

Abstract
Biotechnological applications of enzymes can involve the use of these molecules under nonphysiological conditions. Thus, it is of interest to understand how environmental variables affect protein structure and dynamics and how this ultimately modulates enzyme function. NADH oxidase (NOX) from Thermus thermophilus exemplifies how enzyme activity can be tuned by reaction conditions, such as temperature, cofactor substitution, and the addition of cosolutes. This enzyme catalyzes the oxidation of reduced NAD(P)H to NAD(P)+ with the concurrent reduction of O2 to H2O2, with relevance to biosensing applications. It is thermophilic, with an optimum temperature of approximately 65°C and sevenfold lower activity at 25°C. Moderate concentrations (≈1M) of urea and other chaotropes increase NOX activity by up to a factor of 2.5 at room temperature. Furthermore, it is a flavoprotein that accepts either FMN or the much larger FAD as cofactor. We have used nuclear magnetic resonance (NMR) titration and 15N spin relaxation experiments together with isothermal titration calorimetry to study how NOX structure and dynamics are affected by changes in temperature, the addition of urea and the substitution of the FMN cofactor with FAD. The majority of signals from NOX are quite insensitive to changes in temperature, cosolute addition, and cofactor substitution. However, a small cluster of residues surrounding the active site shows significant changes. These residues are implicated in coupling changes in the solution conditions of the enzyme to changes in catalytic activity.
Keywords: nuclear magnetic resonance, spin relaxation, NMR titration, isothermal titration calorimetry, thermophile, urea activation, enzyme cofactor substitution
Introduction
Enzymes have many industrial applications, including biocatalysis, bioremediation, and biosensing.1–6 In many instances, this involves employing enzymes under conditions that are quite different from those found physiologically in terms of temperature and cosolutes. An important challenge in the rational use and optimization of enzymes in biotechnology is to understand at a molecular level how changes in an enzyme's environment affect its structure and dynamics and how this governs catalysis. For example, moderate concentrations of denaturants such as urea or chaotropic anions in the Hofmeister series increase the catalytic activity of some enzymes.7–11 This effect has been linked to changes in the structure of the active site7 and modulation of protein dynamics,8,11 yet many questions remain regarding the molecular mechanisms underlying this phenomenon. Conversely, thermophilic enzymes generally have lower activity than mesophilic ones at ambient temperatures, and only regain activity as the temperature is raised.12 Nevertheless thermophilic enzymes are associated with several practical advantages; they can perform at higher temperatures, are less prone to microbial contamination, have lower viscosity, and heat treatment simplifies purification.13,14 Furthermore, the substitution of one cofactor for another and the engineering of non-natural cofactors can improve the cost efficiency of biocatalysis and expand the repertoire of available enzymatic reactions.15–17 Thus, a better understanding of active site adaptation to cofactor substitution and thermal and denaturant activation of enzymes is desirable in order to elucidate design principles for improved biocatalysts.
NADH oxidase (NOX) from the thermophilic bacterium Thermus thermophilus is a dimer of 27 kDa monomers that catalyzes the oxidation of reduced nicotinamide adenine dinucleotide (NAD(P)H) with the concurrent reduction of oxygen to hydrogen peroxide in a two electron transfer mechanism. It is functional with either FMN or FAD cofactors bound in a 1:1 monomer:cofactor ratio.18,19 FAD is significantly larger than FMN, as illustrated in Figure 1, differing by the addition of an adenosine monophosphate group. The physiological role of NOX is still not well understood although some homologous NOX enzymes may be engaged in the defense against oxidative stress.20 NOX has high thermal and chemical stability, relatively low molecular mass, broad pH activity, and the ability to accept NADH and NADPH as substrates, making it attractive for biosensor applications.21 In addition, it belongs to a protein family that performs valuable chemical modifications of prodrugs22 and explosive contaminants such as 2,4,6 trinitrotoluene (TNT).6 NOX is most active at approximately 65°C, near the physiological temperature of T. thermophilus, and exhibits approximately sevenfold lower activity at 20°C.11 The activity of NOX is enhanced in the presence of low concentrations of denaturants, such as urea and chaotropic anions of the Hofmeister series.19 Conversely, NOX activity is diminished by kosomotropic agents that promote hydrophobic interactions. For example, the maximum turnover rate is about 2.5-fold higher with the addition of 1M urea and threefold lower with 1M Na2SO4 at 20°C.11
Figure 1.
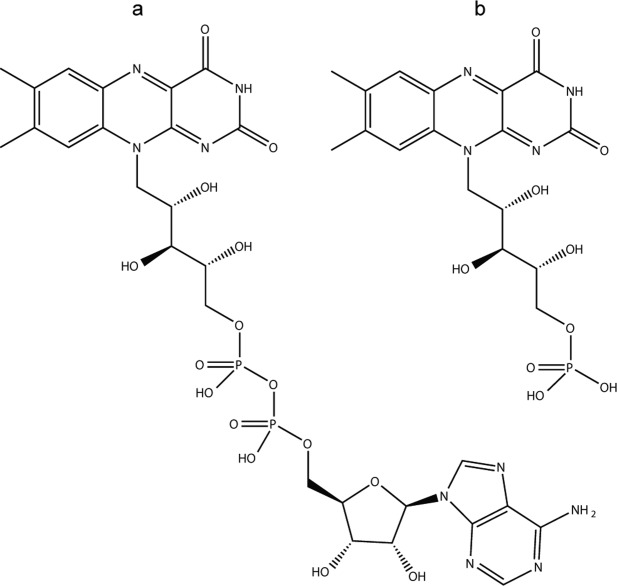
NOX cofactor structures. flavin adenine dinucleotide (FAD) (a) and flavin mononucleotide (FMN) (b).
