

Significance
This work elucidates the interdependence of gene regulation, metabolism, and environmental cues during clostridial acetone-butanol-ethanol (ABE) fermentation. It also demonstrates the necessity of the integrative view for quantitative understanding of that complex process. Therefore, this work advances our fundamental knowledge concerning ABE fermentation. In addition, the work provides a quantitative tool for generating new hypotheses and for guiding strain design and protocol optimization, which facilitates the development of next-generation biofuels. More broadly, by using ABE fermentation as an example, the work further sheds light on systems biology toward an integrated and quantitative understanding of complex microbial physiology.
Keywords: integrated modeling, ABE fermentation, clostridial physiology, systems biology, metabolic engineering
Abstract
Microbial metabolism involves complex, system-level processes implemented via the orchestration of metabolic reactions, gene regulation, and environmental cues. One canonical example of such processes is acetone-butanol-ethanol (ABE) fermentation by Clostridium acetobutylicum, during which cells convert carbon sources to organic acids that are later reassimilated to produce solvents as a strategy for cellular survival. The complexity and systems nature of the process have been largely underappreciated, rendering challenges in understanding and optimizing solvent production. Here, we present a system-level computational framework for ABE fermentation that combines metabolic reactions, gene regulation, and environmental cues. We developed the framework by decomposing the entire system into three modules, building each module separately, and then assembling them back into an integrated system. During the model construction, a bottom-up approach was used to link molecular events at the single-cell level into the events at the population level. The integrated model was able to successfully reproduce ABE fermentations of the WT C. acetobutylicum (ATCC 824), as well as its mutants, using data obtained from our own experiments and from literature. Furthermore, the model confers successful predictions of the fermentations with various network perturbations across metabolic, genetic, and environmental aspects. From foundation to applications, the framework advances our understanding of complex clostridial metabolism and physiology and also facilitates the development of systems engineering strategies for the production of advanced biofuels.
Microbial metabolism is a means by which a microbe uses nutrients and generates energy to live and reproduce. As one of the most fundamental cellular characteristics, it typically involves complex biochemical processes implemented through the orchestration of metabolic reactions and gene regulation, as well as their interactions with environmental cues (1–3). One representative example of such complex processes is solvent production by Clostridium acetobutylicum, a Gram-positive, anaerobic bacterium that is considered to be one of the most prominent species for industrial biofuel production (4).
Solvent [acetone-butanol-ethanol (ABE)] fermentation of the species involves two physiological phases (5–8): During the first phase, the bacterium grows exponentially, and organic acids (acetic acid and butyric acid) are produced with the release of energy—the acidogenic phase. This process causes a dramatic drop in extracellular pH. In response to the substantial decrease of the pH, cells enter the stationary phase, and the organic acids formed are reassimilated to produce solvents including acetone, butanol, and ethanol—the solventogenic phase—thereby helping the bacterium to relieve the stress as a strategy for survival. Solventogenesis is subsequently accompanied by the onset of sporulation.
From a system-level perspective, solvent production by C. acetobutylicum is indeed an extraordinarily complex process that consists of genetic regulation, metabolic shift, and cellular signal integration (7, 9, 10). As illustrated in Fig. 1, there is a core gene regulatory network, centering on the master regulator Spo0A (10). Spo0A governs the expression of a set of functional genes coding for metabolic enzymes that are essential in ABE fermentation (e.g., adc, ctfA/B, and adhE) (11). This genetic regulation via Spo0A therefore leads to metabolic shift by altering the availability of the respective enzymes. Meanwhile, the solvents, acids, and other metabolites are released and thus alter the intracellular and extracellular environments, which in turn provides triggering signals for reprogramming the expression profiles of the genetic network (12). The three parts, metabolic reactions, gene regulation, and environmental cues, therefore constitute an interconnected, multipart system that represents a great degree of biological complexity. Supporting this fact, a recent microarray study has shown that there are at least 245 genes that are differentially expressed during the phase transition (13).
Fig. 1.

A system-level view of the acetone-butanol-ethanol (ABE) fermentation of C. acetobutylicum. ABE fermentation is a complex process that is implemented through the orchestration of metabolic reactions, gene regulation, and environmental cues. Using a modular construction concept, the entire system can be decomposed into three functional modules, with one regulating another to form an integrated global system. Details of individual modules are described in SI Appendix, Figs. S1–S4.
Due to the natural solvent production capability, acid and solvent tolerance, and versatility in consuming various sugars by C. acetobutylicum, there has been considerable interest in studying the metabolism of the bacterium over the past few decades, with a special focus on the end-point behavior of ABE fermentation. These efforts include optimization of fermentation conditions (14) and construction of new strains (15), as well as computational modeling using stoichiometric and kinetic approaches (16–18). However, despite many invaluable studies, advances in optimizing solvent production have been limited, largely due to the underappreciation of the complexity of the interconnected processes and the lack of a global understanding of fermentation. Recently, omics-based attempts have been made to reveal the global characteristics of ABE fermentation (19); however, still missing is a system-level, quantitative picture of the underlying metabolism.
