosis might be differentially regulated in neurons and dividing cells [93]. A recent transcriptomic meta-analysis seeking genes that are differentially regulated in cancer and AD identified Pin1 along with Wnt, P53 and a number of novel candidates [94]. Similarly, common pathogenic processes such as oxidative stress and damage [95], metabolic dysregulation [96, 97] as well as inflammation and immune dysregulation [1], may have different manifestations and effects in each cell type. Thus, further invesigation of the genetic and metabolic overlap between cancer and AD may yield important pathophysiological and therapeutic insights for both diseases.
7. Therapeutic implications of Pin1
Pin1-specific inhibitors offer the potential of a novel anti-cancer strategy that can disrupt many oncogenic pathways at the same time. As Pin1 knockout mice develop normally until adulthood and display little phenotypes for about two-thirds of lifespan in mice, it is possible that specifically targeting Pin1 may not have general toxic effects. Juglone, one of the few known Pin1 inhibitors, binds to and inactivates a cysteine residue in the Pin1 active site [98]. While it has anti-cancer activity, juglone inhibits many other proteins and thus lacks the necessary specificity [99]. Many of the anti-proliferative effects of EGCG, the major flavonoid in green tea, seem to be mediated by Pin1 inhibiting effects [100]. This compound reduced the growth of human breast cancer cells in mouse xenografts [101]. Although EGCG is also not a specific Pin1-inhibitor, further study of its mechanism will likely provide additional insights for drug design. A number of features make Pin1 an attractive target for the design of small molecule inhibitors, including its well-defined active site and very high specificity as well as the potential to inactivate numerous oncogenes and to activate many tumor suppressors. A number of labs have been actively pursuing molecules that can inactivate either the PPIase or the WW domain [102]. Hopefully, these efforts will soon identify specific Pin1 inhibitors that can be used in clinical trials.
The inverse association between cancer and AD brings up obvious concerns about drugs that modulate Pin1 activity. In the case of Pin1 inhibitors for cancer, the solution may be to design drugs that do not cross the blood brain barrier. Because premature aging in knockout mice occurs only after long-term loss of Pin1, short-term anti-cancer therapy may not have acute effects. Proteasome inhibitors are commonly used in the treatment of cancer. Although they can cause Parkinson’s disease in mouse models, therapy-related Parkinsonism has not been reported in cancer patients.
Upregulation of Pin1 could be a viable strategy for neuroprotection if it could be limited to neurons, due its oncogenic potential in cycling cells. Another approach would be to determine the cause of Pin1 inactivation early in the pathogenesis of the disease and prevent it. Our current approach to designing Pin1-based therapy for neurodegenerative disease is based on our data that cis, but not trans, pT231 is an extremely early pathogenic conformation that can lead to AD [54]. Unlike Aβ plaques, tauopathy positively correlates with memory loss in AD patients [103, 104]. However, immunotherapy against tau has fallen far behind partially because misfolded tau was traditionally thought to be intracellular and thus inaccessible to antibodies. It is now known that toxic tau is actively transported from cells and can spread and transmit tauopathy between neurons [105–108] and that antibodies against p-tau induced by active or passive immunization reduce p-tau aggregates and ameliorate memory loss in AD mouse models [109–113]. We are now using conformation-specific antibodies to develop therapies that specifically target the pathogenic cis-conformation of tau, while leaving healthy-tau unharmed. Given that AD takes at least a decade to develop, immunotherapies specifically targeting the early pathogenic p-tau conformations is an exciting approach to arresting disease progression.
Both cancer and AD are broad terms to describe what are in reality a diverse group of diseases with a similar phenotype. As the effects of Pin1-based therapies might depend on the specific type of cancer or its history of development and the specific nature of an individual’s neurodegenerative disease or neural biochemistry, more work is needed to determine when and in which patients targeting Pin1 would make therapeutic sense.
8. Conclusion
The interesting story of Pin1 and its role in cancer and AD illustrates the very different priorities of cycling cells and neurons. Pin1 plays a key role in coordinating and controlling the cell cycle, which is critical to the survival of most tissues. Upregulation of Pin1 promotes cancer through many simultaneous pathways and effective Pin1 inhibitors targeted to cycling cells would be powerful anticancer therapy. The neuron survives not by division but through endurance, and its priority is somatic maintenance in the face of the forces of oxidation, somatic mutation, accumulation of waste products and other aging-related phenomena. Here Pin1 plays a neuroprotective role, helping to put misfolded proteins back to work, avoiding the accumulation of insoluble proteins, and responding to oxidative stress. More research is needed to better understand why Pin1 gets inactivated in AD and how this might be prevented. In the meantime, the ability to identify and target the toxic form of p-tau offers the possibility of developing neuroprotective therapies.