X-ray crystallographic structures have been determined for NOX in complex with both FMN and FAD21 however there are no structural data for the apo form of the enzyme. Previous studies have also examined the temperature and denaturation activation of NOX by molecular dynamics simulations and fluorescence spectroscopy,11,23–25 but high-resolution experimental information is lacking. We have investigated the effects of temperature and urea on NOX as well as its interactions with FMN and FAD at atomic resolution using a combination of nuclear magnetic resonance (NMR) titration experiments, 15N spin relaxation NMR experiments, and isothermal titration calorimetry (ITC). We find that FMN binds NOX approximately 1250-fold more tightly than FAD, suggesting that FMN is the preferred cofactor. The NMR spectrum of the apoenzyme is quite similar to those of the FMN-bound and FAD-bound holo forms, suggesting that the enzyme retains its fold in the absence of cofactors. The 15N spin relaxation measurements report the amplitudes of NH bond vector motions on the ns to ps timescale. We find that the addition of 1.2M urea at 50°C results in some redistribution of flexibility, including several residues in the active site. However, the overall level of dynamics remains relatively constant for the protein as a whole. Series of 15N/1H NMR correlation experiments were collected as the temperature and concentrations of FAD, FMN, and urea were varied. These measurements identify the regions of the protein whose conformations and interactions are most affected by changing these environmental variables. Interestingly, most NMR signals from NOX are relatively insensitive to these perturbations. However, the peaks from a few small clusters of residues surrounding the active site shift significantly in response to multiple environmental factors. These structurally malleable sites of the protein are likely involved in the temperature and/or denaturant activation of the enzyme, as well as its ability to accommodate different cofactors. Interestingly, the side chain indole 15N/1H signal of W47 shows among the largest peak movements in response to urea addition and cofactor substitution. This side chain has been previously suggested to play a key role in gating substrate access to the active site and mediating denaturant activation of the enzyme.23
Results
Tryptophan side chain assignments
Previously, we assigned 165 of the 190 backbone peaks expected in 1H–15N HSQC correlation spectra for nonproline residues of NOX, using standard triple-resonance experiments.26 For this study, we had particular interested in the NH signals from the indole side chains of the tryptophan residues in NOX, since the side chain of W47 is believed to play a key role in catalysis.11,23 NOX contains four tryptophans, two buried, W47 and W52, and two solvent exposed, W131 and W204. Only three 1H–15N signals are observed in the characteristic tryptophan indole region of 1H–15N correlation spectra obtained for NOX (downfield in both 1H and 15N). Two of these were tentatively assigned to the side chains of W131 and W204 based on NOE correlations between indole and backbone protons, but the remaining indole peak was unassigned. In order to confirm these two assignments and to determine the identity of the third indole peak, we employed site-directed mutagenesis. The conservative Trp to Phe substitution was made for each tryptophan residue (W47F, W52F, W131F, W204F). Indole regions of 1H–15N HSQC correlation spectra of the apo forms of the WT and three of the mutants are shown in Figure 2. The Phe side chain is aromatic and has similar size to that to that of Trp but produces no 1H/15N correlation peak, therefore the expected outcome of a Trp to Phe mutation is the disappearance of a single indole resonance from the 1H/15N correlation spectrum with minimal additional spectral perturbation. Indeed, one of the three wild-type Trp NH indole side chain peaks is clearly absent in each of the HSQC spectra collected for the W47F, W131F, W204F mutants. These results agreed with our tentative assignments for the W204 and W131 side chains and indicated that the third indole peak arises from the W47 side chain. Since the backbone signal for W47 is absent, this assignment is consistent with the fact that no NOE connectivity was found between this third side chain peak and a corresponding backbone signal. In the case of the W52F mutant, the HSQC of the apo form resembles that of an unstructured protein,27 with many proton shifts clustered around 8.5 ppm. (Supporting Information Fig. S4). This is consistent with the observation that W52F has diminished flavin binding affinity.28 W52 is deeply embedded in NOX structure at the beginning of the first β-strand of the four-stranded antiparallel β-sheet located in the core of the protein and adjacent to R53, which is involved in an intermolecular salt-bridge network according to molecular dynamics (MD) simulations.23 These interactions provide some rationalization for the intolerance of the protein to substitutions at this position.
Figure 2.
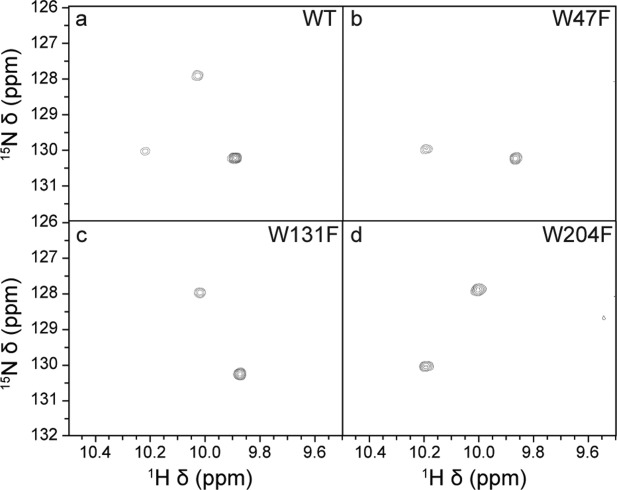
Indole side-chain regions of NOX 1H/15N NMR spectra. Wild-type NOX (a), and the mutants W47F (b), W131F (c), and W204F (d) recorded at 50°C and 18.8 T (800 MHz 1H. Larmor frequency) in the absence of cofactor.
FMN and FAD binding
NOX accepts either flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD), which bind in a 1:1 cofactor:monomer ratio.29 The structural difference between these two molecules is the addition of an adenosine monophosphate group in FAD (Fig. 1). We characterized the binding of FMN and FAD to NOX using isothermal titration calorimetry (ITC), which yields equilibrium dissociation constants, KD, binding enthalpies, ΔH, and binding entropies, ΔS, of the interaction. The ITC data are well fit by the one-set-of-sites binding model (Fig. 3), which implies that cofactors bind to the two subunits of NOX essentially independently. FMN binds NOX much more tightly than does FAD, with KD values of 0.4 ± 0.2 μM and 0.54 ± 0.03 mM for FMN and FAD, respectively. These results support the idea that FMN is the preferred, or physiological, NOX cofactor.21 Interestingly, although FAD binds much less tightly than does FMN, the reaction enthalpies for the two ligands are very similar with ΔH values of –10.1 ± 0.4 and –9.4 ± 0.7 kcal/mol for FAD and FMN, respectively. The weaker binding in the case of FAD is due to a strongly unfavourable entropy term, ΔS = −16 ± 1 cal mol−1 K−1, compared to a negligible binding entropy for FMN, ΔS = 0.2 ± 3 cal/(mol K).
