

Abstract
A large number of cancer stem cells (CSCs) have been isolated and identified; however, none has been cultured in an unlimited manner in vitro without losing tumorigenicity and multipotency. In this study, we successfully clonogenically cultured a newly identified CD34+ liver CSC (LCSC) on feeder cells up to 22 passages (to date) without losing CSC property. Cloned CD34+ LCSC formed a round packed morphology and it could also be cryopreserved and recultured. Stem cell markers, CD34, CD117, and SOX2; normal liver stem cell markers, alpha fetoprotein, CK19, CK18, and OV6; putative CSC markers, CD44, CD133, EpCAM, and CD90; as well as CD31 were expressed in cloned CD34+ LCSC. SOX2 was the major factor in maintaining this LCSC before colonization, and interestingly, OCT4, SOX2, NAONG, Klf4, c-Myc, and Lin28 were upregulated in association with symmetric self-renewal for colony growth of CD34+ LCSC on feeder cells. Gene expression patterns of in vitro differentiation were consistent with our in vivo finding; furthermore, the tumorigenicity of cloned CD34+ LCSC was not different from uncloned CD34+ LCSC sorted from parental PLC. These results show that our cloned CD34+ LCSC maintained CSC property, including self-renewal, bipotency, and tumorigenicity after long-term culture, demonstrating that this LCSC can be cultured in an unlimited manner in vitro. Thus, establishing pure population of CSCs isolated from the patients will provide an opportunity to explore the mechanisms of tumorigenesis and cancer development, and to identify unique biomarkers presenting potential indicators of drug efficacy against CSCs for establishment of a novel strategy for cancer therapy.
Introduction
Cancer stem cells (CSCs) are a subpopulation of tumor cells that possess self-renewal capacity, the ability to initiate tumors, and provide new insight into our understanding of tumor initiation, maintenance, recurrence, and metastasis [1,2]. These cells exhibit unlimited proliferation capacity, the ability to self-renew, and to generate progeny of differentiated cells that constitute a heterogeneous tumor population [3,4]. It has been reported that some CSCs derive from their corresponding adult stem cells [5]; for example, a liver CSC (LCSC) has been shown to be derived from the enhanced self-renewal of normal liver stem cells [6]. Therefore, oncogenic mutations occurring in such a favorable background may change the restricted regulated growth property of normal stem cells into the aberrant uncontrolled growth of cancer cells.
CSCs were first identified in leukemia [7,8] and have also been demonstrated in several solid tumors, including breast, prostate, brain, melanoma, pancreatic, colon, lung, liver carcinomas [9–16], and in cancer cell lines [17–20]. These cells can be expanded in vitro as tumor spheres [21–26] and could reproduce human tumor xenografts when transplanted into immunodeficient mice. However, no report has shown that CSCs can be clonogenically cultured and expanded on feeder cells or using feeder-free conditions for long-term culture without losing tumorigenicity and multipotency. In this study, we successfully clonogenically cultured and expanded newly identified CD34+ LCSC [20] on mouse embryonic fibroblasts (MEF) using our defined culture conditions for up to 22 passages (to date) without losing tumorigenicity or multipotency.
Materials and Methods
Cell lines and cell culture
Hepatoma cell line, PLC/PRF/5 (PLC), was purchased from ATCC (www.atcc.org). Human embryonic stem cell (hESC) line, H9, was purchased from WiCell (www.wicell.com). The cell culture conditions for growing and expanding these line cells were according to the instructions as per provider.
Culture and expansion of the CD34+ LCSC with feeder conditions
Hepatoma cell line, PLC, was stained with the mouse anti-human CD34 antibody conjugated with PE (BD Science), and the CD34+ population was analyzed by BD FACScan (BD Science). Subsequently, the CD34+ cells were sorted from PLC and seeded on MEF (GlobeStem), as the feeder cells under our defined medium consists of Dulbecco's modified Eagle's medium/F12 medium supplemented with basic fibroblast growth factor (4 ng/mL), epidermal growth factor (10 ng/mL), 1× ITS (all from Invitrogen), nicotinamide (1.08 mg/mL), l-ascorbic acid-2 phosphate (0.29 mg/mL), l-proline (30 μg/mL), hydrocortisone (10 nM) (all from Sigma-Aldrich), 1× antibiotic/antimycotic (Invitrogen), and 0.1% bovine serum albumin. CD34+ colony cells were split and passaged between 10 and 14 days depending on colony size and confluence.
