

Significance
Alternative splicing is the main mechanism that drives protein diversity; however, little is known about the physiological cues that control splicing. Here, we show that the stiffness of the extracellular matrix mediates protein splicing in cells both in vitro and in vivo. Alternative splicing mediated by matrix stiffness occurs through the phosphorylation of splicing regulatory factors, serine/arginine rich (SR) proteins, and depends on the PI3K signaling pathway. Because the SR family of proteins are conserved among both vertebrates and invertebrates and are known to also be involved in genome stabilization, translation, and mRNA export, these results suggest a previously unidentified mechanism by which cells can respond and adapt to their mechanical microenvironment in both healthy and diseased states.
Keywords: alternative splicing, extracellular matrix, matrix stiffness, angiogenesis, cancer progression
Abstract
Alternative splicing of proteins gives rise to different isoforms that play a crucial role in regulating several cellular processes. Notably, splicing profiles are altered in several cancer types, and these profiles are believed to be involved in driving the oncogenic process. Although the importance of alternative splicing alterations occurring during cancer is increasingly appreciated, the underlying regulatory mechanisms remain poorly understood. In this study, we use both biochemical and physical tools coupled with engineered models, patient samples, and a murine model to investigate the role of the mechanical properties of the tumor microenvironment in regulating the production of the extra domain-B (EDB) splice variant of fibronectin (FN), a hallmark of tumor angiogenesis. Specifically, we show that the amount of EDB-FN produced by endothelial cells increases with matrix stiffness both in vitro and within mouse mammary tumors. Matrix stiffness regulates splicing through the activation of serine/arginine rich (SR) proteins, the splicing factors involved in the production of FN isoforms. Activation of the SR proteins by matrix stiffness and the subsequent production of EDB-FN are dependent on intracellular contractility and PI3K-AKT signaling. Notably, matrix stiffness-mediated splicing is not limited to EDB-FN, but also affects splicing in the production of PKC βII and the VEGF 165b splice variant. Together, these results demonstrate that the mechanical properties of the microenvironment regulate alternative splicing and establish a previously unidentified mechanism by which cells can adapt to their microenvironment.
Differential expression of protein isoforms through alternative splicing is a key element to generating protein diversity and can result in widely different cell phenotypes and behaviors (1–3). Interestingly, tumors exhibit several major differences in protein isoform expression patterns compared with healthy tissue (2, 4), and some of these changes are thought to favor oncogenesis (1). Notably, splicing events are regulated at the pre-mRNA level by splicing regulatory factors that include a family of serine/arginine rich (SR) proteins (5). Elevated SR protein levels have been associated with cancer (5); however, the physiological cues that drive splicing are not well defined.
Among the various alternatively spliced proteins present in tumors, the splice variant of fibronectin (FN) that includes the extra domain-B (EDB) type III repeat has been of particular interest in the cancer community because inclusion of the EDB fragment has been proposed as a way to identify and target tumor vasculature (6). EDB-FN is produced by endothelial cells (ECs) in tumors and may favor angiogenesis (7–9). Splicing of FN is regulated by several SR proteins, including SRp40 and SRp20, in several different cell types (10–12). Although the presence of EDB-FN is fairly unique to tumor vasculature, the mechanisms governing its expression within the tumor microenvironment are not clear.
The splicing activity of SR proteins that leads to the expression of various protein isoforms including EDB-FN is tightly regulated by phosphorylation (13–15), and this phosphorylation has been shown to be mediated by AKT (14, 16). Interestingly, matrix stiffness can regulate AKT phosphorylation state (17–19). More specifically, recent data indicate that AKT activation and downstream phosphorylation of AKT substrates can occur through changes in the mechanical properties of the tissue in the tumor microenvironment (17). Importantly, tumor tissue stiffens during cancer due to increased collagen deposition and the action of cell-secreted enzymes that cross-link collagen fibrils (20, 21), and this stiffening is believed to be correlated with tumor aggressiveness and metastatic potential (18, 22–25). Elevated extracellular matrix (ECM) stiffness affects several cell behaviors associated with tumors including enhanced cell contractility, response to growth factors, and cell migration (18, 22, 24, 26, 27). Although stiffness has been shown to drive greater AKT activation, AKT activity drives SR protein phosphorylation, and SR proteins mediate splicing, it has not yet been determined whether stiffness-driven signaling can result in SR protein-mediated splicing events during tumorigenesis.
In this study, we used a multidisciplinary approach, with in vitro and in vivo models and biochemical and physical tools, to study the relationship between matrix stiffness and FN splicing in ECs. Here, we demonstrate, for the first time to our knowledge, that EDB-FN expression increases with matrix stiffness, both in vitro and in mouse mammary tumors. Our data indicate that matrix stiffness regulates the dynamics of EDB-FN splicing through Rho-associated protein kinase (ROCK) mediated contractility and downstream SR protein phosphorylation. Moreover, phosphorylation of SR proteins correlates with in vivo PI3K/AKT pathway activity in response to tissue stiffness, whereas in vitro inhibition of PI3K prevents SR protein phosphorylation and EDB-FN splicing. Taken together, our data show, for the first time to our knowledge, that the stiffness of the tumor microenvironment modulates intracellular splicing events.
Results
Extracellular Matrix Stiffness Regulates Protein Splicing.
Expression of the EDB-FN splice variant is known to be characteristic of new angiogenic vessels within the tumor vasculature (9). As such, tumor vasculature of human patient samples stained strongly for EDB, whereas significantly less EDB was evident in normal tissue (Fig. 1A). Moreover, a strong EDB-FN signal was evident in tissue extracts from spontaneous mammary tumors formed in a polyoma middle T antigen (PyMT) mouse model of breast cancer, whereas EDB-FN was not evident in normal mammary gland tissue extract from a WT mouse (Fig. 1B).
Fig. 1.
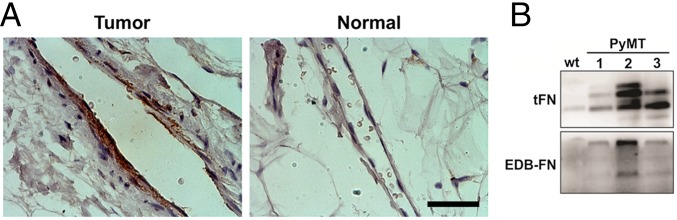
EDB-FN is expressed in human breast tumor vasculature. (A) Immunohistochemical staining of human breast tissue for EDB-FN showing strong EDB-FN staining in the tumor vasculature and no immunoreactivity for EDB-FN in the normal tissue vasculature (same exposure settings). (Scale bar, 50 μm.) (B) Western blotting on total protein extracts from normal mammary tissue of WT and cancerous tissue of PyMT mice showing the presence of EDB-FN in the tumor.