Here, we present a system-level computational framework for the analysis and exploitation of the solvent metabolism of C. acetobutylicum. By adopting a modular construction strategy, we partitioned the entire system into three functional modules that correspond to metabolic reactions, gene regulation, and environmental cues, and then constructed and characterized them individually. Subsequently, we assembled the modules into an integrated model and further trained the model using experimental data from both the literature and our own fermentation experiments. To validate our framework and further illustrate its power, we systematically performed in silico network perturbations over the individual modules of the model and compared the results with a variety of literature reports.
Results and Discussion
The Metabolic Reaction Module.
Acknowledging the complexity of the process, we used a modular construction concept to decompose the system into three functional parts: i.e., metabolic reactions, gene regulation, and environmental cues. The first part is the metabolic network—the cellular infrastructure for solvent biosynthesis. As illustrated in SI Appendix, Fig. S1, acetate and butyrate are formed via multiple enzymatic reactions and reassimilated later to produce acetone, butanol, and ethanol. The enzymes of the acid synthesis pathway are constitutively expressed (green) whereas those solventogenic enzymes (red) are controlled by the phosphorylated Spo0A (Spo0A∼P). In addition, there are internal product inhibitions (blue lines) that negatively regulate metabolite levels. Mathematically, we chose single-cell Spo0A∼P concentration as the input of this module and concentrations of the acids and solvents as the output. Because the total fermented metabolites in a culture are the sum of metabolites produced by individual cells, we described the kinetics of the overall metabolites in fermentation by considering (i) the kinetics of metabolites within a single cell, (ii) the availability and activity of metabolic enzymes, and (iii) the cellular dynamics of population growth (SI Appendix, section 1.1).
To test the module, we performed two representative simulations: acidogenic fermentation, during which cells remain in the acidogenic phase, and regular ABE fermentation, in which cells transit from acidogenic to solventogenic phases. The acidogenic fermentation (Fig. 2A, Left) was simulated by setting the Spo0A∼P concentration null throughout the entire course. As a result, the cells were able to fully consume glucose in the culture, and cellular growth showed two distinct phases, with one increasing and the other decreasing, attributed to the availability of the carbon source. This fermentation resulted in the production of both acetate and butyrate but not the solvents. For regular ABE fermentation (Fig. 2A, Right), we implemented it by turning on the expression of Spo0A∼P to a saturated level during the fermentation course (at hour 10). Jointly determined by the profiles of both Spo0A∼P and remaining glucose, both the glucose uptake and cellular growth showed distinct three-piecewise profiles. Compared with acidogenic fermentation, regular ABE fermentation exhibited reduced production of acids but increased solvent production, primarily due to Spo0A∼P-induced acid reutilizations, qualitatively consistent with experimental reports (11, 20). Notably, the piecewise linear behaviors of the optical density and product yields are attributed to the lack of consideration of the impacts of metabolites and environmental pH on cellular growth in this module, which will be resolved upon integration with other parts.
Fig. 2.

Construction and characterization of individual modules. (A) The metabolic reaction module. (Left) A simulated ABE fermentation with Spo0A remaining low through the entire course, corresponding to an acidogenic fermentation or a fermentation using the spo0A mutant. The fermentation produces high amounts of acids but no solvents. (Right) A simulated fermentation with Spo0A switched on during fermentation, corresponding to a typical ABE fermentation using the WT strain. Compared with Left, this fermentation has reduced accumulations of acids but increased amounts of solvents. (B–E) The genetic regulation module. (B) Phase diagram of the network dynamics with respect to the positive regulation of σK (αs) and the dephosphorylation of Spo0A∼P (βp). Regardless of the initial conditions, Spo0A∼P concentrations can remain high or low for parameters in the upper left or lower right region, respectively. The parameters in the beaker region generate a bistable activity of Spo0A∼P, whose level may be high or low depending on the initial values of the system. (C) The Spo0A∼P concentration as a function of the strength of the positive regulation of σK (αs) and cellular toxicity (Ct). There are two types of the input–output relationships, with one discontinuous (green) and the other continuous (orange). (D and E) Two representative Spo0A∼P profiles in C. Despite the presence of differential behaviors, both profiles show monotonic increase of the Spo0A∼P (system output) with cellular toxicity (system input). (F–H) The module for environmental cues. (F and G) Computed extra- and intracellular pH values for given levels of acetic acid and butyric acid in MS-MES medium. (H) Computed cellular toxicity during the course of two fermentations that use the WT and adc mutant strains, respectively (27). The colors of the circles indicate toxicity level, and the numbers refer to different time points.