Culture of the CD34+ LCSC with feeder-free conditions
The CD34+ LCSCs were removed from culture with mouse feeder cells, seeded on a commercial extracellular matrix (ECM) plate, which is for the culture of human LCSC (www.celprogen.com) under our defined medium to remove the feeder cells, and expanded and maintained for 2 weeks until use.
Cryopreservation and recovery of the CD34+ LCSC
The cloned CD34+ LCSC was dissociated with collagenase type IV (1 mg/mL; Sigma-Aldrich) plus Dispase (1 mg/mL; Stem Cell Technologies); the cell suspension was frozen with an equal volume of freezing medium [60% of fetal bovine serum, 20% of complete growth medium, and 20% of dimethyl sulfoxide (DMSO)] and then stored at −140°C or in liquid nitrogen for future use. For recovery, the frozen tube with the cells was thawed immediately in 37°C water, after being taken from −140°C or liquid nitrogen; the thawed cells were transferred to the tube with 10 mL of warmed completed growth medium and spun at 300 g for 5 min to remove DMSO. The cell pellet was resuspended with the growth medium and seeded on freshly prepared MEF feeder cells.
In vitro differentiation
The CD34+ LCSCs were moved to the collage I-coated plate from the feeder cells and cultured with the growth medium used for culturing the parental PLC. The differentiated cells were harvested, the total RNA was isolated, and cDNA was generated at days 4, 9, and 13 after differentiation. Expression of liver genes, liver cancer markers, and CD34 and CD31 was evaluated by a quantitative polymerase chain reaction (qPCR), as previously described [27].
Assay of drug resistance by the CD34+ LCSC
The CD34+ LCSCs were removed from culture with mouse feeder cells and were seeded, expanded, and maintained on an aforementioned ECM plate under our defined medium to remove feeder cells. The CD34+ LCSCs were treated with cisplantin at 2 μg/mL at day 7 for 7 days, then cells were harvested and stained with antibodies against CD34, and the percentage of cells positive for CD34 was measured by flow cytometry.
Tumorigenicity by the cloned CD34+ LCSC
To investigate whether the tumorigenicity of the CD34+ LCSC was affected after long-term culturing in vitro, the cloned CD34+ LCSCs from the 5th to 18th passage were sorted to exclude feeder cells, and 1,000 cells were injected subcutaneously into NOD/SCID/IL2rg mice (Jackson Laboratory). The uncloned CD34+ cells sorted directly from the parental PLC were also used to inject into the same mouse model with the same cell number, as previously described [20]. Surgical procedures for transplantation and monitoring the tumor formation and subsequent tumor collection were approved by the Animal Care and Use Administrative Advisory Committee of the University of California Davis.
Immunohistochemistry analysis
The CD34+ LCSC on feeder cells and the xenograft tumor tissues on slides were fixed with 4% paraformaldehyde (PFA) and stained with different primary and secondary antibodies as previously described [27]. All antibodies are listed in the Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/scd).
Generation of cDNA and quantitative reverse transcription-polymerase chain reaction
RNA was extracted from the parental PLC, the uncloned CD34+ LCSC sorted from the parental PLC, the cloned CD34+ cells, and the derivatives of the cloned CD34+ LCSC, as well as hESC using the Qiagen mini RNA kit, and cDNAs were generated, and qPCR was performed as previously described [27]. Primers/probes are listed in Supplementary Table S2.
Cryosection of human xenografts
The xenograft tissue was cut into small pieces and fixed with 4% PFA for 4 h, then embedded in Tissue-Tek O.C.T, and stored at −80°C as previously described [28]. The cryosectioned slides were cut with a 5 μm thickness and immunostained with antibodies against antihuman liver proteins as previously described [20].
Evaluation of transcription factors
Expression of stem cell transcription factors in the parental PLC, the uncloned CD34+ LCSC sorted from the parental PLC, and the cloned CD34+ LCSCs were evaluated by qPCR and compared to those in hESC. Primers/probes are listed in Supplementary Table S2.
Statistics
All data are summarized as mean±standard error of the mean from at least three independent measurements. An unpaired Student t-test was used to analyze the data. P<0.05 was considered statistically significant.
Results
Isolation and clonagenic expansion of the CD34+ LCSC
To clonogenically culture and expand newly identified CD34+ LCSCs in vitro, the CD34+ LCSCs were sorted and seeded as single cells on MEF feeder cells under our aforementioned defined medium. After 7–10 days of culture, colonies were formed with a round packed morphology (Fig. 1A; Supplementary Fig. S1). Multiple morphologies were seen on the MEF feeder cells, suggesting that the CD34+ LCSCs are not a homogenous population.
