


Abstract
We propose a novel mechanism of gene regulation in Mycobacterium tuberculosis where the protein Rv1222 inhibits transcription by anchoring RNA polymerase (RNAP) onto DNA. In contrast to our existing knowledge that transcriptional repressors function either by binding to DNA at specific sequences or by binding to RNAP, we show that Rv1222-mediated transcription inhibition requires simultaneous binding of the protein to both RNAP and DNA. We demonstrate that the positively charged C-terminus tail of Rv1222 is responsible for anchoring RNAP on DNA, hence the protein slows down the movement of RNAP along the DNA during transcription elongation. The interaction between Rv1222 and DNA is electrostatic, thus the protein could inhibit transcription from any gene. As Rv1222 slows down the RNA synthesis, upon expression of the protein in Mycobacterium smegmatis or Escherichia coli, the growth rate of the bacteria is severely impaired. The protein does not possess any significant affinity for DNA polymerase, thus, is unable to inhibit DNA synthesis. The proposed mechanism by which Rv1222 inhibits transcription reveals a new repertoire of prokaryotic gene regulation.
INTRODUCTION
Gene regulation is one of the most important requirements of microorganisms for their survival under a wide variety of fluctuating environmental niche (1). The majority of the genes are regulated during transcription and RNA polymerase (RNAP), the key enzyme for mRNA synthesis, together with different sigma factors and transcriptional regulators orchestrates gene expression in bacteria. The level of expression of a given gene generally depends on the cellular demand: the basal level gene expression could be sometimes activated or inhibited. The wide variety of mechanisms by which transcriptional regulators function have been unravelled over the years. While multiple mechanisms exist for the activation of gene expression, inhibition of transcription in prokaryotes could be classified mainly into four categories: (i) a ‘repressor’ occupies the promoter element, either partially or fully, preventing the binding of RNAP to the promoter (2,3), or binds to DNA beside RNAP blocking the promoter escape of RNAP during transcription initiation (4,5), (ii) a transcription factor binds to downstream DNA, thereby blocking the translocation of RNAP (6,7), (iii) an anti-sigma factor binds to a specific sigma factor, inducing a conformational change to the cognate sigma factor and making it deficient in recognizing the promoter elements (8,9) and (iv) a factor that alters the DNA architecture rendering it inaccessible for RNAP (10–12) or unfavourable for RNAP translocation (13). In majority of the above cases, repressors or anti-sigma factors function at specific promoters, and hence gene-specific transcriptional regulation occurs. In addition to the above four, there are transcription factors or small-molecule effectors that do not require any interaction with DNA for transcription inhibition. Hence, the inhibition of transcription by these factors occurs at any gene. For example, DkSA (14,15), Gfh1 (16,17) and ppGpp (18,19) inhibit transcription by binding at the secondary channel of RNAP and modulating the function of RNAP. Several bacteriophage proteins have been reported to inhibit transcription by different mechanisms. gp2 from the bacteriophage T7 binds to RNAP and induces a conformational change in the polymerase making it deficient for RNA synthesis (20). Bacteriophage HK022 Nun protein binds to the transcription elongation complex (EC) through a nut site and prevents the translocation of RNAP (21).
Rv1222, a Mycobacterium tuberculosis transcriptional factor, was reported to function as an anti-sigma factor for σE. Based on the facts that Rv1222 gene is located immediately downstream of sigE gene, Rv1222 binds to σE of the same bacteria, and exclusively inhibits transcription by σE-RNAP holoenzyme, it has been inferred that Rv1222 is a regulator of sigma E factor (RseA) (22,23). However, our study reveals that Rv1222 is not an anti-sigma factor, but inhibits transcription by a completely different mechanism.
Rv1222 is a small protein (16.25 kD) whose function is not known. Microarray mapping of transposon insertions shows that the protein is nonessential (24). Transcriptome analysis of M. tuberculosis, in dormant state or under conditions leading to dormant state, reveals that the mRNA of Rv1222 is upregulated (25–28).
Here, we show that Rv1222 anchors the RNAP onto DNA and thereby slows down the translocation of RNAP along the DNA and RNA synthesis. The inhibition of transcription requires the simultaneous binding of the protein to both RNAP core and DNA. The interaction of the protein with DNA is not sequence specific, hence the Rv1222-mediated inhibition of transcription can occur at any DNA template. When the interactions between Rv1222 and DNA are abrogated by removing 10 residues from the C-terminus, the protein loses its ability to inhibit transcription. On the other hand, as the protein does not bind to DNA polymerase (DNAP), Rv1222 is unable to anchor DNAP onto DNA. Interestingly, when Rv1222 is expressed in Mycobacterium smegmatis or Escherichia coli, the growth rate of the bacteria is significantly reduced due to reduction in the level of RNA synthesis. The way, Rv1222 inhibits transcription, represents a novel mechanism and reveals a new repertoire of prokaryotic gene regulation.
MATERIALS AND METHODS
Cloning strategies
The M. tuberculosis rv1222 gene was amplified by polymerase chain reaction (PCR) from H37Rv genomic DNA (a kind gift from ATCC, USA) using primers (Supplementary Table S1) and cloned in pET28a(+) and pAcYcDuet vectors using NdeI-HindIII and NcoI-HindIII (NEB), respectively. Rv1222ΔC was created by inserting a stop codon by site-directed mutagenesis, at 10 residues prior to the original stop codon of the protein. For Rv1222 expression in M. smegmatis, rv1222 gene was cloned in pLAM12 vector using restriction enzymes NdeI-EcoRI.
Previously, we purified M. tuberculosis (Mtb) RNAP–σA holoenzyme, by co-expressing all RNAP subunits using two-plasmid expression system (pETDuet-rpoB-rpoC and pAcYc-rpoA-sigA) in E. coli (29). For production of recombinant Mtb RNAP–σE holo, we followed the same strategy as above except sigA gene was replaced by sigE in pAcYcDuet-rpoA-sigA. First M. tuberculosis rpoA gene was cleaved with NcoI-BamHI from pET16b-rpoA (30) and cloned in pAcYc Duet. The M. tuberculosis sigE gene was amplified from Mtb genomic DNA H37Rv using primers (Supplementary Table S1) and subsequently cloned in pAcYcDuet-rpoA using EcoRV-XhoI restriction enzymes.
σE-dependent promoter Bpr (31) was amplified from H37Rv using primers and was cloned in pBluescript SK(+) plasmid using EcoRV restriction site. E. coli lacCONS promoter DNA (32) was amplified from 79 bases oligonucleotide template and cloned in pUC19 using KpnI-BamHI restriction enzymes. The lacCONS promoter was amplified from this construct (pUC19-lacCONS) using primers and subsequently cloned in pFPVmcherry with KpnI-XbaI enzymes. sinP3, rrnA (29) and abrB (33) promoters were prepared by PCR with synthetic primers and template and purified by PAGE elution.
Rv1222 protein purification
Using denaturation/renaturation method
E. coli BL21 (DE3) cells were transformed with pET28-rv1222 and grown in Luria Broth (LB) media overnight at 37°C. 2L LB media was inoculated with 1% of overnight culture and was supplemented with 0.5 mM IPTG after cells reached OD600 0.5 and was further grown for 3 h at 37°C. Harvested cells were suspended in buffer A (100 mM sodium phosphate (pH 7.0), 100 mM NaCl and 2 mM β-mercaptoethanol) containing 0.25% deoxycholic acid, protease cocktail inhibitor (Roche), lysed by sonication and centrifuged. The pellet was washed with buffer A + 0.25% triton-X100 + 1 mg/ml lysozyme and further centrifuged. The pellet was dissolved in buffer B (buffer A+ 8M urea) and loaded on Ni-NTA column (Rv1222 gene fused with 6X-histidine at the N- terminus) pre-equilibrated with buffer B, washed with five column volume of buffer B and eluted with buffer B + 100 mM imidazole. The Rv1222 was purified to near-homogeneity by nickel affinity chromatography under denaturing conditions as judged by 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and Coomassie blue staining. The eluted protein was dialysed against buffer A containing 10 μM ZnCl2 with three changes at an interval of 15 h at 4°C. The dialysed protein was concentrated using concentrator (Amicon Ultra 10K), mixed with equal volume 100% glycerol and stored in −80°C. All assays were performed with this refolded Rv1222 protein.
By expressing the protein in soluble form
The E. coli SoluBL21 (Amsbio) cells were transformed with pET28-rv1222 and were grown in M9 minimal media (HiMedia) overnight. One litre fresh M9 media was inoculated with 1% of overnight cultures and was supplemented with 0.5 mM IPTG after cells reached OD600 0.5 and was further grown overnight at 37°C. Cells were harvested, lysed by sonication and purified by Ni-NTA chromatography using buffer A as above. In vitro transcription assay shows that the activity of this Rv1222 is similar to the activity of Rv1222 purified by denaturation/renaturation method (Supplementary Figure S1).
Purification of Mtb RNAP core, Mtb RNAP–σA holo, Mtb RNAP–σE holo and Mtb σA
Mtb RNAP core, Mtb RNAP–σA holo, Mtb RNAP–σE holo and Mtb σA were purified following the protocol as in (29).
Purification Bs RNAP core and Bs σA
The proteins were purified essentially as in (34).
