

Significance
This paper provides a comprehensive analysis of sex differential gene expression in haplodiploid jewel wasps. Between two closely related species, 75% of genes display differential expression, despite males having half the genetic complement of females, with no sex chromosomes. These differences are not directly mediated by sex-specific methylation because almost no sex differences in methylation were observed. Genes with sex-specific expression show low frequency of methylation. However, the majority of female-biased genes are methylated (in both sexes), whereas male-biased ones are mostly nonmethylated in either sex. We conclude that female-biased genes are more likely to be recruited from conserved methylated genes over evolutionary time, whereas most male-biased genes are from genes after recent duplication events that are not methylated.
Keywords: sex-biased expression, DNA methylation, RNA-seq, WGBS-seq, fatty acid desaturase
Abstract
There is extraordinary diversity in sexual dimorphism (SD) among animals, but little is known about its epigenetic basis. To study the epigenetic architecture of SD in a haplodiploid system, we performed RNA-seq and whole-genome bisulfite sequencing of adult females and males from two closely related parasitoid wasps, Nasonia vitripennis and Nasonia giraulti. More than 75% of expressed genes displayed significantly sex-biased expression. As a consequence, expression profiles are more similar between species within each sex than between sexes within each species. Furthermore, extremely male- and female-biased genes are enriched for totally different functional categories: male-biased genes for key enzymes in sex-pheromone synthesis and female-biased genes for genes involved in epigenetic regulation of gene expression. Remarkably, just 70 highly expressed, extremely male-biased genes account for 10% of all transcripts in adult males. Unlike expression profiles, DNA methylomes are highly similar between sexes within species, with no consistent sex differences in methylation found. Therefore, methylation changes cannot explain the extensive level of sex-biased gene expression observed. Female-biased genes have smaller sequence divergence between species, higher conservation to other hymenopterans, and a broader expression range across development. Overall, female-biased genes have been recruited from genes with more conserved and broadly expressing “house-keeping” functions, whereas male-biased genes are more recently evolved and are predominately testis specific. In summary, Nasonia accomplish a striking degree of sex-biased expression without sex chromosomes or epigenetic differences in methylation. We propose that methylation provides a general signal for constitutive gene expression, whereas other sex-specific signals cause sex-biased gene expression.
Sexual dimorphism (SD) between males and females arises through more than one mechanism in evolution. Fundamentally, SD arises from differential selection acting on the two sexes. However, it is also shaped by genetic mechanisms of sex determination. In diploid animals with chromosomal sex determination, the genetic structure of SD is influenced by the sex differences in the composition of sex chromosomes, differential expression of the sex-determination gene networks, and sex chromosome–autosome interactions (1, 2). In contrast, species with haplodiploid sex determination do not have sex chromosomes, but rather, except for dosage, they have identical chromosomal complements. Furthermore, although male embryos start out as haploid, somatic cells often undergo endopolyploidization in both males and females, which can result in similar ploidy levels in particular somatic tissues for both sexes (3, 4).
Sex-biased expression is one of the underlying molecular mechanisms for SD. Nearly all genome-wide analyses of sex-biased gene expression have been conducted in organisms with sex chromosomes. Studies in insects and mammals with sex chromosomes have identified a substantial number of sex-biased genes, and their genome distribution is not random (5–7). In both Drosophila and mouse, there is an excess of female-biased genes and a deficit of male-biased genes on X chromosomes (8–10), which is thought to be partly due to the different strength of sexually antagonistic selection for X-linked genes in males and females (11–13). In hymenopteran species, which have haplodiploid sex determination, unfertilized eggs develop into males whereas fertilized eggs develop into females; however, the underlying genetic mechanisms of sex determination can differ considerably between species (14, 15). For example, in honeybee and many ant species, sex is determined at a single complementary sex-determining locus (csd): heterozygotes develop into females and hemizygous individuals develop into males (14). Inbreeding leads to production of diploid males. In contrast, in species with routine inbreeding, such as Nasonia, sex determination appears to involve an interaction among maternal and zygotic gene products (16–19). Haplodiploid species pose interesting questions for SD at the gene expression level: How does the degree and profile of sex-biased expression compare with other insect species with sex chromosomal sex determination? What is the genome-wide distribution of sex-biased expression in a system that lacks sex chromosomes? How does evolution shape the sex-biased expression profile in a haplodiploid system? What is the relationship between sex-biased gene expression and epigenetic mechanisms such as DNA methylation? To address these questions and understand the conservation of sex-biased gene expression, we examined two parasitoid wasp species, Nasonia vitripennis and Nasonia giraulti.
In addition to gene expression, epigenetic modifications such as DNA methylation also contribute to SD in mammals (20, 21). In insects, little is known about the epigenetic differences between the two sexes and how they relate to the sex-biased expression because most sex-biased expression studies have been conducted in Drosophila, which lacks CpG methylation (22), with only low levels of methylation on adenines (6mA) in embryonic stage (23). Among social insects (bees and ants) various experiments have suggested a role for DNA methylation in regulating changes in gene expression level and splicing (24, 25). In addition, a recent study suggests that differences in DNA methylation may be associated with ploidy regulation in the ant Solenopsis invicta (26).
Wasps of the genus Nasonia are nonsocial parasitoids that are emerging as a model for studies of DNA methylation and epigenetics. These insects are genetically tractable with a short generation time, ease of laboratory rearing, highly inbred lines, and closely related cross-fertile species (27). Their phylogenetic placement also makes Nasonia useful for comparative genomic studies with the social hymenoptera. As such, Nasonia can be used to address questions concerning the extent to which DNA methylation has evolved taxon-specific functions and what features are conserved across the hymenoptera and other insects with DNA methylation. All three DNA methyltranferases are present in the Nasonia genome (28), and DNA methylation is critical for many biological processes including early embryonic development (29). Recently, we analyzed the DNA methylome in N. vitripennis at a base-pair resolution using whole-genome bisulfite sequencing (WGBS-seq) (30). In this current study, we quantify the gene expression level in adult whole-body samples of males and females from two closely related Nasonia species, N. vitripennis and N. giraulti, using RNA sequencing and also compare genome-wide DNA methylation in males and females of both species by WGBS-seq with >20× haploid genome coverage. We combined these data with information on sequence divergence, recombination intensity, and other genomic data to investigate the genetic and epigenetic architecture of sexual dimorphism in gene expression.
Results and Discussion
In studies of sex-biased expression, the quantitative degree of sex bias is often neglected, with all sex-biased genes treated as a group regardless of the degree of bias. However, genes showing different patterns and levels of sex bias are likely subject to different evolutionary processes modulating their expression and sex specificity. Therefore, for the purposes of this study we distinguish three general categories of sex-biased genes. Sex-biased (SB) genes are those that show a significant difference in expression between the sexes [false discovery rate (FDR) >0.05]. These genes can be broadly expressed in both sexes, but the level of expression is significantly different between the sexes. The second category (SB+) contains genes that show both a high expression level in both sexes and also a strong sex bias, with fragments per kilobase of exon per million fragments mapped (FPKM) >4 in both sexes and expressed at least eightfold more highly in one sex. The third category encompasses sex-specific (SS) genes, which are expressed in one sex but show very low or no expression in the other sex; criteria for the SS gene category are FPKM <0.4 in one sex and >2 in the other. We tried a series of different cutoffs for all categories based on actual read alignments in the Integrative Genomics Viewer (IGV) as well as the correlation with presence/absence in expression tiling array data, and they all gave similar patterns to those described in SI Appendix, Methods. These categories help us to reveal different types and patterns of sex-biased gene expression and reveal different processes acting on these categories. Table 1 shows the numbers of genes falling into the three categories in both Nasonia species (N. vitripennis and N. giraulti), and Fig. 1 shows overlap in genes between the species in each category.
