

Abstract
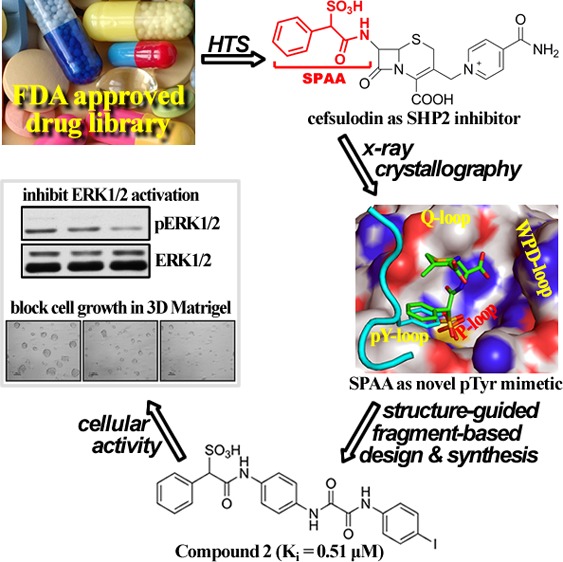
Protein tyrosine phosphatases (PTPs) are potential therapeutic targets for many diseases. Unfortunately, despite considerable drug discovery efforts devoted to PTPs, obtaining selective and cell permeable PTP inhibitors remains highly challenging. We describe a strategy to explore the existing drug space for previously unknown PTP inhibitory activities. This led to the discovery of cefsulodin as an inhibitor of SHP2, an oncogenic phosphatase in the PTP family. Crystal structure analysis of SHP2 interaction with cefsulodin identified sulfophenyl acetic amide (SPAA) as a novel phosphotyrosine (pTyr) mimetic. A structure-guided and SPAA fragment-based focused library approach produced several potent and selective SHP2 inhibitors. Notably, these inhibitors blocked SHP2-mediated signaling events and proliferation in several cancer cell lines. Thus, SPAA may serve as a new platform for developing chemical probes for other PTPs.
Keywords: Protein tyrosine phosphatase, pTyr mimetics, SHP2 inhibitors, fragment-based library, anticancer agents
Proper level of protein tyrosine phosphorylation is vital for cell growth and survival. Aberrant tyrosine phosphorylation, due to imbalance of activities of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs), is linked to numerous human diseases and offers enormous opportunities for therapeutic intervention. The success for such targeted approach has been well established by the more than two-dozen PTK inhibitors already used in clinic.1 However, acquired resistance to PTK inhibitors limit durable responses. Therefore, there is great potential to modulate disease progression at the level of PTPs. To this end, the Src homology 2 (SH2) domain containing PTP2 (SHP2), encoded by the PTPN11 gene, is a positive signal transducer, required for most receptor PTK-mediated Ras/ERK1/2 activation.2,3 In addition, considerable evidence indicates that SHP2 is a bona fide oncoprotein as activating SHP2 mutations are found in leukemia and solid tumors.4,5 Moreover, given the obligatory requirement of SHP2 in growth factor-mediated pathways, thwarting SHP2 activity may also prove effective for cancers caused by abnormal activation of receptor PTKs, some of which respond poorly to kinase inhibitor monotherapy. Hence there is strong interest in developing small molecule SHP2 inhibitors as novel anticancer agents.6
However, the conserved PTP active site (i.e., pTyr-binding pocket)7 makes it difficult to develop isozyme-specific inhibitors. One useful paradigm for the design of potent and selective PTP inhibitors is to engage both the active site and unique peripheral binding pockets by tethering appropriately functionalized moieties to a nonhydrolyzable pTyr mimetic.8,9 Application of this strategy has enabled the development of a number of small molecule PTP probes.10 Unfortunately, most existing pTyr mimetic-containing PTP inhibitors lack appropriate cellular efficacy, which represents a major obstacle in developing PTP-based therapeutics. Consequently, there is continued interest in identifying novel pTyr mimetics with more acceptable pharmacological properties.
To address the bioavailability issue, we sought to explore the existing drug space for previously unknown PTP inhibitory scaffolds. Since known drugs are already used in humans with acceptable pharmacokinetic features and tolerable side effects, such a strategy may supply promising starting points for PTP-based drug development. To this end, we screened the Johns Hopkins Drug Library11 and identified cefsulodin (Figure 1A), a third generation β-lactam antibiotic, as a reversible and competitive SHP2 inhibitor with an IC50 value of 16.8 ± 2.0 μM and a Ki of 6.6 ± 0.2 μM (Figures S1–S3, and more details in Supporting Information). Cefsulodin is also quite selective for SHP2, displaying, with one exception of SHP1, >10-fold selectivity against all PTPs examined (Table 1). As reported before,12 cefsulodin also exhibits inhibitory activity against PTP1B, although with much less potency compared to SHP2 (Table 1).