Figure 3.
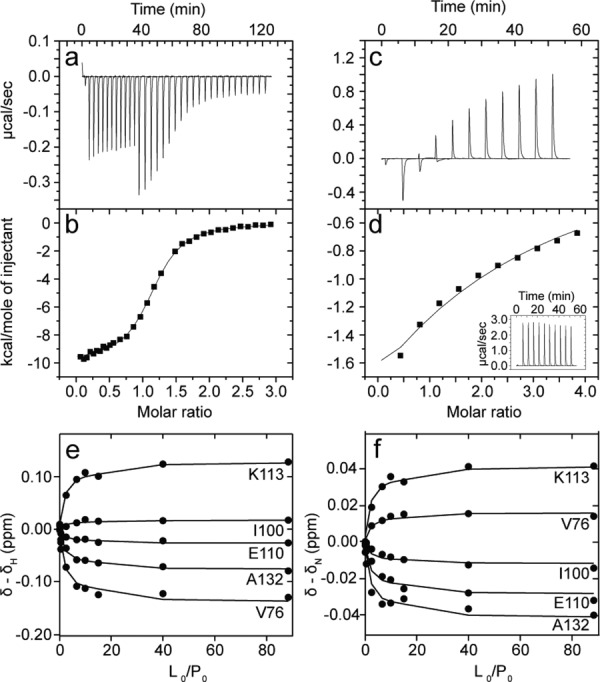
NOX cofactor titrations with FMN (a, b) and FAD (c–f). Raw differential power ITC isotherms for NOX with FMN (a) and FAD (c). Integrated ITC injection heats plotted as a function of the cofactor:monomer molar ratio for FMN (b) and FAD (d). Data were fit to a 1 set of identical sites binding model. FAD injections into buffer alone are shown in the inset to panel (e). NOX 1H (e) and 15N (f) chemical shift displacements plotted as a function of the ligand:protein molar ratio. Lines correspond to fits according to Eq. 2.
We also studied the interaction of the cofactors with NOX by performing NMR titrations, collecting series of 1H/15N HSQC protein spectra as FMN or FAD was incrementally added to the sample. Many peaks shifted gradually as cofactors were added and exhibited some broadening at intermediate points in the titration, characteristic of binding kinetics that are on the intermediate to fast timescale regimes. Under these conditions, the shifts in peak position as a function of ligand concentration can be analyzed to yield binding constants.30 In the case of FMN, the affinity is too high to reliably extract a binding constant from the NMR titration data, as the protein concentrations required for NMR are much greater than the KD. In the case of FAD, the shifts in peak position were analyzed to yield an estimate of the equilibrium binding constant, KD = 0.6 ± 0.3 mM, that matches the ITC-derived value well, as shown in Figure 3.
Interestingly several peaks that are present in the spectra of NOX bound to either FMN or FAD are missing in the absence of cofactors. These peaks reappear gradually as cofactor is added. The affected residues cluster around the active site and include residues A46, and Q105 on the re face and G72, A74, and G159 on the si face of the cofactor flavin ring as well as S193, H194, and L196 in the C-terminal tail of NOX which is also involved in cofactor binding [Fig. 4(a)]. There are two likely causes of these peak disappearances. Firstly, the NMR experiments were performed at 50°C and pH 7.2, in order to match previous studies of NOX catalysis.11 Under these conditions, an unprotected amide hydrogen is expected to exchange with those of water with a rate on the order of 300 s−1, and is therefore not expected to be detected in 1H NMR experiments.31,32 Amide groups that are strongly hydrogen bonded or buried deeply in the protein interior are typically protected from solvent exchange, with rates reduced by factors on the order of 104 to 108.33 Even in unfolded proteins, hydrogen exchange can occur more than tenfold more slowly for amides whose surroundings partially exclude water molecules, for instance due to hydrophobic collapse.33,34 Removal of the cofactor could enhance the hydrogen exchange rates of the residues listed above, leading to their disappearance from NMR spectra. Notably the amide hydrogens of A46 and G72 participate in intramolecular hydrogen bonds in the bound form, according to the X-ray crystal structure.21 Enhanced hydrogen exchange rates for these residues would imply that removing the cofactor results in conformational changes that weaken or break these intramolecular hydrogen bonds. For the others, removal of the cofactor could lead to greater access to water molecules. Alternatively, the disappearance of some or all of these eight resonances could be due to dynamical broadening caused by ms-μs timescale dynamics that are present in the NOX active site when cofactor is absent and are quenched when cofactor is present. In either case, A46, G72, and possibly other residues experience a change in conformation and/or flexibility when cofactor is removed.
Figure 4.