Because increased tissue stiffness is a hallmark of most solid tumor progression and has been shown to affect fibronectin assembly and deposition (28, 29), we assessed whether tissue stiffness was affecting the expression of the EDB-FN splice variant within PyMT tumors. To that end, we decreased tumor tissue stiffness by preventing ECM cross-linking by treating the mice with the lysyl oxidase inhibitor β-aminopropionitrile (BAPN) (Fig. 2A) (17). Notably, we observed a significant reduction of EDB-FN in tumor extract from PyMT mice that had been treated with BAPN compared with the control (Fig. 2 B and C). In addition, immunostaining of frozen sections also showed a reduction of EDB-FN in BAPN-treated mice (Fig. 2D). Quantification of the EDB-FN content of those tumors indicated a lower EDB content (Fig. 2E). In addition, EDB-FN fiber-like structures were shorter in BAPN mice. In contrast, total FN levels were only minimally affected (Fig. 2 B and F and Fig. S1).
Fig. 2.

Effects of BAPN modulation of in vivo stiffness on EDB-FN in mammary tumors. (A) Mechanical measurements performed on tumor tissue from PyMT mice by unconfined compression reveal that BAPN significantly lowers tissue stiffness compared with the mock treatment (water only, Ctrl). (B) Western blotting of cancerous tissue total protein extracts from PyMT mice given either the mock treatment or treated with BAPN showing EDB-FN and total FN. GAPDH was used as loading control. (C) Corresponding densitometric quantification showing both EDB-FN normalized against GAPDH content and the ratio of EDB-FN to total FN. (D) Confocal images of EDB-FN stained frozen tissue sections with or without BAPN treatment and (E) the corresponding quantification of the average EDB-FN fiber-like structures length and content. (F) Representative confocal images of total FN and CD31 with or without BAPN treatment. Plots are mean ± SE, Student t test: *P < 0.05, **P < 0.01. (Scale bars, 30 μm.)
Fig. S1.
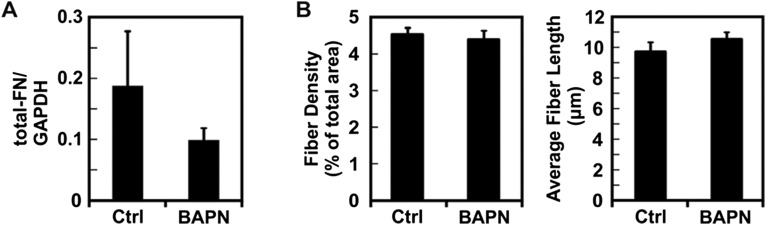
Effects of BAPN treatment on total FN levels in vivo. (A) Quantification of total FN levels observed by Western blot of PyMT tumors from mice treated with BAPN or with the mock treatment (Ctrl). (B) Quantification of the average total FN fiber-like structure length and content from PyMT frozen tumor sections. Plots are mean ± SE.
To confirm that matrix stiffness was driving FN splicing, we implemented an in vitro system. ECs were seeded on compliant (E = 1 kPa) and stiff (E = 10 kPa) polyacrylamide substrates, stained, and imaged for both total FN and EDB-FN (Fig. 3A). Both total FN and EDB-FN showed a visible increase on stiffer matrices (Fig. 3B). Similar to what we observed in vivo, quantification of EDB-FN levels revealed increased inclusion of the EDB variant on stiff matrices compared with the amount of total FN (Fig. 3C). Indeed, normalizing the ratio of EDB-FN to total FN as determined by Western blotting indicated that there was a 50% increase in EDB-FN relative to total FN with increasing matrix stiffness (Fig. 3D). To further confirm our observations, quantitative real-time RT-PCR was used to quantify the mRNA expression of EDB-FN. Our results indicate that EDB-FN mRNA expression increased ∼2.5-fold on stiff matrices (Fig. 3E). When taken together with the in vivo data, these results clearly demonstrate that increasing matrix stiffness stimulates the inclusion of the EDB domain in FN.
Fig. 3.

Expression of the EDB-FN splice variant increases with ECM stiffness in vitro. ECs were plated on compliant (E = 1 kPa) and stiff (E = 10 kPa) substrates (100,000 cells/mL), grown over 3 d, and subjected to immunofluorescence staining or Western blot. (A) Immunofluorescent staining of both EDB-FN and total FN increases with increasing substrate stiffness (images were acquired with the same exposure settings). (Scale bar, 50 μm.) (B) Representative Western blot of total protein extracts showing EDB-FN and total FN on compliant and stiff substrates. GAPDH was used as loading control. (C) Densitometric quantification of EDB-FN and total FN normalized against GAPDH content. (D) Corresponding ratio of EDB-FN to total FN showing a 30% increase in EDB-FN expression with increasing stiffness (three independent experiments). (E) EDB-FN expression as determined by quantitative real-time RT-PCR indicates a significant increase in mRNA expression with increasing stiffness (three independent experiments). Plots are mean ± SE, Student t test: *P < 0.05, ***P < 0.001.
There are several other splice variants known to be up-regulated in tumors apart from EDB-FN (1). Among the numerous proteins that are regulated by splicing and are also associated with tumor progression, the PKC βII and VEGF 165b splice variant are of particular interest because they are, respectively, pro- and antiangiogenic (30, 31). We assayed for the expression of the PKC βII isoform as a function of stiffness. Similar to what was observed for EDB-FN, PKC βII expression was lower in tumor tissue from BAPN-treated mice (Fig. S2A). Interestingly, the antiangiogenic VEGF 165b isoform expression was higher in tumor tissue from BAPN-treated mice (Fig. S2B). Of note, both PKC βII and VEGF 165b expression modulation was observed in all of the cells present within the tumor and not found exclusively in ECs. In vitro experiments using polyacrylamide substrates recapitulated these results, where PKC βII expression by ECs was increased more than twofold on stiffer matrices, whereas VEGF 165b expression was decreased in these conditions (Fig. S3 C and D). Notably, these data indicate that matrix stiffness-mediated splicing is not limited solely to EDB-FN or solely to ECs, and it may constitute a fundamental mechanotransduction mechanism within multiple different cell types.
Fig. S2.

ECM stiffness regulates the expression of the PKC βII and VEGF 165b isoforms. (A) Representative confocal images of frozen tissue sections from PyMT tumors with or without BAPN treatment and stained for PKC βII showing the overall increase of the PKC βII isoform with tumor tissue stiffness. (B) Representative confocal images of frozen tissue sections from PyMT tumors with or without BAPN treatment stained for VEGF 165b and total VEGF showing the overall decrease of the VEGF 165b isoform in stiffer tumor tissue. (C) Representative Western blot of total protein extract from ECs in culture on compliant and stiff substrates with the corresponding densitometric quantification showing increased PKC βII isoform expression. Actin was used as loading control. (D) Representative Western blot of total protein extract from ECs in culture on compliant and stiff substrates with the corresponding densitometric quantification showing increased VEGF 165b isoform expression. GAPDH was used as loading control. Plots are mean ± SE, Student t test: ***P < 0.001. (Scale bars, 30 μm.)
Fig. S3.

EDB-FN splice variant expression is mediated by cell contractility. (A) Western blot of total protein extract from cells plated on collagen-coated substrates and treated with or without the RhoA inhibitor CT04 showing a decrease in EDB-FN compared with control. Plots are mean ± SE, Student t test: *P < 0.05, ***P < 0.001. (B) Representative images of EDB-FN and total fibronectin of ECs cultured overnight on collagen-coated substrates and treated with or without CT04. Images were acquired using the same exposure settings. (C) Representative images of EDB-FN and total fibronectin of ECs cultured overnight on collagen-coated substrates and treated with blebbistatin or ML-7. Images were acquired using identical exposure settings. (Scale bars, 30 μm.)