Purification of σE
E. coli BL21 (DE3) cells containing pET30-sigE (gift from Dr Rodrigue) were grown in 1L LB (containing 50 μg/ml Kanamycin) at 37°C till OD600 reached 0.5. Protein production was induced by adding 0.5 mM IPTG, followed by growing them for 3 h at 37°C. Cells were harvested by centrifugation (6000 rpm, 10 min, 4°C), resuspended in 20 ml buffer (50 mM Tris-Cl, 200 mM KCl, 10 μM ZnCl2, 5 mM βME, 1 mM PMSF, 20% Glycerol) and disrupted by sonication. The lysates were spun at 25,000 rpm for 30 min at 4°C. The supernatant was loaded onto Ni-NTA column pre-equilibrated with the above buffer and protein was eluted at 400 mM imidazole. The protein was further purified on MonoQ HR10/10 in Akta purifier (GE Healthcare) using a 0.2–1.0 M NaCl gradient in buffer (20 mM Tris-Cl and 5% Glycerol). The purified protein was concentrated and kept at −80°C after adding equal volume of glycerol.
Purification of Ec holo RNAP
E. coli RNAP holo was purified as in (35).
EMSA assay
The forward primer for Bpr DNA fragment was labelled using 32P γ ATP and T4 polynucleotide kinase (NEB) following manufacturers’ protocol. The promoter DNA fragment was then amplified by PCR and was precipitated with equal volume of isopropanol and 0.1 volume of 3M Na-acetate (pH 5.3). Two sets of EMSA assays were performed. In the first set, 100 nM σE was incubated respectively with 0, 100, 200 and 400 nM Rv1222 in Tx buffer [45 mM Tris-Cl (pH 8), 5 mM MgCl2, 70 mM KCl, 1 mM DTT, 10% Glycerol, 1.5 mM MnCl2] at 37°C for 5 min and the samples were added with 100 nM RNAP and 20 nM 32P-labelled DNA and further incubated at 37°C for 15 min to form open complex. In the second set, 100 nM RNAP–σE holo was incubated respectively with 0, 100, 200 and 400 nM Rv1222 in Tx buffer at 37°C for 5 min and then added with 20 nM 32P-labelled DNA, following incubation at 37°C for 15 min. In both cases, heparin was added to the samples at 0.5 μg/μl before resolving on 5% PAGE in 1X TBE buffer (89 mM Tris base, 89 mM Boric acid, 2 mM EDTA) for 1 h at 4°C and then examined by phosphor imaging (Typhoon Trio + GE Healthcare).
In vitro transcription assay
Two sets of in vitro transcription assays were performed with RNAP–σE holo. In the first set, 100 nM RNAP–σE holo was incubated respectively with 0, 100, 200 and 400 nM Rv1222 in 10 μl Tx buffer at 37°C for 5 min and then was added with 50 nM Bpr promoter DNA fragment following an incubation at 37°C for 15 min to form open complex. In the second set, 100 nM RNAP–σE holo was incubated 50 nM Bpr promoter DNA fragment in 10 μl Tx buffer 37°C for 15 min to form open complex and then added with Rv1222 (0, 100, 200 and 400 nM) for 5 min at 37°C. Heparin was added to the samples at 0.5 μg/μl before transcription was initiated with NTP (125 μM ATP, GTP, CTP and 20 μM α32P-CTP (0.4 μCi)) as in Banerjee et al. After 5 min, the reactions were terminated by addition of 2.5 μl of (Formamide Loading Buffer (FLB): 80% formamide, 10 mM EDTA, 0.01% Bromophenol Blue, 0.01% Xylene Cyanol), resolved in 12% Urea-PAGE and was scanned by storage phosphor scanner (Typhoon Trio + GE healthcare).
In vitro transcription assay using 100 nM Mtb RNAP–σA holo and 50 nM rrnA promoter DNA fragment was performed essentially as above except Rv1222 was incubated with open complex for 5 min at 37°C before heparin was added. The transcription reactions were terminated after 5 min following the addition of NTP unless stated otherwise.
In vitro transcription assay using 100 nM Ec RNAP–σ70 holo and 50 nM lacCONS DNA was performed essentially as in Mukhopadhyay et al. (35), except Rv1222 was incubated with open complex for 5 min at 37°C before heparin was added. The transcription reactions were terminated after 5 min following the addition of NTP unless stated otherwise.
For nuclease activity assay, P32-labelled transcripts (81 nt) were formed using in vitro transcription assay with 100 nM Ec RNAP–σ70 holo and 50 nM T7A1 promoter DNA fragment. Rv1222 was added after the formation of transcripts and incubated for 5 min before resolving on 12% Urea-PAGE.
Single-round transcription assays to monitor the rate of RNA synthesis in the absence and presence of Rv1222 by Mtb RNAP or Ec RNAP were performed as above except that the reaction was stopped at different time points before resolving on 12% Urea-PAGE.
In vitro transcription assay using Bs RNAP–σA holo was performed as following: 100 nM Bs RNAP core was incubated 200 nM Bs σA in buffer [18 mM Tris (pH 8.0), 10 mM NaCl, 8 mM βME, 10 mM MgCl2] and incubated on ice for 30 min to form holo enzyme. Fifty nanomolar abrB promoter DNA was added to the holoenzyme to form open complex at 37°C for 20 min and further added with Rv1222 (0, 100, 200 and 400 nM) and incubated at 37°C for 5 min. Transcription was initiated with NTP (final concentration: 250 μM of ATP, GTP, UTP and 25 μM of α32P-CTP (0.4 μCi) at 37°C for 5 min.
Approximate IC50 of transcription inhibition for each of the three RNAPs were estimated as follows: the intensity on radioactive band of the run-off transcript at each Rv1222 concentration was quantified from the phosphor imaging of the gels and the mean values of the intensities from two or three replicates were plotted against the concentration of Rv1222.
In vitro transcription assay using 100 nM Mtb RNAP core and 50 nM tailed-template DNA was performed essentially as in Gnatt et al. except that 100 nM Mtb RNAP core was incubated with varying concentrations of Rv1222 and then was added with 50 nM tailed-template DNA fragment. Transcription was initiated with NTP (final concentration: 250 μM of ATP, GTP, UTP and 25 μM of α32P-CTP (0.2 μCi)) at 37°C for 5 min.
In vitro transcription assay with 0.2U of T7 RNA Polymerase (Bio-Bharati India Ltd) and 100 nM T7 promoter DNA fragment was performed as in (36).
In vitro transcription assay using Ec RNAP core and Kool NC-45™ Template was performed as per manufacturers’ protocol (epicentre).
Fluorescence anisotropy assays
Labelling of Rv1222 with TMR
Cysteine labelling was performed with Rv1222 protein having three cysteine residues at positions 70, 73 and 109. One hundred micromolar purified protein in 200 μl was reduced as in (37). The sample was reacted with 5-fold molar excess of tetramethylrhodamine (TMR)-6-maleimide in buffer [100 mM sodium phosphate (pH 7.3), 200 mM NaCl, 1 mM EDTA]. The protein sample was centrifuged for 10 min at 13 000 rpm and then loaded onto 10 ml BioGel P6 column (Bio-Rad) pre-equilibrated with buffer C to remove the free dye. The labelled Rv1222 was eluted in void volume, mixed with equal volume of 100% glycerol and kept at −80°C. The activity of the TMR labelled Rv1222 derivative was confirmed by its inhibition of in vitro transcription by RNAP. The labelling efficiency of the protein was 0.85.
Fluorescence anisotropy measurements
Twenty nanomolar of TMR labelled Rv1222 in 60 μl Tx buffer was titrated with increasing concentrations of RNAP and/or DNA at 37°C and fluorescence intensities and anisotropy measurements were monitored with Ex at 540 nm and Em at 580 nm on a PTI Fluorescence Master QM400 System fitted with automatic polarizers. Normalized fluorescence anisotropy increments ΔA/Ao, where A and Ao are the anisotropy value of Rv1222 at x nM RNAP [or DNA] or zero RNAP [or DNA], respectively, and ΔA = A–Ao, were plotted against Rv1222 concentration using Sigma Plot software. The dissociation constants (Kd) of the bindings for RNAP or DNA were determined by fitting the curves with single parameters hyperbolic function (f = aX/(Kd+X), where f is the concentration of complex and X is the concentration of Rv1222). To monitor the binding affinity of Rv1222 to RNAP and DNA simultaneously, first equal concentration of Mtb RNAP core and Bpr DNA fragment were incubated together for 10 min at 25°C before titrating with Rv1222. The dissociation constant (X0) for binding of Rv1222 to both RNAP and DNA simultaneously was determined by fitting the curve with sigmoidal function (f = a/(1+exp(-(X–X0)/b), where f is the concentration of complex, X is the concentration of Rv1222 and X0 is the half saturation constant).
In vivo recombinant reporter assays
E. coli BL21 (DE3) cells were transformed with three sets of two plasmids: (i) pFPVmCherry-lacCONS + pAcYc (for control), (ii) pFPVmCherry-lacCONS + pAcYc Rv1222 and (iii) pFPVmCherry-lacCONS + pAcYc Rv1222ΔC. The co-transformed cells were grown in 50 ml LB media at 37°C with antibiotics (35 μg/ml Chloramphenicol and 100 μg/ ml Ampicillin) until OD600 reached 0.4 and induced by the addition of 0.5 mM IPTG and were grown further for 14 h at 37°C. Cells were diluted to make up equal number of cells in each sample and fluorescence properties of the cells were analysed by FACS Aria (Becton Dickinson) (38).
Effect of Rv1222 on bacterial growth
M. smegmatis
M. smegmatis MC2155 cells containing pLAM12 or pLAM12-Rv1222 were grown at 37°C in Middlebrook 7H9 broth (HiMedia) with 0.2% glycerol and 0.05% tween 80 and 20 μg/ml kanamycin for 18 h. Fifty millilitre fresh 7H9 media supplemented with 0.2% acetamide was inoculated with 0.2% of the saturated culture and growth of the cells were monitored for 18 h by measuring the OD600 at 1 h interval.