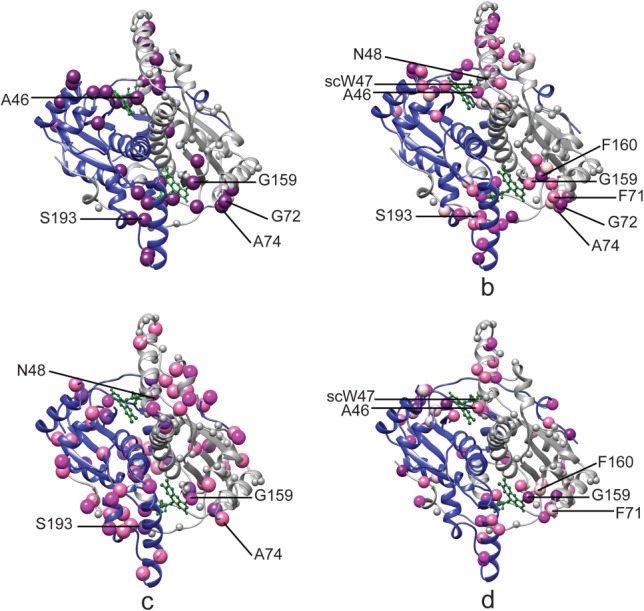
NOX chemical shift perturbations by environmental variables. Backbone ribbon diagrams of NOX21 (PDB 1NOX), with the two subunits colored in blue and white and the FMN cofactor illustrated in green balls and sticks. In (a), dark purple spheres correspond to residues whose 1H/15N peaks are missing in spectra of apo NOX but not in the presence of FMN or FAD. In (b–d) spheres correspond to residues with larger than average 1H/15N chemical shift perturbations (ppm) in response to changing environmental variables: FMN vs. FAD [>0.25 dark purple; 0.17–0.25 purple; 0.11–0.17 dark pink; 0.10–0.11 light pink] (b) 25–50°C [|∂δN/∂T| > 17 ppb/K dark pink; ∂δH/∂T < −4.5 ppb/K light pink; both [|∂δN/∂T| > 17 ppb/K, and ∂δH/∂T < −4.5 ppb/K dark purple] (c) and 0 vs. 2M urea [>0.55 dark purple; 0.45–0.45 purple; 0.35–0.45 dark pink; 0.32–0.35 light pink] (d). Small white spheres on the white monomer indicate the locations of residues lacking crosspeaks in 1H/15N correlation NMR spectra. Malleable residues listed in Table2 are labeled on the structures. Image generated with UCSF chimera.69
We then compared the HSQC spectra of NOX saturated with FMN and FAD (Supporting Information Fig. S5) in order to pinpoint regions of the protein that are most affected by cofactor substitution. The 1H and 15N chemical shift differences between corresponding peaks in the two spectra were used to calculate Δδ, where
| 1 |
The positions of most peaks do not change significantly with the substitution of FAD for FMN. The average Δδ value is only 0.082 ppm with a standard deviation of 0.009 ppm. Some of the largest shifts are exhibited by residues G190, Y191, and S193 with Δδ = 0.56, 0.20, and 0.20 ppm, respectively [Fig. 4(b)]. A second cluster of residues consisting of R109, E110, Q112, and K113, in helix F, and Q105, in the loop preceding helix F also have high Δδ values (0.19, 0.18, 0.24, and 0.12 ppm, respectively). Both of these groups of residues abut the additional adenine that distinguishes FAD from FMN cofactor (Supporting Information Fig. S9). Ring currents in the aromatic adenine are expected to alter the chemical shifts of nearby 1H nuclei in an orientation-specific manner.35 Therefore, these changes in peak position likely reflect the formation of new contacts with the larger FAD cofactor and/or conformational rearrangements. Signals for A46 and N48 and the W47 side chain (scW47) also exhibit large backbone chemical shift variations with Δδ values of 0.20, 0.19, and 0.17 ppm, respectively, as do A70, F71, G72, Q73, A74, and G159, F160, D161. These clusters of residues surround the flavin ring of the cofactor, whose position is conserved in the FMN- and FAD-containing structures (Supporting Information Fig. S9). These changes in chemical shift therefore are likely to reflect rearrangement of active site rather than the formation of new protein-cofactor contacts.
Temperature variation
In order to gain some insight into how raising the temperature increases the activity of NOX, we have examined temperature-dependent changes in spectra of the protein. 1H–15N HSQC correlation spectra were recorded for FMN-saturated NOX at temperatures ranging from 25 to 50°C in 5°C intervals [Fig. 5(a)]. Both 1H and 15N chemical shifts exhibited linear dependences on temperature [Fig. 5(a)]. The temperature coefficients (slopes of δ vs. T plots) range from −13.85 to 0.66 ppb/K for 1H and −41.20 to 19.07 ppb/K for 15N with means values of −2.86 and −5.48 ppb/K respectively. No significant correlation is evident between the 1H and 15N temperature coefficients of the same peaks (r = 0.18). Amide groups involved in stable secondary structure typically have proton temperature coefficients less negative than −4.5 ppb/K. Amides with 1H temperature factors more negative than −4.5 ppb/K are most likely not hydrogen bonded36 and are indicated with black bars in Figure 5(d). As expected, most residues with 1H temperature factors less than –4.5 ppb/K lie in loops and at the N- and C-termini of the protein. However, several residues with quite negative 1H temperature factors are located within elements of secondary structure, for instance in helices D, E, F, and G. One of these, A74, lies in the active site, on si side of the cofactor flavin ring. In these cases, the highly negative coefficients likely indicate some degree of temperature-dependent structural rearrangement. Interestingly, at pH 7.2 and 50°C, unprotected amide hydrogens are expected to exchange with those of water sufficiently rapidly (≈300 s−1) so that their NMR signals are predicted to be extremely weak or invisible.31,32 The fact that many of the amide 15N/1H groups lack hydrogen bonds in the X-ray crystal structure and yet produce strong signals in NMR spectra suggests that they are sterically protected from solvent hydrogen exchange by their surroundings in the molecule.33,34
Figure 5.
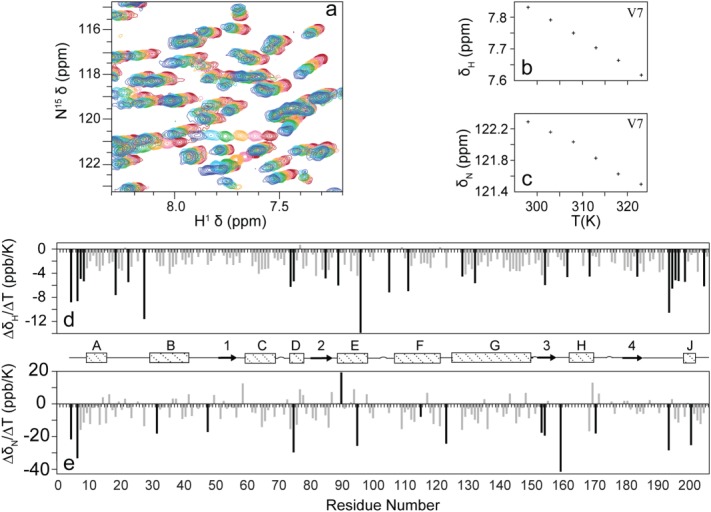
Effect of temperature on NOX NMR spectra. Overlay of subregion of NOX 1H–15N HSQC correlation spectra obtained over temperatures spanning 25°C (blue) to 50°C (red) (a) representative temperature-dependent changes in 1H (b) and 15N (c) chemical shifts obtained for the backbone amide resonance of Val 7. Temperature coefficients for 1H (d) and 15N (e) plotted as a function of residue number. 1H temperature coefficients more negative than −4.5 ppb/K and 15N temperature coefficients greater in magnitude than 177 ppb/K are indicated by black bars. Secondary structure elements21 were taken as αA (10–16), αB (30–42), β1 (52–57), αC (60–69), αD (74–78), β2 (81–87), αE (89–98), αF (107–121), αG (125–149), β3 (153–157), αH (162–169), β4 (179–184), αJ (198–201).