Highlight.
Pin1 regulates the function of a subset of phosphoproteins by catalyzing cis-trans isomerization.
Pin1 is activated and thereby promotes tumorigenesis by turning on multiple cancer pathways.
Pin1 is inhibited and thereby promotes against Alzheimer’s disease by acting on tau and APP.
Pin1 opens exciting preventive and therapeutic horizons for both cancer and neurodegeneration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tabares-Seisdedos R, Rubenstein JL. Inverse cancer comorbidity: a serendipitous opportunity to gain insight into CNS disorders. Nature reviews Neuroscience. 2013;14:293–304. doi: 10.1038/nrn3464. [DOI] [PubMed] [Google Scholar]
- 2.Lu KP. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem Sci. 2004;29:200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 4.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 5.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 6.Pawson T, Scott JD. Protein phosphorylation in signaling--50 years and counting. Trends Biochem Sci. 2005;30:286–290. doi: 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Lee MS, Tsai LH. Cdk5: one of the links between senile plaques and neurofibrillary tangles? J Alzheimers Dis. 2003;5:127–137. doi: 10.3233/jad-2003-5207. [DOI] [PubMed] [Google Scholar]
- 8.Lu KP, Liou YC, Vincent I. Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer’s disease. Bioessays. 2003;25:174–181. doi: 10.1002/bies.10223. [DOI] [PubMed] [Google Scholar]
- 9.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 10.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld J, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism. Science. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 11.Lu PJ, Zhou XZ, Shen M, Lu KP. A function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 12.Zhou XZ, Lu PJ, Wulf G, Lu KP. Phosphorylation-dependent prolyl isomerization: A novel signaling regulatory mechanism. Cell Mol Life Sci. 1999;56:788–806. doi: 10.1007/s000180050026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura K, Greenwood A, Binder L, Bigio EH, Denial SJ, Nicholson L, Zhou XZ, Lu KP. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell. 2012;149:232–244. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Driver JA, Zhou XZ, Lu KP. Regulation of protein conformation by Pin1 offers novel disease mechanisms and therapeutic approaches in Alzheimer’s disease. Discovery medicine. 2014;17:93–99. [PMC free article] [PubMed] [Google Scholar]
- 15.Lee TH, Chen CH, Suizu F, Huang P, Schiene-Fischer C, Daum S, Zhang YJ, Goate A, Chen RH, Zhou XZ, Lu KP. Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol Cell. 2011;42:147–159. doi: 10.1016/j.molcel.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liou YC, Zhou XZ, Lu KP. Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci. 2011;36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 18.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nature Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 19.Lee TH, Pastorino L, Lu KP. Peptidyl-prolyl cis-trans isomerase Pin1 in aging, cancer and Alzheimer’s disease. Expet Rev Mol Med. 2011;13:e21. doi: 10.1017/S1462399411001906. [DOI] [PubMed] [Google Scholar]
- 20.Liou YC, Zhou XZ, Lu KP. The prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci. 2011;36:501–514. doi: 10.1016/j.tibs.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic peptidyl-prolyl isomerase Pin1 suggests that substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 22.Lu KP, Liou YC, Zhou XZ. Pinning down the proline-directed phosphorylation signaling. Trends Cell Biol. 2002;12:164–172. doi: 10.1016/s0962-8924(02)02253-5. [DOI] [PubMed] [Google Scholar]
- 23.Atchison FW, Capel B, Means AR. Pin1 regulates the timing of mammalian primordial germ cell proliferation. Development. 2003;130:3579–3586. doi: 10.1242/dev.00584. [DOI] [PubMed] [Google Scholar]