EDB-FN Splice Variant Expression Is Mediated by Cell Contractility.
The ability of cells to adapt to ECM stiffness is known to rely on cell contractility (27). Cellular contractility has been found to typically increase on stiffer matrices and is required for proper activation of several signaling pathways that control gene expression (32–34). As such, we investigated whether cell contractility mediated by the Rho-ROCK pathway could regulate EDB-FN expression. ECs were plated on stiff matrices (E = 10 kPa) and treated with the ROCK inhibitor Y27632. Notably, both Western blotting and immunofluorescent staining revealed that ROCK inhibition greatly reduced the amount of EDB-FN present (Fig. 4 A and B). We confirmed this observation by using a siRNA-based approach to knock down ROCK in ECs (Fig. 4C). Depletion of ROCK also led to decreased levels of observable EDB-FN (Fig. 4 C and D). Furthermore, direct inhibition of RhoA with the C3 transferase resulted in lower EDB-FN expression (Fig. S3 A and B). Inhibition of cell contractility with either blebbistatin or the myosin light chain kinase inhibitor ML-7 show a similar trend (Fig. S3C). Overall, these results indicate that expression of the EDB-FN isoform by ECs depends on Rho-ROCK–mediated cell contractility.
Fig. 4.

EDB-FN splice variant expression is mediated by cell contractility. (A) Western blot of total protein extract from cells plated on 10-kPa substrates and treated with or without the ROCK inhibitor Y27632 showing a decrease in EDB-FN compared with control. Corresponding densitometric quantification showing both EDB-FN normalized against GAPDH content and the ratio of EDB-FN to total FN. (B) Representative images of EDB-FN and total fibronectin of EC cells cultured overnight on 10-kPa substrates and treated with or without Y27632. (C) Western blot for ROCK, EDB-FN, and total FN expression after treatment with siRNA against ROCK compared with control. Corresponding densitometric quantification showing both EDB-FN normalized against GAPDH content and the ratio of EDB-FN to total FN. (D) Fluorescent staining for EDB-FN after treatment with siRNA against ROCK on stiff (E = 10 kPa) substrates. Images were acquired using the same exposure settings. Plots are mean ± SE, Student t test: *P < 0.05, **P < 0.01. Images were acquired using the same exposure settings. (Scale bars, 30 μm.)
ECM Stiffness Regulates SRp40 Phosphorylation in Culture and in Vivo.
In light of the above data implicating ECM stiffness and cell contractility as modulators of EDB-FN splicing, and because SRp40 is one of the main regulators of EDB-FN expression (11, 12), we extended our analysis to the SR proteins. Remarkably, phosphorylation levels of SR proteins increased with matrix stiffness in vitro (Fig. 5 A and B). Inhibition of cell contractility with the Y27632 inhibitor significantly lowered the phosphorylation of several SR proteins, including SRp40 (Fig. 5C). Immunostaining of PyMT tumors showed a similar effect of BAPN on SR protein phosphorylation levels, where decreasing tumor stiffness decreased SR protein phosphorylation (Fig. 5D). These data indicate that both matrix stiffness and cell contractility influence the activation of SR proteins involved in splicing regulation.
Fig. 5.

The interplay of ECM stiffness and cell contractility regulates SRp40 phosphorylation. (A) Fluorescent staining for phosphorylated SR protein of ECs plated on compliant (E = 1 kPa) and stiff (E = 10 kPa) substrates. (B) Western blot of whole protein extract from ECs plated on either compliant (E = 1 kPa) or stiff (E = 10 kPa) substrates showing an increase in SR protein phosphorylation with increasing substrate stiffness. (C) Western blot of whole protein extract from EC cells plated on stiff (E = 10 kPa) substrates showing that Y27632-mediated inhibition of cell contractility prevent SR protein phosphorylation compared with control (Ctrl). Actin was used as a loading control. (D) Representative image of frozen tissue sections stained for phosphorylated SR protein and CD31 showing that the BAPN treatment lowered SR protein phosphorylation compared with the mock treatment (Ctrl) (images were acquired with identical exposure settings). (Scale bars, 30 μm.)
PI3K Is Required for SRp40-Mediated Splicing of EDB-FN.
There are several kinases that are known to phosphorylate SR proteins (14, 15). Among those, AKT has previously been reported as being involved in SRp40-mediated FN splicing (35). Additionally, recent in vivo evidence points to a prominent role of the PI3K/AKT pathway in regulating cell response to matrix stiffness (17). Therefore, we assessed the extent to which matrix stiffness-mediated EDB-FN splicing occurs through PI3K involvement. Using an antibody against phosphorylated substrates of AKT, we confirmed in vivo that lowering tumor tissue stiffness with BAPN reduced the activity of the PI3K/AKT pathway (Fig. 6A). Similarly, we detected higher phosphorylation levels of AKT when EC were seeded on stiffer matrix (Fig. S4). We then inhibited PI3K using the pharmacological inhibitor wortmannin in ECs seeded on stiff matrices and assayed for the presence of EDB-FN after 20 h (Fig. 6 B and C). Notably, EDB-FN expression was decreased in wortmannin-treated samples. In agreement with the decreased level of EDB-FN observed at 20 h, both SR protein phosphorylation and AKT phosphorylation decreased after 1-h wortmannin treatment (Fig. 6 D–F and Fig. S5). Together, these data indicate that EDB-FN splicing occurs through the involvement of the PI3K/AKT pathway most likely downstream of cell contractility, thus further describing the mechanism by which microenvironmental mechanics regulates alternative splicing.
Fig. 6.

PI3K-AKT signaling is required for splicing of EDB-FN. (A) Representative image of frozen tissue sections stained for phosphorylated AKT substrates and VE-cadherin showing that the BAPN treatment influences PI3K/AKT signaling pathway activation. (Scale bar, 50 μm.) (B) Confocal images of EDB-FN and total FN on EC cells plated on 10-kPa substrates and kept in culture for 24 h with or without (Ctrl) wortmannin (Wort). (C) Western blot of whole protein extract from EC cells seeded on 10-kPa substrates showing that wortmannin (Wort)-mediated inhibition of PI3K decreases EDB-FN compared with control. Corresponding densitometric quantification showing both EDB-FN normalized against GAPDH content and the ratio of EDB-FN to total FN. (D) Representative image of ECs stained for phosphorylated SR proteins and AKT phosphorylation on Ser-473 showing that 1-h treatment with wortmannin prevents both SR protein and AKT phosphorylation. (Scale bar, 30 μm.) (E) Corresponding quantification of SR protein phosphorylation computed from at least 18 different fields of view from three independent experiments. (F) Western blot of whole protein extract from EC cells plated on stiff (E = 10 kPa) substrates showing that wortmannin (Wort)-mediated inhibition of PI3K lowered SR protein phosphorylation compared to control (Ctrl). Actin was used as a loading control. Plots are mean ± SE, Student t test: *P < 0.05, **P < 0.01. Images were acquired with identical exposure settings.