The Gene Regulation Module.
With recent advances in molecular studies of the acidogenesis–soventogenesis switch, the picture of the underlying genetic network has started to emerge—it centers on the master regulator Spo0A and its phosphorylated form Spo0A∼P and possesses a positive feedback loop mediated by multiple sigma factors (e.g., σF and σK) (SI Appendix, Fig. S3) (21). Here, the environmental signals, such as external pH and undissociated acids, can trigger the production and phosphorylation of Spo0A (22). Meanwhile, phosphorylated Spo0A (Spo0A∼P) controls the expression of downstream genes, including adc, ctfA/B, and adhE, that are essential for the acidogenic-to-solventogenic transition. Therefore, in this module, environmental signals serve as the system inputs, and Spo0A∼P serves as the output. Based on the above information, we constructed a simple feedback-based kinetic model. Here, the concentrations of the four key molecules (Spo0A, Spo0A∼P, σF, and σK) were adopted as the model variables, and their kinetics were described using differential equations (SI Appendix, section 1.2). In addition, we introduced a Hill function (SI Appendix, Eq. S31) to describe the response of the gene regulatory network to environmental cues and proposed a quantitative metric, cellular toxicity (see SI Appendix, section 1.3.1 for details), as a measure of overall deleterious effects from the cues.
To evaluate the appropriateness of our model, we used nonlinear dynamics tools to analyze its dynamic properties. Fig. 2B shows the phase diagram of the system dynamics with respect to the strengths of the positive regulation of σK (αs) and the dephosphorylation of Spo0A∼P (βp). The results suggest that the network can be locked in single states or act as a bistable switch (21), depending on parameter regimes. To further examine whether the genetic network responds appropriately to environmental cues, we plotted a three-dimensional profile of Spo0A∼P concentration that increases monotonically with cellular toxicity (indicator of environmental cues) for all αs values (Fig. 2C). Interestingly, the profile of the Spo0A∼P level may be either discontinuous with a sudden shift (green line) or continuous (orange line), depending on the strength of positive feedback (αs). The two differential responses are further illustrated in Fig. 2 D and E. To date, the detailed profile of Spo0A∼P remains unknown (discontinuous or continuous), due to the lack of single-cell Spo0A expression data. However, despite the possible presence of differences in details, this module showed a positive correlation of Spo0A∼P with the environmental cues and successfully mimicked the Spo0A production response during the acidogenic–solventogenic transition as in previous experimental studies (11, 23).
The Module for Environmental Cues.
Although often overlooked, the biochemical events of the metabolic and gene regulatory networks often cause bidirectional interactions with both intra- and extracellular environments (SI Appendix, Fig. S4): Molecules synthesized via metabolic reactions are released into the intracellular compartment and further possibly diffuse across the cell membrane to the extracellular milieu, causing the change of the metabolite concentrations in the environments; dissociation of molecules can further alter environmental pH; conversely, the molecular composites in the environment, including protons and undissociated forms of organic acids, may be toxic to the cells and thereby induce a cellular stress response and alteration of gene expression. Therefore, the environmental cues serve as the mediator for metabolic reactions and gene regulation, perceiving the information from the former and transmitting it to the latter, bridging the two fundamental processes.
To establish a quantitative description of the functions of environmental cues in ABE fermentation, we first introduced a unified metric, cellular toxicity, to account for the overall effects of environmental cues on gene regulation. We chose cellular toxicity as a measure of environmental effects due to the following reasons: A subset of metabolites (e.g., organic acids and solvents) were shown to be toxic to the cells, and a high level of those molecules reduces and even fully inhibits cell growth (24); meanwhile, from a physiological viewpoint, the solvent production of C. acetobutylicum is a survival strategy responding to environmental stress; additionally, a subset of stress response genes (e.g., groEL-groES) are indeed activated when cells transit from acidogenic into solventogenic phases to increase their tolerance (25).