- 24.Bao L, Sauter G, Sowadski J, Lu KP, Wang D. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–1737. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Lu KP. Pin1 is overexpressed in breast cancer and potentiates the transcriptional activity of phosphorylated c-Jun towards the cyclin D1 gene. EMBO J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee TH, Chen CH, Suizu F, Huang P, Schiene-Fischer C, Daum S, Zhang YJ, Goate A, Chen RW, Lu KP. Death associated protein kinase 1 phosphoylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol Cell. 2011;22:147–159. doi: 10.1016/j.molcel.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckerdt F, Yuan J, Saxena K, Martin B, Kappel S, Lindenau C, Kramer A, Naumann S, Daum S, Fischer G, Dikic I, Kaufmann M, Strebhardt K. Polo-like kinase 1-mediated phosphorylation stabilizes Pin1 by inhibiting its ubiquitination in human cells. J Biol Chem. 2005;280:36575–36583. doi: 10.1074/jbc.M504548200. [DOI] [PubMed] [Google Scholar]
- 28.Rangasamy V, Mishra R, Sondarva G, Das S, Lee TH, Bakowska JC, Tzivion G, Malter JS, Rana B, Lu KP, Kanthasamy A, Rana A. Mixed-lineage kinase 3 phosphorylates prolyl-isomerase Pin1 to regulate its nuclear translocation and cellular function. Proc Natl Acad Sci U S A. 2012;109:8149–8154. doi: 10.1073/pnas.1200804109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CH, Chang CC, Lee TH, Luo ML, Huang P, Liao PH, Wei S, Li FA, Chen RH, Zhou XZ, Shih HM, Lu KP. SENP1 desumoylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2013;73:3951–3962. doi: 10.1158/0008-5472.CAN-12-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacLachlan TK, Somasundaram K, Sgagias M, Shifman Y, Muschel RJ, Cowan KH, El-Deiry WS. BRCA1 effects on the cell cycle and the DNA damage response are linked to altered gene expression. The Journal of biological chemistry. 2000;275:2777–2785. doi: 10.1074/jbc.275.4.2777. [DOI] [PubMed] [Google Scholar]
- 31.Hamdane M, Dourlen P, Bretteville A, Sambo AV, Ferreira S, Ando K, Kerdraon O, Begard S, Geay L, Lippens G, Sergeant N, Delacourte A, Maurage CA, Galas MC, Buee L. Pin1 allows for differential Tau dephosphorylation in neuronal cells. Molecular and cellular neurosciences. 2006;32:155–160. doi: 10.1016/j.mcn.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 32.Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Uchida T, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 33.Lee TH, Tun-Kyi A, Shi R, Lim J, Soohoo C, Finn G, Balastik M, Pastorino L, Wulf G, Zhou XZ, Lu KP. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat Cell Biol. 2009;11:97–105. doi: 10.1038/ncb1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liou YC, Ryo R, Huang HK, Lu PJ, Bronson R, Fujimori F, Uchidafl U, Hunter T, Lu KP. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc Natl Acad Sci USA. 2002;99:1335–1340. doi: 10.1073/pnas.032404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slee EA, O’Connor DJ, Lu X. To die or not to die: how does p53 decide? Oncogene. 2004;23:2809–2818. doi: 10.1038/sj.onc.1207516. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani F, Tocco F, Girardini J, Smith P, Gasco M, Lu X, Crook T, Del Sal G. The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat Struct Mol Biol. 2007;14:912–920. doi: 10.1038/nsmb1306. [DOI] [PubMed] [Google Scholar]
- 37.Wulf GM, Liou YC, Ryo A, Lee SW, Lu KP. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. The Journal of biological chemistry. 2002;277:47976–47979. doi: 10.1074/jbc.C200538200. [DOI] [PubMed] [Google Scholar]
- 38.Lee HW, Blasco MA, Gottlieb GJ, WnHorner J, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 39.Brenkman AB, de Keizer PL, van den Broek NJ, van der Groep P, van Diest PJ, van der Horst A, Smits AM, Burgering BM. The peptidyl-isomerase Pin1 regulates p27kip1 expression through inhibition of Forkhead box O tumor suppressors. Cancer Res. 2008;68:7597–7605. doi: 10.1158/0008-5472.CAN-08-1059. [DOI] [PubMed] [Google Scholar]
- 40.Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB, Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 41.Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Markesbery WR, Zhou XZ, Lu KP, Butterfield DA. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol Aging. 2006;27:918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Li QF, Wu CT, Duan HF, Sun HY, Wang H, Lu ZZ, Zhang QW, Liu HJ, Wang LS. Activation of sphingosine kinase mediates suppressive effect of interleukin-6 on human multiple myeloma cell apoptosis. British journal of haematology. 2007;138:632–639. doi: 10.1111/j.1365-2141.2007.06711.x. [DOI] [PubMed] [Google Scholar]