Table 1.
Summary of number of genes in sex-biased categories in two Nasonia species
| Sex-biased expression category | Criteria | N. vitripennis (%) | N. giraulti (%) | Overlap (%) |
| Covered genes in RNA-seq | FPKM > 1 at least one replicate | 13,050 | 12,618 | 11,815 |
| Female-biased genes | q < 0.05; female-biased | 5,095 (39) | 4,403 (35) | 3,960 (34) |
| Male-biased genes | q < 0.05; male-biased | 5,332 (41) | 5,162 (41) | 3,930 (33) |
| Extremely female-biased genes | FPKM > 4 in males; female/male > 8 | 302 (2.3) | 210 (1.7) | 152 (1.3) |
| Extremely male-biased genes | FPKM > 4 in females; male/female > 8 | 70 (0.5) | 78 (0.6) | 25 (0.2) |
| Female-specific genes | FPKM < 0.4 in males; FPKM > 2 in females | 321 (2.5) | 295 (2.3) | 164 (1.4) |
| Male-specific genes | FPKM < 0.4 in females; FPKM > 2 in males | 296 (2.3) | 185 (1.5) | 126 (1.1) |
Fig. 1.
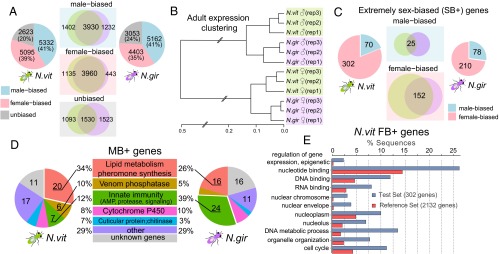
Distribution and functional category enrichment of sex-biased genes in N. vitripennis and N. giraulti. (A) Pie charts of male-biased (blue), female-biased (pink), and unbiased genes (gray) for N. vitripennis and N. giraulti, respectively. The overlaps for these three categories between two species are shown in the Venn diagrams (Middle). (B) Clustering of N. vitripennis and N. giraulti adult whole-body expression profiles (based on 11,815 expressed genes in both species) using Hierarchical Cluster Analysis (hclust package in R). (C) Pie charts of highly expressed and extremely sex-biased (SB+) genes (male-biased in blue and female-biased in pink) for N. vitripennis and N. giraulti, respectively. The overlaps between two species for extremely male- and female-biased genes are shown in the Venn diagrams (Middle). (D) Significantly enriched functional categories for highly expressed and extremely male-biased (MB+) genes in N. vitripennis (Left) and N. giraulti (Right). (E) Significantly overrepresented GO terms for N. vitripennis highly expressed and extremely female-biased (FB+) genes (adjusted P value < 0.05).
Among the key findings are the following: (i) There is extensive sex-biased gene expression in Nasonia (∼75% of genes are differentially expressed between the sexes in adults). (ii) However, there are few or no differences in DNA methylation between males and females, indicating that methylation is not a major contributor to sex differences at the gene expression level. (iii) The frequencies of methylated genes differ between male- versus female-biased genes (although their methylation status does not differ between males and females). (iv) Evolutionary rates are quite different among sex-bias categories and consistent with evolutionary constraints on more broadly expressed female-biased genes. (v) Sex-biased genes differ in their distribution across the chromosomes and relative to recombination rates. Results are described in more detail below.
Substantial Sex-Biased Expression Is Found in N. vitripennis and N. giraulti.
Using an FDR < 0.05 as a cutoff for genes with FPKM > 1 in at least one sample (SI Appendix, Methods and Table S1), significant sex-biased gene expression was found in 80% (10,427/13,050) of N. vitripennis and 76% (9,565/12,618) of N. giraulti genes (Table 1), indicating substantial sex-biased expression in a haplodiploid species with no sex chromosomes. This percentage is even greater than the proportion of sex-biased genes in Drosophila, which have qualitatively distinct sex chromosomes (31). Strikingly, extremely strong sex differences were also found in these two Nasonia species: 429 genes showed >100-fold and 60 showed >1,000-fold difference in N. vitripennis (SI Appendix, Fig. S1). Sex-biased expression is conserved in Nasonia, and the majority of sex-biased genes overlap between the two species (Fig. 1A). Nine sex-biased genes were selected for quantitative RT-PCR (qRT-PCR) validation, and all of them were confirmed with excellent quantitative fidelity (SI Appendix, Methods, Fig. S2, and Table S2).
The high level of sex-biased expression is further revealed when the expression data are clustered by sex and species. Despite the fact that the two species diverged ∼1 million years ago (28), samples did not cluster by species, but rather by sex; i.e., males of the two species clustered together, as did females (Fig. 1B and SI Appendix, Fig. S3) (P value < 2.2 × 10−16, test of the equivalence of two correlation coefficients using Fisher Z-transformation). In contrast, a much stronger male and female expression correlation (r2 = 0.80–0.85) was found in Drosophila species (32). As seen in Table 1, genes in the SB category constitute the majority of those showing a sex bias (e.g., ∼5,400 male-biased and ∼5,100 female-biased in N. vitripennis). Similar numbers are found in N. giraulti.
A striking pattern in Nasonia is the large number of genes with extremely high fold expression differences between the two sexes, i.e., the SB+ and SS categories (Table 1). For the SB+ genes, we found 70 highly expressed and extremely male-biased genes (MB+ genes) in N. vitripennis and 78 in N. giraulti, with an overlap of 25 MB+ genes in the two species (Fig. 1C). The relatively low overlap between the species (∼16%) suggests rapid evolution of gene regulation for extreme male-biased expression. Remarkably, although the ∼70 MB+ genes make up only 0.5% of all genes, their summed expression accounts for 10% of total transcript abundance in N. vitripennis males and 12% in N. giraulti males. This contrasts with females, where the highly expressed and extremely female-biased (FB+) and female-specific (FS) genes account for only 0.3% of total expression in adult N. vitripennis females (0.5% in N. giraulti females).
Gene Ontology (GO) term analysis revealed that MB+ genes are highly enriched for immunity genes (33, 34) and sex-pheromone synthetic enzymes, including key enzymes in Nasonia male sex-pheromone pathways, such as acyl-CoA desaturase and short-chain dehydrogenase/reductase (Fig. 1D). Venom acid phosphatases, the cytochrome P450 family, and cuticular proteins are also enriched (Fig. 1D), suggesting the function of these extremely male-biased genes is for detoxification, defense, and mate choice.
In fact, the most highly expressed gene among adult Nasonia is a key enzyme for sex-pheromone production. Nasvi2EG017727 has the highest expression level in the entire dataset, with FPKM = 49,357 in N. vitripennis males (Fig. 2A). This MB+ gene is already highly expressed in females (FPKM = 61), but the male expression level is more than 800-fold higher (confirmed by qRT-PCR, SI Appendix, Fig. S2A). Surprisingly, 5% of all uniquely mapped reads in males are located at this locus, whose abundance equals that of all of the rest of the MB+ genes combined. Several differentially spliced forms were found only in males (Fig. 2A).
Fig. 2.