Figure 1.
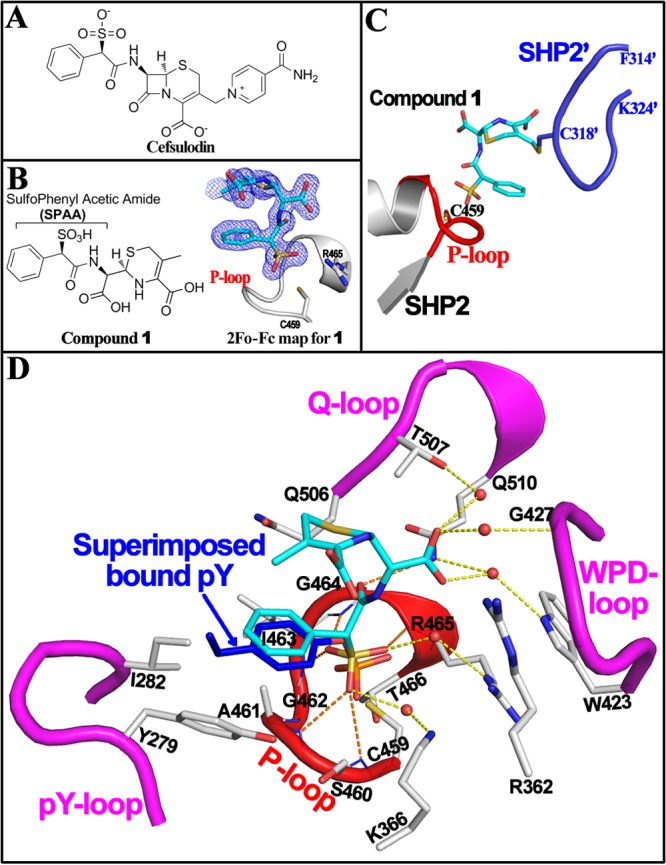
Crystal structure of SHP2 with cefsulodin derived compound 1 identifies SPAA as a novel pTyr mimetic. (A) Structure of cefsulodin. (B) Structure and electron density of compound 1 bound at SHP2 active site. (C) Under crystallization conditions, the bound compound 1 (cyan) is covalently attached to C318 in a flexible loop (blue) from a nearby symmetric SHP2. (D) Interaction details of compound 1 (cyan) with SHP2. Residues within 5 Å distance of compound 1 were shown in stick (gray). H-bonds or water-bridged H-bonds between compound 1 and SHP2 were indicated by dash line. Four loops forming SHP2 active site pocket were highlighted in red or magenta. A PTP1B·pTyr-peptide structure (PDB#: 1EE0) was superimposed onto the SHP2·1 structure, and the bound pTyr (blue) was found to overlap well with SPAA.
Table 1. IC50 Values (μM) of Cefsulodin and SPAA Derived SHP2 Inhibitors against a Panel of PTPs.
| enzyme | cefsulodin | 2 | 3 | 4 |
|---|---|---|---|---|
| SHP2 | 16.8 ± 2.0 | 1.5 ± 0.1 | 1.4 ± 0.04 | 2.3 ± 0.04 |
| SHP1 | 21.0 ± 3.0 | 7.3 ± 1.3 | 4.8 ± 1.1 | 6.0 ± 0.02 |
| PTP1B | 190 ± 18 | 9.3 ± 2.0 | 3.0 ± 0.06 | 7.0 ± 1.0 |
| LYP | >200 | >50 | >50 | >50 |
| HePTP | 250 ± 20 | >50 | >50 | >50 |
| Meg2 | 350 ± 20 | >50 | >50 | >50 |
| PTPα | >200 | >50 | >50 | >50 |
| PTPβ | >200 | >50 | >50 | >50 |
| PTPε | >200 | >50 | >50 | >50 |
| PTPγ | >200 | >50 | >50 | >50 |
| PTPμ | >200 | >50 | >50 | >50 |
| LAR | >200 | >50 | >50 | >50 |
| VHR | >200 | >50 | >50 | >50 |
| CDC14A | 300 ± 50 | >50 | >50 | >50 |
| LMWPTP | >200 | >50 | >50 | >50 |
| PP5 | >200 | >50 | >50 | >50 |
To define the molecular basis of SHP2 inhibition by cefsulodin and to guide the design of cefsulodin-based SHP2 inhibitors, we determined the crystal structure of SHP2 in complex with cefsulodin at 1.6 Å resolution (PDB#: 4RDD). Data collection and structure refinement statistics are summarized in Table S1. Unexpectedly, compound 1, but not cefsulodin, was unambiguously identified in the SHP2 active site (Figure 1B). Detailed structural, kinetic, and mass spectrometry analyses suggest that, upon binding of cefsulodin to SHP2 in the crystalline state, a covalent adduct was formed between 1 and SHP2 due to displacement of the isonicotinamide tail in cefsulodin by C318 from a nearby symmetry-related SHP2 molecule, followed by hydrolysis of the β-lactam ring (Figure 1C; Figures S4–S6, and more details on Page 7 in Supporting Information).