E. coli
E. coli BL21 cells harbouring plasmid pAcYc-Rv1222 was grown overnight at 37°C in LB containing 35 μg/ml chloramphenicol. Fifty millilitre fresh LB media was inoculated with 1% of the saturated culture and treated with different IPTG concentrations (25 μM, 50 μM, 100 μM). OD600 of the cells was monitored at 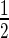 h interval. For control, identical assays were performed with E. coli harbouring the plasmid pAcYc Rv1222ΔC at identical IPTG concentration.
h interval. For control, identical assays were performed with E. coli harbouring the plasmid pAcYc Rv1222ΔC at identical IPTG concentration.
In vivo32P-labelling of RNA
Each of two separate 120 ml 7H9 media was inoculated with M. smegmatis (one with pLAM12 and other with pLAM12-Rv1222) as above. When OD600 of the cells reached 0.2, 25 μl of P32-orthophosphoric acid (5Ci/ml) was added to the culture and cells were grown for another hour before 5 ml of cell culture was aliquoted and pelleted at 30 min intervals. OD600 of the cells was measured at each of the time points. RNA was isolated from the sample of each aliquot using RNA kit (Agilent technologies). Amount of 32P-labelled RNA in each sample was measured by liquid scintillation counter (Perkin Elmer Tri-Carb 2800TR). Radioactive count of each sample RNA was divided by the OD600 of cells at each time point to get the relative amount of P32-labelled RNA per cell (in arbitrary units).
Fe-BABE foot-printing
lacCONS promoter DNA fragment was labelled with 32P γATP at 5′ of upstream end as described above for Bpr promoter DNA.
Preparation of Rv1222 derivatives
The three cysteine residues of Rv1222 at positions 70, 73 and 109 were mutated to alanine or glycine residue to create a no-cysteine Rv1222 derivative. Previously, Cys 70 and Cys 73 residues were shown to be essential for the function of Rv1222 by Barik et al. Contrary to this observation, we found that the no-Cys derivative of Rv1222 remains active in inhibition of transcription. Using this protein derivative, we generated two single-cysteine derivatives of Rv1222 by incorporating a cysteine residue at position A23C (near N-terminus) and G133C (near C-terminus) respectively using site-directed mutagenesis (Stratagene Inc). Each of the single-Cys derivatives of Rv1222 was purified by denaturation/renaturation following the protocol as described above. Activities of the Rv1222 derivatives were confirmed by assessing the ability of each protein to inhibit transcription by in vitro transcription assay.
Labelling of Rv1222 with Fe-BABE
The single-Cys Rv1222 derivatives were treated and reacted with Fe-BABE (ThermoFischer) in 1:5 molar ratio as per manufacturers’ protocol. The unreacted Fe-BABE was removed by passing the reaction samples through P6 column that was pre-equilibrated with buffer [20 mM Tris (pH 8.0), 5% glycerol and 0.2 M NaCl]. The protein was distributed in small aliquots and stored at −80°C.
Fe-BABE-mediated protein–DNA foot-printing assay
First, we tested the ability of each of Fe-BABE labelled Rv1222 derivatives to induce cleavage on the DNA. Each of Fe-BABE labelled Rv1222 derivatives (800nM) was incubated with 1 μM DNA in buffer [50 mM Tris-Cl (pH 8), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, 5% Glycerol, 5 μg/ml BSA] for 15 min at 37°C. The cleavage reactions were initiated as described below. As only Rv1222 derivative labelled at G133C (C-terminus end) is able to produce cleavage on DNA, we performed subsequent Fe-BABE-mediated protein–DNA foot-printing assay (39,40) using this protein derivative.
Ec RNAP holo (2 μM) was incubated with 1 μM 32P-labelled DNA in the above buffer at 37°C for 10 min to form open complex (RPo). Four hundred nanomolar of Rv1222 was added to RPo and incubated further for 15 min at 37°C. Twenty five micromolar each of ATP, GTP, UTP was added to sample and kept for 15 min to form stalled EC with 15 nt RNA (EC15). The cleavage reactions were initiated by adding 0.22 mg/ml ascorbate and 0.1 mM H2O2 to each sample. After incubation for 30 s at 37°C, reactions were quenched with 0.3 mg/ml thiourea. Then 70 μl sterile H2O, 10 μl 3M sodium acetate (pH 5.2) and 200 μl chilled ethanol were added to each sample and kept at −20°C for 1 h. The samples were centrifuged at 13,000 rpm for 15 min, washed with 200 μl chilled 70% ethanol and dried in vacuum. The samples were dissolved in 10 μl of FLB [95% formamide, 50 mM EDTA, 0.01% xylene cyanol], denatured by heating at 95°C. Samples (equal counts) were resolved by electrophoresis on 8% Urea-PAGE in 1X TBE and scanned by storage phosphor imager in Typhoon Trio+ (GE Healthcare). As control reaction, unlabelled Rv1222 was incubated with RPo as above and the cleavage reaction was performed. In another control, Fe-BABE labelled Rv1222 was incubated only with the DNA fragment (no RPo formed) prior to cleavage reaction.
In vitro replication assay
The single-stranded DNA template and the Cy5-labelled primer was annealed in buffer [50 mM Tris (pH 7.5) and 100 mM NaCl] by heating to 95°C followed by cooling to 25°C. Fifty nanomolar annealed DNA template was incubated increasing concentrations of Rv1222 (0, 100 nM, 200 nM, 400 nM) and for 5 min at 37°C. 0.5 U of Klenow (Thermo Scientific) and 0.25 mM dNTP mix were added to the sample and incubation was carried on for another 1 min (41). The reactions were stopped by adding 2 μl FLB (80% formamide, 10 mM EDTA). The reaction samples were then run in 12% Urea-PAGE and the gel was scanned in Typhoon Trio+ (GE Healthcare) scanner at Cy5 channel. To assess the effect of Rv1222 on the kinetics of DNA synthesis, the assay was performed as above except the reactions were stopped at different time points.
RESULTS
Rv1222 does not function as an anti-sigma E
Previous reports suggest that M. tuberculosis (Mtb) Rv1222 protein functions as an anti-sigma factor for σE. In principle, binding of an anti-sigma factor with sigma inhibits transcription either (i) by preventing the association of the sigma factor with RNAP (42) or (ii) by preventing the association of RNAP to the promoter DNA by inducing a conformational change in sigma. In both cases, as RNAP cannot bind to DNA, the polymerase does not initiate open-complex formation. However, once the RNAP promoter open complex is formed, anti-sigma is not able to act on sigma and thus is unable to inhibit transcription. To test how Rv1222 inhibits transcription, we performed EMSA and in vitro transcription assays. In both the assays, we performed two sets of experiments. For EMSA, in the first set, we incubated Rv1222 and σE to form a complex before adding to RNAP and subsequently formed the open complex (Figure 1A). In the second set, we incubated RNAP and σE to from the holo enzyme, and then added Rv1222 to the RNAP holo before forming the open complex (Figure 1B). Heparin was added to the samples to remove any non-specific RNAP–DNA complexes other than open complex (RPo). In both the cases, Rv1222 did not inhibit open-complex formation. For the in vitro transcription assay, in the first set, we incubated RNAP–σE holo with Rv1222 and added promoter DNA fragment to form an open complex, followed by transcription initiation (Figure 1C). In the second set, we first formed the open complex and then added Rv1222 before transcription initiation (Figure 1D). In both the cases, Rv1222 inhibited transcription with similar efficiency (full gels of Figure 1C and D were shown in Supplementary Figure S2A). To eliminate the possibility that the inhibition of transcription could be due to the presence of RNase in the preparation of Rv1222, first P32-labelled RNA was formed by in vitro transcription assay and subsequently was incubated with Rv1222 at identical conditions used in Figure 1C or D. As no degradation of the transcript was observed in the presence of Rv1222, it was confirmed that the inhibition of transcription by Rv1222 was not due to the presence of any RNase in the preparation (Supplementary Figure S2B).
Figure 1.
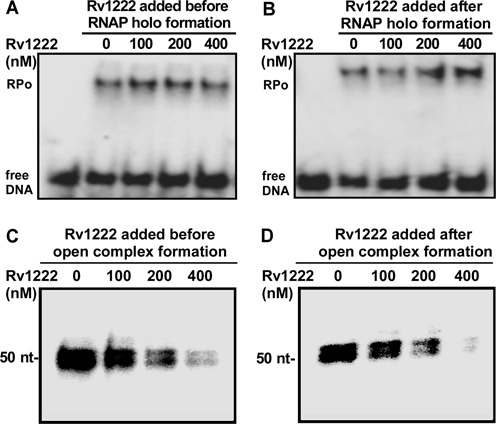
Rv1222 does not inhibit open-complex formation (RPo) but inhibits transcription by Mtb RNAP–σE holo. Upper panel: EMSA assay with P32-labelled Bpr promoter DNA fragment. (A) Rv1222 and 100 nM σE were incubated together to form a complex, then 100 nM Mtb RNAP core was added and subsequently open complex was formed following addition of Bpr promoter DNA fragment. Heparin (0.5 μg/μl) was added to the complex before running on 5% PAGE. Lane 1: free DNA, Lanes 2–5: RPo formation in presence of Rv1222. (B) 100 nM Mtb core RNAP and 100 nM σE were incubated to form RNAP–σE holo, then Rv1222 was added to the holo and subsequently open complex was formed. Lane 1: free DNA, Lanes 2–5: RPo formation in presence of Rv1222. Lower panel: in vitro transcription assay. (C) 100 nM RNAP–σE holo was incubated with Rv1222 before 50 nM Bpr promoter DNA was added to the mixture to form open complex, challenged by heparin and followed by transcription initiation. Lanes 1–4: increasing concentrations of Rv1222. Run-off transcript size is 50 nt. (D) The open complex was formed first by incubating 100 nM RNAP–σE holo and 50 nM promoter DNA fragment and then Rv1222 was added before transcription initiation. Lanes 1–4: increasing concentrations of Rv1222.
