

Abstract
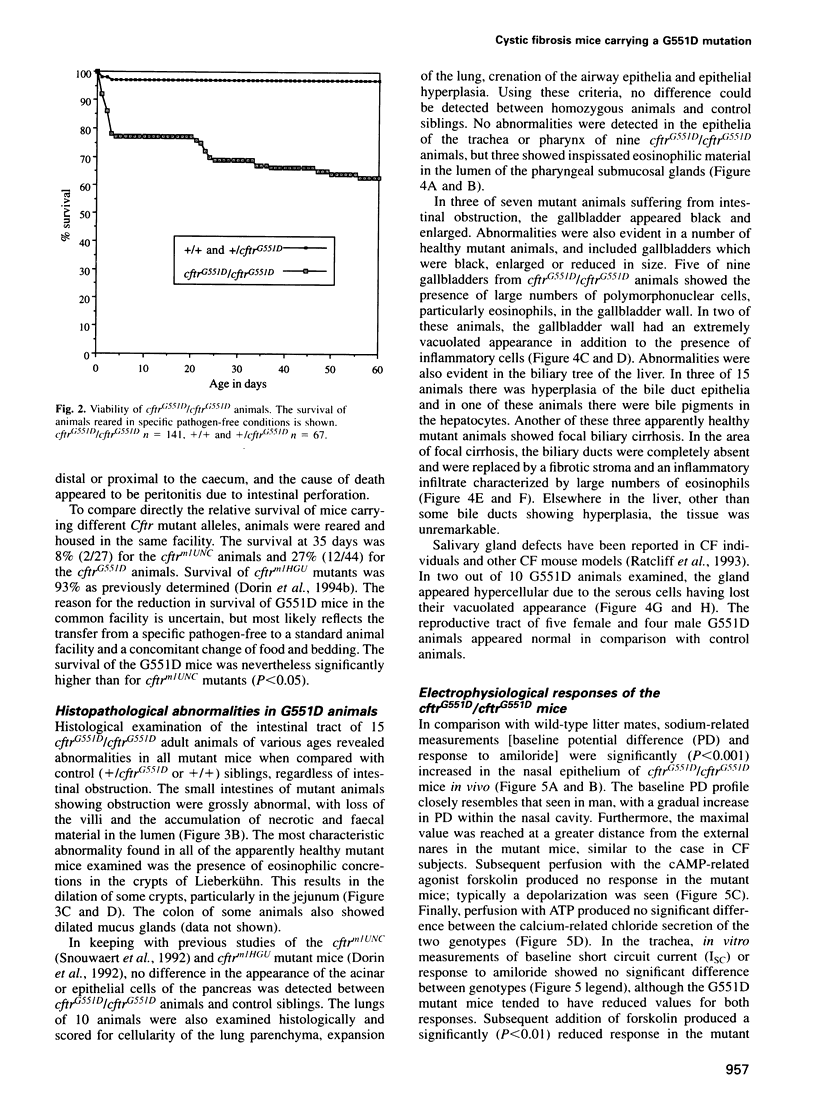
We have generated a mouse carrying the human G551D mutation in the cystic fibrosis transmembrane conductance regulator gene (CFTR) by a one-step gene targeting procedure. These mutant mice show cystic fibrosis pathology but have a reduced risk of fatal intestinal blockage compared with 'null' mutants, in keeping with the reduced incidence of meconium ileus in G551D patients. The G551D mutant mice show greatly reduced CFTR-related chloride transport, displaying activity intermediate between that of cftr(mlUNC) replacement ('null') and cftr(mlHGU) insertional (residual activity) mutants and equivalent to approximately 4% of wild-type CFTR activity. The long-term survival of these animals should provide an excellent model with which to study cystic fibrosis, and they illustrate the value of mouse models carrying relevant mutations for examining genotype-phenotype correlations.