Fig. S4.
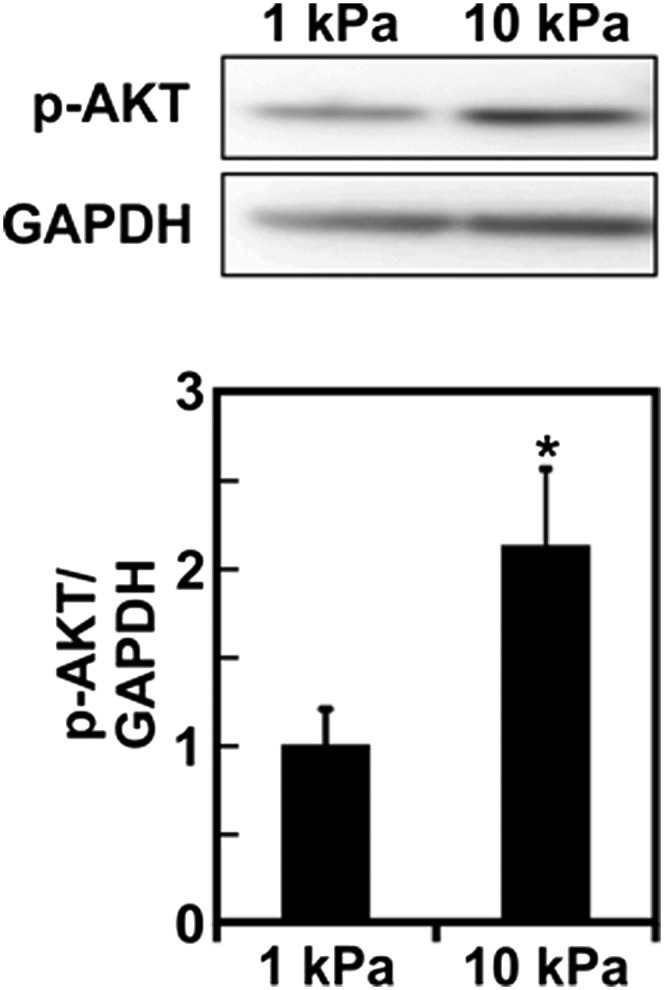
Substrate stiffness regulates AKT activation. Western blot of whole protein extract from ECs plated on either compliant (E = 1 kPa) or stiff (E = 10 kPa) substrates showing an increase in AKT phosphorylation with increasing substrate stiffness with the corresponding densitometric quantification. GAPDH was used as the loading control. Plots are mean ± SE, Student t test: *P < 0.05.
Fig. S5.

Quantification of SR protein phosphorylation. (A) To perform single cell quantification of the SR phosphorylation signal, a threshold was applied on the DAPI-stained nucleus to obtain a binary image in ImageJ. The binary image was then used to select overlay regions that were then applied on the p-SR channel. The average signal intensity was measured for each region of interest. (Scale bar, 30 μm.) (B) Representative zoomed-in confocal images of cells seeded on 10-kPa substrates with or without a 1-h wortmannin treatment showing the overlay regions.
Discussion
Here, we provide, to our knowledge, the first direct evidence that alternative splicing is regulated by ECM stiffness. Specifically, both in vitro and in vivo data indicate that the stiffness of the microenvironment regulates EDB-FN splicing. ECM stiffness mediates splicing by regulating SR protein phosphorylation and requires both cell contractility and activation of the PI3K/AKT pathway. Moreover, our in vivo data suggest that regulation of alternative splicing by ECM stiffness likely occurs in many cell types and may not be unique to only ECs or the EDB isoform of FN. As such, ECM stiffness may be a significant mediator of the production of the diverse array of protein isoforms present in a broad range of tissues and disease states.
FN is one of the most well-characterized ECM proteins. Alternative splicing of FN has been studied extensively, in part, because it is tightly linked to angiogenesis (9, 36–39). In ECs, the splicing events resulting in the inclusion of either the EDA or the EDB domains are correlated with tumor angiogenesis (36). It has been shown that both of these events can be regulated by SRp40 (11, 12). However, the physiological cues that mediate this splicing are largely unknown. Our data indicate that matrix stiffness can control SR protein phosphorylation, including that of SRp40, which regulates the splicing of numerous other proteins including proangiogenic molecules such as VEGF, notably by limiting the expression of the antiangiogenic VEGF 165b splice variant in favor of more proangiogenic forms (40–43). Moreover, the SR protein response to matrix stiffness is controlled by a signaling pathway that involves PI3K/ATK and requires cellular contractility. Indeed, inhibition of either cell contractility or PI3K prevented SR protein phosphorylation and inclusion of the EDB domain of fibronectin. Interestingly, AKT signaling has previously been implicated in SRp40-mediated splicing of the PKC family (14, 16), suggesting that AKT is likely to be involved in a similar way in both stiffness-mediated PKC and EDB-FN splicing. In addition, the regulation of PI3K/AKT pathway by matrix stiffness is increasingly well documented in a number of different experimental tumor models (17–19, 44). Taken together, our data uncover a fundamental mechanism regulating EDB-FN expression in ECs. Indeed, considering the ubiquitous expression of EDB-FN in many aggressive solid tumors (7, 45–47) and the altered splicing patterns found in tumors (2, 4), the present findings are most likely representative of a general mechanism that extends beyond ECs and EDB-FN.
Interestingly, each of the hallmarks of cancer is associated with a switch in protein isoform expression patterns (2, 48). It has been suggested that tumor progression may be associated with coordinated control of splicing events (2, 31, 40). Efforts to uncover the regulators of the splicing machinery have identified several signaling pathways associated with growth factors and tumor progression (11, 49). Importantly, our results define a previously unidentified mechanism by which alternative splicing is regulated in cells through changes in the mechanical properties of the ECM. This regulatory mechanism could act through either direct activation of integrin signaling pathways or indirectly through the modulation of other pathways. In the case of direct activation, mechanical cues at ECM-cell adhesion sites can be converted directly into biochemical signals (50). Notably, there is compelling evidence implicating ECM stiffness in the regulation of PTEN and PI3K activation in epithelial cells (17). For the indirect case, we and others have shown that the interplay between the ECM stiffness and cell contractility can modulate growth factor-induced signaling pathways (27, 32, 51). Notably, both myosin light chain kinase and Rho-ROCK mediated contractility have been shown to be important in matrix stiffness regulation of growth factor signaling (51). Because our data link splicing and Rho/ROCK-mediated contractility and given that growth factors are known to regulate alternative splicing (16, 40), the interplay between ECM stiffness and growth factors could lead to a wide variety of splicing patterns.