The highest expressed and extremely male-biased gene Nasvi2EG017727 is a fatty acid desaturase responsible for male-derived sex pheromone synthesis. (A) IGV browser screenshot showing Nasonia adult female (Top) and male (male) RNA-seq coverage (on log scale), exon–exon junctions, and read alignments in Nasvi2EG017727 gene region. (B) Neighbor-joining phylogenetic tree for the fatty acid desaturase family in Nasonia (N. vitripennis, blue), honey bee (Apis mellifera, red), ant (Camponotus floridanus, green) and fly (D. melanogaster, black). Male- and female- biased genes with greater than fourfold sex differences in Nasonia were plotted with a blue/red box indicating the degree of sex-biased expression. (C–F) Barplot of RNA-seq expression level (log2FPKM) in N. vitripennis and N. giraulti males and females for selected Nasonia genes in fatty acid desaturase family: Nasvi2EG017727 (C), Nasvi2EG021420 (D), Nasvi2EG009037 (E), and Nasvi2EG002222 (F). Statistical significance was calculated using edgeR (ns: q-value > 0.05; ***q-value < 0.001).
This gene belongs to a 15-member fatty acid desaturase gene family (Fig. 2B), at least one of which confers the Δ12-desaturase activity and is critical for the ability to synthesize linoleic acid (C18:2Δ9,12) from oleic acid (C18:1Δ9) in Nasonia (Fig. 2B and SI Appendix, Table S3) (35). Linoleic acid is an essential unsaturated fatty acid in other insects that lack Δ12-desaturase activity, and in Nasonia it is the precursor of the male-derived sex pheromone (4R,5R)- and (4R,5S)-5-hydroxy-4-decanolides, which is highly attractive to virgin females (36, 37). Male mating success positively correlates with the amount of pheromone that they produce (38), which likely explains the high levels of expression of these genes. We checked the expression profile in the 15 desaturase genes and found an interesting sex-biased pattern: five genes displayed eightfold or more sex-biased expression and the remaining 10 genes are either unbiased or only slightly biased, with a less-than-fourfold sex difference (Fig. 2B and SI Appendix, Table S3). Interestingly, although Nasvi2EG017727 is 800-fold male-biased in expression (Fig. 2C), its closest paralog, Nasvi2EG021420 (90.6% protein sequence identity), is unbiased (Fig. 2D). Of the other two male-biased genes, Nasvi2EG009037 has an eightfold sex difference in N. vitripennis only (Fig. 2E) and Nasvi2EG002222 is 10-fold male-biased in N. giraulti only (Fig. 2F). As the only extremely male-biased gene in this gene family in both species, it is highly likely that Nasvi2EG017727 is responsible for male-specific Δ12-desaturase activity, which is a key process in male sex pheromone synthesis (36) (SI Appendix, Table S3). The two with species-specific sex biases (Nasvi2EG009037 and Nasvi2EG002222) could be involved in species-specific modifications of the male sex pheromone.
This MB+ gene can be used to illustrate how selection may act upon sex-biased genes that still maintain expression in both sexes. Nasvi2EG017727 was likely subject to strong sexually antagonistic selection. Suppose the expression level in the ancestral species is FPKM = 60. If selection in the male favors expression to be FPKM = 50,000 for male-specific sex-pheromone synthesis, up-regulating the expression level to 50,000 in both sexes could be easy to achieve, but this extremely high expression might be toxic to females or a waste of cellular energy. Therefore, such selection pressures can produce elevated expression levels in males, but because there is (apparently) also a necessary function in females, the gene cannot become entirely male-specific. Thus, we expect these SB+ genes to be more evolutionarily constrained than SS genes, which are expressed almost exclusively in one sex. Although Nasvi2EG017727 might be considered to be involved in species divergence in pheromones, there is no evidence for directional selection in Nasonia; it has a nonsynonymous substitution (dN)/synonymous substitution (dS) ratio of 0.109 between the two species, which is more consistent with purifying selection after the establishment of extremely male-biased expression. Instead, intense selection is likely to be acting on the cis-regulation of this gene.
In Drosophila melanogaster, sex pheromones are also dimorphic between the two sexes, with high levels of dienes in females and monoenes in males (39). Δ9-Desaturases play important roles in the synthesis and sexual dimorphism of the Drosophila sex pheromone: the desat1 (desaturase 1) in D. melanogaster is involved in sex pheromone production in both sexes (40); desat2 (desaturase 2) is female-specific in African populations for the synthesis of an African-specific subtype of the sex pheromone in females (41); desatF (desaturase F) is also female-specific and is required for a second desaturation step to produce dienes in females (42) with lineage-specific rapid evolution (43). Thus, the Drosophila sex pheromone evolution seemed to be female-driven by female-specific desaturase genes, but this is not the case in Nasonia. On the phylogenetic tree of the insect desaturase family, Drosophila desat genes are on the same branch with genes that in Nasonia are either unbiased or weakly male-biased (Fig. 2B). In contrast, the Nasonia sex pheromone gene Nasvi2EG017727 is extremely male-biased and was derived from a recent Nasonia lineage-specific expansion in this family, which is not present in honeybee or ant (Fig. 2B). This is consistent with the facts that Drosophila lacks Δ12-desaturase activity and all fly desat genes are Δ9 desaturases. The Δ12-desaturase ability has been demonstrated only in Nasonia among hymenopterans (35), suggesting the potential gain of novel function and extremely male-biased expression pattern after recent gene duplication.
The patterns in FB+ genes are quite different from MB+ genes. Numbers of FB+ genes are about threefold greater than MB+ genes in these species (302 in N. vitripennis and 210 in N. giraulti). In each species, >50% of FB+ (152) are shared between the species (Fig. 1C), which is significantly greater than the 35% MB+ shared between the species (P < 0.05, χ2 test). The finding suggests that the sex-specific functions of FB+ are more conserved than are MB+ genes. In terms of functional characterization, FB+ genes are significantly enriched for nuclear-located genes that are responsible for epigenetic regulation of gene expression (Fig. 1E), which might be relevant to female reproductive functions such as egg production and/or maternal epigenetic programming of early embryonic development. SB+ genes may have important or essential functions in the lower expressing sex, whereas the extremely high expression in the high-expressing sex could reflect processes that are targets for sexually antagonistic selection in Nasonia. Therefore, SB+ genes are likely to have faced sexually antagonistic selection, resulting in extremely sex differential expression.
In contrast to SB+ genes, SS genes are much less likely to suffer from antagonistic selection between the sexes because they have very low or zero expression in the alternate sex. Among SS genes, there are 200–300 male-specific (MS) and FS genes in N. vitripennis and N. giraulti, half of which are shared between the two species (SI Appendix, Fig. S4). Thus, genes in the SS category changed relatively quickly between Nasonia species. FS genes were enriched for genes that respond to stress and extracellular stimulus (SI Appendix, Fig. S4). MS genes are mostly unannotated (likely due to their rapid sequence evolution) and thus failed to provide informative BLAST matches in Blast2GO (44); therefore, they did not show significant functional enrichments. Although SS genes could have a large fold change due to the extremely low expression in one sex, the average absolute expression difference is much lower than for SB+ genes. The 200–300 SS genes comprise ∼1.5–2.3% of all expressed genes and account for just 0.7% of the entire transcriptome abundance.
Little or No Methylation Differences Between the Sexes.