Mathematically, we proposed cellular toxicity as a function of intracellular levels of undissociated acids, solvents, and pH (SI Appendix, Eq. S36). The underlying reasons are that these variables constitute the major factors causing growth suppression and solvent production, as suggested by previous studies (26), and thus serve as the triggering factors for solvent production. To further bridge the impact of metabolic reactions on environmental cues and the influence of environmental cues on gene regulation, we modeled three key steps associated with environmental cues—dissociation of organic acids, diffusion of metabolites, and pH change (SI Appendix, sections 1.3.2–1.3.4). As a result, we were able to obtain both intra- and extracellular pH values and metabolite concentrations of C. acetobutylicum cultures under given conditions. SI Appendix, Fig. S5 shows transient dynamics of intra- and extracellular metabolites, as well as corresponding pH values for a given dose of acids. SI Appendix, Fig. S6 shows the impact of cell density on the steady-state distributions of these variables. To examine the effectiveness of our pH model and further illustrate its power, we computed the pH values of nine simple buffer compositions (SI Appendix, Fig. S7), showing a good agreement with experiments. We also used the model to calculate the external and internal pH values of a more complex MS-MES medium (27) mixed with given levels of acetic and butyric acid (Fig. 2 F and G). With the above modeling, cellular toxicity can be subsequently acquired. Fig. 2H shows the computed time evolution of the cellular toxicity of two clostridial cultures, one using the WT ATCC 824 strain and the other using an adc mutant (27). Beyond the specific datasets, the model can also be used to compute cellular toxicity of arbitrary cultures of C. acetobutylicum (SI Appendix, Fig. S8).
Module Integration and Whole Model Training.
Upon systematic modeling, validation, and calibration of the individual modules above, we assembled them into an integrated framework via their input–output interconnections as illustrated in Fig. 1. We then examined the plausibility of using the resulting framework to understand complete ABE fermentation by C. acetobutylicum. Specifically, we aimed to reproduce the temporal fermentation patterns of the WT ATCC 824 strain, as well as its ctfA and adc mutants (27). For the WT strain, the computational simulation was straightforward and implemented by numerically integrating the equations of the three modules simultaneously. To simulate fermentations with the mutants, we first performed in silico gene knockout assays by setting null for the enzyme concentrations (CtfA and Adc, respectively) (SI Appendix, section 2.1) and then numerically integrated the modified equation sets. Parameters were chosen to minimize the discrepancy between in silico pH-uncontrolled fermentations and experimental data (27). Fig. 3A shows the comparison of the simulated patterns (blue, green, and red lines) with experimental data (blue circles, green squares, and red triangles) for the fermentations using the WT, ctfA mutant, and adc mutant, respectively, suggesting that the integrated model was indeed able to successfully reproduce complex ABE fermentation profiles.
Fig. 3.
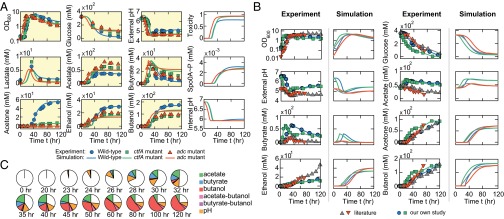
Calibrations and primary tests of the integrated model. (A) Comparison of simulated ABE fermentations with the data from previous experimental reports. Combining the modules for metabolic reactions, gene regulation, and environmental cues, the integrated model was able to reproduce the pH-uncontrolled fermentations of the WT (blue lines), ctfA-knockout (green lines), and adc-knockout (red lines) strains. The corresponding experimental results (blue circles, green squares, and red triangles) were adapted from previous studies (27). (B) Additional comparisons of experimental and computational metabolite profiles for the pH-uncontrolled fermentations that use the WT strain. The experimental data with blue circles and green squares were obtained from our own fermentations; the data with gray triangles (28) and red inverted triangles (29) were adapted from previous studies. (C) Computed time evolution of the cellular toxicity during the pH-uncontrolled WT fermentation in A (corresponding to the blue lines).
To examine whether the consistency between our simulations and the experiments was specific to the dataset we adopted for parameter fitting, we applied the same model, without any modifications or additional parameter fitting, directly to two new sets of fermentation data from the literature (gray and red triangles in Fig. 3B). In addition, we experimentally performed two pH-uncontrolled ABE fermentations using the WT C. acetobutylicum ATCC 824 strain (blue circles and green squares in Fig. 3B). Altogether, the comparisons of our model predictions and the experimental data from the literature, as well as our own fermentation assays, demonstrated that our framework and the associated parameter set are versatile and not limited to specific datasets. Additionally, by leveraging the model’s capability in revealing fermentation dynamics, we traced the temporal patterns of cellular toxicity, the measure of environmental effects, for the pH-uncontrolled, WT fermentation in Fig. 3A. As depicted in Fig. 3C, the model was able to show the time evolution, as well as the composition of cellular toxicity, over the course of fermentation, illustrating the combinatorial feature of toxicity from multiple sources as suggested by experiments (9, 24).
Systematic Network Perturbations Across Different Parts.
To further validate our integrated framework and also to illustrate its power in predicting complex fermentation and physiological processes, we performed a set of in silico network perturbations over different parts of the model, including the metabolic network, genetic network, and environmental cues, and used the resulting variants to conduct computational fermentations for a systematic comparison with experimental findings. The same set of parameters identified for the WT model in the above section was used throughout all of the network perturbation assays to ensure consistency of the modeling.