The fact that Rv1222 does not inhibit open-complex formation but inhibits transcription when added after open-complex formation clearly indicates that the protein does not function as an anti-sigma factor as suggested previously.
Rv1222 inhibits transcription by both RNAP holo and RNAP core from M. tuberculosis, E. coli and B. subtilis
As Rv1222 does not function as an anti-sigma factor, but still inhibits RNAP–σE holo, we investigated whether the protein would inhibit transcription by an RNAP holo enzyme that contains a sigma factor other than σE. Using in vitro transcription assay, we tested the effect of Rv1222 on Mtb RNAP–σA holo in which the σA, the principal sigma factor of M. tuberculosis was associated with RNAP core (Figure 2A). The data showed that Rv1222 inhibited transcription by Mtb RNAP–σA holo. Subsequently, we performed the same assay using the E. coli (Ec) RNAP–σ70 holo (Figure 2B) and Bacillus subtilis (Bs) RNAP–σA holo (Figure 2C). The data further showed that Rv1222, despite being a transcription factor of M. tuberculosis, inhibited transcription by both E. coli and B. subtilis RNAP and inhibited transcription by all three RNAP with similar efficiencies. Approximate values of IC50 of transcription inhibition by Mtb, Bs and Ec RNAP were ∼77 nM, ∼62 nM, ∼70 nM respectively as estimated from two to three replicates of the assays. The results further confirm that the inhibition of transcription by Rv1222 is not σE specific. Since the structure and sequence of RNAP core is conserved among the different bacterial species (43), it is likely that Rv1222 targets RNAP core enzyme for transcriptional inhibition and not any specific sigma factor.
Figure 2.
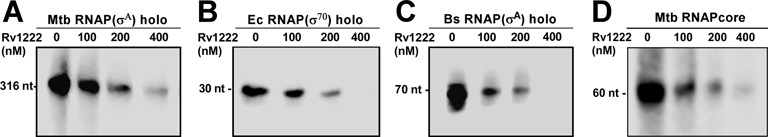
Rv1222 inhibits transcription by RNAP from M. tuberculosis, E. coli, B. subtilis. (A) 100 nM Mtb RNAP–σA holo and 50 nM rrnA promoter. Concentrations of Rv1222 were indicated. Run-off transcripts 316 nt. (B) 100 nM Ec RNAP–σ70 holo, and 50 nM lacCONS promoter. Run-off transcript sizes were 30 nt. (C) 100 nM Bs RNAP holo and 50 nM abrB promoter. Run-off transcript sizes were 70 nt. (D) 100 nM Mtb core RNAP and 50 nM tailed-template DNA. Transcript sizes were 60 nt.
To test whether Rv1222 targets RNAP core enzyme for transcription inhibition, we performed in vitro transcription assay with the Mtb RNAP core and a tailed-template DNA fragment (a double-stranded DNA fragment that contains a single-strand overhang of ∼10 bases) (44,45). RNAP core is able to bind the overhang junction and to perform transcription from this DNA. The assay data showed that Rv1222 inhibited transcription by RNAP core (Figure 2D) with efficiency similar to RNAP holo (IC50∼75 nM). To further validate this observation, we performed another complimentary fluorescence based in vitro transcription assay using Ec RNAP core and Kool-template DNA (46). The data also confirmed that Rv1222 was able to inhibit transcription by Ec RNAP core (Supplementary Figure S3) and did not require any sigma factor for its function.
Rv1222 binds to RNAP core and DNA
Since Rv1222 targets RNAP core, and not any sigma factor for transcriptional inhibition, we argued that the protein should bind to RNAP core with high affinity and should exhibit little or no affinity towards the sigma factors, namely σE and σA. To test this hypothesis, we monitored the binding affinities of Rv1222 to Mtb RNAP core and different sigma factors by fluorescence anisotropy assay (47). We used TMR labelled Rv1222 protein for this assay (Figure 3). The binding affinities of Rv1222 to RNAP core (Figure 3A), σE and σA were estimated to be ∼60 ± 10 nM, ∼270 ± 45 nM and ∼690 ± 270 nM respectively (Figure 3A and also Supplementary Figure S4). Data showed that the protein binds to RNAP core with at least 5-fold higher affinity compared to both the sigma factors. Interestingly Rv1222 displayed ∼2-fold higher affinity for the σE over σA. This comparatively higher affinity for σE is consistent with the western blot data obtained by previous groups (22,23) demonstrating σE–Rv1222 interaction. The fact that Rv1222 showed binding affinities to sigma factors could be explained by the possible ionic interaction among the proteins. Rv1222 is a highly positively charged protein (pI = 8.68) whereas the sigma factors are negatively charged in neutral buffer condition. Therefore, it is possible that the sigma factors contain negatively charged residues at the surface of the proteins whereas Rv1222 contains positively charged residues at its surface.
Figure 3.
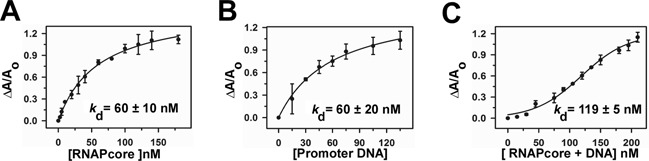
Rv1222 simultaneously binds to RNAP and DNA: fluorescence anisotropy assay. Twenty nanomolar TMR labelled Rv1222 was added with (A) Mtb RNAP core, (B) DNA, (C) Mtb RNAP core + DNA. Fluorescence anisotropy of the labelled protein was monitored at Ex 530 and Em 580 nm. Each data set represents mean of three replicates.
As Rv1222 is a positively charged protein, we speculated that the protein might bind to the DNA fragment due to its negative charge. To test this idea, we investigated whether the protein possessed any affinity for DNA (Figure 3B). We used two DNA fragments: one containing a promoter element and another containing no promoter element. Our anisotropy assay with the labelled Rv1222 showed that the protein bound to these DNA fragments with similar affinities (for promoter DNA fragment, Kd = 60 ± 20 nM, Figure 3B; for promoterless DNA fragment, Kd = 52 ± 8 nM, Supplementary Figure S5). When the binding assays were performed at high salt (200 mM NaCl, instead of 100 mM NaCl), Rv1222 completely lost its ability to bind to DNA (Supplementary Figure S6), whereas the protein retained its affinity to RNAP. Therefore, we conclude that interaction of Rv1222 to DNA is non-specific, possibly ionic, whereas its interaction to RNAP is specific. As Rv1222 exhibits affinity for RNAP and DNA, on the other hand RNAP is known to bind DNA fragment, we expected that the presence of RNAP would alter the binding affinity of Rv1222 to the DNA fragment. The anisotropy assay showed that Rv1222 binds to RNAP core and DNA simultaneously (nature of the binding curve is sigmoidal indicating simultaneous binding of ligands (Figure 3C), whereas for individual ligand binding, the nature of the curve is hyperbolic (Figure 3A and B) and the presence of RNAP increased the binding affinity of Rv1222 to DNA (Kd = 119 ± 5 nM).
C-terminal tail of Rv1222 is critical for its interaction with DNA
To identify which part of the protein interacts with DNA, we performed DNA–protein foot-printing assay with Fe-BABE labelled Rv1222. We generated two single Cys derivatives of the protein: the position of cysteine is either at residue23 (close to N-termini) or at residue133 (close to C-termini) (Figure 4A). The protein derivatives were subsequently labelled with Fe-BABE and were subjected to DNA–protein foot-printing assay. Both the labelled Rv1222 derivatives were active in inhibiting transcription (Supplementary Figure S7). We observed only the C-terminus-labelled protein derivative, not the N-terminus-labelled protein derivative, produced the Fe-BABE induced cleavage on the DNA (Supplementary Figure S8). The reason could be due to the proximity of the Fe-BABE labelling site of the protein to its DNA binding site. Thus, the result indicates that the protein may contain DNA binding determinant near or at the C-terminus. Sequence analysis of the protein by BindN+ (freeware) predicted that 12 residues at the C-terminal tail of Rv1222 could contain determinants for DNA binding. The protein contains five positively charged residues at the C-terminal tail. Indeed, when the 10 C-terminal residues were deleted from Rv1222, the resultant protein (Rv1222ΔC) lost its ability to bind DNA (Figure 4B). As a result, Rv1222ΔC lost its ability to inhibit transcription, although retained its affinity for RNAP (Kd = 72 ± 26 nM) (Figure 4C and D). Thus, we conclude that the positively charged C-terminal tail of Rv1222 is critical, or possibly responsible for its interaction with DNA. The result showing the loss of ability of the protein to bind DNA at high salt (Supplementary Figure S6) indicates that the interactions among Rv1222 and DNA are weak and possibly ionic. At this salt concentration, although Rv1222 retains its affinity to RNAP, the protein loses its ability to inhibit transcription (Supplementary Figure S6). However, the possibility that deletion of the C-terminus tail alters putative DNA binding site of the protein, if any, could not be excluded. Nevertheless, our data clearly show that the interaction of Rv1222 to DNA is essential for the inhibition of transcription.