FIG. 1.

Clonogenically culturing and expanding CD34+ liver cancer stem cells (LCSC). (A) CD34+ LCSC formed colonies with round and packed morphology after culturing on mouse embryonic fibroblast feeder cells under our defined condition, and multiple morphologies were observed on the feeder cells. (B) CD34+ LCSC could be cryopreserved and then recovered on feeder cells, and multiple morphologies were also shown similar to those before freezing. (C) CD34+ LCSC could be expanded on a commercial extracellular matrix (ECM) plate to remove feeder cells and maintained feeder free for a short period employing our culture conditions. CD34+ LCSC also maintained multiple morphologies on ECM. Scale bar, 100 μm. Color images available online at www.liebertpub.com/scd
The CD34+ LCSC could not only be expanded and maintained on the feeder cells but could also be cryopreserved and then recovered on the feeder cells. Figure 1B showed colonies of the CD34+ LCSC after culture following recovery from frozen cells at the 12th passage (total 22 passages). Multiple morphologies were also shown, similar to those before freezing (Fig. 1A).
The CD34+ LCSCs could be expanded on a commercial ECM (Celprogen) plate, which is for culturing LCSC, and maintained for a short period to remove the feeder cells under our culture conditions. The CD34+ LCSCs also maintained multiple morphologies (Fig. 1C).
Characterization of the CD34+ LCSC
To characterize the CD34+ LCSC, colonies on the feeder cells were immune-stained with antibodies against specific markers (Supplementary Table S1). Stem cell markers, CD34, CD117 (c-kit), and SOX2; normal liver stem cell markers, alpha fetoprotein (AFP), CK19, CK18, and OV6; liver cancer markers, CD44, CD133, EpCAM, and CD90; as well as CD31 were expressed in the cloned CD34+ LCSC (Fig. 2). Stem cell markers, OCT4, NAONG, and SOX2, which are all expressed in ESC, and induced pluripotent stem cells (iPSC), which culture and expand as clonal growth, are factors that are associated with pluripotency and self-renewal, a unique property of stem cells. However, only SOX2 could be detected in the cloned CD34+ LCSC by immunochemistry, suggesting that SOX2 is highly expressed in the CD34+ LCSC and is a significant factor in regulating this LCSC (Fig. 2).
FIG. 2.
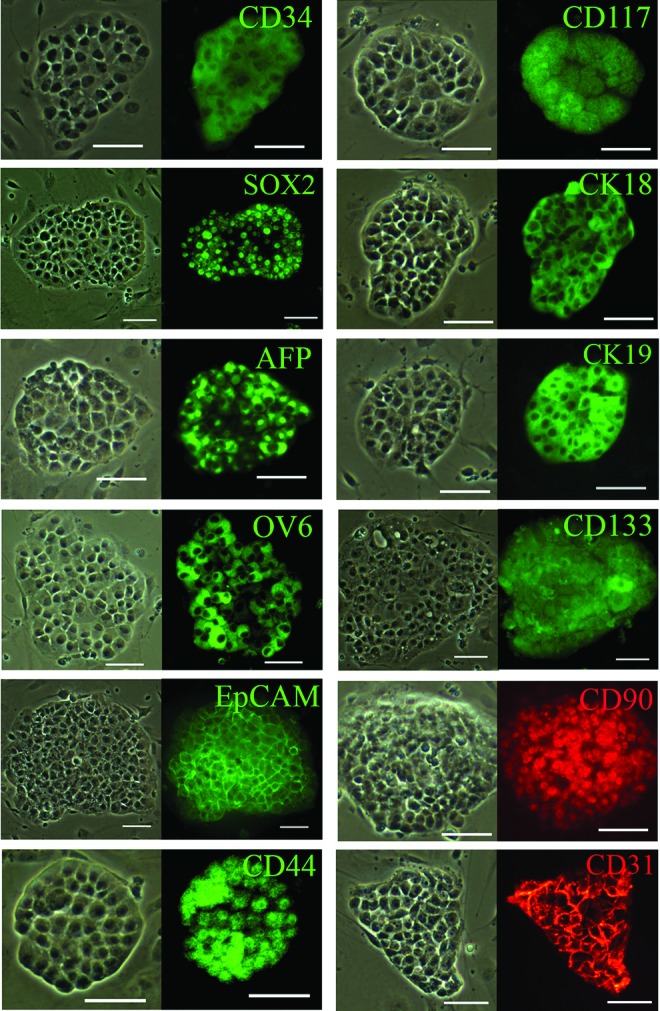
Characterization of cloned CD34+ LCSC. Stem cell markers (CD34, CD117, and SOX2), normal liver stem cell markers [CK18, alpha-fetoprotein (AFP), CK19, and OV6], putative cancer stem cell markers (CD133, EpCAM, CD90, and CD44), as well as CD31 were expressed by CD34+ LCSC on feeder cells, as determined by immunohistochemistry. Scale bar, 100 μm. Color images available online at www.liebertpub.com/scd
Expression of transcription factors in the CD34+ LCSC under different conditions
Stem cell transcription factors, OCT4, NAONG, SOX2, c-Myc, Klf4, and Lin28, which are related to pluripotency and self-renewal, as well as CD34, were evaluated in the parental PLC line, the uncloned CD34+ LCSC sorted from the parental PLC, and the cloned CD34+ LCSC by qPCR (Supplementary Table S2). When compared to the parental PLC line, SOX2 was upregulated, expression of OCT4 and NAONG was not changed, and c-Myc, Klf4, and Lin28 were downregulated in the sorted CD34+ cells. When compared to the sorted CD34+ LCSC, all six stem cell transcription factors, SOX2, OCT4, NAONG, c-Myc, Klf4, and Lin28, were upregulated in the cloned CD34+ LCSC (Fig. 3A). These factors are associated with stem cell self-renewal; thus, it appears that the upregulation of these genes was required for the clonal growth of the CD34+ LCSC on MEF feeder cells (Fig. 3A).