In addition, we have used the temperature dependence of 15N chemical shifts reported for ubiquitin37 as an indication of what is to be expected for a stably folded protein. For ubiquitin, all 15N temperature factors are smaller in magnitude than 17 ppb/K. Many 15N temperature factors in NOX exceed this value [black bars in Fig. 5(e)], which likely indicates that these residues experience temperature-dependent conformational changes. Interestingly, about half of these lie near the ends of elements of secondary structure or at the ends of loops, closely following or preceding secondary structure. Studies of β-turn and coiled-coil α-helical peptides have found that termini of the structured regions partially unfold or fray as the temperature is raised.38,39 Thus, it is possible that the elevated 15N temperature coefficients for residues near the ends of secondary structure regions similarly correspond to some degree of fraying. Several peaks in the active site show unusually large 15N temperature coefficients. For example, G159, on the si face of the cofactor flavin ring and S193, which is located in the C-terminal region of the protein and interacts directly with the FMN phosphate have a large negative 15N temperature coefficients of –41.2 and –28.3 ppb/K, indicative of some degree of conformational adjustment in the active site as the temperature is raised.
Urea titrations
It was previously shown that adding low concentrations of denaturants increases NOX activity. This is believed to result from local conformational changes and/or increased dynamics in the active site.19 NMR titrations were carried out to explore the impact of urea on NOX at both 20 and 50°C. The extent of urea activation is much greater at 20°C than at 50°C. For example, the maximum catalytic rate increases approximately 2.5-fold with the addition of 1M urea at 20°C, while the increase at 50°C is roughly 5%.11 However, the spectral properties of NOX are far more favourable at 50°C, as described below. At both 20 and 50°C, 1H–15N HSQC spectra were collected for FMN-saturated NOX with increasing concentrations of urea up to 2M. In both cases, peaks throughout the spectra shift with increasing urea concentrations. However, there are distinct differences between the two datasets. At 50°C, the spectra remain sharp and well-resolved throughout the titration series. In contrast at 20°C, there was pronounced broadening and weakening of signals, with approximately 15% of peaks disappearing by 1.2M urea and the majority of signals gone or unresolvable by 2M urea (Supporting Information Fig. S6). This is unlikely to be due to increasing tumbling correlation time as the viscosity of 1.2 molar urea is only 5% greater than that of pure water.40 It is also unlikely to be due to global unfolding of the protein, as the enzyme is equally active at 0 and 4M urea,11 and global unfolding would be expected to be more prevalent at the higher temperature. Also unlikely is increased exchange of amide hydrogens with those of solvent, as the intrinsic rate of exchange is expected to be roughly 100-fold slower at 20°C compared to 50°C.31 Furthermore, it is not likely to be caused by protein aggregation, as denaturants typically reduce rather than increase protein self-association.41 The most likely explanation, therefore, is that the addition of low concentrations of urea induces pervasive ms–μs timescale dynamics throughout the enzyme. These motions are either absent at 50°C, or the timescale has been shifted to a faster regime in which spectral broadening is no longer evident.
Comparing spectra obtained with 0 and 2M urea at 50°C, Δδ values range from 0.028 ppm for A132 to 1.74 ppm for scW47, with a mean of 0.3 ppm and a standard deviation of 0.3 ppm. 75% of peaks shift by less than 0.30 ppm, so any Δδ value greater than 0.30 can be considered a relatively large shift. These peak movements may reflect either urea-induced conformational rearrangements of the enzyme or direct interactions between the protein and urea molecules. As urea is not aromatic, it will not produce long-range ring-current effects per se. Only nuclei directly interacting with urea molecules, for instance by hydrogen bonding, or those that experience changes in local protein conformation are expected to exhibit chemical shift perturbations. According to the X-ray crystal structure, 25 of the 33 15N/1H pairs with Δδ > 0.3 are physically occluded from interacting directly with urea (see “Methods”). Thus, chemical shift perturbations for these residues probably reflect protein conformational changes rather than direct binding of urea, assuming that the steric accessibility is similar in the solution and crystal forms of the enzyme. The large Δδ value for the side chain of W47 is notable as it has previously been implicated in urea-modulated gating of the active site. Many of the residues exhibiting the largest changes in chemical shift are located near the active site of the protein [Fig. 4(d)]. For instance, in helix F, which is also implicated in gating substrate access to the active site, E110, K113, A115, Q117, and R118 exhibit Δδ values of 0.49, 0.41, 0.36, 0.52, and 0.38 ppm, respectively. In addition, residues L158, G159, F160 which are located at the upper edge of the cofactor exhibit large Δδ values of 0.36, 1.60, and 0.37 ppm.
The peak shifts observed at 50°C are largely recapitulated at 20°C, although pervasive broadening is the most pronounced spectral change with urea addition at the lower temperature. Of the 37 peaks that shift furthest with the addition of urea at 50°C, 12 are absent from the spectrum at 20°C and 0 M urea. A further 4 disappear at 1.2 M urea. 17 are within the top 25% of Δδ values obtained at 20°C. Notably, only 4 out of 37 peaks with the largest shifts at 50°C are present at 20°C and do not show large urea-dependent changes.