Figure 4.
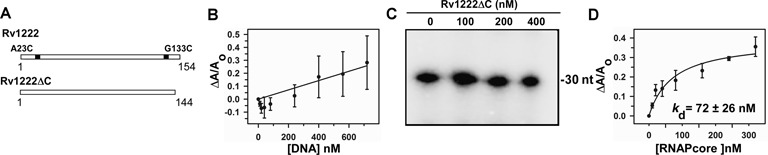
C-terminal tail of Rv1222 is critical for DNA binding and inhibition of transcription. (A) Schematic representation of Rv1222 WT and Rv1222ΔC. A23C and G133C were the positions in two single-cysteine protein derivatives at which Fe-BABE was conjugated. (B) 20 nM TMR labelled Rv1222ΔC was added with DNA. Fluorescence anisotropy of the labelled protein was monitored at Ex 530 nm and Em 580 nm. Each data set represents mean of three replicates. (C) Rv1222ΔC does not inhibit transcription: in vitro transcription assay. Ec RNAP holo and lacCONS promoter DNA fragments were used. Rv1222ΔC was incubated with the open complex before initiation of transcription. Run-off transcript sizes were 30 nt. (D) Binding of Rv1222ΔC to RNAP core: Same as B except that labelled Rv1222ΔC was added with Mtb RNAP core instead of DNA.
Rv1222 binds DNA adjacent to RNAP in the open complex (RPo) and stalled EC
To test the location of Rv1222 on DNA in the context of open complex (RPo) and stalled EC (containing 15 nt RNA), we performed protein–DNA foot-printing assay with Fe-BABE labelled Rv1222 (40). We used sub-stoichiometric level of labelled Rv1222 (400 nM) than DNA (1 μM) and RNAP (2 μM), so that majority of the labelled protein remained bound at the DNA adjacent to RNAP while avoiding sites of DNA where there is no RNAP (Figure 5A). This is evident from comparison of the results of Supplementary Figures S5A and S8. In Supplementary Figure S8, Fe-BABE labelled Rv1222 produces nicks on free DNA at multiple sites (lane 3) as the concentration of the protein (800 nM) was 2-fold higher than DNA concentration as compared to Figure 5A in which nicks were observed on DNA at sites adjacent to RNAP (lanes 3 and 4).
Figure 5.
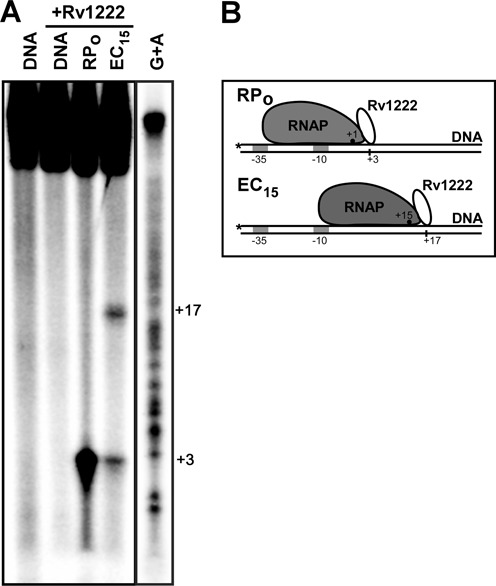
Location of Rv1222 in open complex (RPo) and stalled-EC (EC15). (A) Fe-BABE induced protein–DNA foot-printing assay. Four hundred nanomolar of Rv1222 labelled with Fe-BABE at G133C (near C-termini) was used in this assay. Two micromolar Ec RNAP and 1 μM lacCONS15 promoter were used to form open complex. In both cases (lanes 3 and 4), Rv1222 was added to the mixture after open-complex formation. For formation of stalled EC at +15 (EC15, lane 4), transcription was initiated with ATP, GTP and UTP (no CTP), so that RNAP could synthesize 15 nt RNA. Controls: lane 1, DNA, RNAP were incubated, no Fe-BABE labelled Rv1222 was used; Lane 2, Fe-BABE labelled Rv1222 and DNA were incubated, no RNAP was used. DNA sequencing ladder was run on the same gel, but its contrast was different from the actual figure. (B) Model of transcriptional complexes showing the location of Rv1222. Vertical lines on the template DNA show the positions of Fe-BABE induced nick. *represents the positions of labelling by P32 on DNA. Filled dots within RNAP represent the active centre. As the active centre of RNAP shifted upon transition from open complex to EC, the position of Fe-BABE induced nick is also shifted, showing the movement of Rv1222 with RNAP along the DNA.
In both open complex and stalled EC (EC15), Rv1222 was added after open-complex formation. For the formation of stalled EC, a modified lacCONS15 DNA fragment (35) was used in which the first cytosine base is located at +16 in the non-template strand from the transcription start site. Thus, upon initiation of transcription with ATP, GTP and UTP on this DNA template, RNAP synthesized 15 nt RNA and stalled (EC15). We observed a Fe-BABE induced nick at +3 position (with respect to transcription start site) on the DNA template in the open complex, in which the RNAP active centre is located at +1, whereas in the EC, the position of the nick was shifted to +17 position, in which the RNAP active centre is located at +15 (Figure 5B). This 14 nt shift in the nick site closely corresponds to the translocation of RNAP on DNA template by 15 nt. Since Rv1222 was added before transcription initiation and sub-stoichiometric amount of labelled Rv1222 was used for this assay, it is likely that Rv1222 binds to the open complex and translocates with the elongating RNAP that stalls at +15 and subsequently produces a nick on DNA at +17. However, the possibility of re-binding of Rv1222 to the stalled EC cannot be completely ruled out from this assay. In this context, it is important to note that Rv1222 inhibits transcription elongation when added after the formation of stalled EC (Supplementary Figure S9). In the stalled EC, in addition to the nick at +17, Rv1222 also produced a nick at +3 on DNA that corresponds to the nick for open complex (lane 4, Figure 5). The result indicates that, as expected, only a part of the open complexes formed were able to initiate transcription and form ECs. The result also suggests that Rv1222 binds to RNAP at a site that faces downstream DNA.
Rv1222 slows down RNA synthesis by RNAP in vitro and in vivo
As Rv1222 was found to bind to RNAP and DNA simultaneously, it could be possible that the protein anchors RNAP onto DNA and thus could exert frictional force as RNAP translocates along the DNA during transcription. If this is the case, we might expect Rv1222 not to arrest or prohibit transcription, rather influence RNAP to slow down RNA synthesis. To test this hypothesis, we performed single-round in vitro transcription assay in the presence and absence of Rv1222 and monitored the amount of transcripts produced at each fixed time point. As Rv1222 neither affects the open-complex formation (Figure 1A and B), we first formed open complex prior to incubation with Rv1222 and followed by transcription initiation. Heparin was added to the sample to ensure single-round transcription. We performed the assay with Mtb RNAP as well as Ec RNAP. For Mtb RNAP, in the absence of Rv1222, the amount of transcripts was saturated at 5 min, whereas in the presence of 200 nM Rv1222, the time required for saturation increased to 20 min (Figure 6A and B). The total amount of transcript at saturation level in both the cases remained the same. In the presence of 400 nM Rv1222, the saturation level of transcripts could not be reached at the maximum time point we tested, however, the amount was increasing with time. For Ec RNAP, the result obtained was similar to that with Mtb RNAP, except that, the time taken by Ec RNAP to produce saturation level transcripts in the presence of Rv1222 was less than that of Mtb RNAP (Figure 6C and D). Overall, the result indicates that Rv1222 reduces the amount of RNA synthesis by RNAP in a time-dependent manner and the reduction does not depend on promoter sequence and the amount of RNA synthesis decreases with the concentration of Rv1222 present.
Figure 6.
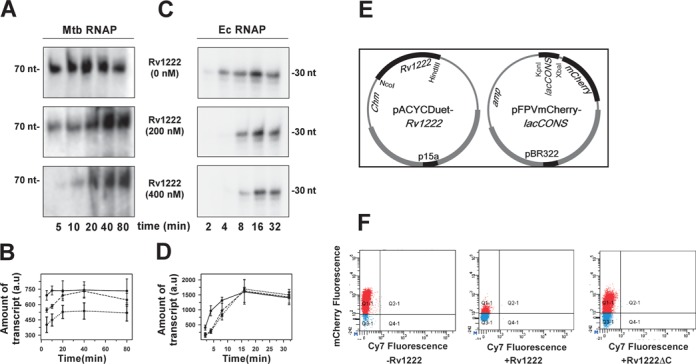
Rv1222 inhibits transcription in vitro (in a time-dependent manner) and in vivo. (A) In vitro transcription reactions were performed with 100 nM Mtb RNAP and 50 nM sinP3 promoter DNA and 32P-labelled NTP. Rv1222 was added to the reaction mixture after open-complex formation, before addition of NTP. Heparin was added to the reaction mixtures to ensure single-round transcription. The reactions were stopped at the indicated times after the addition of NTP. (B) The intensities of the bands at different time points as obtained from the in vitro transcription assay with Mtb RNAP were plotted against time. ——-Rv1222, - -- - - - 200 nM Rv1222, ····· 400 nM Rv1222. (Data is representative of three independent experiments.) (C) Same as A for Ec RNAP. (D) Same as B for Ec RNAP. (E) Strategy for promoter activity assay: pAcYc Duet plasmid, pAcYc Rv1222 and pAcYc Rv1222ΔC were co-transformed with pFPVmCherry-lacCONS plasmid in E. coli BL21 (DE3) cells and grown at 37°C up to OD (at 600 nm) 0.4, induced with 0.5 mM IPTG, and further grown for 14 h. (F) FACS data: cells were diluted to make up equal number of cells in each sample. Aliquots of cells from above assay were scanned at mCherry (610 nm) and Cy7 (760 nm) fluorescence channels; first panel (control): pFPVmCherry-lacCONS + pAcYcDuet; second panel: pFPVmCherry-lacCONS + pAcYc-Rv1222 Duet; third panel: pFPVmCherry-lacCONS + pAcYc-Rv1222ΔC Duet. The percentage of fluorescent cells for the panels –Rv1222, +Rv1222 and +Rv1222ΔC are 94%, 14% and 84%, respectively. Cy7 channel was used as reference at which the cells display minimum auto fluorescence.