Overall, matrix stiffness-mediated alternative splicing could potentially provide cells with a mechanism to adapt to mechanical stimulation. Changes in signaling proteins expression due to altered splicing patterns in response to altered ECM mechanical properties could result in changes in cell phenotype. For example, each of the PKC isoforms has been implicated in affecting actin dynamics and cell motility (52–55), suggesting that alternative splicing due to matrix stiffness could contribute to the known changes in cell motility that occur on compliant matrices compared with stiff matrices (24, 26). Stiffness-mediated down-regulation of the antiangiogenic isoform of VEGF, which would result in the alternative splicing of more proangiogenic isoforms (31), could play an important role in regulating angiogenesis. In addition, adipocyte differentiation has been shown to be independently driven by SRp40-mediated splicing (41) and tissue stiffness (56), hinting at a possible role of splicing in tissue stiffness induced differentiation. Therefore, our findings have important ramifications for the fundamental understanding of how changes in the mechanical microenvironment ultimately regulate cell-ECM interactions in normal physiological responses, such as tissue morphogenesis, and abnormal responses, like tumorigenesis.
Materials and Methods
Mouse Studies.
All mice were maintained following a protocol approved by the Cornell University Institutional Animal Care and Use Committee. MMTV-PyMT transgenic and WT control mice, both on the FVB strain background, were obtained from the Jackson Laboratory. To alter tumor stiffness in vivo, mice were treated beginning at 4 wk of age with BAPN (3 mg/kg body weight; Sigma) in the drinking water (n = 12 per group). After 8 wk of treatment, mice were euthanized by CO2 asphyxiation and necropsied. Mammary tumors were collected and snap frozen in liquid nitrogen.
Mechanical Testing.
The mechanical properties of mammary gland tumors from PyMT mice were assayed using unconfined compression on a Bose Enduratec 3200 (Bose) equipped with a 250 × g load cell with a load capacity of 5.11 N. Freshly isolated tumor samples were snap frozen and allowed to thaw only immediately before performing the mechanical testing (57, 58). The tissue sample was kept in PBS for 15 min to allow rehydration, after which a 3- or 4-mm cylindrical tissue section was excised using a biopsy punch (58). The tissue section height was measured and then subjected to a 3–15% strain in 3% increments (59). The elastic modulus was computed from the stress-strain relaxation curves using a poroviscoelastic model and a custom Matlab code (The Mathworks) (59, 60).
Cell Culture.
Bovine aortic endothelial cells (BAECs) and human umbilical vein endothelial cells (HUVECs) were maintained and plated as described previously (61, 62). BAECs were maintained in medium supplemented with 10% (vol/vol) FetalClone III (Fisher), and 1% each of penicillin–streptomycin, MEM amino acids (Invitrogen), and MEM vitamins (Mediatech). HUVECs were maintained and plated at 37 °C and 5% (vol/vol) CO2 in Medium 200 with 5% (vol/vol) FBS and 2% (vol/vol) low serum growth supplement (LSGS). For inhibitor studies, the cells were allowed to attach and spread for 2 h before adding the inhibitors at concentration of 10 μM for Y27632 or 2 μM for wortmannin. For the wortmannin treatment, an equivalent amount of DMSO vehicle was added to the controls.
Polyacrylamide Gel Synthesis and Stiffness Characterization.
Polyacrylamide (PA) gels were prepared and characterized as described previously (61, 63, 64). The range of substrate stiffness used (0.2–10 kPa) was chosen to mimic the stiffness of vascular and tumor tissue as described previously (63, 65, 66).
Fibronectin Fiber Quantification.
Evaluation of the fibronectin fiber lengths was performed using 40× confocal images as described previously (67). Briefly, 10 confocal images acquired in frozen sections from two different mice were analyzed using the Tubeness plugin in the ImageJ software. A threshold was applied to the resulting images, before fibronectin fragment length measurements.
Statistical Tests.
Parametric one-way or two-way ANOVAs with post hoc Tukey’s honest significance test were performed where appropriate. P < 0.05 was considered statistically significant.
All reagents and a detailed description of additional protocols used in this study, including Western blotting, immunofluorescence of fixed samples, immunohistochemistry and immunofluorescence on tissue sections, quantitative real-Time PCR, and ROCK knockdown experiments are described in SI Materials and Methods.
SI Materials and Methods
Reagents.
Primary antibodies used were as follows: mouse monoclonal anti-fibronectin (A17, ab26245) and mouse monoclonal anti-human EDB-fibronectin (BC-1, ab154210; Abcam); primary goat polyclonal anti-VE-cadherin (C19, sc-6458), rabbit polyclonal anti CD31 (sc-1506), rabbit polyclonal anti-ROCK (H-85, sc-5560), rabbit polyclonal anti PKC βII (C-18, sc-210), rabbit polyclonal anti-fibronectin (H300, sc-9068), and goat polyclonal anti-fibronectin (N-20, sc-6953; Santa Cruz); mouse monoclonal anti–EDB-fibronectin from Richard O. Hynes (Massachusetts Institute of Technology, Cambridge, MA); mouse monoclonal anti-pan phosphoepithope SR proteins (1H4, MABE50), and primary mouse monoclonal anti-GAPDH (MAB374; Millipore); and primary rabbit polyclonal anti-S451 AKT (D9E, #4060; Cell Signaling) and primary rabbit polyclonal anti-phospho AKT substrate (RXXS*/T*) (#9614; Cell Signaling). Secondary antibodies used were as follows: Alexa 488-donkey anti-mouse IgG, Alexa 594-goat anti-mouse IgG, Alexa-594-goat anti-rabbit IgG (Invitrogen) and HRP-goat anti-rabbit IgG and anti-mouse IgG (Rockland). FBS, LSGS, and Fetal Clone III (FCIII) were purchased from Gibco (Life Technologies); PI3K inhibitor wortmannin, myosin inhibitor blebbistatin, myosin light chain kinase inhibitor ML-7, and ROCK inhibitor Y27632 were purchased from Calbiochem. RhoA inhibitor C3 transferase (CT04) was purchased from Cytoskeleton. siRNA against ROCK1 (sc-29473) and control siRNA (sc-37007) were purchased from Santa Cruz. All other chemicals were from Sigma-Aldrich.
Western Blotting.
ECs were plated on 1- and 10-kPa PA gels and kept in culture for either 3 d or 24 h when inhibitors were used and rinsed with ice-cold PBS. Total proteins were extracted with preheated (90 °C) 2× sample buffer (52). For tumor samples, whole frozen tumors were ground using a liquid nitrogen-cooled mortar and pestle. The tumor was then collected with ice-cold RIPA buffer. The lysate was harvested, mixed, and spun at 14,000 × g at 4 °C, and the supernatant was removed for analysis. Laemmli buffer (1×) was added to the supernatant, and samples were heated for 5 min at 95 °C. Ten to 20 μg was subjected to SDS/PAGE with a Mini-PROTEAN Tetra System (BioRad) and electrotransferred onto a PVDF membrane. Membranes were blocked with 5% (wt/vol) milk (Nestlé) in Tris-buffered saline (TBS)-polyoxyethylene (20) sorbitan monolaurate (Tween; JT Baker) or 5% (wt/vol) BSA. Membranes were then incubated overnight 1:500 in TBS-Tween with an anti-fibronectin primary antibody [A17, H300, or BC1 for EDB-FN (Santa Cruz Biotechnology); or anti-EDB-FN (Hynes Laboratory) for mouse tumor samples] at 4 °C, and 1:2,000 in TBS-Tween with 0.1% milk with an HRP-conjugated secondary antibody for 1 h at room temperature. Samples were images with a Las-4000 imaging system (Fujifilm Life Science) after addition of Immobilon Chemiluminescent Substrate (Millipore). Samples were stripped with Restore Stripping Buffer (Thermo Scientific), reblocked with milk, incubated 1:1,000 in TBS-Tween with an anti-GAPDH primary antibody at room temperature for 1 h, incubated 1:2,000 in TBS-Tween with 0.1% milk with an HRP-conjugated secondary antibody for 45 min at room temperature, and reimaged. Densitometry was performed with ImageJ and expressed as the fold change of the ratio of the protein of interest to GAPDH or actin. Data were the result of three independent experiments.