For the metabolic module, we conducted computational knockout assays for the genes pta, ctfA/B, and adhE that are all critical in producing acids and solvents. The mutations of these genes block the carbon fluxes to acetate and all of the solvents (acetone, butanol, and ethanol) but leave the butyrate formation pathway intact, which will lead to an expectation of abolished acetate and solvent production but enhanced butyrate production. We implemented the corresponding network perturbation by setting zero for the concentrations of Pta, CtfA/B, and AdhE in the model. With the modified model, we conducted an in silico fermentation assay with pH controlled above 5.0 (Fig. 4A). Supporting our expectation and agreeing with the experimental data (Fig. 4A, Left) (30), the simulated fermentation (Fig. 4A, Right) gave rise to excessive amounts of butyrate but minimal acetate and the solvents, although cell growth remained normal. The butyrate-producing phenotype was also observed for the same strain when pH was controlled above 6.0 (SI Appendix, Fig. S10). Interestingly, compared with the case of pH ≥ 6.0, the glucose uptake in the pH ≥ 5.0 assay was severely impaired despite the fact that there is a much lower butyrate production (Fig. 4B, Top). We therefore asked whether cellular toxicity can interpret this observation. Indeed, our calculation shows a higher toxicity in the case of pH ≥ 5.0 (Fig. 4B, Bottom), primarily attributed to the toxicity of the undissociated butyric acid. This result thereby shows a consistency with our initial assumption that it is undissociated acids (not total acids) that primarily contribute to the cellular toxicity.
Fig. 4.

Perturbations of the metabolic reaction network. (A) Comparison of the simulated and experimental fermentation patterns for a pta-ctfB-adhE knockout strain (30), where both have an enhanced butyrate accumulation but minimal productions of acetate and all of the solvents. pH is controlled above 5.0. (B) Computed glucose consumptions, total and undissociated butyrate, and cellular toxicities of the fermentations using the pta-ctfB-adhE mutant, for the cases when pH is controlled above 5.0 and 6.0.
With respect to the perturbation of the genetic regulation module, we computationally knocked out and overexpressed the master regulator gene spo0A. We chose spo0A as the perturbation target because of its central role in controlling cellular phase transition from acidogenesis to solventogenesis. The knockout was implemented by setting the concentration of Spo0A to zero; the spo0A overexpression was implemented by assigning a higher production rate (31). Simultaneously, we increased the specific maintenance rate (SI Appendix, Eq. S26) to mimic the metabolic burden placed by plasmid maintenance (32). To examine how the network perturbations impact ABE fermentation, we compared the levels of metabolites produced by the mutants with their references—for the knockout mutant, the WT served as its control whereas, for the overexpression strain, the WT loaded with an empty plasmid vector was recruited. We found that, in contrast to the WT fermentation (blue lines, Fig. 5), the spo0A mutant (green lines, Fig. 5) failed to initiate the solventogenic transition and to fully use glucose, leading to a phenomenon similar to acid crash (20). In addition, the spo0A overexpression strain (red lines, Fig. 5) shows a higher solvent productivity than its control (gray lines, Fig. 5). Notably, both spo0A overexpression stain and its control exhibited a delayed but prolonged fermentation as a consequence of host–plasmid interaction (32). All of the in silico results are qualitatively consistent with experimental data.
Fig. 5.

Comparison of the computational and experimental fermentation profiles for the strains with genetic network perturbations. The fermentations of the spo0A-knockout (green lines), spo0A overexpression (red lines), and their control (blue and gray lines) strains were simulated with the integrated model and further compared with corresponding experimental results reported in previous studies [green squares (33), red triangles (34), blue circles (33), and gray triangles (34)]. Notice that the experimental data do not include the glucose consumptions of the WT and spo0A-mutant strains and the ethanol yields for the spo0A overexpression strain and its control, due to the lack of information in the original literature.
Regarding the environmental cues, we decided to alter the environmental pH as an approach for network perturbation. This perturbation was motivated by the importance of environmental pH control on ABE fermentation (20, 35): Improper pH settings may cause acid crash, leading to incomplete sugar utilization and abolished solvent production because of poor transition from acidogenesis to solventogenesis; in contrast, optimal pH control may accelerate sugar utilization and result in enhanced solvent production enabled by good transition from acidogenesis into solventogenesis. Computationally, environmental pH control can be achieved by discarding the pH equation (SI Appendix, Eq. S45) in the original model and, instead, assigning a constant value, when needed, to the external pH. The right columns of Fig. 6 show the computational temporal profiles of the target metabolites for the cases of pH controlled above 5.0 (blue lines), 5.5 (green lines), and 6.0 (red lines): the first has minimal acid accumulations but high solvent production, representing a good solvent fermentation; the last has increased acid accumulations but minimal production of solvents, similar to an acidogenic fermentation; and the middle has intermediate levels of acids and solvents. All of these pH-controlled fermentations are qualitatively consistent with experimental reports (Fig. 6, Left). Again, we compared our modeling results with additional experiments performed by multiple groups (SI Appendix, Fig. S11), supporting the generic nature of our model.