Full text
PDF


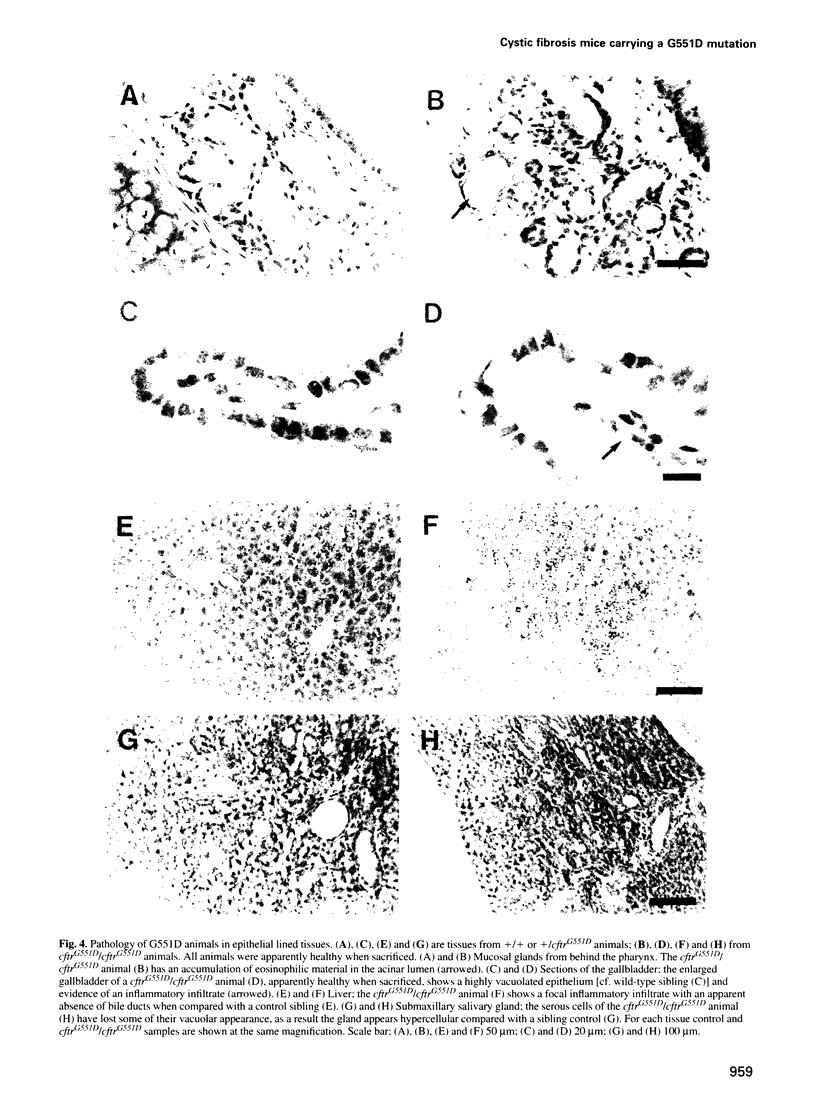




Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Alton E. W., Middleton P. G., Caplen N. J., Smith S. N., Steel D. M., Munkonge F. M., Jeffery P. K., Geddes D. M., Hart S. L., Williamson R. Non-invasive liposome-mediated gene delivery can correct the ion transport defect in cystic fibrosis mutant mice. Nat Genet. 1993 Oct;5(2):135–142. doi: 10.1038/ng1093-135. [DOI] [PubMed] [Google Scholar]
- Becq F., Jensen T. J., Chang X. B., Savoia A., Rommens J. M., Tsui L. C., Buchwald M., Riordan J. R., Hanrahan J. W. Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proc Natl Acad Sci U S A. 1994 Sep 13;91(19):9160–9164. doi: 10.1073/pnas.91.19.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson D. J., Dorin J. R., McLachlan G., Ranaldi V., Lamb D., Doherty C., Govan J., Porteous D. J. Lung disease in the cystic fibrosis mouse exposed to bacterial pathogens. Nat Genet. 1995 Apr;9(4):351–357. doi: 10.1038/ng0495-351. [DOI] [PubMed] [Google Scholar]
- Delaney S. J., Rich D. P., Thomson S. A., Hargrave M. R., Lovelock P. K., Welsh M. J., Wainwright B. J. Cystic fibrosis transmembrane conductance regulator splice variants are not conserved and fail to produce chloride channels. Nat Genet. 1993 Aug;4(4):426–431. doi: 10.1038/ng0893-426. [DOI] [PubMed] [Google Scholar]
- Dorin J. R., Dickinson P., Alton E. W., Smith S. N., Geddes D. M., Stevenson B. J., Kimber W. L., Fleming S., Clarke A. R., Hooper M. L. Cystic fibrosis in the mouse by targeted insertional mutagenesis. Nature. 1992 Sep 17;359(6392):211–215. doi: 10.1038/359211a0. [DOI] [PubMed] [Google Scholar]
- Dorin J. R., Stevenson B. J., Fleming S., Alton E. W., Dickinson P., Porteous D. J. Long-term survival of the exon 10 insertional cystic fibrosis mutant mouse is a consequence of low level residual wild-type Cftr gene expression. Mamm Genome. 1994 Aug;5(8):465–472. doi: 10.1007/BF00369314. [DOI] [PubMed] [Google Scholar]
- Drumm M. L., Wilkinson D. J., Smit L. S., Worrell R. T., Strong T. V., Frizzell R. A., Dawson D. C., Collins F. S. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991 Dec 20;254(5039):1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- Gregory R. J., Rich D. P., Cheng S. H., Souza D. W., Paul S., Manavalan P., Anderson M. P., Welsh M. J., Smith A. E. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol. 1991 Aug;11(8):3886–3893. doi: 10.1128/mcb.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb B. R., Pickles R. J., Ye H., Yankaskas J. R., Vick R. N., Engelhardt J. F., Wilson J. M., Johnson L. G., Boucher R. C. Inefficient gene transfer by adenovirus vector to cystic fibrosis airway epithelia of mice and humans. Nature. 1994 Oct 27;371(6500):802–806. doi: 10.1038/371802a0. [DOI] [PubMed] [Google Scholar]