Fast timescale NOX dynamics with 2M urea
We investigated the effects of urea on the ns–ps timescale motions of NOX using NMR 15N spin relaxation experiments. We recorded 15N R1, R1ρ, and steady-state {1H}–15N NOE NMR measurements on an FMN-saturated NOX sample at 50°C in the presence of 1.2M urea. The relaxation data were analyzed according to the Lipari–Szabo model-free formalism42 which yields an order parameter, S2, for each residue. The order parameters reflect the mobility of individual NH bond vectors and can take values between 0 and 1. NH bond vectors that are completely rigid in the molecular frame lead to S2 values of 1, while bond vectors that undergo completely unrestricted dynamics on the ns–ps timescale in the molecular frame lead to S2 values of 0. Values of S2 obtained in the presence of 1.2M urea were compared with those previously obtained in absence of urea (Fig. 6).26 Overall, the same trends in flexibility are observed in presence and absence of urea. The more flexible regions of the protein include residues V28, located in a loop connecting helices A and B, a cluster of residues, E188, G190, Y191, and S193, located in a long loop that connects the fourth β-strand and the short C-terminal helix J and forms part of the cofactor binding site, and W204, located at the extreme C-terminus of the protein. The root-mean-squared deviation between the two datasets is just 0.036. Nevertheless, some redistribution of flexibility is apparent when NOX is in presence of urea. Among others, NH bonds of residues A40, G141, G184 experience more mobility (ΔS2 = –0.12 ± 0.05, –0.09 ± 0.07, –0.17 ± 0.08) while those of I100 and A121 are more rigid (ΔS2 = 0.10 ± 0.04, 0.11 ± 0.05) with the addition of urea. Although the urea-induced changes in fast-timescale dynamics are limited to a small number of residues, it is it noteworthy that I100 and A121 lie in the active-site cleft of the enzyme, and their rigidification could be related to urea-induced changes in activity occurring there. The more distal residues are less likely to be directly involved, although in special cases, changes in fast-timescale dynamics can have long-range effects on enzyme catalysis.43
Figure 6.
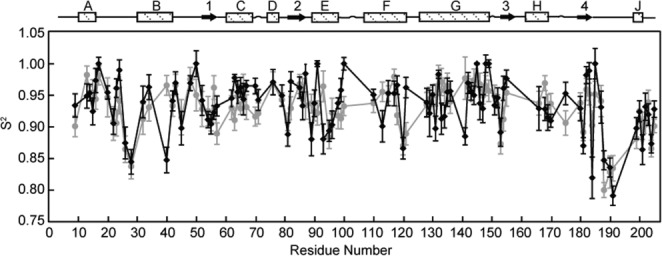
Effect of urea on fast timescale motions in NOX. Lipari-Szabo order parameters, S2, reflecting motions on the ns–ps timescale, obtained for NOX in the presence (black) and absence (gray) of 1.2M urea, plotted as a function of residue number.
Discussion
NOX from the thermophilic bacterium Thermus thermophilus, exhibits several characteristics that are of interest from the point of view of understanding how enzyme function can be tailored through solution conditions and the selection of cofactors and cosolutes in biotechnological applications. NOX is capable of accepting either FMN or the much larger FAD molecule as cofactors.21 Similarly to many other thermozymes, it exhibits significantly greater activity at 65°C than it does at 20°C.11 Its activity can be partially rescued at ambient temperatures through the addition of moderate concentrations of denaturants such as urea,11,19 a useful characteristic it shares with several other enzymes.7–10 We have used solution NMR spectroscopy and ITC to study the interactions of NOX with FMN and FAD, the denaturant urea, and to probe the effects of temperature on this enzyme.
We find that the spectra of apo- and both FMN- and FAD-saturated NOX are similar, suggesting that the protein maintains its overall fold even in the absence of cofactor. This places it in contrast to some other proteins,44 such as some c-type cytochromes,45 iron–sulfur containing proteins,46 and acetyl transferases,47 which require cofactor binding to adopt the holo-configuration. We found evidence for some enhanced protein mobility in the absence of cofactor, as signals for some residues in the NOX active site are absent in the apo-spectrum, possibly due to weakened hydrogen bonding that enhances solvent exchange or ms–μs timescale motions that broadened the signals beyond detection. Interestingly, the FMN-binding protein flavodoxin also retains its structure but exhibits enhanced dynamics in the absence of cofactor.48 In addition, a comparison of the spectra obtained for FMN- and FAD-saturated NOX provides an indication of how the protein adjusts conformationally to the larger cofactor. X-ray crystallographic structures of the two forms indicated that the only structural changes accompanying cofactor substitution occur in the C-terminal region of the protein, comprising residues E189 to H194 (Supporting Information Fig. S9).21 To some extent the NMR data agree with this observation as large chemical shift changes are observed for G190, Y191, and S193, although these perturbations could also be due to aromatic ring current shifts from the adenine moiety of FAD. Interestingly, comparable chemical shift perturbations are also observed for other residues in and around the active site, including those facing both sides of the flavin ring. This suggests that there may be structural adjustments in NOX accompanying cofactor substitution in solution at 50°C that are not evident in the crystal lattice at 12°C.
Our ITC binding data for FMN and FAD are consistent with cofactors binding independently to the two subunits of NOX, which agrees with the observation that subunits undergo only minor changes in structure or dynamics when binding cofactor. Our ITC results indicate that FAD binds about 3 orders of magnitude less tightly than FMN. The heats of binding are essentially identical, with ΔH values of –10.1 ± 0.4 and –9.4 ± 0.7 kcal/mol, for FAD and FMN, respectively. The reduction in affinity is due to a large entropic penalty for FAD binding [ΔS = −16 ± 1 cal/(mol K)], while the binding entropy for FMN binding is close to zero [ΔS = 0.2 ± 3 cal/(mol K)]. This unfavorable binding entropy of FAD compared to FMN can be largely rationalized in terms of conformational restriction of the cofactor. The X-ray crystal of the FAD-bound enzyme shows that the additional adenine moiety is sandwiched in between the helix E/helix F loop and the C-terminal region of the protein (Supporting Information Fig. S9). Thus, binding of FAD is associated with the restriction of up to seven additional rotatable bonds not present in FMN. In fact, the difference in binding entropies for FMN and FAD determined by ITC closely approaches the entropic penalty of restricting 7 independently rotatable bonds from 3 rotamers to 1 (assuming an entropic cost of – R × ln{3} cal/(mol K) per bond where R is the ideal gas constant). This is clearly an oversimplification, as it ignores the enthalpies and entropies of additional FAD/protein contacts and protein conformational rearrangements. Nevertheless, the difference in binding entropies between FMN and FAD is of a magnitude than can be explained largely in terms of differences in the sizes and number of degrees of conformational freedom for the two cofactors. In other words, FAD binds less tightly than FMN because the additional adenine moiety is essentially trapped in the active site cleft. It suffers an entropic penalty upon binding while forming few, if any, favorable compensatory interactions with the protein.