Fig. 6.

ABE fermentation profiles upon the perturbations of the environmental cues (pH control). Fermentations with external pH controlled above 5.0 (blue lines), 5.5 (green lines), and 6.0 (red lines) were simulated using the integrated model. For comparison, experimental results are also presented with the blue circles (36), green squares (36) and red triangles (35) corresponding to the fermentations with external pH controlled above 5.0, 5.5, and 6.0, respectively.
The systematic in silico perturbations above, along with the comparisons with multisource experimental data, demonstrated that the model is capable of predicting the complex physiological processes of C. acetobutylicum, affirming the necessity of integrating all of the three aspects for modeling ABE fermentation.
Conclusions
In this paper, we present an integrated computational framework of clostridial ABE fermentation that combines metabolic reactions, gene regulation, and environmental cues. Although valuable attempts have been made previously (18), to our knowledge, this work is the first study that explicitly integrates the interdependent three aspects into clostridial ABE fermentation and demonstrates the necessity of this integration for a systematic understanding of the complex process. It is also, to our knowledge, the first study that successfully integrates a large volume of seemingly heterogeneous experimental data with different strains (both the WT and its mutants) under various settings (e.g., pH control and medium variation) and from multiple laboratories into a consistent picture.
Notably, although the framework involves a system-level integration of molecular and cellular events, it was not intended to include (and it was also practically impossible to include) every single process associated with clostridial physiology and ABE fermentation. For instance, redox and energy balances were not explicitly modeled in our framework; instead, we introduced a global parameter to describe the impact of the overall cellular state on enzyme activity (SI Appendix, section 1.1.2). This approximation is reasonable for this specific system, given the experimental evidence showing relative minor cofactor [both NAD(P)H/NAD(P)+ and ATP/ADP] variations across the entire course of fermentation (37). However, the impacts of cofactors can be exaggerated in some scenarios, particularly when the balances are significantly perturbed. To investigate this issue, we have extended our model to incorporate cofactor kinetics and their modulation to metabolic reactions and further used the extended model to conduct a case study (SI Appendix, section 3.2). In the future, it will be valuable to more systematically study the roles of cofactors in ABE fermentation.
This work advances our fundamental understanding of ABE fermentation by elucidating the system-level orchestration of gene regulation, metabolism, and environmental cues, identifying the multiscale link between single-cell molecular events and macroscopic batch fermentations and providing a mechanistic scheme for computing the environmental cues. The work also provides a powerful tool for generating new hypotheses and for guiding strain design and protocol optimization, facilitating the development of next-generation biofuels. More broadly, the modular model development approach used in the study can serve as a general strategy for modeling microbial physiology that involves multiple subnetworks; additionally, by using ABE fermentation as an example, our study demonstrates the necessity and power of an integrated and quantitative view for understanding physiological processes, which resonates with the emerging trend of quantitative biology toward microbial physiology (38). Therefore, our work also advances the study of quantitative microbial physiology in general.
Methods
The integrated model was developed by decomposing the system into three functional modules, constructing and characterizing each individually, and assembling them back. Differential-algebraic equations were used for model development. Custom-tailored MATLAB (MathWorks) codes were developed to implement computational simulations. Gene knockouts and overexpression were implemented by altering the concentrations of the corresponding protein appropriately. The model was further extended to consider cofactors. For experimental fermentation, the WT C. acetobutylicum ATCC 824 spore stock was used to perform anaerobic pH-uncontrolled fermentations in MS-MES medium. Temperature was controlled at 37 °C and agitation was carried out at 55 rpm. Cell growth was measured by optical density in the fermentation broth at A600. The pH profiles were recorded using the NBS BioCommand software. Metabolites were quantified by high performance liquid chromatography. Details of the computational modeling and experimental fermentation are described in SI Appendix.
Supplementary Material
Acknowledgments
We thank Professor E. Terry Papoutsakis for providing the C. acetobutylicum ATCC 824 strain. This work was supported by T.L.’s startup fund from the University of Illinois at Urbana–Champaign. V.C. was supported by the Council of Higher Education (YOK) of Turkey.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423143112/-/DCSupplemental.