FIG. 3.

Comparison of expression patterns of stem cell transcription factors among three cell types. (A) Expression of stem cell transcription factors (SOX2, OCT4, NAONG, c-Myc, Klf4, Lin28) and stem cell marker, CD34, was compared among the parental PLC (PLC), the uncloned CD34+ LCSC sorted from the parental PLC (sorted CD34+), and cloned CD34+ LCSC (cloned CD34+) employing quantitative polymerase chain reaction (qPCR). (B, C) Expression of these six stem cell transcription factors was evaluated and compared among the sorted CD34+ LCSC (sorted CD34+), the cloned CD34+ LCSC (cloned CD34+), and human embryonic stem cell using qPCR. All values are mean±standard error of the mean (SEM) from three independent experiments. *P<0.05; **P<0.01.
When compared to the CD34+ LCSC, we found that OCT4, SOX2, and NAONG were extremely highly expressed in hESC (Fig. 3B); these three factors regulate hundreds and thousands of genes and maintain the pluripotency of ESC and iPSC [29,30], whereas our CD34+ LCSCs are bipotent stem cells, which only differentiate into hepatic and cholangiocytic lineages in vitro and in vivo [20]. Thus, it is not surprising that these pluripotent genes are more highly expressed in ESC and iPSC. Another three factors, c-Myc, Klf4, and Lin28, which are related to pluripotency and self-renewal [31,32], were also highly expressed in hESC (Fig. 3C). Interestingly, only Klf4 was highly expressed in the CD34+ LCSC when compared to its expression in hESC. This is likely because Klf4 is associated with cancer initiation and development [33]; thus, the total Klf4 is high in cancer cells and our CSC (Fig. 3C).
Assay of drug resistance by the CD34+ LCSC
Drug resistance is a CSC characteristic; the change of the percentage of the CD34+ LCSC under drug treatment is the parameter to indicate the drug sensitivity. The percentage of the CD34+ LCSC on feeder cells depended on total colony density; to avoid high density causing spontaneous differentiation or low density resulting in low yield of the CD34+ LCSC for analysis, normally we maintained around 200 colonies in each well of the six-well culture plates. Flow cytometry results showed that 45% of cells were positive for CD34 under such circumstances (Fig. 4A). The CD34+ cells could be enriched to 88% on ECM culture within 3 days after moving from the feeder cell culture (Fig. 4B); however, the percentage of the CD34+ cells decreased to 57% without the support of MEF feeder cells at day 14 of ECM culture (Fig. 4C). When the CD34+ cells were treated with cisplatin at 2 μg/mL for 7 days from day 7 on ECM culture after their removal from MEF feeder cells, the percentage of the CD34+ cells was markedly increased to 71% when compared to those without treatment with cisplatin, as determined by flow cytometry (Fig. 4D). This indicated that the CD34+ LCSCs were resistant to the antitumor agent.
FIG. 4.