We next contrasted the methylome of adult males and females by whole-genome bisulfite sequencing (WGBS-seq) with >25× haploid genome coverage and >15× average CpG coverage (SI Appendix, Methods, Figs. S5–S6, and Tables S4–S6). The strains used are highly inbred, which also reduces calling errors due to sequence heterogeneity (30). As previously described, most of Nasonia methylated CpG sites (mCpGs) were located on exons in the first 1 kb of the coding region of the gene (30). Gene methylation status (methylated vs. nonmethylated genes) was determined by the same criteria in our recent N. vitripennis methylome study (>10% mCpGs in the 5′-most 1-kb coding region) (30) (SI Appendix, Methods). In contrast to clustering of gene expression profiles by sex, CpG methylation shows a significantly higher correlation within species (Spearman’s rho = 0.78∼0.90) relative to between species (Spearman’s rho = 0.62∼0.65; P value < 2.2 × 10−16, test of the equivalence of two correlation coefficients using Fisher Z-transformation) (Fig. 3A).
Fig. 3.

Clustering of DNA methylation profile and candidate differentially methylated genes between sexes and between the two Nasonia species. (A) Clustering of adult whole-body methylation profile for methylated CpGs using Hierarchical Cluster Analysis (hclust package in R). (B) Significant candidate differentially methylated genes between the two sexes in N. vitripennis (Top Left) and N. giraulti (Top Right). (Bottom) Distribution of percentage of differentially methylated sites in sex-differentially methylated genes in N. giraulti. (C) Significant candidate differentially methylated genes between N. vitripennis and N. giraulti. (Top) Number of N. vitripennis-specific methylated genes and N. giraulti-specific methylated genes in a pie chart. (Bottom) Distribution of percentage of differentially methylated sites in candidate species differentially methylated genes.
We found very little evidence for sex differences in DNA methylation in either species. From the initial analysis, we found ∼80 candidates (or 0.7% of ∼12,000 covered genes) with putative sex differential methylation (sex-DM genes, Fig. 3B), but the percentage of methylation differences is small (<50%, Fig. 3B). When we checked at the individual CpG level, there were few sex-differentially methylated CpG sites (sex-DMCpGs), and these are not replicated in the two species and do not cluster in genes (SI Appendix, Fig. S7 and SI Text). We selected four sex-DM genes for validation in independent biological replicates, and we were able to confirm only one, but with a much lower magnitude of methylation difference (SI Appendix, Table S7 and Figs. S8–S11). Therefore, the number of likely sex-DM genes and sites within the genome is remarkably small and unlikely to play a major role in the extensive sex bias in gene expression found in Nasonia.
DNA methylation is also very conserved between species, with only ∼150 (or 1.4%) differentially methylated genes (species-DM genes, Fig. 3C) between N. vitripennis and N. giraulti. We selected six candidate species-DM genes for validation using the PyroMark assay, and all were confirmed (SI Appendix, Table S7 and Figs. S13–S18).
Many lines of evidence now suggest that an important role of DNA methylation in insects is to stabilize gene expression across development and tissues (45). However, studies in social insects have implicated the dynamics of DNA methylation in caste determination (46, 47) and epigenetic changes in gene expression between tissues and sexes, as well as in differential splicing (reviewed in ref. 24). Support for a role of DNA methylation in differential gene expression of social insects includes pharmacological disruptions of DNA methylation and RNA interference knockdowns of DNA methyltransferases that alter gene expression (48). However, it could be argued that these effects might represent by-products of genome-wide disruption in DNA methylation, rather than a specific targeted role of methylation in development and differentiation by sex or caste. Studies in social Hymenoptera also indicate that changes in DNA methylation occur between the sexes and correlate with changes in gene expression (26). This finding contrasts with our own, which finds no clear-cut differences in DNA methylation between adult males and females, despite the extensive differences in gene expression. We did not observe sex differential methylation even in well-known sex-differentially spliced genes in Nasonia including doublesex (49). Weak and variable sex differences in methylation often occur at the boundary of the clustered methylated CpG regions (SI Appendix, Figs. S8–S9 and SI Text), which might be due to variability at the edge of strong CpG methylation clusters or local bisulfite conversion artifacts. Our results indicate that DNA methylation does not directly cause the massive sex-biased expression observed in Nasonia, and solid evidence to the contrary would require manipulation of DNA methylation of some targeted candidate genes.
Glastad et al. (26) have implicated methylation changes with ploidy level in the ant S. invicta. The study took advantage of diploid male production to show that global methylation patterns of diploid males were more similar to that of diploid queens and workers than to that of haploid males. In contrast, we found no significant differences in methylation between males and females of Nasonia. Interpretation of these results is complicated by the fact that endopolyploidization of somatic cells can occur in Hymenoptera (3). Therefore, differences between Nasonia and Solenopsis may be driven in part by the differences in ploidy levels in different tissues. The observation that methylation level may play a role in ploidy regulation is intriguing and warrants further investigation.
Male-Biased and Female-Biased Genes Differ in Methylation Frequency and Expression Breadth.
Overall, 38% of expressed genes (FPKM > 1 in at least one replicate) covered by our WGBS-seq data are methylated in the Nasonia genomes. Although methylation does not play a direct role in sex-biased gene expression (i.e., methylation barely differs between the sexes), sex-biased gene categories do differ considerably in their frequencies of methylation. Both MS and FS genes were almost all nonmethylated (Fig. 4 A–C). In contrast, among SB genes with twofold or more sex difference, only 6% of MB genes were methylated (Fig. 4A), whereas 67% of the FB genes were methylated (Fig. 4B, P value < 2.2 × 10−16, χ2 test). The methylation difference was even higher for SB+ genes, with only 3% of MB+ genes methylated in contrast to 80% of FB+ genes (Fig. 4C), both of which deviate dramatically from the genome average (38%). In addition, the range of MB fold change is much larger for nonmethylated genes (up to 1,070-fold) than for methylated genes (up to 150-fold), with only 3 (or 0.5%) methylated vs. 81 (or 3.2%) nonmethylated genes having a 10-fold or more male-biased expression. The finding suggests that only nonmethylated genes can be extremely male-biased (SI Appendix, Fig. S19–S20).
Fig. 4.
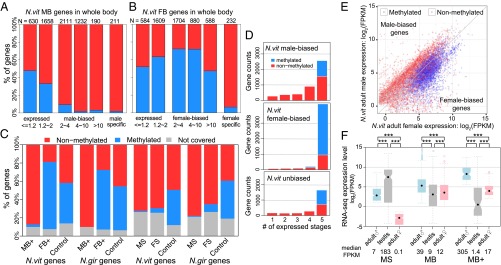
Sex-biased expression, DNA methylation, and expression breadth. (A and B) Barplot of percentage methylated genes in male-biased/male-specific (A) and female-biased/female-specific genes (B) in N. vitripennis adult whole-body samples binned by fold difference between the two sexes. (C) Stacked barplot of percentage of methylated (blue), nonmethylated (red), and uncovered genes (gray). (Left) Extremely male-biased (MB+) and female-biased (FB+) highly expressed genes in N. vitripennis and N. giraulti with control unbiased genes matching their expression level. (Right) Male- (MS) and female-specific (FS) genes in N. vitripennis and N. giraulti with control unbiased genes matching their expression level. (D) Stacked barplot of the number of expressed developmental stages in females (from 1 to 5) for methylated (blue) and nonmethylated (red) male-biased (Top), female-biased (Middle), and unbiased genes (Bottom) in Nasonia whole-adult samples. (E) Scatterplot of male expression level (log2 FPKM) on y axis against female expression level (log2 FPKM) on x axis for N. vitripennis adult whole-body samples, color-coded by DNA methylation status (blue: methylated genes; red: nonmethylated genes). (F) Boxplot of RNA-seq expression level (log2 FPKM) for male-specific (MS), male-biased (MB) and extremely male-biased highly expressed genes (MB+) in male whole-body (blue), male testis (gray), and female whole-body samples (pink). Statistical significance was calculated by Mann–Whitney U test (***P value < 0.001).