All our in vitro data suggested that Rv1222 reduces the amount of RNA synthesis by RNAP. We therefore expected that the protein would also reduce transcription in vivo and that reduction would also not be specific for any particular promoter. To test this hypothesis we developed an in vivo reporter assay in E. coli involving two plasmids (38): one contained an mCherry expression cassette (48) under the control of lacCONS promoter, while the second compatible plasmid contained Rv1222 ORF under the control of an inducible promoter (Figure 6E). In the absence of Rv1222, RNAP would produce mCherry mRNA from the lacCONS promoter and subsequently express the mCherry protein. In the presence of Rv1222, the amount of promoter activity would be reduced as the protein slows down the mRNA synthesis by RNAP. As expected, we observed a significant decrease in the percentage of cells (14%) that produces mCherry fluorescence when Rv1222 was expressed, as compared to the percentage of cells (94%) that lacks Rv1222 expression (Figure 6F). On the other hand, when Rv1222ΔC was expressed, the percentage of cells that produce mCherry fluorescence was relatively unaffected (84%) and was close to that observed in the absence of Rv1222. This result further corroborates the fact that Rv1222ΔC loses its ability to inhibit transcription.
Rv1222 does not inhibit DNAP or T7 RNAP
Since we find that Rv1222 binds to DNA non-specifically, there is a possibility that the DNA, upon binding with Rv1222 randomly, presents a ‘bumpy road’ to molecule that moves along the DNA slowing down the movement of the molecule. As DNAP moves along DNA during DNA synthesis (49) like RNAP during RNA synthesis, Rv1222 bound DNA would slow down not only RNA synthesis but also DNA synthesis. We performed in vitro replication assays with E. coli DNAP (Klenow fragment) to investigate whether Rv1222 had any effect on DNA synthesis in a dose-dependent manner or on kinetics of DNA synthesis. The results showed that Rv1222 neither affected the DNA synthesis by DNAP (Figure 7A) nor the rate of DNA synthesis (Figure 7C). This is due the inability of Rv1222 to efficiently bind DNAP (Kd = 245 ± 78 nM, as compared to Kd for RNAP = 60 ± 10 nM, Figure 7B). Similarly, when in vitro transcription assay was performed with RNAP from bacteriophage T7, Rv1222 was unable to inhibit the polymerase (Figure 7D). The binding assay of Rv1222 to T7 RNAP revealed that the protein exhibits very little affinity to this polymerase (Figure 7E), consistent with the fact that T7 RNAP does not share sequence homology with the bacterial RNAP. This observation suggests that the binding of Rv1222 to DNA could not be the sole reason for inhibition of transcription.
Figure 7.
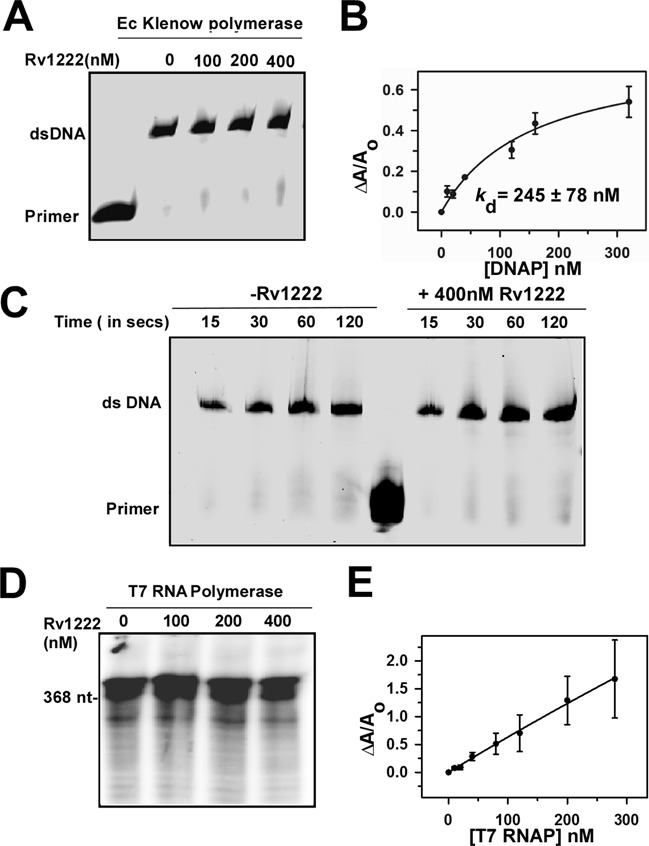
Rv1222 does not inhibit DNA synthesis. (A)In vitro replication assay: 0.5U of Ec Klenow DNAP, Cy5-labelled primer and 65 base DNA fragments were used in the assay. Rv1222 was incubated with DNAP before the replication reactions were initiated with dNTP. Reactions were stopped after 1min and products were separated on 12% Urea-PAGE. The gel was scanned on a Typhoon Trio+ at Cy5 channel. (B) Binding of 20 nM TMR labelled Rv1222 to Klenow polymerase. (C) Effect of Rv1222 on the kinetics of DNA replication: Same as A except the reactions were stopped at the indicated time interval. (D) In vitro transcription assay with T7 RNAP: 0.2 U of T7 RNAP was used with 100 nM of T7 promoter containing DNA fragment. (E) Binding of 20 nM TMR labelled Rv1222 to T7 RNAP by fluorescence anisotropy.
Rv1222 slows down the growth rate of bacteria
As Rv1222 reduces the level of mRNA synthesis in vivo, the protein would be responsible for reducing the amount of total mRNA in the cell. As transcription is one of the key cellular processes, any decrease in total mRNA level within the cell would be reflected in a slow growth of the cell. We investigated this hypothesis by introducing Rv1222 into M. smegmatis as well as in E. coli and monitored cell growth. The plasmid pLAM12 containing the Rv1222 gene under an acetamide inducible promoter was inserted in M. smegmatis and the growth rate of the cells was monitored. We observed a 4-fold decrease in the growth rate when Rv1222 was induced, as compared to the growth rate of M. smegmatis cells that did not have any expression of Rv1222 (Figure 8A). To investigate whether the reduced growth rate of the cell was due to reduction of the mRNA levels, we quantified the mRNA levels of the cell at different time points of growth. For this assay, we grew M. smegmatis in a media that contained P32-sodium phosphate. Upon 32P-phosphate uptake by the cell, newly synthesized NTP would incorporate the radiolabelled phosphate, which subsequently would be incorporated into the RNA (50). In this assay, we intended to measure the in vivo level of RNA under different levels of Rv1222 in the cells. However, as the growth rate of the cell changed upon increase in Rv1222 expression, we estimated the relative amount of radiolabelled RNA per cell at each time point during the cell growth by normalizing it against the OD600. When Rv1222 was expressed, the level of total RNA per cell was significantly reduced (Figure 8B). Although from this assay we cannot rule out whether other possible biological pathways, if any, are involved in the reduction of growth rate by Rv1222, it is very likely that the observed slow growth rate of the cell in the presence of Rv1222 is due to reduction in the level of RNA synthesis. To monitor the effect of Rv1222 expression on the growth rate of E. coli, we inserted a plasmid containing an IPTG inducible rv1222 gene into the bacteria. As IPTG concentration was increased, the growth rate of the bacteria is reduced. At 100 μM IPTG concentration the growth rate was reduced by ∼75% upon expression of Rv1222 (Figure 8C). This reduction was changed to 20% upon expression of Rv1222ΔC (Figure 8D). This indicates that deletion of the C-terminal part severely impairs the ability of Rv1222 to inhibit bacterial growth, consistent with our observation that Rv1222ΔC does not affect RNA synthesis. We conclude that the expression of Rv1222 in the bacteria significantly affects the growth rate of the cells.
Figure 8.
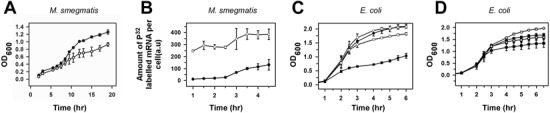
Expression of Rv1222 in bacteria slows down its growth. (A) M. smegmatis containing plasmid pLAM12-Rv1222 (for expression of Rv1222) or plasmid pLAM12 (for control) were grown in 7H9 media (with kanamycin 20 μg/ml) at 37°C for 20 h. Cell growths (OD600) were monitored at regular intervals. Data are representative of three independent experiments. Filled circle: cells without Rv1222; Open circle: cells with Rv1222. (B) Amount of synthesis of 32P-labelled RNA in vivo in M. smegmatis with or without expression of Rv1222. Saturated cultures of M. smegmatis containing pLAM12 and pLAM12-Rv1222 respectively were diluted to OD600 0.1 in 7H9 Media and allowed to grow till OD600 0.2 at 37°C. After addition P32–Na-phosphate, 5 ml aliquots of cells were pelleted at regular time intervals. Density of cells (OD600) in each aliquot was measured at each interval. Total RNA from each sample was isolated using RNA isolation kit. Amount of radioactive RNA in the RNA samples was estimated using scintillation counter. Amount of P32-labelled RNA/O.D was plotted at each time interval. Experiments were repeated thrice. Filled circle: cells with Rv1222; Open circle: cells without Rv1222. (C) E. coli cells harbouring the pAcYc Rv1222 plasmid were grown in the presence of 35 μg/ml Chloramphenicol. Overnight cultures were subcultured at 1:100 dilution with the addition of varying concentrations (‘open circle’ uninduced, ‘closed circle’ 25 μM, ‘open square’ 50 μM, ‘filled square’ 100 μM) of IPTG and cell growth was monitored. (D) Same as C except pAcYc Rv1222ΔC plasmid was used.