Immunofluorescence of Fixed Samples.
Endothelial cells on PA gels were processed for fluorescence imaging as described previously (63) with anti-FN primary antibodies (A17 or BC1 for EDB-FN only, Santa Cruz Biotechnology). Fluorescent images were acquired with a Zeiss Axio Observer Z1m or a Zeiss 710 confocal microscope, and z-stacks were reconstructed with Zen software (v. 2009; Carl Zeiss).
Immunohistochemistry and Immunofluorescence on Tissue Sections.
Formalin-fixed, paraffin-embedded, invasive ductal carcinoma and patient-matched normal human breast tissue from New York Presbyterian Hospital-Weill Cornell Medical College was used for this study in accordance with protocols approved by the Institutional Review Board at Weill Cornell Medical College. For each tumor case, 4-μm-thick sections were deparaffinized in xylene and rehydrated in graded alcohols, and antigen retrieval was performed in a 10 mM citrate buffer (pH 6). Envision+System-HRP (DAB; Dako) was used for immunostaining according to the manufacturer’s protocol. The sections were incubated overnight at 4 °C with mouse anti-human EDB-FN (BC1; Abcam) at a 1:200 dilution. Primary antibody was omitted for negative control. Slides were incubated with labeled polymer-HRP anti-mouse secondary antibodies and visualization was performed using a substrate-DAB+Chromogen solution. Slides were counterstained with Harris hematoxylin.
Snap frozen mouse tumor samples were embedded within OCT compound and processed in a cryostat to obtain either 8- or 30-μm-thick sections. For EDB-FN staining, the sections were air dried and fixed for 5 min in cold acetone. The sections were then washed two times in PBS for 10 min and air dried. Otherwise, the tumor samples were immediately fixed after sectioning in 4% (wt/vol) paraformaldehyde for 10 min at room temperature. The sections were then washed two times in PBS as above and kept in PBS. Before the staining procedure, air-dried sections were rehydrated for 5 min in PBS. All sections were blocked for 1 h using the Vectashield mouse-block (Vector Laboratories). The sections were washed once with PBS containing 2% (vol/vol) FBS and 0.01% Tween × 100 for 5 min. The sections were incubated overnight at 4 °C with mouse anti-EDB-FN (Hynes Laboratory), with goat anti-FN and rabbit anti-CD31 or with mouse anti-pSR proteins and rabbit anti-CD31. Primary antibody was omitted for the negative control. Slides were incubated for 1 h at room temperature with Alexa488-donkey anti-mouse, Alexa594-donkey anti-rabbit, Alexa488-donkey anti-rabbit, or Alexa594-donkey anti-goat as needed. Slides were mounted in DAPI containing Vectashield mounting media. Fluorescent images were acquired with a Zeiss 710 confocal microscope.
Quantitative Real-Time PCR.
HUVECs were plated on 1- and 10-kPa PA gels, rinsed with PBS, and collected after the addition of 0.25% trypsin-EDTA (Gibco) for 10 min at 37 °C after 3 d. Cells were pelleted and rinsed with PBS, and total RNA was purified with an RNeasy Plus Mini Kit (Qiagen). To generate cDNA, 1 μg of total RNA per sample was mixed with 80 μM random primers (Invitrogen, Life Technologies), 10 mM deoxynucleotide solution mix (New England Biolabs), and nuclease-free water, and heated for 5 min at 75 °C. After the addition of 40 U/μL RNase inhibitor and 200 U/μL M-MuLV reverse transcriptase in M-MuLV reaction buffer (New England Biolabs), cDNA was synthesized in an iCycler thermal cycler (BioRad). cDNA was diluted 1:50 in deionized water, prepared for quantitative real-time PCR by the addition of 300 nM forward primer, 300 nM reverse primer (Integrated DNA Technologies), and 1× iQ SYBR Green Supermix (BioRad), and analyzed with a My iQ Real-Time PCR Detection System (BioRad). The primers used for EDB-FN were 5′-CCTGGAGTACAATGTCAGTG-3′ (forward) and 5′-GGTGGAGCCCAGGTGACA-3′ (reverse) (68). Primers for the reference gene GAPDH were 5′-CATGAGAAGTATGACAACAGCCT-3′ (forward) and 5′-AGTCCTTCCACGATACCAAAGT-3′ (reverse). The results were expressed as a fold change in mRNA expression of EDB-FN relative to GAPDH on 10-kPa gels compared with 1-kPa gels using the Livak method. Data were the results of three independent experiments.
ROCK Knockdown.
ROCK-mediated cellular contractility was disrupted by plating BAECs in the presence of 10 μM Y27632 (65). Samples were fixed at 4–48 h after plating and stained to localize FN, actin, and nuclei as described above. ROCK was also disrupted with RNAi. BAECs were treated with an siRNA against bovine ROCK (Invitrogen) or a control siRNA (Santa Cruz Biotechnology). To knock down ROCK, BAECs in six-well plates were incubated with 20 pmol siRNA in 5 μL Lipofectamine 2000 in Optimem for 5 h. Transfected cells were plated on PA substrates 24 h after transfection, and samples were fixed at 24-h time points over the next 3 d and stained as described above. Separate experiments determined the expression of ROCK with Western blotting over the same 3-d period as described above but using an anti-ROCK primary antibody. Similarly, HUVECs were treated as described above for BAECs with siRNA against human ROCK or a control siRNA (Santa Cruz Biotechnology) and stained to localize EDB-FN on the third day after plating as described above. Western blotting was performed at this same time point as described above but using an anti-ROCK primary antibody (H-85; Santa Cruz Biotechnology).
Supplementary Material
Acknowledgments
We thank Lawrence J. Bonassar for providing access to the Enduratec system and Brandon Borde for useful input and discussions. The anti–EDB-FN was a gift from Richard Hynes (Howard Hughes Medical Institute, Massachusetts Institute of Technology). This work was supported in part by National Institutes of Health (NIH) Award 1R01HL127499 (to C.A.R.-K.), National Science Foundation (NSF) Awards 1055502 and 1435755 (to C.A.R.-K.), NIH Award R01 CA163255 (to R.S.W.), an NSF Graduate Science, Technology, Engineering and Mathematics (STEM) fellowship (to J.P.C. and B.N.M.), the Morgan Family fellowship (to B.N.M.), and NIH Training Grant T32 GM008500 (to Y.L.N.A.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505421112/-/DCSupplemental.