What can explain these differences in methylation status among the different SB gene categories? In our previous work we have shown in Nasonia that methylated genes tend to be expressed in all developmental stages and that methylation is negatively correlated with expression variability across development (30). Methylated genes are also more conserved at longer evolutionary timescales (30). Furthermore, duplicated genes that had lost their methylation since the divergence of Apis and Nasonia showed greater developmental stage specialization, suggesting that nonmethylated genes tend to evolve into specialized genes (30). The low frequency of methylated genes among MS and FS genes is therefore consistent with evidence that nonmethylated genes show greater tissue- and stage-specific differentiation in gene expression.
Sex-biased expression can be viewed as a form of expression variability, so we determined whether methylation and expression breadth are also associated with sex-biased expression, using tiling expression data available for five developmental stages of female N. vitripennis (30) (SI Appendix, Methods). The majority of FB genes (90%) are expressed in all five stages, compared with only half of MB genes (56%) (Fig. 4D). Unbiased genes fall in between (67%). This is due in part because of the fact that 67% of FB genes are methylated, and methylated genes tend to be expressed in all stages. However, when we focused only on nonmethylated genes, a larger proportion of FB genes (72%) were still expressed in all five stages than were MB genes (44%) (Fig. 4D). Therefore, we concluded that FB genes are more likely to be expressed in multiple developmental stages than MB genes and more likely to have been recruited from methylated standard housekeeping genes over evolutionary time, as also indicated by GO analysis. On the other hand, MB genes are more likely to originate from faster-evolving nonmethylated genes (Fig. 4E). This finding suggests an interesting theme: Nasonia males could be viewed as a specialized tissue or reproductive organ, given the fact that nonmethylated genes tend to show more stage specialization, which is consistent with the male’s short life span and their specialized function in courtship and mating.
Male Sex-Specific Genes, but Not Male Sex-Biased Genes, Are Driven by Testis Expression.
It is known that genes expressed in testis often have the fastest evolutionary rates in many species (5), and Nasonia MS genes are enriched for genes showing extremely high expression in testis (Fig. 4F). Therefore, male-biased genes could in general be primarily due to biased expression in testes. This is not the case, as MB genes are not highly enriched for testis-specific genes as many show higher expression in females than in testis (Fig. 4F). Furthermore, MB+ genes are hardly expressed at all in testis (median FPKM = 1.4; Fig. 4F). Although data are not available to us on ovary-specific expression, female SS genes (mean expressed stages = 2.82) do show a much higher level of specialization than FB genes (mean expressed stages = 4.57) during development [P value < 2.2 × 10−16, Mann–Whitney U (MWU) test), and FB+ genes are expressed in significantly more life stages (mean expressed stages = 4.77) than FB genes (P value = 0.0003, MWU test). These results suggest that the majority of SS genes can be explained by gonad differences between the sexes, which are more likely to have sex-specific functions in sex-specific tissues. In contrast, SB genes are not enriched for highly expressed genes in gonads, and SB+ genes are actually underrepresented in gonad expression.
Evolutionary Rates Differ Among Sex-Biased Gene Categories.
Rapid evolution of sex-biased genes has been commonly observed in Drosophila and other species (5). Based on the nucleotide divergence between N. vitripennis and N. giraulti in coding regions, sex-specific (both MS and FS) genes have the highest nucleotide substitution rate among all gene categories (sex-biased and unbiased; P value < 2.2 × 10−16, MWU test; Fig. 5A). The dS rates for sex-specific genes were homogeneous with other functional categories (P value > 0.05, MWU test; Fig. 5B), and their greater divergence is due to dN rates (P value < 2.2 × 10−16, MWU test; Fig. 5C). The rapid evolution of SS genes could be due to the relaxation of purifying selection in the nonexpressing sex and acceleration of adaptive protein sequence evolution in the high-expressing sex.
Fig. 5.

Evolution of sex-biased genes. (A) Boxplot of nucleotide divergence in coding regions between N. vitripennis and N. giraulti for genes in the following sex-biased expression categories: MB: male-biased; FB: female-biased; UB: unbiased genes; MS: male-specific; FS: female-specific. (Left) All genes. (Right) Nonmethylated genes only. (Middle) Methylated (blue) and nonmethylated genes (red) for all categories combined. Statistical significance was calculated by Mann–Whitney U test (ns: P value > 0.05; **P value < 0.01; ***P value < 0.001). (B and C) Boxplot of synonymous (dS) (B) and nonsynonymous (dN) (C) substitution rate between N. vitripennis and N. giraulti for genes in the following sex-biased expression categories: same as in A. Methylated (blue) and nonmethylated genes (red) for all categories combined were also plotted. Statistical significance was calculated by Mann–Whitney U test (ns: P value > 0.05; *P value < 0.05; **P value < 0.01; ***P value < 0.001). (D) Histogram of dN/dS estimates for genes in the following sex-biased expression categories: MB: male-biased; FB: female-biased; MS: male-specific; FS: female-specific; UB: unbiased genes; ALL: all expressed genes; MB+: highly expressed and extremely male-biased genes; FB+: highly expressed and extremely female-biased genes. (E) Stacked barplot of percentage of sex-biased and control genes in the following categories: Ortholog1 (orange)—single-copy genes in Nasonia with an ortholog in Apis; Ortholog2 (green) —paralogous genes in Nasonia resulting from an ancient gene duplication event before the split of Nasonia and Apis; Paralog (blue) —paralogous genes in Nasonia resulting from a recent gene duplication event along the Nasonia lineage; None (gray)—Nasonia-specific single-copy genes with no ortholog found in Apis. (Left) Highly expressed and extremely male-biased (MB+) and female-biased (FB+) genes in N. vitripennis and N. giraulti with control unbiased genes matching their expression level. (Right) Male- and female-specific genes in N. vitripennis and N. giraulti with control unbiased genes matching their expression level.
For SB genes, MB genes evolved faster than FB genes or unbiased genes (P value < 0.001, MWU test; Fig. 5A). This is not just because methylated genes have lower nucleotide substitution rates than nonmethylated genes (P value < 2.2 × 10−16, MWU test; Fig. 5A) and most male-biased genes are nonmethylated (Fig. 4). Excluding the methylated genes from the analysis yields the same pattern (Fig. 5A). MB genes have more significant divergence at nonsynonymous sites (Fig. 5C) compared with FB genes, and although they also differ in synonymous rates (Fig. 5B), the dN/dS ratio for sex-specific and MB+ genes was significantly elevated compared with other sex-biased categories (Fig. 5D). In addition, sex-specific and MB+ genes had a larger proportion with dN/dS > 1 (P value < 0.001, MWU test; Fig. 5D and SI Appendix, Fig. S21).
Such patterns are consistent with there being a role of sex differential selection and sexual selection driving the substitution rates. One explanation for the differences in divergence is that FB genes are more constrained due to their broader expression. The data suggest that FB genes are recruited from “house-keeping” genes that are broadly expressed across development, but then become female-biased in adults. If this is correct, we expect to see a correlation between expression breadth and rate of evolution for both FB and unbiased genes. As predicted, we observed a much higher negative correlation between expression breadth (number of expressed stages) and rate of evolution for FB and unbiased genes than for MB genes (Spearman’s rho −0.30 vs. −0.21; SI Appendix, Fig. S22). In addition, genes with reduced expression breadth (fewer number of expressed stages) showed greater rates of dN/dS (Spearman’s rho = −0.23, P value < 2.2 × 10−16; SI Appendix, Fig. S23).