DISCUSSION
Previously, Dona et al., using a combination of western blot assay and in vivo pulled down assay and also in vitro transcription assay, showed that Rv1222 binds to σE. In another work, Barik et al. showed that Rv1222 inhibits transcription from a σE-dependent promoter. These results led to the interpretation that Rv1222 functions as an anti-σE. Here, we show that Rv1222 exhibits a moderate binding affinity to σE and also inhibits transcription from a σE-dependent promoter, consistent with the previous observation. However, if Rv1222 was an anti-sigma factor for σE, it would inhibit the open-complex formation and would not inhibit transcription when added after open-complex formation. Our results show that Rv1222 does not affect the open-complex formation, rather inhibits transcription when added after open-complex formation. These observations argue the previous conclusion and indicate that the protein may not function as an anti-σE. We further show that Rv1222 inhibits transcription by RNAP core and RNAP holoenzymes of three different bacteria: E. coli, B. subtilis and M. tuberculosis, in association with their respective principal sigma factors. Therefore, our results unequivocally establish that inhibition of transcription by Rv1222 does not involve the binding of the protein to σE. This inhibition occurs at the RNA synthesis step including promoter escape and transcription elongation, but not at the open-complex formation step. The C-terminal tail of Rv1222 that contains positively charged residues is critical, possibly responsible for DNA binding. The protein derivative in which this tail is deleted fails to bind DNA and is consequently inefficient for transcription inhibition. Thus, binding of Rv1222 to RNAP alone cannot be the sole reason for the inhibition of transcription. On the other hand, Rv1222 does not bind to DNAP or T7 RNAP and does not inhibit these polymerases. Thus, the binding of Rv1222 on DNA also cannot be the sole reason for the observed inhibition of transcription by Rv1222. Therefore, inhibition of transcription requires simultaneous binding of Rv1222 to the RNAP core and to DNA. Based on our experimental data we propose a model in which Rv1222 anchors RNAP onto DNA and thereby restricts the translocation of RNAP along DNA during RNA synthesis. Our model predicts that Rv1222 does not completely prevent transcription, but slows down the synthesis of mRNA by RNAP. This is a novel global mechanism for transcriptional regulation and is not restricted to any specific promoter. This is different from the mechanism by which HK022 Nun protein anchors the RNAP and completely prevents its translocation at specific DNA site (21).
When Rv1222 was overexpressed in M. smegmatis or E. coli, the growth rate of the bacteria is significantly reduced. The level of synthesis of mRNA in M. smegmatis is reduced when Rv1222 is expressed in the cell. As transcription is one of the essential steps in the bacterial growth cycle, it is plausible that slowing down of mRNA synthesis would result in a slow growth of the bacterium. Thus, our results indicate that Rv1222 could be a determinant for the growth rate of mycobacterium. However, to confirm whether Rv1222 is responsible for slow rate of RNA synthesis, further study is required involving generating a knock out mutant of M. tuberculosis strain that lacks the expression of Rv1222 (under progress). It is also possible that the level of Rv1222 increases when the bacteria enter the dormant stage, making the bacteria extremely slow growing consistent with the observations that the mRNA level of M. tuberculosis Rv1222 is upregulated in dormant state or under growth conditions leading to dormant state. On the other hand, the protein is degraded by ClpC1P2 protease upon PknB-dependent phosphorylation (23). Thus, there may be a possibility that proteolysis of Rv1222 in dormant state may restore the bacteria in its active state. To test whether the level of Rv1222 is a possible determinant in transition of the bacteria from active to dormant state, further investigation is required to monitor the level of expression of the protein in the two different states, e.g. active and dormant state. We note that the Rv1222 is present in various mycobacteria, e.g. M. tuberculosis, M. leprae, M. bovis, M. marinum and M. smegmatis and with high sequence homology. We hypothesize that Rv1222 in these mycobacteria function by a similar mechanism as the protein from M. tuberculosis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank R. Landick (UW-Madison), R. H. Ebright (Waksman Institute), S. Wigneshweraraj (Imperial College London) for suggestions; R. Sur (University of Calcutta), A. B. Dutta and P. Parrack (Bose Institute) for critically reading the manuscript and comments.
FUNDING
Department of Biotechnology, India Grant [BT/PR 5345/MED/29/648/2012] to JM; CSIR India Fellowships to P.R., R.B., and R.K.P. Funding for open access charge: Institutional support.
Conflict of interest statement. None declared.
REFERENCES
- 1.Cases I., de Lorenzo V., Ouzounis C.A. Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 2003;11:248–253. doi: 10.1016/s0966-842x(03)00103-3. [DOI] [PubMed] [Google Scholar]
- 2.Schlax P.J., Capp M.W., Record M.T. Jr. Inhibition of transcription initiation by lac repressor. J. Mol. Biol. 1995;245:331–350. doi: 10.1006/jmbi.1994.0028. [DOI] [PubMed] [Google Scholar]
- 3.Schiering N., Tao X., Zeng H., Murphy J.R., Petsko G.A., Ringe D. Structures of the apo- and the metal ion-activated forms of the diphtheria tox repressor from Corynebacterium diphtheriae. Proc. Natl Acad. Sci. U.S.A. 1995;92:9843–9850. doi: 10.1073/pnas.92.21.9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caslake L.F., Ashraf S.I., Summers A.O. Mutations in the alpha and sigma-70 subunits of RNA polymerase affect expression of the mer operon. J. Bacteriol. 1997;179:1787–1795. doi: 10.1128/jb.179.5.1787-1795.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zwieb C., Kim J., Adhya S. DNA bending by negative regulatory proteins: Gal and Lac repressors. Genes Dev. 1989;3:606–611. doi: 10.1101/gad.3.5.606. [DOI] [PubMed] [Google Scholar]
- 6.Belitsky B.R., Sonenshein A.L. Roadblock repression of transcription by Bacillus subtilis CodY. J. Mol. Biol. 2011;411:729–743. doi: 10.1016/j.jmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He B., Zalkin H. Repression of Escherichia coli purB is by a transcriptional roadblock mechanism. J. Bacteriol. 1992;174:7121–7127. doi: 10.1128/jb.174.22.7121-7127.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hughes K.T., Mathee K. The anti-sigma factors. Annu. Rev. Microbiol. 1998;52:231–286. doi: 10.1146/annurev.micro.52.1.231. [DOI] [PubMed] [Google Scholar]
- 9.Helmann J.D. Anti-sigma factors. Curr. Opin. Microbiol. 1999;2:135–141. doi: 10.1016/S1369-5274(99)80024-1. [DOI] [PubMed] [Google Scholar]
- 10.Atlung T., Ingmer H. H-NS: a modulator of environmentally regulated gene expression. Mol. Microbiol. 1997;24:7–17. doi: 10.1046/j.1365-2958.1997.3151679.x. [DOI] [PubMed] [Google Scholar]
- 11.Tupper A.E., Owen-Hughes T.A., Ussery D.W., Santos D.S., Ferguson D.J., Sidebotham J.M., Hinton J.C., Higgins C.F. The chromatin-associated protein H-NS alters DNA topology in vitro. EMBO J. 1994;13:258–268. doi: 10.1002/j.1460-2075.1994.tb06256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimizu M., Miyake M., Kanke F., Matsumoto U., Shindo H. Characterization of the binding of HU and IHF, homologous histone-like proteins of Escherichia coli, to curved and uncurved DNA. Biochim. Biophys. Acta. 1995;1264:330–336. doi: 10.1016/0167-4781(95)00173-5. [DOI] [PubMed] [Google Scholar]
- 13.Tapias A., Lopez G., Ayora S. Bacillus subtilis LrpC is a sequence-independent DNA-binding and DNA-bending protein which bridges DNA. Nucleic Acids Res. 2000;28:552–559. doi: 10.1093/nar/28.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furman R., Danhart E.M., NandyMazumdar M., Yuan C., Foster M.P., Artsimovitch I. pH dependence of the stress regulator DksA. PloS One. 2015;10:e0120746. doi: 10.1371/journal.pone.0120746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lennon C.W., Ross W., Martin-Tumasz S., Toulokhonov I., Vrentas C.E., Rutherford S.T., Lee J.H., Butcher S.E., Gourse R.L. Direct interactions between the coiled-coil tip of DksA and the trigger loop of RNA polymerase mediate transcriptional regulation. Genes Dev. 2012;26:2634–2646. doi: 10.1101/gad.204693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tagami S., Sekine S., Kumarevel T., Hino N., Murayama Y., Kamegamori S., Yamamoto M., Sakamoto K., Yokoyama S. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. Nature. 2010;468:978–982. doi: 10.1038/nature09573. [DOI] [PubMed] [Google Scholar]