Analysis of drug resistance and in vitro differentiation. (A) The percentage of CD34+ cells on mouse embryonic fibroblasts feeder cell culture. (B) The percentage of CD34+ cells at day 3 on ECM after being moved from feeder cell culture. (C) The percentage of CD34+ cells at day 14 on ECM culture after being moved from feeder cell culture. (D) The percentage of CD34+ cells at day 14 on ECM culture with treatment by cisplatin at 2 μg/mL from day 7 after being moved from feeder cell culture. (E) Expression of liver genes [albumin (ALB), AFP, CK19, and CK7], liver cancer markers (CD133, EpCAM, CD44, and CD90), as well as CD31 and CD34 was evaluated by qPCR at days 4, 9, and 13 after the differentiation of cloned CD34+ LCSC and compared to undifferentiated cloned CD34+ LCSC (CD34+). All values are mean±SEM from three independent experiments. *P<0.05; **P<0.01.
In vitro differentiation
To access in vitro differentiation of the cloned CD34+ LCSC, the differentiated cells were harvested at days 4, 9, and 13 after differentiation. Expression of liver genes albumin, AFP, CK19, and CK7; liver cancer markers CD133, CD44, CD90, and EpCAM, as well as CD34 and CD31 were evaluated by qPCR (Supplementary Table S2). Expression of all genes, except CD34, was increased over time (Fig. 4E); these results show that expression of these genes was consistent with our in vivo finding [20].
Tumorigenicity by the cloned CD34+ LCSC
To evaluate whether the tumorigenicity of the cloned CD34+ LCSC was affected, the uncloned CD34+ cells sorted from the parental PLC and the cloned CD34+ LCSCs at the 5th and the 18th passage growing on feeder cells were injected into mice. The number of cells required and the timing of the tumor formation were not different between the four groups (Fig. 5A, B). The results of hematoxylin and eosin staining for human xenografts produced by the transplantation of the cloned CD34+ LCSC and CD34+ PLC exhibited the typical histologic features of human liver carcinomas (HLCs). There are polygonal cells with pleomorphic nuclei in cells with distinct cell borders, nucleus:cytoplasm (N:C) ratios are increased, and nuclei are hyperchromatic with prominent nucleoli. The presence of plump endothelial cell lining the sinusoids is another hallmark of the histopathological features of hepatocellular carcinomas (HCC) (single black arrow, Fig. 5B; Supplementary Figs. S2A, S4A, and S5B). In any high-grade malignancy, HLC may contain a different size foci of necrosis (double black arrows, Fig. 5B; Supplementary Figs. S2B, S4A, and S6A) and dilated blood vessels (single white arrow, Fig. 5B; Supplementary Figs. S2B, S4A, and S6B). These xenografts behaved like typical HLCs, in which they exhibited a nested pattern with large tumor nodules separated by thick fibrous bands (double white arrows, Fig. 5B; Supplementary Figs. S3A, S4A, and S7A). The neoplastic cells in HCC can synthesize and store various components of hepatocytes, such as lipids, bile, alpha-1 antitrypsin (α1-AT), AFP, and other cytoplasmic constituents, which also occurred in the xenografts (black head arrow, Fig. 5B; Supplementary Figs. S3B, S4A, and S7B). Immunochemistry results showed that the tissues of human xenografts expressed human liver-specific proteins, hepatocyte-specific antigen—Hep Par 1, CK19, α1-AT, and liver cancer marker, EpCAM (Fig. 5C; Supplementary Figs. S4B), further confirming that the tumor tissues were HLC xenografts. These in vitro and in vivo results indicate that the cloned CD34+ LCSC maintained their differentiation capacity after long-term culture.
FIG. 5.