The crystal structure of the SHP2·1 complex (Figure 1D) showed that the sulfonic acid is in close proximity to the catalytic P-loop and forms multiple hydrogen bonds with the backbone amides of S460, A461, I463, G464, and R465, as well as two water-mediated hydrogen bonds with the R465 and K366 side chains. The α-benzene ring is located within a hydrophobic pocket constituted by A461, I463, I282, and Y279, which normally functions to recognize and stabilize the tyrosine ring of PTP substrates during catalysis. Superimposition of the SHP2·1 structure with a previously reported PTP1B·pTyr-peptide structure13 revealed that the sulfophenyl acetic amide (SPAA) motif overlaps very well with pTyr in the phosphopeptide (Figure 1D). In addition, the carboxylic acid derived from the β-lactam ring opening makes multiple water bridged polar interactions with W423 and G427 in the WPD loop as well as T507 and Q510 in the Q-loop.
The observed mode of interaction between SHP2 and 1 as well as structural comparison with the PTP1B·pTyr-peptide complex identify SPAA as a unique pTyr mimetic (Figure 2A), which could be employed for the design of novel sulfonic acid based PTP inhibitors. Unlike classical pTyr mimetics phosphonodifluoromethyl phenylalanine (F2Pmp) and sulfamic acid (Figure 2B), in which the peripheral site binding groups are usually placed opposite to the acid moieties on the benzene ring, the novelty of SPAA is that it allows molecular diversity to be introduced from the same side of the benzene ring where the sulfonic acid resides (Figure 2A), thereby enabling exploration of previously uncharted binding pockets outside of the PTP active site.
Figure 2.
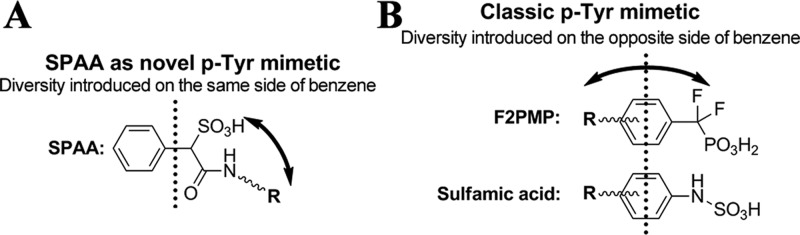
SPAA is a novel pTyr mimetic. (A) The distinctive feature of SPAA as a novel pTyr mimetic. (B) Structures and common feature of representative classic pTyr mimetics.
To demonstrate the utility of SPAA for PTP inhibitor development, we prepared SPAA fragment-based focused libraries using appropriately functionalized linkers to introduce structural diversity (Figure 3A). As a proof-of-concept, four focused libraries were designed, in which a benzene ring was chosen as the linker and amide bond formation served as a vehicle for structural diversification. Lib-1 to Lib-4 were key precursors incorporating SPAA and linkers, and the libraries were synthesized by coupling the available carboxylic acid on the oxalyl moiety (Figure 3A) with a set of 192 amines (Figure S7) that differ in size, charge, lipophilicity, and other drug-like properties to afford a total of 768 compounds. Lib-1 and Lib-2 differed by the position on the benzene ring the oxalic acid is attached to, while Lib-3 and Lib-4 were designed to explore flexibility on either side of the benzene linker. The libraries were assembled directly in 96-well plates by standard HBTU amide coupling chemistry and screened at both ∼10 and ∼1 μM against SHP2.