- 17.Laptenko O., Kim S.S., Lee J., Starodubtseva M., Cava F., Berenguer J., Kong X.P., Borukhov S. pH-dependent conformational switch activates the inhibitor of transcription elongation. EMBO J. 2006;25:2131–2141. doi: 10.1038/sj.emboj.7601094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ross W., Vrentas C.E., Sanchez-Vazquez P., Gaal T., Gourse R.L. The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol. Cell. 2013;50:420–429. doi: 10.1016/j.molcel.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perederina A., Svetlov V., Vassylyeva M.N., Tahirov T.H., Yokoyama S., Artsimovitch I., Vassylyev D.G. Regulation through the secondary channel–structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 20.Mekler V., Minakhin L., Sheppard C., Wigneshweraraj S., Severinov K. Molecular mechanism of transcription inhibition by phage T7 gp2 protein. J. Mol. Biol. 2011;413:1016–1027. doi: 10.1016/j.jmb.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vitiello C.L., Gottesman M.E. Bacteriophage HK022 Nun protein arrests transcription by blocking lateral mobility of RNA polymerase during transcription elongation. Bacteriophage. 2014;4:e32187. doi: 10.4161/bact.32187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dona V., Rodrigue S., Dainese E., Palu G., Gaudreau L., Manganelli R., Provvedi R. Evidence of complex transcriptional, translational, and posttranslational regulation of the extracytoplasmic function sigma factor sigmaE in Mycobacterium tuberculosis. J. Bacteriol. 2008;190:5963–5971. doi: 10.1128/JB.00622-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barik S., Sureka K., Mukherjee P., Basu J., Kundu M. RseA, the SigE specific anti-sigma factor of Mycobacterium tuberculosis, is inactivated by phosphorylation-dependent ClpC1P2 proteolysis. Mol. Microbiol. 2010;75:592–606. doi: 10.1111/j.1365-2958.2009.07008.x. [DOI] [PubMed] [Google Scholar]
- 24.Murry J.P., Sassetti C.M., Lane J.M., Xie Z., Rubin E.J. Transposon site hybridization in Mycobacterium tuberculosis. Methods Mol. Biol. 2008;416:45–59. doi: 10.1007/978-1-59745-321-9_4. [DOI] [PubMed] [Google Scholar]
- 25.Murphy D.J., Brown J.R. Identification of gene targets against dormant phase Mycobacterium tuberculosis infections. BMC Infect. Dis. 2007;7:84. doi: 10.1186/1471-2334-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hampshire T., Soneji S., Bacon J., James B.W., Hinds J., Laing K., Stabler R.A., Marsh P.D., Butcher P.D. Stationary phase gene expression of Mycobacterium tuberculosis following a progressive nutrient depletion: a model for persistent organisms. Tuberculosis (Edinb.) 2004;84:228–238. doi: 10.1016/j.tube.2003.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voskuil M.I., Visconti K.C., Schoolnik G.K. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis (Edinb.) 2004;84:218–227. doi: 10.1016/j.tube.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Schnappinger D., Ehrt S., Voskuil M.I., Liu Y., Mangan J.A., Monahan I.M., Dolganov G., Efron B., Butcher P.D., Nathan C., et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the Phagosomal Environment. J. Exp. Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee R., Rudra P., Prajapati R.K., Sengupta S., Mukhopadhyay J. Optimization of recombinant Mycobacterium tuberculosis RNA polymerase expression and purification. Tuberculosis (Edinb.) 2014;94:397–404. doi: 10.1016/j.tube.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Jacques J.F., Rodrigue S., Brzezinski R., Gaudreau L. A recombinant Mycobacterium tuberculosis in vitro transcription system. FEMS Microbiol. Lett. 2006;255:140–147. doi: 10.1111/j.1574-6968.2005.00071.x. [DOI] [PubMed] [Google Scholar]
- 31.Song T., Song S.E., Raman S., Anaya M., Husson R.N. Critical role of a single position in the -35 element for promoter recognition by Mycobacterium tuberculosis SigE and SigH. J. Bacteriol. 2008;190:2227–2230. doi: 10.1128/JB.01642-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Margeat E., Kapanidis A.N., Tinnefeld P., Wang Y., Mukhopadhyay J., Ebright R.H., Weiss S. Direct observation of abortive initiation and promoter escape within single immobilized transcription complexes. Biophys. J. 2006;90:1419–1431. doi: 10.1529/biophysj.105.069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strauch M.A., Perego M., Burbulys D., Hoch J.A. The transition state transcription regulator AbrB of Bacillus subtilis is autoregulated during vegetative growth. Mol. Microbiol. 1989;3:1203–1209. doi: 10.1111/j.1365-2958.1989.tb00270.x. [DOI] [PubMed] [Google Scholar]
- 34.Yang X., Lewis P.J. Overproduction and purification of recombinant Bacillus subtilis RNA polymerase. Protein Expr. Purif. 2008;59:86–93. doi: 10.1016/j.pep.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Mukhopadhyay J., Kapanidis A.N., Mekler V., Kortkhonjia E., Ebright Y.W., Ebright R.H. Translocation of sigma(70) with RNA polymerase during transcription: fluorescence resonance energy transfer assay for movement relative to DNA. Cell. 2001;106:453–463. doi: 10.1016/s0092-8674(01)00464-0. [DOI] [PubMed] [Google Scholar]
- 36.Arnaud-Barbe N., Cheynet-Sauvion V., Oriol G., Mandrand B., Mallet F. Transcription of RNA templates by T7 RNA polymerase. Nucleic Acids Res. 1998;26:3550–3554. doi: 10.1093/nar/26.15.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim Y., Ho S.O., Gassman N.R., Korlann Y., Landorf E.V., Collart F.R., Weiss S. Efficient site-specific labeling of proteins via cysteines. Bioconjug. Chem. 2008;19:786–791. doi: 10.1021/bc7002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banerjee R., Rudra P., Saha A., Mukhopadhyay J. Recombinant reporter assay using transcriptional machinery of mycobacterium tuberculosis. J. Bacteriol. 2015;197:646–653. doi: 10.1128/JB.02445-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samaha R.R., Joseph S., O'Brien B., O'Brien T.W., Noller H.F. Site-directed hydroxyl radical probing of 30S ribosomal subunits by using Fe(II) tethered to an interruption in the 16S rRNA chain. Proc. Natl Acad. Sci. U.S.A. 1999;96:366–370. doi: 10.1073/pnas.96.2.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Owens J.T., Miyake R., Murakami K., Chmura A.J., Fujita N., Ishihama A., Meares C.F. Mapping the sigma70 subunit contact sites on Escherichia coli RNA polymerase with a sigma70-conjugated chemical protease. Proc. Natl Acad. Sci. U.S.A. 1998;95:6021–6026. doi: 10.1073/pnas.95.11.6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lacenere C., Garg M.K., Stoltz B.M., Quake S.R. Effects of a modified dye-labeled nucleotide spacer arm on incorporation by thermophilic DNA polymerases. Nucleosides Nucleotides Nucleic Acids. 2006;25:9–15. doi: 10.1080/15257770500377714. [DOI] [PubMed] [Google Scholar]
- 42.Minakhin L., Camarero J.A., Holford M., Parker C., Muir T.W., Severinov K. Mapping the molecular interface between the sigma(70) subunit of E. coli RNA polymerase and T4 AsiA. J. Mol. Biol. 2001;306:631–642. doi: 10.1006/jmbi.2001.4445. [DOI] [PubMed] [Google Scholar]
- 43.Ebright R.H. RNA polymerase: structural similarities between bacterial RNA polymerase and eukaryotic RNA polymerase II. J. Mol. Biol. 2000;304:687–698. doi: 10.1006/jmbi.2000.4309. [DOI] [PubMed] [Google Scholar]
- 44.Mukhopadhyay J., Das K., Ismail S., Koppstein D., Jang M., Hudson B., Sarafianos S., Tuske S., Patel J., Jansen R., et al. The RNA polymerase ‘switch region’ is a target for inhibitors. Cell. 2008;135:295–307. doi: 10.1016/j.cell.2008.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadesch T.R., Chamberlin M.J. Studies of in vitro transcription by calf thymus RNA polymerase II using a novel duplex DNA template. J. Biol. Chem. 1982;257:5286–5295. [PubMed] [Google Scholar]
- 46.Daubendiek S.L., Kool E.T. Generation of catalytic RNAs by rolling transcription of synthetic DNA nanocircles. Nat. Biotechnol. 1997;15:273–277. doi: 10.1038/nbt0397-273. [DOI] [PubMed] [Google Scholar]
- 47.Owen B., McMurray C. Rapid method for measuring DNA binding to protein using fluorescence anisotropy. Protocol Exchange. 2009 doi:10.1038/nprot.2009.80. [Google Scholar]
- 48.Carroll P., Schreuder L.J., Muwanguzi-Karugaba J., Wiles S., Robertson B.D., Ripoll J., Ward T.H., Bancroft G.J., Schaible U.E., Parish T. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PloS One. 2010;5:e9823. doi: 10.1371/journal.pone.0009823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steitz T.A. DNA polymerases: structural diversity and common mechanisms. J. Biol. Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 50.Kapatral V., Bina X., Chakrabarty A.M. Succinyl coenzyme A synthetase of Pseudomonas aeruginosa with a broad specificity for nucleoside triphosphate (NTP) synthesis modulates specificity for NTP synthesis by the 12-kilodalton form of nucleoside diphosphate kinase. J. Bacteriol. 2000;182:1333–1339. doi: 10.1128/jb.182.5.1333-1339.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.