Tumorigenicity by cloned CD34+ LCSC. (A) The ability of the cells to form tumors in vivo was evaluated by the injection of the same numbers of sorted CD34+ PLC and cloned CD34+ LCSCs from the 5th passage (p5) to the 18th passage (p18) into NOD/SCID/IL2rg mice. (B) The tumors were formed within 2 months, and the number and the timing of tumor formation were not different between these four groups. Hematoxylin and eosin staining of human xenografts produced by the injection of cloned CD34+ LCSC into mice, exhibited the typical histologic features of human liver carcinomas, including polygonal cells with pleomorphic nuclei in cells with distinct cell borders; the increased nucleus:cytoplasm ratios; the presence of endothelial cells lining the sinusoids (single black arrow); small and large foci of necrosis (double black arrows) and dilated blood vessels (single white arrow); nested growth pattern with large tumor nodules separated by a thick fibrous band (double white arrows); various components of hepatocytes such as lipids and other cytoplasmic constituents (black head arrow). (C) Human liver-specific proteins, hepatocyte-specific antigen-Hep Par1, CK19, alpha 1-antitrypsin (α1-AT), and liver cancer marker, EpCAM were expressed in the tissues of aforementioned human xenografts. The specificity of primary antibodies was evaluated by employing isotype controls. Scale bar, 100 μm. Color images available online at www.liebertpub.com/scd
Discussion
A large number of CSCs have been isolated and identified from solid tumors [9–16] and cancer cell lines [17–20]. To maintain the tumorigenicity and stem cell properties in vitro, sphere formation is the only current approach to culture and expand these CSCs [21–26] for a short period. It has been shown that CSCs could be cultured on the feeder cells [34]; however, the cells still grew as spheres on the feeder cells [34], and in addition, autologous intratumoral fibroblasts were required as the feeder cells [34]. Thus, it appears to still be a challenge to culture CSC in vitro without losing their tumorigenicity and multipotency. With stem cell technologies in conjunction with employing our defined culture medium, we could clonogenically culture and expand newly identified CD34+ LCSC [20] on MEF feeder cells for prolonged culture. The cloned CD34+ LCSC could be passaged at least 22 generations (to date) without losing CSC properties, including symmetric self-renewal-based colony formation, multipotency, and tumorigenicity. In addition, the cloned CD34+ LCSC could also be cryopreserved and recultured with these properties. The cloned CD34+ LCSCs have multiple morphologies in culture that differ from colonies of ESC and iPSC, which are uniform (Fig. 1A). In our previous report, 12 subpopulations of these CD34+ cells could independently form HLC xenografts in mice and they functioned as tumor-initiating cells [20]. Clonal growth is one of the stem cell features and thus their capacity for colony formation as described in this study demonstrates that these subpopulations of CD34+ cells have the capacity to perform symmetric self-renewal, further confirming that they are CSC populations. Furthermore, the finding of having multiple morphologies in culture also demonstrates that these CD34+ LCSCs from one origin represent a heterogeneous population, also confirming the results from our previous study in which the CD34+ LCSC could form three types of HLC xenografts and each type of HLC xenograft consisted of a heterogeneous population [20].
It is well known that OCT4, SOX2, and NAONG control pluripotency and self-renewal in ESC and iPSC [29,30]; however, only SOX2 was highly expressed in the sorted CD34+ LCSC (Figs. 2A and 3A). Thus, it appears that SOX2 might be the major factor to regulate CD34+ LCSC self-renewal in the parental PLC culture. In the routine culture of the parental PLC line, which lacks the conditions to establish and maintain an appropriate stem cell niche, the CD34+ LCSCs undergo self-renewal by choosing an asymmetric division, which produces a stem cell and a progenitor cell; thus, they do not form colonies. Colony growth of stem cells requires an appropriate and stable stem cell niche plus appropriate intrinsic and extrinsic factors. The feeder cells providing the niche environment, growth factors, and cytokines secreted by the feeder cells, as well as growth factors and reagents provided with our defined culture medium, consisted of the extrinsic factors required for colony growth of stem cells. After expanding and maintaining on the feeder cells, OCT4, SOX2, NAONG, c-Myc, Klf4, and Lin28 were upregulated; these factors are required as intrinsic factors for colony growth of stem cells. With the support of these necessary intrinsic and extrinsic factors, the CD34+ LCSC could undergo symmetric self-renewal to form colonies on the feeder cells. In our routine culturing of colony growth-based stem cells, including CSC, ESC, and iPSC, the changes of extrinsic factors usually produce some bad colonies, and the reason leading to the failure of colony growth and maintenance is that stem cells choose asymmetric self-renewal or undergo spontaneous differentiation by these changes.
As stem cell transcription factors, expression of OCT4, SOX2, NAONG, c-Myc, Klf4, and Lin28 varies in different types of stem cells and culture conditions of the same stem cells (growth pattern). The expression of all these factors was increased in the cloned CD34+ LCSC (clonal growth) when compared to the uncloned CD34+ LCSC sorted from the parental PLC (spread growth). Generally, the greater the stem cell potency is, the higher these gene expressions are. Five of six factors were highly expressed in pluripotent stem cells hESC when compared to those in our CD34+ LCSC; it is reasonable that their expressions are lower in bipotent stem cells. Interestingly, only Klf4 expression was higher in the CD34+ LCSC than in hESC and this is also not surprising because Klf4 is associated with cancer initiation and development [31], the total Klf4 product is high in cancer cells and CSCs; thus, this suggests that Klf4, c-Myc, and a proto-oncogene gene [35] are potentially highly expressed in another type of CSC.