References
- 1.Ghigna C, Valacca C, Biamonti G. Alternative splicing and tumor progression. Curr Genomics. 2008;9(8):556–570. doi: 10.2174/138920208786847971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33(46):5311–5318. doi: 10.1038/onc.2013.533. [DOI] [PubMed] [Google Scholar]
- 3.Irimia M, Blencowe BJ. Alternative splicing: Decoding an expansive regulatory layer. Curr Opin Cell Biol. 2012;24(3):323–332. doi: 10.1016/j.ceb.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Brinkman BM. Splice variants as cancer biomarkers. Clin Biochem. 2004;37(7):584–594. doi: 10.1016/j.clinbiochem.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 5.Long JC, Caceres JF. The SR protein family of splicing factors: Master regulators of gene expression. Biochem J. 2009;417(1):15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 6.Neri D, Bicknell R. Tumour vascular targeting. Nat Rev Cancer. 2005;5(6):436–446. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- 7.Castellani P, et al. Differentiation between high- and low-grade astrocytoma using a human recombinant antibody to the extra domain-B of fibronectin. Am J Pathol. 2002;161(5):1695–1700. doi: 10.1016/S0002-9440(10)64446-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zardi L, et al. Transformed human cells produce a new fibronectin isoform by preferential alternative splicing of a previously unobserved exon. EMBO J. 1987;6(8):2337–2342. doi: 10.1002/j.1460-2075.1987.tb02509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khan ZA, et al. EDB fibronectin and angiogenesis—A novel mechanistic pathway. Angiogenesis. 2005;8(3):183–196. doi: 10.1007/s10456-005-9017-6. [DOI] [PubMed] [Google Scholar]
- 10.Lim LP, Sharp PA. Alternative splicing of the fibronectin EIIIB exon depends on specific TGCATG repeats. Mol Cell Biol. 1998;18(7):3900–3906. doi: 10.1128/mcb.18.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han F, et al. Transforming growth factor-beta1 regulates fibronectin isoform expression and splicing factor SRp40 expression during ATDC5 chondrogenic maturation. Exp Cell Res. 2007;313(8):1518–1532. doi: 10.1016/j.yexcr.2007.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du K, Peng Y, Greenbaum LE, Haber BA, Taub R. HRS/SRp40-mediated inclusion of the fibronectin EIIIB exon, a possible cause of increased EIIIB expression in proliferating liver. Mol Cell Biol. 1997;17(7):4096–4104. doi: 10.1128/mcb.17.7.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saitoh N, et al. The distribution of phosphorylated SR proteins and alternative splicing are regulated by RANBP2. Mol Biol Cell. 2012;23(6):1115–1128. doi: 10.1091/mbc.E11-09-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel NA, et al. Molecular and genetic studies imply Akt-mediated signaling promotes protein kinase CbetaII alternative splicing via phosphorylation of serine/arginine-rich splicing factor SRp40. J Biol Chem. 2005;280(14):14302–14309. doi: 10.1074/jbc.M411485200. [DOI] [PubMed] [Google Scholar]
- 15.Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: Both hyper- and hypophosphorylation inhibit splicing. Mol Cell Biol. 1999;19(10):6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel NA, et al. Insulin regulates alternative splicing of protein kinase C beta II through a phosphatidylinositol 3-kinase-dependent pathway involving the nuclear serine/arginine-rich splicing factor, SRp40, in skeletal muscle cells. J Biol Chem. 2001;276(25):22648–22654. doi: 10.1074/jbc.M101260200. [DOI] [PubMed] [Google Scholar]
- 17.Mouw JK, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20(4):360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. 2012;23(5):781–791. doi: 10.1091/mbc.E11-06-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schrader J, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53(4):1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levental KR, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DuFort CC, Paszek MJ, Weaver VM. Balancing forces: Architectural control of mechanotransduction. Nat Rev Mol Cell Biol. 2011;12(5):308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim JH, Asthagiri AR. Matrix stiffening sensitizes epithelial cells to EGF and enables the loss of contact inhibition of proliferation. J Cell Sci. 2011;124(Pt 8):1280–1287. doi: 10.1242/jcs.078394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plodinec M, et al. The nanomechanical signature of breast cancer. Nat Nanotechnol. 2012;7(11):757–765. doi: 10.1038/nnano.2012.167. [DOI] [PubMed] [Google Scholar]
- 24.Pathak A, Kumar S. Transforming potential and matrix stiffness co-regulate confinement sensitivity of tumor cell migration. Integr Biol (Camb) 2013;5(8):1067–1075. doi: 10.1039/c3ib40017d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bordeleau F, Alcoser TA, Reinhart-King CA. Physical biology in cancer. 5. The rocky road of metastasis: The role of cytoskeletal mechanics in cell migratory response to 3D matrix topography. Am J Physiol Cell Physiol. 2014;306(2):C110–C120. doi: 10.1152/ajpcell.00283.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peyton SR, Putnam AJ. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J Cell Physiol. 2005;204(1):198–209. doi: 10.1002/jcp.20274. [DOI] [PubMed] [Google Scholar]
- 27.Paszek MJ, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Carraher CL, Schwarzbauer JE. Regulation of matrix assembly through rigidity-dependent fibronectin conformational changes. J Biol Chem. 2013;288(21):14805–14814. doi: 10.1074/jbc.M112.435271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubow KE, et al. Crosslinking of cell-derived 3D scaffolds up-regulates the stretching and unfolding of new extracellular matrix assembled by reseeded cells. Integr Biol (Camb) 2009;1(11-12):635–648. doi: 10.1039/b914996a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, et al. Centrosomal PKCbetaII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res. 2008;68(16):6831–6839. doi: 10.1158/0008-5472.CAN-07-6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nowak DG, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: A novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285(8):5532–5540. doi: 10.1074/jbc.M109.074930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huynh J, Bordeleau F, Kraning-Rush CM, Reinhart-King CA. Substrate stiffness regulates PDGF-induced circular dorsal ruffle formation through MLCK. Cell Mol Bioeng. 2013;6(2):138–147. doi: 10.1007/s12195-013-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer-ter-Vehn T, et al. Contractility as a prerequisite for TGF-beta-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci. 2006;47(11):4895–4904. doi: 10.1167/iovs.06-0118. [DOI] [PubMed] [Google Scholar]
- 34.Helfman DM, Pawlak G. Myosin light chain kinase and acto-myosin contractility modulate activation of the ERK cascade downstream of oncogenic Ras. J Cell Biochem. 2005;95(5):1069–1080. doi: 10.1002/jcb.20498. [DOI] [PubMed] [Google Scholar]
- 35.Phanish MK, et al. The regulation of TGFβ1 induced fibronectin EDA exon alternative splicing in human renal proximal tubule epithelial cells [published online ahead of print June 24, 2014] J Cell Physiol. 2014 doi: 10.1002/jcp.24703. [DOI] [PubMed] [Google Scholar]