Gene duplication followed by sex-biased specialization is one obvious way that SD can evolve (5, 50–52). To check whether subfunctionalization after gene duplication plays a role in generating the sex-biased expression pattern in Nasonia, we used the ortholog/paralogy identifications available from the Nasonia Official Gene Set 2 (SI Appendix, Methods and Fig. 5E): Ortholog1—single-copy genes in Nasonia with an ortholog in Apis; Ortholog2—paralogous genes in Nasonia with the gene duplication before the split of Nasonia and Apis; Paralog—paralogous genes in Nasonia with the gene duplication event along the Nasonia lineage; None—Nasonia-specific single-copy genes with no ortholog found in Apis, which could represent novel genes in Nasonia, fast-evolving genes that share less than 20% protein sequence identity, or genes lost in Apis.
Consistent with our expectation, we found that extremely MB+ genes are significantly more likely to have a recently duplicated paralog (Paralog category) than they are to be single-copy genes (Ortholog1 category) (P value < 2.2 × 10−16, χ2 test; Fig. 5E), suggesting that these MB+ genes emerged following relatively recent gene duplications, more often than the FB+ or unbiased genes. Those MB+ genes are enriched for key enzymes in Nasonia male-specific pheromone synthesis as well as genes involved in detoxification and defense (Fig. 1D). In contrast, extremely FB+ genes do not have a significantly different composition of the four gene categories compared with control genes with matched expression levels (P value = 0.35, χ2 test; Fig. 5E). Consistent with our model, they are enriched for house-keeping functions in the nucleus, which is likely to be shared among other hymenopterans. This could be the reason why a larger proportion of FB genes are from ancient duplications in the ancestral species (Fig. 5E).
For SS genes, nearly 40% of both male- and female-specific genes do not have a detectable ortholog in Apis (Fig. 5E), which is consistent with the higher nucleotide substitution rate for these genes (Fig. 5A) and/or their recent origin. Furthermore, >80% of FS genes with detectable orthologs are in the Paralog category, resulting from recent duplications in the Nasonia lineage (Fig. 5E). However, MS genes do not show such an enrichment. The reasons for this difference are unclear, but could be consistent with the view that female-specific genes tend to be recruited from more broadly expressed essential genes, and therefore female-specific expression cannot readily evolve until a gene duplication has occurred.
Sex-Biased Genes Are Nonrandomly Distributed on Chromosomes.
We scanned along the chromosomes tallying the sex-biased expression status and found local clusters of sex-biased genes in both parallel and antiparallel orientations (Fig. 6A and SI Appendix, Fig. S24). Among the 1,008 adjacent sex-biased gene pairs (more than twofold sex differences and <500 bp apart), 26% (260) have reciprocal sex-biased expression pattern (one is male-biased and the other is female-biased). Of these close neighboring genes, 55% have female-biased genes methylated and male-biased genes nonmethylated. The exact nature of these chromosomal domains remains to be investigated.
Fig. 6.

Chromosome location of sex-biased genes and recombination intensity. (A, Top) Genetic distance (cM) in black and local recombination intensity (cM/Mb) in red along Nasonia chromosome 2. A green bar at marked positions was drawn at the bottom of the graph to demonstrate local marker density. (Bottom) Fold differences of male-biased (MB), female-biased (FB), male-specific (MS), female-specific (FS), and unbiased genes (UB). The pericentromeric region, where there is virtually no recombination, is labeled by a yellow box. (B) Barplot of percentage of female-biased plus female-specific (FB and FS, Top), unbiased (UB, Middle), and male-biased plus male-specific (MB and MS, Bottom) genes on five Nasonia chromosomes. Expected percentage of genes for each category expected under a random, uniform distribution was drawn as a dotted horizontal line. Statistical significance was calculated from permutation tests (ns: P value > 0.05; *P value < 0.05; **P value < 0.01; ***P value < 0.001).
In Drosophila, MB genes are depleted on the X chromosome and FB genes are enriched on the X (9). We plotted the recombination intensity along the five Nasonia chromosomes and found that FB genes were under-represented in the pericentromeric regions (P value < 0.001, permutation test; Fig. 6B) where there is extremely low recombination (Fig. 6A). Also, the local clusters of FB genes seemed to track with the recombination intensity peaks (Fig. 6A). FB genes have a higher average recombination intensity than MB genes (P value = 0.008, MWU test; SI Appendix, Fig. S22). However, when methylated genes were excluded from the analysis, there was no significant difference in recombination intensity between FB and MB genes (P value = 0.63, MWU test; SI Appendix, Fig. S25). Therefore, the pattern is primarily driven by an enrichment of methylated genes in regions of higher recombination (P value = 3.1 × 10−6, MWU test; SI Appendix, Fig. S25). We do not know why methylated genes are underrepresented in regions of low recombination. One possibility is that they tend to be more broadly expressed across development and may be more essential, and thus relocation of these genes over long evolutionary timescales reduces the genetic load imposed by the Hill–Robertson effect in regions of low recombination (53).
Conclusion.
Nasonia present an opportunity to investigate both the patterns of sex differential gene expression and its relationship to DNA methylation. Results show that ∼75% of genes display sex differences in expression; however, there are very few differences in DNA methylation between the sexes, indicating that methylation is not an important factor in SD in this species. The finding is especially striking given that there are no differences in gene presence/absence between the sexes; due to haplodiploidy, males and females have the same gene complement (i.e., there are no sex chromosomes). Distinguishing different levels of sex bias reveals that male-biased and female-biased genes evolve from different ontologies. By performing the analysis in two Nasonia species, we were able to see that the sex differences, both in expression and in methylation status, are well conserved. It would appear that sexually antagonistic selection has played a role in driving the requisite differences in expression. Female-biased genes are more often recruited from standard house-keeping genes that are more likely to be methylated and to show expression broadly across development. Male-biased genes tend to be younger, nonmethylated, and not predominated by genes expressed in testes. Based on our findings in Nasonia, we posit a two-component process: methylation is a signal for broad constitutive and stable expression (30, 45), whereas sex-biased expression is achieved by modulation through other epigenetic signals (e.g., sex-specific transcription factor-binding sites). Further study will reveal whether observed differences between Nasonia and social insects regarding methylation and gene expression are due to divergence among these taxa in how the DNA methylation toolkit is used.
Materials and Methods
RNA-seq Data Analysis.
Total RNA samples were extracted from a pool of 10, 24-h adult whole-body samples from N. vitripennis (strain AsymCX) and N. giraulti (strain R16A) males and females, using Qiagen RNeasy Plus mini kit. RNA-seq libraries were prepared and sequenced on an Illumina HiSeq2000 instrument following standard Illumina protocols (SI Appendix, Methods). Approximately 90% of the reads were uniquely mapped to the reference genomes (28) using TopHat v2.0 (54) (SI Appendix, Table S1). Per gene reads counts were summarized by Cufflinks v2.1.0 (55) and fed into the edgeR package in Bioconductor (56, 57). Normalization was performed, and total expression levels (FPKM) were calculated using edgeR (SI Appendix, Methods). Differentially expressed genes between the two sexes and the two species were detected by edgeR using a 5% FDR q-value < 0.05. Testis RNA-seq data from N. vitripennis were downloaded from Sequence Read Archive with accession no. SRX325702 (58) and analyzed using the same pipeline.