To expand the CD34+ LCSC without the feeder cells, a commercial ECM that is designed to culture LCSCs was employed; however, the percentage of CD34+ population decreased over time. The CD34+ LCSCs were maintained and expanded on the ECM for a very short period. Matrigel, which has been successfully used for culturing ESC/iPCS under feeder-free conditions, was also employed to establish feeder-free conditions for culturing and expanding the CD34+ LCSC; however, the CD34+ LCSC failed to culture and expand as colony growth on Matrigel under our defined conditions, rather the cells differentiated. Thus, more work is necessary to establish feeder-free conditions for the maintenance of the cloned CD34+ LCSC.
Differentiation potential is a basic property of stem cells; our cloned CD34+ LCSC could readily form HLC xenografts in mice, and the number and the timing of the tumor formation were similar to the CD34+ LCSC, which are directly sorted from the parental PLC line [20]. During in vitro differentiation, the derivatives of our cloned CD34+ LCSC also expressed hepatocyte markers, cholangiocyte markers, liver cancer markers, and CD31; thus, the in vitro gene expression pattern was consistent with the results of in vivo gene expression shown in our previous study [20] and hence these results indicate that the cloned CD34+ LCSC maintained their tumorigenicity and multipotency after long-term culture.
In summary, we successfully clonogenically cultured and expanded newly identified CD34+ LCSC [20] for a prolonged period on the feeder cells employing our defined medium without losing tumorigenicity and multipotency, demonstrating that this LCSC can be cultured and expanded in an unlimited manner in vitro as normal stem cells, such as ESC and iPSC, do; thus, a large number of CSC can potentially be isolated from the patients and cultured in an unlimited manner for research and clinical studies. Therefore, establishing pure populations of CSCs from the patients will allow for better genomic, epigenomic, transcriptomic, proteomic, and metabolomic analysis and thus provide an opportunity to explore the mechanisms of tumorigenesis and cancer development, and will also allow for a global comparison between CSCs and normal stem cells to identify specific biomarkers for the CSC population. These specific biomarkers from CSCs may represent potential indicators of drug efficacy against CSCs and the establishment of novel drug development assays. Furthermore, identifying cell-specific surface markers of CSCs and dissecting and characterizing signaling pathways that impact on CSC self-renewal and differentiation will not only provide an opportunity to explore the mechanism of transformation of CSC but will also provide the potential for developing therapeutics aimed at CSC treatment.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant DK075415 (to M.A.Z.) and the GlaxoSmithKline Research Fund of the Korean Association for the Study of the Liver (to S.C.P., and J.R.E.).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Reya T, Morrison SJ, Clark MF. and Weissman IL. (2001). Stem cell, cancer and cancer stem cells. Nature 414:105–111 [DOI] [PubMed] [Google Scholar]
- 2.Pardal R, Clarke MF. and Morrison SJ. (2003). Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 3:895–902 [DOI] [PubMed] [Google Scholar]
- 3.Eramo A, Lotti F, Sette G, Pilozzi E, Biffonil M, Di Virgilio A, Conticello C, Ruco L, Peschle C, et al. (2008). Identification and expression of the tumorigenic lung cancer stem cell population. Cell Death and Differ 15:504–514 [DOI] [PubMed] [Google Scholar]
- 4.Vermeulen L, Todaro M, de Sousa F, Sprick MR, Kemper K, Perez Alea M, Richel DJ, Stassi G. and Medema JP. (2008). Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci U S A 105:13427–13432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, et al. (2006). A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 12:410–416 [DOI] [PubMed] [Google Scholar]
- 6.Chiba T, Zheng Y, Kita K, Yokosuka O, Saisho H, Onodera M, Miyoshi H, Nakano M, Zen Y, et al. (2007). Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology 133:937–950 [DOI] [PubMed] [Google Scholar]
- 7.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, et al. (1994). A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367:645–648 [DOI] [PubMed] [Google Scholar]
- 8.Bonnet D. and Dick JE. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3:730–737 [DOI] [PubMed] [Google Scholar]
- 9.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ. and Clarke MF. (2003). Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, et al. (2006). Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 25:1696–1708 [DOI] [PubMed] [Google Scholar]