- 43.Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 44.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Küllertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 45.Zita MM, Marchionni I, Bottos E, Righi M, Del Sal G, Cherubini E, Zacchi P. Postphosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors function. The EMBO journal. 2007;26:1761–1771. doi: 10.1038/sj.emboj.7601625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet neurology. 2011;10:187–198. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 48.Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, Huang HK, Bronson R, Bing G, Li X, Hunter T, Lu KP. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424:556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 49.Chen CH, Lib W, Sultana R, You MH, Kondo A, Shahpasand K, Kim BO, Luo ML, Nechama M, Lin YM, Yao Y, Lee TH, Zhou XZ, Swomley AM, Butterfield DA, Zhang Y, Lu KP. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol Dis. 2014 doi: 10.1016/j.nbd.2014.12.027. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim BM, You MH, Chen CH, Lee S, Hong Y, Hong Y, Kimchi A, Zhou XZ, Lee TH. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis. 2014;5:e1237. doi: 10.1038/cddis.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 52.Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, Clerici M, Arosio B, Crovella S. Pin1 promoter polymorphisms are associated with Alzheimer’s disease. Neurobiol Aging. 2007;28:69–74. doi: 10.1016/j.neurobiolaging.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 53.Pastorino L, Ma SL, Balastik M, Huang P, Pandya D, Nicholson L, Lu KP. Alzheimer’s disease-related loss of Pin1 function influences the intracellular localization and the processing of AbetaPP. J Alzheimers Dis. 2012;30:277–297. doi: 10.3233/JAD-2012-111259. [DOI] [PubMed] [Google Scholar]
- 54.Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L, Zhou XZ, Lu KP. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell. 2012;149:232–244. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 56.Iqbal K, Liu F, Gong CX, del Alonso AC, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009;118:53–69. doi: 10.1007/s00401-009-0486-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cruz JC, Tsai LH. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- 59.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 60.Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci. 2006;26:10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 62.Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson L, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 63.Ma SL, Pastorino L, Zhou XZ, Lu KP. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3beta (GSK3beta) activity: novel mechanism for Pin1 to protect against Alzheimer disease. The Journal of biological chemistry. 2012;287:6969–6973. doi: 10.1074/jbc.C111.298596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lim J, Balastik M, Lee TH, Nakamura K, Liou YC, Sun A, Finn G, Pastorino L, Lee VM, Lu KP. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. The Journal of clinical investigation. 2008;118:1877–1889. doi: 10.1172/JCI34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma SL, Tang NL, Tam CW, Lui VW, Lam LC, Chiu HF, Driver JA, Pastorino L, Lu KP. A PIN1 polymorphism that prevents its suppression by AP4 associates with delayed onset of Alzheimer’s disease. Neurobiol Aging. 2012;33:804–813. doi: 10.1016/j.neurobiolaging.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bornens M. Centrosome composition and microtubule anchoring mechanisms. Curr Opin Cell Biol. 2002;14:25–34. doi: 10.1016/s0955-0674(01)00290-3. [DOI] [PubMed] [Google Scholar]
- 67.Doxsey S. Duplicating dangerously: linking centrosome duplication and aneuploidy. Mol Cell. 2002;10:439–440. doi: 10.1016/s1097-2765(02)00654-8. [DOI] [PubMed] [Google Scholar]