Analysis of DNA Methylation from WGBS-seq Data.
Genomic DNAs were extracted from the same strains using the DNeasy Blood and Tissue Kit (Qiagen). WGBS-seq libraries were made following Illumina protocols (SI Appendix, Methods and Table S4). Illumina reads were aligned to the converted N. vitripennis reference genome (v1.0) using BWA (59) and allowing up to four mismatches per read. CpG methylation level and gene methylation status were summarized for covered CpGs in the genome (SI Appendix, Methods). A detailed protocol can be found in our previous publication (30).
Validation of Differentially Expressed and Differentially Methylated Genes.
Selected sex-biased and sex-specific candidate genes (SI Appendix, Table S2) were validated by qRT-PCR with SYBR Green using a Roche LightCycler 480 Real-Time PCR System (SI Appendix, Methods). CpG methylation levels in differentially methylated candidates were validated by bisulfite sequencing and the PyroMark Assay. Detailed protocols could be found in ref. 30 and SI Appendix, Methods.
Recombination Intensity and Phylogenetic and Evolutionary Analysis.
Local recombination intensity was estimated based on the current Nasonia linkage map (60). For Nasvi2EG017727, protein sequences from insect species were obtained from OrthoDB7 (61) and aligned using ClustalW (62). An unrooted neighbor-joining tree (Fig. 2B) was constructed in MEGA5 (63). Synonymous and nonsynonymous substitution rates between N. vitripennis and N. giraulti were estimated using codeml in PAML 4.7 (64) for 12,469 perfectly aligned OGS2 genes with no indels (SI Appendix, Methods).
Supplementary Material
Acknowledgments
We thank Amanda Manfredo and Li (Grace) Chi for assistance with qRT-PCR and Pyrosequencing experiments, Amanda Avery for bisulfite sequencing experiments, and Rachel Edwards for preparation of samples. This work was supported in part by NIH Grant R01 GM64590; the Cornell Center for Comparative and Population Genomics and the Meinig Family Professorship (A.G.C.); and National Science Foundation Division of Integrative Organismal Systems Grant 1250790, NIH Grant R01 GM098667, and the Nathaniel and Helen Wisch Professorship (to J.H.W.).
Footnotes
The authors declare no conflict of interest.
Data deposition: All RNA-seq data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE61156). WGBS-seq data for N. vitripennis male and N. giraulti male/female samples also have been deposited in the GEO database (accession no. GSE61158).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1510338112/-/DCSupplemental.
References
- 1.Mank JE. Sex chromosomes and the evolution of sexual dimorphism: Lessons from the genome. Am Nat. 2009;173(2):141–150. doi: 10.1086/595754. [DOI] [PubMed] [Google Scholar]
- 2.Williams TM, Carroll SB. Genetic and molecular insights into the development and evolution of sexual dimorphism. Nat Rev Genet. 2009;10(11):797–804. doi: 10.1038/nrg2687. [DOI] [PubMed] [Google Scholar]
- 3.Aron S, de Menten L, Van Bockstaele DR, Blank SM, Roisin Y. When hymenopteran males reinvented diploidy. Curr Biol. 2005;15(9):824–827. doi: 10.1016/j.cub.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 4.Wertheim B, Beukeboom LW, van de Zande L. Polyploidy in animals: Effects of gene expression on sex determination, evolution and ecology. Cytogenet Genome Res. 2013;140(2-4):256–269. doi: 10.1159/000351998. [DOI] [PubMed] [Google Scholar]
- 5.Ellegren H, Parsch J. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 2007;8(9):689–698. doi: 10.1038/nrg2167. [DOI] [PubMed] [Google Scholar]
- 6.Rinn JL, Snyder M. Sexual dimorphism in mammalian gene expression. Trends Genet. 2005;21(5):298–305. doi: 10.1016/j.tig.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Meiklejohn CD, Coolon JD, Hartl DL, Wittkopp PJ. The roles of cis- and trans-regulation in the evolution of regulatory incompatibilities and sexually dimorphic gene expression. Genome Res. 2014;24(1):84–95. doi: 10.1101/gr.156414.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meisel RP, Malone JH, Clark AG. Disentangling the relationship between sex-biased gene expression and X-linkage. Genome Res. 2012;22(7):1255–1265. doi: 10.1101/gr.132100.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parisi M, et al. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299(5607):697–700. doi: 10.1126/science.1079190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao G, et al. A long-term demasculinization of X-linked intergenic noncoding RNAs in Drosophila melanogaster. Genome Res. 2014;24(4):629–638. doi: 10.1101/gr.165837.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vicoso B, Charlesworth B. Evolution on the X chromosome: Unusual patterns and processes. Nat Rev Genet. 2006;7(8):645–653. doi: 10.1038/nrg1914. [DOI] [PubMed] [Google Scholar]
- 12.Cox RM, Calsbeek R. Sexually antagonistic selection, sexual dimorphism, and the resolution of intralocus sexual conflict. Am Nat. 2009;173(2):176–187. doi: 10.1086/595841. [DOI] [PubMed] [Google Scholar]
- 13.Rice WR. Sex chromosomes and the evolution of sexual dimorphism. Evolution. 1984;38(4):735–742. doi: 10.1111/j.1558-5646.1984.tb00346.x. [DOI] [PubMed] [Google Scholar]
- 14.Beukeboom LW. Sex determination in Hymenoptera: A need for genetic and molecular studies. BioEssays. 1995;17(9):813–817. doi: 10.1002/bies.950170911. [DOI] [PubMed] [Google Scholar]
- 15.Heimpel GE, de Boer JG. Sex determination in the hymenoptera. Annu Rev Entomol. 2008;53:209–230. doi: 10.1146/annurev.ento.53.103106.093441. [DOI] [PubMed] [Google Scholar]
- 16.van de Zande L, Verhulst EC. Genomic imprinting and maternal effect genes in haplodiploid sex determination. Sex Dev. 2014;8(1-3):74–82. doi: 10.1159/000357146. [DOI] [PubMed] [Google Scholar]
- 17.Verhulst EC, Lynch JA, Bopp D, Beukeboom LW, van de Zande L. A new component of the Nasonia sex determining cascade is maternally silenced and regulates transformer expression. PLoS ONE. 2013;8(5):e63618. doi: 10.1371/journal.pone.0063618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verhulst EC, Beukeboom LW, van de Zande L. Maternal control of haplodiploid sex determination in the wasp Nasonia. Science. 2010;328(5978):620–623. doi: 10.1126/science.1185805. [DOI] [PubMed] [Google Scholar]
- 19.Beukeboom LW, van de Zande L. Genetics of sex determination in the haplodiploid wasp Nasonia vitripennis (Hymenoptera: Chalcidoidea) J Genet. 2010;89(3):333–339. doi: 10.1007/s12041-010-0045-7. [DOI] [PubMed] [Google Scholar]
- 20.Menger Y, Bettscheider M, Murgatroyd C, Spengler D. Sex differences in brain epigenetics. Epigenomics. 2010;2(6):807–821. doi: 10.2217/epi.10.60. [DOI] [PubMed] [Google Scholar]
- 21.McCarthy MM, et al. The epigenetics of sex differences in the brain. J Neurosci. 2009;29(41):12815–12823. doi: 10.1523/JNEUROSCI.3331-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raddatz G, et al. Dnmt2-dependent methylomes lack defined DNA methylation patterns. Proc Natl Acad Sci USA. 2013;110(21):8627–8631. doi: 10.1073/pnas.1306723110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang G, et al. N(6)-methyladenine DNA modification in Drosophila. Cell. 2015;161(4):893–906. doi: 10.1016/j.cell.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 24.Yan H, et al. Eusocial insects as emerging models for behavioural epigenetics. Nat Rev Genet. 2014;15(10):677–688. doi: 10.1038/nrg3787. [DOI] [PubMed] [Google Scholar]
- 25.Lyko F, Maleszka R. Insects as innovative models for functional studies of DNA methylation. Trends Genet. 2011;27(4):127–131. doi: 10.1016/j.tig.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Glastad KM, Hunt BG, Yi SV, Goodisman MA. Epigenetic inheritance and genome regulation: Is DNA methylation linked to ploidy in haplodiploid insects? Proc Biol Sci. 2014;281(1785):20140411. doi: 10.1098/rspb.2014.0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Werren JH, Loehlin DW. 2009. The parasitoid wasp Nasonia: an emerging model system with haploid male genetics. Cold Spring Harb Protoc 2009(10):pdb.emo134.