References
- 1.Lee JW, et al. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Biol. 2012;8(6):536–546. doi: 10.1038/nchembio.970. [DOI] [PubMed] [Google Scholar]
- 2.Mukhopadhyay A, Redding AM, Rutherford BJ, Keasling JD. Importance of systems biology in engineering microbes for biofuel production. Curr Opin Biotechnol. 2008;19(3):228–234. doi: 10.1016/j.copbio.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Stephanopoulos G, Alper H, Moxley J. Exploiting biological complexity for strain improvement through systems biology. Nat Biotechnol. 2004;22(10):1261–1267. doi: 10.1038/nbt1016. [DOI] [PubMed] [Google Scholar]
- 4.Lee SY, et al. Fermentative butanol production by Clostridia. Biotechnol Bioeng. 2008;101(2):209–228. doi: 10.1002/bit.22003. [DOI] [PubMed] [Google Scholar]
- 5.Lütke-Eversloh T, Bahl H. Metabolic engineering of Clostridium acetobutylicum: Recent advances to improve butanol production. Curr Opin Biotechnol. 2011;22(5):634–647. doi: 10.1016/j.copbio.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 6.Jang Y-S, et al. Enhanced butanol production obtained by reinforcing the direct butanol-forming route in Clostridium acetobutylicum. MBio. 2012;3(5):e00314–12. doi: 10.1128/mBio.00314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paredes CJ, Alsaker KV, Papoutsakis ET. A comparative genomic view of clostridial sporulation and physiology. Nat Rev Microbiol. 2005;3(12):969–978. doi: 10.1038/nrmicro1288. [DOI] [PubMed] [Google Scholar]
- 8.Long S, Jones DT, Woods DR. The relationship between sporulation and solvent production in Clostridium acetobutylicum P262. Biotechnol Lett. 1984;6(8):529–534. [Google Scholar]
- 9.Wang Q, Venkataramanan KP, Huang H, Papoutsakis ET, Wu CH. Transcription factors and genetic circuits orchestrating the complex, multilayered response of Clostridium acetobutylicum to butanol and butyrate stress. BMC Syst Biol. 2013;7(1):120. doi: 10.1186/1752-0509-7-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dürre P, et al. Transcriptional regulation of solventogenesis in Clostridium acetobutylicum. J Mol Microbiol Biotechnol. 2002;4(3):295–300. [PubMed] [Google Scholar]
- 11.Harris LM, Welker NE, Papoutsakis ET. Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J Bacteriol. 2002;184(13):3586–3597. doi: 10.1128/JB.184.13.3586-3597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alsaker KV, Papoutsakis ET. Transcriptional program of early sporulation and stationary-phase events in Clostridium acetobutylicum. J Bacteriol. 2005;187(20):7103–7118. doi: 10.1128/JB.187.20.7103-7118.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimmler C, et al. Genome-wide gene expression analysis of the switch between acidogenesis and solventogenesis in continuous cultures of Clostridium acetobutylicum. J Mol Microbiol Biotechnol. 2011;20(1):1–15. doi: 10.1159/000320973. [DOI] [PubMed] [Google Scholar]
- 14.Kharkwal S, Karimi IA, Chang MW, Lee D-Y. Strain improvement and process development for biobutanol production. Recent Pat Biotechnol. 2009;3(3):202–210. doi: 10.2174/187220809789389117. [DOI] [PubMed] [Google Scholar]
- 15.Lütke-Eversloh T. Application of new metabolic engineering tools for Clostridium acetobutylicum. Appl Microbiol Biotechnol. 2014;98(13):5823–5837. doi: 10.1007/s00253-014-5785-5. [DOI] [PubMed] [Google Scholar]
- 16.Dash S, Mueller TJ, Venkataramanan KP, Papoutsakis ET, Maranas CD. Capturing the response of Clostridium acetobutylicum to chemical stressors using a regulated genome-scale metabolic model. Biotechnol Biofuels. 2014;7(1):144. doi: 10.1186/s13068-014-0144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shinto H, et al. Kinetic modeling and sensitivity analysis of acetone-butanol-ethanol production. J Biotechnol. 2007;131(1):45–56. doi: 10.1016/j.jbiotec.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Mayank R, Ranjan A, Moholkar VS. Mathematical models of ABE fermentation: Review and analysis. Crit Rev Biotechnol. 2013;33(4):419–447. doi: 10.3109/07388551.2012.726208. [DOI] [PubMed] [Google Scholar]
- 19.Jang Y-S, et al. Proteomic analyses of the phase transition from acidogenesis to solventogenesis using solventogenic and non-solventogenic Clostridium acetobutylicum strains. Appl Microbiol Biotechnol. 2014;98(11):5105–5115. doi: 10.1007/s00253-014-5738-z. [DOI] [PubMed] [Google Scholar]
- 20.Maddox IS, et al. The cause of “acid-crash” and “acidogenic fermentations” during the batch acetone-butanol-ethanol (ABE-) fermentation process. J Mol Microbiol Biotechnol. 2000;2(1):95–100. [PubMed] [Google Scholar]