- 68.Ryo A, Liou YC, Wulf G, Nakamura N, Lee SW, Lu KP. Pin1 is an E2F target gene essential for the Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ryo A, Nakamura N, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nature Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- 70.Ayala G, Wang D, Wulf G, Frolov A, Le R, Wheeler T, Sowadski JM, Lu KP, Bao L. Pin1 is a novel prognostic marker in prostate cancer. Cancer Research. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 71.Sasaki T, Ryo A, Uemura H, Ishiguro H, Inayama Y, Yamanaka S, Kubota Y, Nagashima Y, Harada M, Aoki I. An immunohistochemical scoring system of prolyl isomerase Pin1 for predicting relapse of prostate carcinoma after radical prostatectomy. Pathol Res Pract. 2006;202:357–364. doi: 10.1016/j.prp.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 72.Rippmann JF, Hobbie S, Daiber C, Guilliard B, Bauer M, Birk J, Nar H, Garin-Chesa P, Rettig WJ, Schnapp A. Phosphorylation-dependent proline isomerization catalyzed by Pin1 is essential for tumor cell survival and entry into mitosis. Cell Growth Differ. 2000;11:409–416. [PubMed] [Google Scholar]
- 73.Wulf G, Garg P, Liou YC, Iglehart D, Lu KP. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004;23:3397–3407. doi: 10.1038/sj.emboj.7600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takahashi K, Akiyama H, Shimazaki K, Uchida C, Akiyama-Okunuki H, Tomita M, Fukumoto M, Uchida T. Ablation of a peptidyl prolyl isomerase Pin1 from p53-null mice accelerated thymic hyperplasia by increasing the level of the intracellular form of Notch1. Oncogene. 2007;26:3835–3845. doi: 10.1038/sj.onc.1210153. [DOI] [PubMed] [Google Scholar]
- 75.Tao LJ, Chen YS, Yao L, Zou B, Tao LS, Kong J, Liu YQ, Cao Q, Yin CJ. promoter polymorphism (−842 G>C) contributes to a decreased risk of cancer: Evidence from meta-analysis. Oncology letters. 2014;8:1360–1366. doi: 10.3892/ol.2014.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hung AY, Koo EH, Haass C, Selkoe DJ. Increased expression of beta-amyloid precursor protein during neuronal differentiation is not accompanied by secretory cleavage. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:9439–9443. doi: 10.1073/pnas.89.20.9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takagi K, Ito S, Miyazaki T, Miki Y, Shibahara Y, Ishida T, Watanabe M, Inoue S, Sasano H, Suzuki T. Amyloid precursor protein in human breast cancer: an androgen-induced gene associated with cell proliferation. Cancer science. 2013;104:1532–1538. doi: 10.1111/cas.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li ZH, Xiong QY, Tu JH, Gong Y, Qiu W, Zhang HQ, Wei WS, Hou YF, Cui WQ. Tau proteins expressions in advanced breast cancer and its significance in taxane-containing neoadjuvant chemotherapy. Medical oncology. 2013;30:591. doi: 10.1007/s12032-013-0591-y. [DOI] [PubMed] [Google Scholar]
- 79.Smoter M, Bodnar L, Grala B, Stec R, Zieniuk K, Kozlowski W, Szczylik C. Tau protein as a potential predictive marker in epithelial ovarian cancer patients treated with paclitaxel/platinum first-line chemotherapy. Journal of experimental & clinical cancer research: CR. 2013;32:25. doi: 10.1186/1756-9966-32-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teng BL, Hacker KE, Chen S, Means AR, Rathmell WK. Tumor suppressive activity of prolyl isomerase Pin1 in renal cell carcinoma. Molecular oncology. 2011;5:465–474. doi: 10.1016/j.molonc.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yeh ES, Means AR. PIN1, the cell cycle and cancer. Nature reviews Cancer. 2007;7:381–388. doi: 10.1038/nrc2107. [DOI] [PubMed] [Google Scholar]
- 82.Corsellis J. Mental Illness and the Ageing Brain. Oxford University Press; London: 1962. [Google Scholar]
- 83.Tirumalasetti F, Han L, Birkett DP. The relationship between cancer and Alzheimer’s disease. J Am Geriatr Soc. 1991;39:840. doi: 10.1111/j.1532-5415.1991.tb02713.x. [DOI] [PubMed] [Google Scholar]
- 84.DeSouky AL. The relationship between cancer and Alzheimer’s disease. J Am Geriatr Soc. 1992;40:1075. doi: 10.1111/j.1532-5415.1992.tb04490.x. [DOI] [PubMed] [Google Scholar]
- 85.Yamada M, Sasaki H, Mimori Y, Kasagi F, Sudoh S, Ikeda J, Hosoda Y, Nakamura S, Kodama K. Prevalence and risks of dementia in the Japanese population: RERF’s adult health study Hiroshima subjects. Radiation Effects Research Foundation. J Am Geriatr Soc. 1999;47:189–195. doi: 10.1111/j.1532-5415.1999.tb04577.x. [DOI] [PubMed] [Google Scholar]
- 86.Driver JA. Understanding the link between cancer and neurodegeneration. Journal of Geriatric Oncology. 2012;3:58–67. [Google Scholar]
- 87.Musicco M, Adorni F, Di Santo S, Prinelli F, Pettenati C, Caltagirone C, Palmer K, Russo A. Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology. 2013;81:322–328. doi: 10.1212/WNL.0b013e31829c5ec1. [DOI] [PubMed] [Google Scholar]