- 36.Astrof S, et al. Direct test of potential roles of EIIIA and EIIIB alternatively spliced segments of fibronectin in physiological and tumor angiogenesis. Mol Cell Biol. 2004;24(19):8662–8670. doi: 10.1128/MCB.24.19.8662-8670.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coltrini D, et al. Impact of VEGF-dependent tumour micro-environment on EDB fibronectin expression by subcutaneous human tumour xenografts in nude mice. J Pathol. 2009;219(4):455–462. doi: 10.1002/path.2626. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto-Uoshima M, Yan YZ, Schneider G, Aukhil I. The alternatively spliced domains EIIIB and EIIIA of human fibronectin affect cell adhesion and spreading. J Cell Sci. 1997;110(Pt 18):2271–2280. doi: 10.1242/jcs.110.18.2271. [DOI] [PubMed] [Google Scholar]
- 39.White ES, Muro AF. Fibronectin splice variants: Understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life. 2011;63(7):538–546. doi: 10.1002/iub.493. [DOI] [PubMed] [Google Scholar]
- 40.Nowak DG, et al. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci. 2008;121(Pt 20):3487–3495. doi: 10.1242/jcs.016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper DR, et al. Long non-coding RNA NEAT1 associates with SRp40 to temporally regulate PPARγ2 splicing during adipogenesis in 3T3-L1 cells. Genes (Basel) 2014;5(4):1050–1063. doi: 10.3390/genes5041050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wee CD, Havens MA, Jodelka FM, Hastings ML. Targeting SR proteins improves SMN expression in spinal muscular atrophy cells. PLoS ONE. 2014;9(12):e115205. doi: 10.1371/journal.pone.0115205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan XB, et al. Alternative splicing in exon 9 of glucocorticoid receptor pre-mRNA is regulated by SRp40. Mol Biol Rep. 2010;37(3):1427–1433. doi: 10.1007/s11033-009-9529-z. [DOI] [PubMed] [Google Scholar]
- 44.Umesh V, Rape AD, Ulrich TA, Kumar S. Microenvironmental stiffness enhances glioma cell proliferation by stimulating epidermal growth factor receptor signaling. PLoS ONE. 2014;9(7):e101771. doi: 10.1371/journal.pone.0101771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scarpino S, et al. Expression of EDA/EDB isoforms of fibronectin in papillary carcinoma of the thyroid. J Pathol. 1999;188(2):163–167. doi: 10.1002/(SICI)1096-9896(199906)188:2<163::AID-PATH335>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 46.Hauptmann S, et al. Extracellular matrix proteins in colorectal carcinomas. Expression of tenascin and fibronectin isoforms. Lab Invest. 1995;73(2):172–182. [PubMed] [Google Scholar]
- 47.Castellani P, et al. The fibronectin isoform containing the ED-B oncofetal domain: A marker of angiogenesis. Int J Cancer. 1994;59(5):612–618. doi: 10.1002/ijc.2910590507. [DOI] [PubMed] [Google Scholar]
- 48.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 49.Dales JP, et al. Hypoxia inducible factor 1alpha gene (HIF-1alpha) splice variants: Potential prognostic biomarkers in breast cancer. BMC Med. 2010;8:44. doi: 10.1186/1741-7015-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogel V, Sheetz MP. Cell fate regulation by coupling mechanical cycles to biochemical signaling pathways. Curr Opin Cell Biol. 2009;21(1):38–46. doi: 10.1016/j.ceb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng Y, Lai T, Koh CG, LeDuc PR, Chiam KH. Investigating circular dorsal ruffles through varying substrate stiffness and mathematical modeling. Biophys J. 2011;101(9):2122–2130. doi: 10.1016/j.bpj.2011.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bordeleau F, Galarneau L, Gilbert S, Loranger A, Marceau N. Keratin 8/18 modulation of protein kinase C-mediated integrin-dependent adhesion and migration of liver epithelial cells. Mol Biol Cell. 2010;21(10):1698–1713. doi: 10.1091/mbc.E09-05-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larsson C. Protein kinase C and the regulation of the actin cytoskeleton. Cell Signal. 2006;18(3):276–284. doi: 10.1016/j.cellsig.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 54.Ng T, et al. PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 1999;18(14):3909–3923. doi: 10.1093/emboj/18.14.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oh MA, et al. PKCdelta and cofilin activation affects peripheral actin reorganization and cell-cell contact in cells expressing integrin alpha5 but not its tailless mutant. J Cell Sci. 2007;120(Pt 15):2717–2730. doi: 10.1242/jcs.003566. [DOI] [PubMed] [Google Scholar]
- 56.Zhao W, Li X, Liu X, Zhang N, Wen X. Effects of substrate stiffness on adipogenic and osteogenic differentiation of human mesenchymal stem cells. Mater Sci Eng C Mater Biol Appl. 2014;40:316–323. doi: 10.1016/j.msec.2014.03.048. [DOI] [PubMed] [Google Scholar]
- 57.Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol (Camb) 2011;3(9):910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang SC, et al. Injection molding of chondrocyte/alginate constructs in the shape of facial implants. J Biomed Mater Res. 2001;55(4):503–511. doi: 10.1002/1097-4636(20010615)55:4<503::aid-jbm1043>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 59.Gleghorn JP, Jones AR, Flannery CR, Bonassar LJ. Boundary mode frictional properties of engineered cartilaginous tissues. Eur Cell Mater. 2007;14:20–28, discussion 28–29. doi: 10.22203/ecm.v014a02. [DOI] [PubMed] [Google Scholar]
- 60.Bonnevie ED, Puetzer JL, Bonassar LJ. Enhanced boundary lubrication properties of engineered menisci by lubricin localization with insulin-like growth factor I treatment. J Biomech. 2014;47(9):2183–2188. doi: 10.1016/j.jbiomech.2013.10.028. [DOI] [PubMed] [Google Scholar]
- 61.Califano JP, Reinhart-King CA. Substrate stiffness and cell area drive cellular traction stresses in single cells and cells in contact. Cell Mol Bioeng. 2010;3(1):68–75. doi: 10.1007/s12195-010-0102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular traction stresses increase with increasing metastatic potential. PLoS ONE. 2012;7(2):e32572. doi: 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Califano JP, Reinhart-King CA. A balance of substrate mechanics and matrix chemistry regulates endothelial cell network assembly. Cell Molec Bioengineer. 2008;1(2-3):122–132. [Google Scholar]
- 64.Reinhart-King CA, Dembo M, Hammer DA. Endothelial cell traction forces on RGD-derivatized polyacrylamide substrata. Langmuir. 2003;19(5):1573–1579. [Google Scholar]
- 65.Huynh J, et al. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med. 2011;3(112):112ra122. doi: 10.1126/scitranslmed.3002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peloquin J, Huynh J, Williams RM, Reinhart-King CA. Indentation measurements of the subendothelial matrix in bovine carotid arteries. J Biomech. 2011;44(5):815–821. doi: 10.1016/j.jbiomech.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 67.Bordeleau F, Myrand Lapierre M-E, Sheng Y, Marceau N. Keratin 8/18 regulation of cell stiffness-extracellular matrix interplay through modulation of Rho-mediated actin cytoskeleton dynamics. PLoS ONE. 2012;7(6):e38780. doi: 10.1371/journal.pone.0038780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cseh B, et al. Autocrine fibronectin directs matrix assembly and crosstalk between cell-matrix and cell-cell adhesion in vascular endothelial cells. J Cell Sci. 2010;123(Pt 22):3989–3999. doi: 10.1242/jcs.073346. [DOI] [PubMed] [Google Scholar]