- 11.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD. and Dirks PB. (2004). Identification of human brain tumour initiating cells. Nature 432:396–401 [DOI] [PubMed] [Google Scholar]
- 12.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder ED, et al. (2005). A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res 65:9328–9337 [DOI] [PubMed] [Google Scholar]
- 13.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF. and Simeone DM. (2007). Identification of pancreatic cancer stem cells. Cancer Res 67:1030–1037 [DOI] [PubMed] [Google Scholar]
- 14.O'Brien CA, Pollett A, Gallinger S. and Dick JE. (2007). A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110 [DOI] [PubMed] [Google Scholar]
- 15.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT. and Jacks T. (2005). Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121:823–835 [DOI] [PubMed] [Google Scholar]
- 16.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PWK, Lam CT, Poon RTP, et al. (2008). Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 13:153–166 [DOI] [PubMed] [Google Scholar]
- 17.Kondo T, Setoguchi T. and Taga T. (2004). Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A 101:781–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, Gao Q, Suo Z, Munthe E, Solberg S, Ma L, Wang M, Westerdaal NA, Kvalheim G. and Gaudernack G. (2013). Identification and characterization of cells with cancer stem cell properties in human primary lung cancer cell lines. PLoS One 8:e57020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Almanaa TN, Geusz ME. and Jamasbi RJ. (2013). A new method for identifying stem-like cells in esophageal cancer cell lines. J Cancer 4:536–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park SC, Nguyen NT, Eun JR, Zhang Y, Jung YJ, Tschudy-Seney B, Trotsyuk A, Lam A, Ramsamooj R, et al. (2015). Identification of cancer stem cell subpopulations of CD34+ PLC/PRF/5 that result in three types of human liver carcinomas. Stem Cell Dev 24:1008–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C. and De Maria R. (2006). Identification and expansion of human colon-cancer-initiating cells. Nature 445:111–115 [DOI] [PubMed] [Google Scholar]
- 22.Jung P, Sato T, Merlos-Suárez A, Barriga FM, Iglesias M, Rossell D, Auer H, Gallardo M, Blasco MA, et al. (2011). Isolation and in vitro expansion of human colonic stem cells. Nat Med 17:1225–1227 [DOI] [PubMed] [Google Scholar]
- 23.Cao L, Zhou Y, Zhai B, Liao J, Xu W, Zhang R, Li J, Zhang Y, Chen L, et al. (2011). Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol 11:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA. and Daidone MG. (2005). Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res 65:5506–5510 [DOI] [PubMed] [Google Scholar]
- 25.Stecca B, Santini R, Pandolfi S. and Penachioni JY. (2013). Culture and isolation of melanoma-initiating cells. Curr Protoc Stem Cell Biol 24:3.6.1–3.6.12 [DOI] [PubMed] [Google Scholar]
- 26.Benayoun L. and Shaked Y. (2013). In vitro enrichment of tumor-initiating cells from human established cell lines. Curr Protoc Stem Cell Biol 24:3.7.1–3.7.15 [DOI] [PubMed] [Google Scholar]
- 27.Duan Y, Ma X, Zou W, Wang C, Bahbahan IS, Ahuja TP, Tolstikov V. and Zern MA. (2010). Differentiation and characterization of metabolically functioning hepatocytes from human embryonic stem cells. Stem Cells 28:674–686 [DOI] [PubMed] [Google Scholar]
- 28.Duan Y, Catana A, Meng Y, Yamamoto N, He S, Gupta S, Gambhir SS. and Zern MA. (2007). Differentiation and enrichment of hepatocyte-like cells from human embryonic stem cells in vitro and in vivo. Stem Cells 25:3058–3068 [DOI] [PubMed] [Google Scholar]
- 29.Boyer LA, Lee IT, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM. and Murray HL. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, et al. (2006). The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38:431–440 [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K. and Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 26:663–676 [DOI] [PubMed] [Google Scholar]
- 32.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920 [DOI] [PubMed] [Google Scholar]
- 33.Evans PM. and Liu C. (2008). Roles of Krüpel-like factor 4 in normal homeostasis, cancer and stem cells. Acta Biochim Biophys Sin 40:554–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Y, Hu Y, Zhou J. and Zhang M. (2011). Establishing a lung cancer stem cell culture using autologous intratumoral fibroblasts as feeder cells. Cell Biol Int 35:509–517 [DOI] [PubMed] [Google Scholar]
- 35.Kato GJ. and Dang CV. (1992). Function of the c-Myc oncoprotein. FASEB J 6:3065–3072 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.