- 28.Werren JH, et al. Nasonia Genome Working Group Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327(5963):343–348. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zwier MV, Verhulst EC, Zwahlen RD, Beukeboom LW, van de Zande L. DNA methylation plays a crucial role during early Nasonia development. Insect Mol Biol. 2012;21(1):129–138. doi: 10.1111/j.1365-2583.2011.01121.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, et al. Function and evolution of DNA methylation in Nasonia vitripennis. PLoS Genet. 2013;9(10):e1003872. doi: 10.1371/journal.pgen.1003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Assis R, Zhou Q, Bachtrog D. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol. 2012;4(11):1189–1200. doi: 10.1093/gbe/evs093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meisel RP, Malone JH, Clark AG. Faster-X evolution of gene expression in Drosophila. PLoS Genet. 2012;8(10):e1003013. doi: 10.1371/journal.pgen.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian C, Gao B, Fang Q, Ye G, Zhu S. Antimicrobial peptide-like genes in Nasonia vitripennis: A genomic perspective. BMC Genomics. 2010;11:187. doi: 10.1186/1471-2164-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sackton TB, Werren JH, Clark AG. Characterizing the infection-induced transcriptome of Nasonia vitripennis reveals a preponderance of taxonomically-restricted immune genes. PLoS ONE. 2013;8(12):e83984. doi: 10.1371/journal.pone.0083984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaul B, et al. Oleic acid is a precursor of linoleic acid and the male sex pheromone in Nasonia vitripennis. Insect Biochem Mol Biol. 2014;51:33–40. doi: 10.1016/j.ibmb.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 36.Blaul B, Ruther J. How parasitoid females produce sexy sons: A causal link between oviposition preference, dietary lipids and mate choice in Nasonia. Proc Biol Sci. 2011;278(1722):3286–3293. doi: 10.1098/rspb.2011.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niehuis O, et al. Behavioural and genetic analyses of Nasonia shed light on the evolution of sex pheromones. Nature. 2013;494(7437):345–348. doi: 10.1038/nature11838. [DOI] [PubMed] [Google Scholar]
- 38.Ruther J, Matschke M, Garbe LA, Steiner S. Quantity matters: Male sex pheromone signals mate quality in the parasitic wasp Nasonia vitripennis. Proc Biol Sci. 2009;276(1671):3303–3310. doi: 10.1098/rspb.2009.0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jallon JM, David JR. Variations in cuticular hydrocarbons among the 8 species of the Drosophila melanogaster subgroup. Evolution. 1987;41(2):294–302. doi: 10.1111/j.1558-5646.1987.tb05798.x. [DOI] [PubMed] [Google Scholar]
- 40.Labeur C, Dallerac R, Wicker-Thomas C. Involvement of desat1 gene in the control of Drosophila melanogaster pheromone biosynthesis. Genetica. 2002;114(3):269–274. doi: 10.1023/a:1016223000650. [DOI] [PubMed] [Google Scholar]
- 41.Dallerac R, et al. A delta 9 desaturase gene with a different substrate specificity is responsible for the cuticular diene hydrocarbon polymorphism in Drosophila melanogaster. Proc Natl Acad Sci USA. 2000;97(17):9449–9454. doi: 10.1073/pnas.150243997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chertemps T, Duportets L, Labeur C, Ueyama M, Wicker-Thomas C. A female-specific desaturase gene responsible for diene hydrocarbon biosynthesis and courtship behaviour in Drosophila melanogaster. Insect Mol Biol. 2006;15(4):465–473. doi: 10.1111/j.1365-2583.2006.00658.x. [DOI] [PubMed] [Google Scholar]
- 43.Shirangi TR, Dufour HD, Williams TM, Carroll SB. Rapid evolution of sex pheromone-producing enzyme expression in Drosophila. PLoS Biol. 2009;7(8):e1000168. doi: 10.1371/journal.pbio.1000168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conesa A, et al. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 45.Hunt BG, Glastad KM, Yi SV, Goodisman MA. The function of intragenic DNA methylation: Insights from insect epigenomes. Integr Comp Biol. 2013;53(2):319–328. doi: 10.1093/icb/ict003. [DOI] [PubMed] [Google Scholar]
- 46.Lyko F, et al. The honey bee epigenomes: Differential methylation of brain DNA in queens and workers. PLoS Biol. 2010;8(11):e1000506. doi: 10.1371/journal.pbio.1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herb BR, et al. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat Neurosci. 2012;15(10):1371–1373. doi: 10.1038/nn.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li-Byarlay H, et al. RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc Natl Acad Sci USA. 2013;110(31):12750–12755. doi: 10.1073/pnas.1310735110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oliveira DC, et al. Identification and characterization of the doublesex gene of Nasonia. Insect Mol Biol. 2009;18(3):315–324. doi: 10.1111/j.1365-2583.2009.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Connallon T, Knowles LL. Intergenomic conflict revealed by patterns of sex-biased gene expression. Trends Genet. 2005;21(9):495–499. doi: 10.1016/j.tig.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 51.Parsch J, Ellegren H. The evolutionary causes and consequences of sex-biased gene expression. Nat Rev Genet. 2013;14(2):83–87. doi: 10.1038/nrg3376. [DOI] [PubMed] [Google Scholar]
- 52.Connallon T, Clark AG. The resolution of sexual antagonism by gene duplication. Genetics. 2011;187(3):919–937. doi: 10.1534/genetics.110.123729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78(2):737–756. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim D, et al. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nikolayeva O, Robinson MD. edgeR for differential RNA-seq and ChIP-seq analysis: An application to stem cell biology. Methods Mol Biol. 2014;1150:45–79. doi: 10.1007/978-1-4939-0512-6_3. [DOI] [PubMed] [Google Scholar]
- 57.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akbari OS, Antoshechkin I, Hay BA, Ferree PM. Transcriptome profiling of Nasonia vitripennis testis reveals novel transcripts expressed from the selfish B chromosome, paternal sex ratio. G3 (Bethesda) 2013;3(9):1597–1605. doi: 10.1534/g3.113.007583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Desjardins CA, et al. Fine-scale mapping of the Nasonia genome to chromosomes using a high-density genotyping microarray. G3 (Bethesda) 2013;3(2):205–215. doi: 10.1534/g3.112.004739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Waterhouse RM, Tegenfeldt F, Li J, Zdobnov EM, Kriventseva EV. OrthoDB: A hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41(Database issue) D1:D358–D365. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 63.Tamura K, et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.