Figure 3.

Design and synthesis of SPAA-based SHP2 inhibitors. (A) The structure-guided and fragment-based design. (B) The structures of the top three hits (2 to 4) and a negative control (5).
The top three hits (compounds 2 to 4, Figure 3B) were resynthesized and purified, and their IC50s for SHP2 were determined. Although precursors Lib-1 to Lib-4 showed no inhibitory activity against SHP2 at 50 μM, compounds 2 to 4 exhibited IC50s in the range of 1.4–2.3 μM (Table 1), which are 7–12-fold more potent than that of cefsulodin. Interestingly, compounds 2 to 4 are all from Lib-1, with the oxalic acid handle located at the para position of the benzene linker, indicating that the orientation of the added diversity fragments is important. In addition, compounds 2 to 4 have either biaryl or single aryl group with bulky substituents at the terminal position, highlighting a clear preference for lipophilic moieties. This observation is supported by the substantial loss of activity for compound 5 (IC50 > 50 μM), which has a single benzene at the terminal position. Selectivity profiling against a large panel of human PTPs showed that compound 2 exhibits at least several-fold selectivity against all PTPs tested, including the closest homologous protein, SHP1 (Table 1). Kinetic analysis revealed that compound 2 is a reversible and competitive inhibitor of SHP2 with a Ki value of 0.51 ± 0.03 μM (Figure 4), consistent with the expectation that the SPAA moiety binds the PTP active site.
Figure 4.

Lineweaver–Burk plot for compound 2-mediated inhibition of SHP2 catalyzed hydrolysis of para-nitrophenyl phosphate. Compound 2 concentrations were 0 (●), 0.25 (○), 0.5 (▼), and 0.75 (▽) μM, respectively.
Given the observed potency and selectivity toward SHP2 by the SPAA-based inhibitors, we evaluated their ability to inhibit SHP2-dependent cell signaling and proliferation. Previous studies showed that SHP2 is required for growth of a patient derived nonsmall cell lung cancer (NSCLC) cell line H1975,14 which expresses the secondary gatekeeper T790M/L858R double-mutations in epidermal growth factor receptor (EGFR), rendering the cells resistant to EGFR inhibitors gefitinib and erlotinib.15 As shown in Figure 5A, compounds 2–4 were able to reduce H1975 cell proliferation in a dose-dependent manner, while compound 5 was ineffective as expected, given that it has no inhibition against SHP2 at 50 μM. Similar results were also obtained with human breast cancer cell line MDA-MB-231 (Figure 5B). Thus, compound 2 appeared to be the most efficacious among this series, and it was selected for further mechanistic study.
Figure 5.

Cellular activity of SPAA-based SHP2 inhibitors. MTT assay for compounds 2 to 5 in H1975 lung cancer cell line (A) or in MDA-MB-231 breast cancer cell line (B). Compounds 2–4 significantly (**p < 0.01) reduced cell proliferation in a dose-dependent manner in both cell lines. (C) Compound 2 decreased the EGF-induced ERK1/2 activation dose dependently in H1975 cell. (D) Compound 2 blocked EGF-induced ERK1/2 activation and SHP2-mediated dephosphorylation of paxillin, while the negative control compound 5 failed to exert any effect on ERK1/2 and paxillin phosphorylation. (E) Compound 2 had no effect on PMA-induced ERK1/2 activation. (F) Compound 2 inhibited the growth of the ErbB2 positive SKBR3 cells in 3D Matrigel. (G) Compound 2 inhibited ERK1/2 activation in SKBR3 cells cultured in a 3D Matrigel environment. The results shown in this figure are representatives from at least two independent experiments, and the numbers below the gels are presented as mean ± SD. The quantification and normalization were performed as follows: band intensity was quantified using the ImageJ program, and the ratios of pERK1/2/total ERK1/2 or pPaxillin(Y118)/total Paxillin were calculated and normalized to the reference.