NOX activity is enhanced approximately 2.5-fold in 1M urea and sevenfold at 65°C, compared to the denaturant-free enzyme at 20°C. The NMR data shed light on the mechanisms of urea and temperature activation of NOX. Of particular interest, the side-chain indole NMR signal of W47 shows the largest urea-induced chemical shift perturbations of all NOX 15N/1H resonances at both 20 and 50°C. This residue has been previously implicated in the denaturant activation of the enzyme based on tryptophan fluorescence experiments.11 Furthermore, molecular dynamics (MD) simulations have suggested that W47 controls substrate access to the active site through a “gating” mechanism.21 The residue alternates between an “open” conformation in which its indole side chain contacts helix F and a water molecule bridges its backbone amide and the cofactor, and a “closed” conformation in which its side chain packs more closely against the cofactor and the bridging water molecule is displaced (see Supporting Information Fig. S10 and Fig. 4 of Hritz et al.23). The crystal structure corresponds to the “open” conformation, while the MD simulations indicated that the “closed” conformation is more stable in solution. Urea was found to shift the equilibrium towards the “open” state in MD simulations. Thus urea activation of the enzyme was proposed to be mediated through a shift in the “gating” equilibrium of W47 that facilitates substrate access to the active site. The large urea-dependent changes in the chemical shift we observe for the indole side chain of W47 provide strong, site-specific, experimental support for this mechanism.
The temperature dependence of NOX dynamics has also been addressed by MD simulations.24,28 These studies suggested that urea and temperature activation of the enzyme proceed via different mechanisms, as the “open”/“closed” equilibrium of W47 is largely unaffected by temperature. Our NMR results agree with these findings as the W47 indole resonance does not exhibit large changes in chemical shift as the temperature is varied. The nitrogen temperature factor, dδN/dT = −9.84 ppb/K, is typical for a stably folded protein,37 while the temperature factor of the proton, dδH/dT = −6.24 ppb/K, is characteristic of those lacking hydrogen bonds, as expected.36 NMR chemical shifts are consistent with additional aspects of the molecular dynamics simulations. NOX contains two networks of salt bridges involving D88, R174, E90, R53, D91, E98, and R200, which are believed to help conformationally stabilize helix F. Only interactions of E90 and R128 were found to be disrupted with increasing temperature in the simulations.24 Interestingly, E90 and R128 are the only two residues out of the seven participating in the salt bridge network to show large temperature dependent changes in chemical shift (E90 dδN/dT = 19.1 ppb/K; R128 dδN/dT = −22.0 ppb/K). The NMR data provide further insight into differences in the mechanism of temperature and urea activation of this enzyme. Addition of urea at 20°C is accompanied by pervasive spectral broadening characteristic of motions on the millisecond to microsecond timescales, while increasing the temperature does not produce this effect. Motions on these timescales can play significant roles in the catalytic cycles of enzymes49–51 and are not captured in standard molecular dynamics simulations which typically extend for only tens of nanoseconds. Thus, the NMR results suggest that urea activation may involve extensive millisecond timescale motions in the enzyme, while temperature activation does not.
Finally, an interesting pattern emerges from a comparison of spectra obtained for NOX in the apo form and in the presence of FMN, FAD, urea, and at different temperatures. The majority of 15N/1H NMR cross peaks are not sensitive to changes in the enzyme's environment, or shift in response just one of the variables tested. However, the signals for a small subset of residues (listed in Table2) are sensitive to multiple (two or more) co-solutes or variations in temperature. Backbone 15N/1H chemical shifts are largely determined by aromatic ring-current shifts, amide proton hydrogen bonding and by the dihedral angles of the side chain and backbone of adjacent residues (1–2 residues apart in the primary sequence).52–54 Thus a few specific sites in the protein exhibit a far greater tendency to structurally reconfigure in response to changing environmental conditions than does the rest of the protein. We shall refer to the tendency to undergo local restructuring in response to changing environmental conditions malleability. The first malleable site, containing A46, scW47, and N48 is located in the loop between helix αB and the first beta sheet, adjacent to the re face of the cofactor. All three residues are sensitive to replacing FMN with FAD. Since they are located near the conserved flavin moiety of the cofactor, this is likely due to reorganization of the active site rather than the formation of new contacts with the additional adenine moiety of FAD. Additionally, A46 experiences a conformational and/or dynamical transition upon cofactor removal, N48 exhibits a large 15N temperature factor, while the signals for scW47 and A46 are perturbed by the addition of urea. In the case of scW47, this is consistent with a urea-dependent conformational transition seen in molecular dynamics simulations.23 In the case of A46, the amide 15N and 1H are inaccessible to urea in the X-ray crystal structure, implicating urea-dependent conformational rearrangements at this location. A second malleable site, containing F71, G72, A74, G159, and F160, abuts the oxygen-rich side of the cofactor. Signals for all five residues are displaced upon FMN/FAD substitution. Since they are not adjacent to the additional adenine moiety of FAD, these changes in chemical shift likely reflect conformational rearrangements in the active site rather than the formation of new protein/cofactor contacts. The signals for three of these residues are also absent in the apo-protein, reflecting either ms–μs timescale motions or elevated hydrogen exchange in this state. The amide of G72 is involved in intramolecular hydrogen bonding in the cofactor-bound protein, thus it is clear that this residue experiences a change in conformation and/or flexibility when cofactor is removed. As well, 3 of these cross-peaks shift significantly with urea addition (2 of which correspond to urea-inaccessible residues), and 2 are temperature sensitive. Finally, S193 is sensitive to cofactor addition and temperature variation. All of these residues are within 15 Å of the cofactor which is not surprising since many of the environmental perturbations we tested are expected to target the active site of the enzyme. Nevertheless, there are over 100 nonproline backbone amide groups within 15 Å of the cofactor, therefore we are confident that the hypersensitivity of the 8 residues listed in Table2 is not simply a function of being located near the active site and instead reflects an unusual proclivity for remodeling of local protein structure and interacting with cosolutes. The peaks for these residues shift in different directions depending on the environmental condition being varied. This implies that structural plasticity surrounding the active site is more complicated than a dynamic equilibrium between two different conformations whose populations are shifted by changes in the environment, as all peaks for a given residue would fall on a line in this case.55 Instead, each environmental variable produces a different conformational change, with the largest perturbations recurring at the hypersensitive residues identified here. Interestingly, the majority of malleable residues do not exhibit greater than average ns–ps timescale mobility when evaluated under individual sets of conditions (FMN-saturated, 50°C, 0, and 2M urea). Their conformational plasticity is revealed only when environmental conditions are varied. In this sense, malleability can be distinguished from flexibility in terms of the energy landscape view of protein folding and function.56,57 When a residue is flexible, the corresponding minimum in the energy landscape is relatively broad in the corresponding dimensions, or multiple minima of similar energy are present so that a plurality of conformations is well populated. When a residue is malleable, the minimum in the energy landscape may be either broad or sharp; however, the width or position of the energy minimum shifts significantly in response to environmental variables. Thus flexibility is a property of the energy landscape under a single set of conditions, whereas malleability is a property of how the energy landscape changes as conditions are varied. It has been previously shown that dynamically flexible residues can play key roles in catalysis, and that mutations of these residues can profoundly affect enzyme efficiency.58,43,59 Malleable residues may have a similar relationship to the activation of enzymes (by temperature, denaturant, cofactor substitution, etc.). Targeting these residues for mutagenesis combined with fine-tuning of the environmental conditions may be an avenue for optimizing the efficiency of enzymes in industrial applications.