- Hamosh A., King T. M., Rosenstein B. J., Corey M., Levison H., Durie P., Tsui L. C., McIntosh I., Keston M., Brock D. J. Cystic fibrosis patients bearing both the common missense mutation Gly----Asp at codon 551 and the delta F508 mutation are clinically indistinguishable from delta F508 homozygotes, except for decreased risk of meconium ileus. Am J Hum Genet. 1992 Aug;51(2):245–250. [PMC free article] [PubMed] [Google Scholar]
- Hovell T. M. Remarks on Treatment of Irritative Coughing. Proc R Soc Med. 1917;10(LARYNGOL):99–100. [PMC free article] [PubMed] [Google Scholar]
- Hyde S. C., Gill D. R., Higgins C. F., Trezise A. E., MacVinish L. J., Cuthbert A. W., Ratcliff R., Evans M. J., Colledge W. H. Correction of the ion transport defect in cystic fibrosis transgenic mice by gene therapy. Nature. 1993 Mar 18;362(6417):250–255. doi: 10.1038/362250a0. [DOI] [PubMed] [Google Scholar]
- Koller B. H., Kim H. S., Latour A. M., Brigman K., Boucher R. C., Jr, Scambler P., Wainwright B., Smithies O. Toward an animal model of cystic fibrosis: targeted interruption of exon 10 of the cystic fibrosis transmembrane regulator gene in embryonic stem cells. Proc Natl Acad Sci U S A. 1991 Dec 1;88(23):10730–10734. doi: 10.1073/pnas.88.23.10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan J., Hiestand D., Daram P., Huang Z., Muccio D. D., Hartman J., Haley B., Cook W. J., Sorscher E. J. Cystic fibrosis transmembrane conductance regulator mutations that disrupt nucleotide binding. J Clin Invest. 1994 Jul;94(1):228–236. doi: 10.1172/JCI117311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour S. L., Thomas K. R., Capecchi M. R. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988 Nov 24;336(6197):348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- Prince L. S., Workman R. B., Jr, Marchase R. B. Rapid endocytosis of the cystic fibrosis transmembrane conductance regulator chloride channel. Proc Natl Acad Sci U S A. 1994 May 24;91(11):5192–5196. doi: 10.1073/pnas.91.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh M. J., Smith A. E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993 Jul 2;73(7):1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- Widdicombe J. H., Welsh M. J., Finkbeiner W. E. Cystic fibrosis decreases the apical membrane chloride permeability of monolayers cultured from cells of tracheal epithelium. Proc Natl Acad Sci U S A. 1985 Sep;82(18):6167–6171. doi: 10.1073/pnas.82.18.6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Devor D. C., Engelhardt J. F., Ernst S. A., Strong T. V., Collins F. S., Cohn J. A., Frizzell R. A., Wilson J. M. Molecular basis of defective anion transport in L cells expressing recombinant forms of CFTR. Hum Mol Genet. 1993 Aug;2(8):1253–1261. doi: 10.1093/hmg/2.8.1253. [DOI] [PubMed] [Google Scholar]
- Zhou L., Dey C. R., Wert S. E., DuVall M. D., Frizzell R. A., Whitsett J. A. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994 Dec 9;266(5191):1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]