Since SHP2 phosphatase activity is required for the full activation of the Ras-ERK1/2 pathway,2 we assessed the effect of 2 on EGF-induced ERK1/2 activation in H1975 cells. As expected, 2 effectively reduced the EGF-induced ERK1/2 phosphorylation in a dose-dependent manner (Figure 5C), but the structurally related inactive compound 5 had no appreciable effect on pERK1/2 level (Figure 5D). Importantly, compound 2 increased the phosphorylation level of Y118 in paxillin, which is a substrate for SHP2,16 while 5 had no effect on paxillin phosphorylation (Figure 5D). To provide further evidence that the observed cellular effect of 2 is SHP2 dependent, we examined the effect of 2 on PMA (phorbol 12-myristate 13-acetate)-induced ERK1/2 activation, which is SHP2 independent,17 and instead involve activation of protein kinase C and Raf18 in a Ras-independent manner.19 Thus, SHP2 inhibitors would not be expected to affect PMA-induced ERK1/2 phosphorylation. Indeed, compound 2 had no effect on PMA-induced ERK1/2 phosphorylation (Figure 4E). Taken together, the results indicate that 2 specifically inhibits SHP2-mediated cellular signaling events.
Finally, we evaluated the effect of SHP2 inhibition by 2 on the growth of SKBR3 cells in Matrigel. SKBR3 cells are an ErbB2 positive breast cancer cell line that when grown in Matrigel more accurately reflect the features of human tumors than when grown on plastic.20 These cells through upregulation of ErbB2 strongly activate Ras, which promotes ERK1/2 signaling when grown in Matrigel.21 As shown in Figure 5F,G, SKBR3 cells formed tumor-like growths in Matrigel treated with vehicle, but their growth was dose dependently inhibited by 2. Further, while both control and 2 treated cells showed equivalent levels of total ERK1/2, their levels of phosphorylation were concordantly inhibited by increasing concentrations of 2. Taken together, the results showed that compound 2 can specifically inhibit SHP2-mediated signaling events and the growth of H1975 lung cancer cells, MDA-MB-231, and ErbB2 positive SKBR3 breast cancer cells.
In summary, we describe a novel strategy for the discovery of PTP inhibitors by exploring the existing drug space. In contrast to the traditional approach to drug repurposing, which entails identifying new uses for existing drugs,22,23 this study illustrates another path for drug repurposing, namely, by identifying a successful pharmacophore from an existing drug that could be further elaborated and refined for novel therapeutic targets. By screening a large FDA-approved drug collection we discovered that cefsulodin, a third generation β-lactam cephalosporin antibiotic, exhibits SHP2 inhibitory activity. X-ray crystallography study and structural analyses identify SPAA as a novel nonhydrolyzable pTyr mimetic that enables molecular diversity to be introduced from the same side of the benzene ring where the sulfonic acid resides to capture interactions with previously uncharted regions around the PTP active site. To demonstrate the utility of SPAA as a pTyr mimetic, we employed a structure-guided and fragment-based approach that led to the discovery of several SPAA-containing SHP2 inhibitors with IC50 in the low micromolar range and several-fold selectivity over a panel of human PTPs. Notably, these inhibitors blocked SHP2-mediated signaling and exhibit excellent antiproliferative activity in several cancer cell lines, demonstrating the utility of exploring the existing drug space for the development of PTP inhibitors with favorable pharmacological properties. More importantly, SPAA may serve as a versatile scaffold to develop selective and bioavailable inhibitors for other members of the PTP family.
Glossary
ABBREVIATIONS
- ERK1/2
extracellular-signal-regulated kinase 1/2
- PTK
protein tyrosine kinase
- PTP
protein tyrosine phosphatase
- pTyr
phosphotyrosine
- SH2
Src homology 2
- SPAA
sulfophenyl acetic amide
Supporting Information Available
Characterization of SHP2 inhibition by cefsulodin; stability study of cefsulodin under various conditions; detailed structure analyses of SHP2·cefsulodin complex; structural characterization of the target compounds by NMR and MS; experimental procedures. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00118.
Author Contributions
∥ R.H. and Z.-H.Y. contributed equally to this work
This work was supported by NIH Grants CA152194, CA69202, and CA151765, FAMRI (Flight Attendant Medical Research Institute Fund), and ICTR (UL1 RR 025005).
The authors declare no competing financial interest.