- 21.Al-Hinai MA, Jones SW, Papoutsakis ET. The Clostridium sporulation programs: Diversity and preservation of endospore differentiation. Microbiol Mol Biol Rev. 2015;79(1):19–37. doi: 10.1128/MMBR.00025-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terracciano JS, Kashket ER. Intracellular conditions required for initiation of solvent production by Clostridium acetobutylicum. Appl Environ Microbiol. 1986;52(1):86–91. doi: 10.1128/aem.52.1.86-91.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones SW, et al. The transcriptional program underlying the physiology of clostridial sporulation. Genome Biol. 2008;9(7):R114. doi: 10.1186/gb-2008-9-7-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Tsao GT. Mathematical modeling of inhibition kinetics in acetone-butanol fermentation by Clostridium acetobutylicum. Biotechnol Prog. 1994;10(5):532–538. [Google Scholar]
- 25.Tomas CA, Beamish J, Papoutsakis ET. Transcriptional analysis of butanol stress and tolerance in Clostridium acetobutylicum. J Bacteriol. 2004;186(7):2006–2018. doi: 10.1128/JB.186.7.2006-2018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hüsemann MH, Papoutsakis ET. Solventogenesis in Clostridium acetobutylicum fermentations related to carboxylic acid and proton concentrations. Biotechnol Bioeng. 1988;32(7):843–852. doi: 10.1002/bit.260320702. [DOI] [PubMed] [Google Scholar]
- 27.Lehmann D, et al. Modifying the product pattern of Clostridium acetobutylicum: Physiological effects of disrupting the acetate and acetone formation pathways. Appl Microbiol Biotechnol. 2012;94(3):743–754. doi: 10.1007/s00253-011-3852-8. [DOI] [PubMed] [Google Scholar]
- 28.Wietzke M, Bahl H. The redox-sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum. Appl Microbiol Biotechnol. 2012;96(3):749–761. doi: 10.1007/s00253-012-4112-2. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann D, Lütke-Eversloh T. Switching Clostridium acetobutylicum to an ethanol producer by disruption of the butyrate/butanol fermentative pathway. Metab Eng. 2011;13(5):464–473. doi: 10.1016/j.ymben.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Jang Y-S, Woo HM, Im JA, Kim IH, Lee SY. Metabolic engineering of Clostridium acetobutylicum for enhanced production of butyric acid. Appl Microbiol Biotechnol. 2013;97(21):9355–9363. doi: 10.1007/s00253-013-5161-x. [DOI] [PubMed] [Google Scholar]
- 31.Tracy BP, Gaida SM, Papoutsakis ET. Development and application of flow-cytometric techniques for analyzing and sorting endospore-forming clostridia. Appl Environ Microbiol. 2008;74(24):7497–7506. doi: 10.1128/AEM.01626-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walter KA, Mermelstein LD, Papoutsakis ET. Host-plasmid interactions in recombinant strains of Clostridium acetobutylicum ATCC 824. FEMS Microbiol Lett. 1994;123(3):335–341. [Google Scholar]
- 33.Tomas CA, et al. DNA array-based transcriptional analysis of asporogenous, nonsolventogenic Clostridium acetobutylicum strains SKO1 and M5. J Bacteriol. 2003;185(15):4539–4547. doi: 10.1128/JB.185.15.4539-4547.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alsaker KV, Spitzer TR, Papoutsakis ET. Transcriptional analysis of spo0A overexpression in Clostridium acetobutylicum and its effect on the cell’s response to butanol stress. J Bacteriol. 2004;186(7):1959–1971. doi: 10.1128/JB.186.7.1959-1971.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Monot F, Engasser J-M, Petitdemange H. Influence of pH and undissociated butyric acid on the production of acetone and butanol in batch cultures of Clostridium acetobutylicum. Appl Microbiol Biotechnol. 1984;19(6):422–426. [Google Scholar]
- 36.Lehmann D, Radomski N, Lütke-Eversloh T. New insights into the butyric acid metabolism of Clostridium acetobutylicum. Appl Microbiol Biotechnol. 2012;96(5):1325–1339. doi: 10.1007/s00253-012-4109-x. [DOI] [PubMed] [Google Scholar]
- 37.Amador-Noguez D, Brasg IA, Feng XJ, Roquet N, Rabinowitz JD. Metabolome remodeling during the acidogenic-solventogenic transition in Clostridium acetobutylicum. Appl Environ Microbiol. 2011;77(22):7984–7997. doi: 10.1128/AEM.05374-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T. Interdependence of cell growth and gene expression: Origins and consequences. Science. 2010;330(6007):1099–1102. doi: 10.1126/science.1192588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.