- 88.Ou SM, Lee YJ, Hu YW, Liu CJ, Chen TJ, Fuh JL, Wang SJ. Does Alzheimer’s disease protect against cancers? A nationwide population-based study. Neuroepidemiology. 2013;40:42–49. doi: 10.1159/000341411. [DOI] [PubMed] [Google Scholar]
- 89.Roe CM, Behrens MI, Xiong C, Miller JP, Morris JC. Alzheimer disease and cancer. Neurology. 2005;64:895–898. doi: 10.1212/01.WNL.0000152889.94785.51. [DOI] [PubMed] [Google Scholar]
- 90.Roe CM, Fitzpatrick AL, Xiong C, Sieh W, Kuller L, Miller JP, Williams MM, Kopan R, Behrens MI, Morris JC. Cancer linked to Alzheimer disease but not vascular dementia. Neurology. 2010;74:106–112. doi: 10.1212/WNL.0b013e3181c91873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Driver JA, Beiser A, Au R, Kreger BE, Splansky GL, Kurth T, Kiel DP, Lu KP, Seshadri S, Wolf PA. Inverse association between cancer and Alzheimer’s disease: results from the Framingham Heart Study. BMJ. 2012;344:e1442. doi: 10.1136/bmj.e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Driver JA. Inverse association between cancer and neurodegenerative disease: review of the epidemiologic and biological evidence. Biogerontology. 2014;15:547–557. doi: 10.1007/s10522-014-9523-2. [DOI] [PubMed] [Google Scholar]
- 93.Behrens MI, Lendon C, Roe CM. A common biological mechanism in cancer and Alzheimer’s disease? Current Alzheimer research. 2009;6:196–204. doi: 10.2174/156720509788486608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ibanez K, Boullosa C, Tabares-Seisdedos R, Baudot A, Valencia A. Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS genetics. 2014;10:e1004173. doi: 10.1371/journal.pgen.1004173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lanni C, Racchi M, Memo M, Govoni S, Uberti D. p53 at the crossroads between cancer and neurodegeneration. Free radical biology & medicine. 2012;52:1727–1733. doi: 10.1016/j.freeradbiomed.2012.02.034. [DOI] [PubMed] [Google Scholar]
- 96.Demetrius LA, Driver J. Alzheimer’s as a metabolic disease. Biogerontology. 2013;14:641–649. doi: 10.1007/s10522-013-9479-7. [DOI] [PubMed] [Google Scholar]
- 97.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hennig L, Christner C, Kipping M, Schelbert B, Rucknagel KP, Grabley S, Kullertz G, Fischer G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry. 1998;37:5953–5960. doi: 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- 99.Chao SH, Greenleaf AL, Price DH. Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic acids research. 2001;29:767–773. doi: 10.1093/nar/29.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shirakami Y, Shimizu M, Moriwaki H. Cancer chemoprevention with green tea catechins: from bench to bed. Current drug targets. 2012;13:1842–1857. doi: 10.2174/138945012804545506. [DOI] [PubMed] [Google Scholar]
- 101.Urusova DV, Shim JH, Kim DJ, Jung SK, Zykova TA, Carper A, Bode AM, Dong Z. Epigallocatechin-gallate suppresses tumorigenesis by directly targeting Pin1. Cancer Prev Res (Phila) 2011;4:1366–1377. doi: 10.1158/1940-6207.CAPR-11-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moore JD, Potter A. Pin1 inhibitors: Pitfalls, progress and cellular pharmacology. Bioorganic & medicinal chemistry letters. 2013;23:4283–4291. doi: 10.1016/j.bmcl.2013.05.088. [DOI] [PubMed] [Google Scholar]
- 103.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 104.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 105.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. The Journal of biological chemistry. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo JL, Lee VM. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. The Journal of biological chemistry. 2011;286 doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nature reviews Neuroscience. 2010;11:155–159. doi: 10.1038/nrn2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem. 2011;118:658–667. doi: 10.1111/j.1471-4159.2011.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Experimental neurology. 2010;224:472–485. doi: 10.1016/j.expneurol.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 113.Ubhi K, Masliah E. Recent advances in the development of immunotherapies for tauopathies. Experimental neurology. 2011;230 doi: 10.1016/j.expneurol.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]