Supplementary Material
References
- Cohen P.; Alessi D. R. Kinase Drug Discovery - What’s Next in the Field?. ACS Chem. Biol. 2013, 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel B. G.; Gu H. H.; Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [DOI] [PubMed] [Google Scholar]
- Grossmann K. S.; Rosario M.; Birchmeier C.; Birchmeier W. The Tyrosine Phosphatase Shp2 in Development and Cancer. Adv. Cancer Res. 2010, 106, 53–89. [DOI] [PubMed] [Google Scholar]
- Bentires-Alj M.; Paez J. G.; David F. S.; Keilhack H.; Halmos B.; Naoki K.; Maris J. M.; Richardson A.; Bardelli A.; Sugarbaker D. J.; Richards W. G.; Du J. Y.; Girard L.; Minna J. D.; Loh M. L.; Fisher D. E.; Velculescu V. E.; Vogelstein B.; Meyerson M.; Sellers W. R.; Neel B. G. Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Gelb B. D.. Noonan Syndrome and Related Disorders: Genetics and Pathogenesis. In Annual Review of Genomics and Human Genetics; Annual Reviews: Palo Alto, CA, 2005; Vol. 6, pp 45–68. [DOI] [PubMed] [Google Scholar]
- Butterworth S.; Overduin M.; Barr A. J. Targeting protein tyrosine phosphatase SHP2 for therapeutic intervention. Future Med. Chem. 2014, 6, 1423–1437. [DOI] [PubMed] [Google Scholar]
- Barr A. J.; Ugochukwu E.; Lee W. H.; King O. N. F.; Filippakopoulos P.; Alfano I.; Savitsky P.; Burgess-Brown N. A.; Muller S.; Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puius Y. A.; Zhao Y.; Sullivan M.; Lawrence D. S.; Almo S. C.; Zhang Z. Y. Identification of a second aryl phosphate-binding site in protein-tyrosine phosphatase 1B: A paradigm for inhibitor design. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 13420–13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K.; Keng Y. F.; Wu L.; Guo X. L.; Lawrence D. S.; Zhang Z. Y. Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure. J. Biol. Chem. 2001, 276, 47311–47319. [DOI] [PubMed] [Google Scholar]
- He R. J.; Zeng L. F.; He Y. T.; Zhang S.; Zhang Z. Y. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J. 2013, 280, 731–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong C. R.; Chen X. C.; Shi L. R.; O Liu J.; Sullivan D. J. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat. Chem. Biol. 2006, 2, 415–416. [DOI] [PubMed] [Google Scholar]
- Miller O. J.; El Harrak A.; Mangeat T.; Baret J. C.; Frenz L.; El Debs B.; Mayot E.; Samuels M. L.; Rooney E. K.; Dieu P.; Galvan M.; Link D. R.; Griffiths A. D. High-resolution dose-response screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmiento M.; Puius Y. A.; Vetter S. W.; Keng Y. F.; Wu L.; Zhao Y.; Lawrence D. S.; Almo S. C.; Zhang Z. Y. Structural basis of plasticity in protein tyrosine phosphatase 1B substrate recognition. Biochemistry 2000, 39, 8171–8179. [DOI] [PubMed] [Google Scholar]
- Xu J.; Zeng L. F.; Shen W. H.; Turchi J. J.; Zhang Z. Y. Targeting SHP2 for EGFR inhibitor resistant non-small cell lung carcinoma. Biochem. Biophys. Res. Commun. 2013, 439, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W.; Miller V. A.; Politi K. A.; Riely G. J.; Somwar R.; Zakowski M. F.; Kris M. G.; Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y.; Meng S.; Mei L.; Zhao Z. J.; Jove R.; Wu J. Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J. Biol. Chem. 2004, 279, 8497–8505. [DOI] [PubMed] [Google Scholar]
- Yamauchi K.; Milarski K. L.; Saltiel A. R.; Pessin J. E. Protein-tyrosine-phosphatase SHPTP2 is a required positive effector for insulin downstream signaling. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt B.; Frith D.; Stabel S. Signaling from TPA to map kinase requires protein-kinase-c, Raf and MEK - reconstitution of the signaling pathway in-vitro. Oncogene 1994, 9, 3213–3218. [PubMed] [Google Scholar]
- Ueda Y.; Hirai S.; Osada S.; Suzuki A.; Mizuno K.; Ohno S. Protein kinase C delta activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J. Biol. Chem. 1996, 271, 23512–23519. [DOI] [PubMed] [Google Scholar]
- Muthuswamy S. K.; Xue B. Cell Polarity as a Regulator of Cancer Cell Behavior Plasticity. Annu. Rev. Cell Dev.Biol. 2012, 28, 599–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B.; Lo A. T.; Park C. C.; Gray J. W.; Bissell M. J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburn T. T.; Thor K. B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discovery 2004, 3, 673–683. [DOI] [PubMed] [Google Scholar]
- Chong C. R.; Sullivan D. J. New uses for old drugs. Nature 2007, 448, 645–646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.