Table 2.
Residues with NMR Signals Sensitive to Environmental Conditions
| Apo proteina | Cofactor substitutionb | Temperature variationc | Urea titrationd | Relative reagent accessible surface area (%)e | |
|---|---|---|---|---|---|
| A46 | X | X | X | 0 | |
| scW47 | X | X | 67f | ||
| N48 | X | X | 22 | ||
| F71 | X | X | 0 | ||
| G72 | X | X | 0 | ||
| A74 | X | X | X | 0 | |
| G159 | X | X | X | X | 79 |
| F160 | X | X | 0 | ||
| S193 | X | X | X | 0 |
Backbone 15N/1H resonance absent in spectra of apo-NOX and present in holo-NOX.
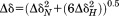 > 0.1 ppm, comparing FMN- and FAD-saturated NOX spectra.
> 0.1 ppm, comparing FMN- and FAD-saturated NOX spectra.
|∂δN/∂T| > 17 ppb/K or ∂δH/∂T < −4.5 ppb/K.
Δδ > 0.3, comparing NOX spectra obtained with 0 and 2M urea.
Amide NH accessibility based on a 2.5 Å probe.
Indole NH accessibility based on a 2.5 Å probe.
Materials and Methods
Construction of mutants
NOX W47F, W52F, W131F, and W204F mutants were constructed using two-step PCR directed mutagenesis (primers listed in Table1). Tryptophan residues at positions 47, 52, 131, and 204 were mutated to phenylalanine by replacing the codon TGG with TTC. In the first step, two separate PCR reactions were performed using the pTNADOX plasmid containing the Nox gene60 as the template and either primers (Nox_fwd and WxF_rev) or (Nox_rev and WxF_fwd), giving rise to two amplicons. The amplicons were mixed and subjected to a second PCR with only primers Nox_fwd and Nox_rev. Primers Nox_fwd and Nox_rev contained EcoRI and HindIII sites, respectively. The amplicons were purified, digested with EcoRI and HindIII and ligated into the pTNADOX plasmid which had been treated with the same restriction enzymes. High fidelity Phusion polymerase (NEB, New England Biolabs, CA) was used for amplification and the mutants were verified by DNA sequencing. Only one step PCR was done for mutant 204, using primers Nox_fwd and W204F_rev, since the mutation is located close to the 3′ end of Nox gene.
Table 1.
Primers Used for Mutagenesis
| Primer | Sequence |
|---|---|
| NOX_fwd | 5′-ACTCCCgaattcATGGAGGCGACCCTT-3′ |
| W47_fwd | 5′-GCCCTCGGCCTTCAACCTCCAGC-3′ |
| W52_fwd | 5′-CCTCCAGCCCTTCCGGATCGTGG-3′ |
| W131_fwd | 5′-GCGAAAGGCCTTCGCCTCCGGG-3′ |
| NOX_rev | 5′-TCCTCaagcttCCCTAGCGCCAGAGGA-3′ |
| W47_rev | 5′-GCTGGAGGTTGAAGGCCGAGGGC-3′ |
| W52_rev | 5′-CCACGATCCGGAAGGGCTGGAGG-3′ |
| W131_rev | 5′-CCCGGAGGCGAAGGCCTTTCGC-3′ |
| W204_rev | 5′-TCCTCaagcttCCCTAGCGGAAGAGGA-3′ |
Mutated codons are underlined in bold. Lowercase letters indicate restriction endonuclease sites (fwd, EcoRI; rev, HindIII).
Protein expression and purification
The protein was overexpressed in Escherichia coli bacteria19 (NovaBlue, EMD Biosciences, San Diego CA), previously transformed with the pTNADOX plasmid encoding Thermus thermophilus NOX under the control of a tac promoter.60 Cells were grown at 37°C in Luria–Bertani broth up to an OD600 of 0.7 a.u., pelleted by centrifugation (4500g, 10 min, RT), washed once in M9 salts, and resuspended in M9 minimal media containing ammonium chloride as the exclusive nitrogen source. Unlabelled ammonium chloride was used to produce samples for ITC experiments and 15NH4Cl [200 mg/L (Sigma-Aldrich, St. Louis, MO, USA)] was used for NMR samples. Protein expression was induced with 200 mg/mL isopropyl β-d-thiogalactoside (IPTG, EMD Biosciences, San Diego, CA, USA) 1 h after the transfer. Bacteria were harvested by centrifugation (4500g, 10 min, RT) after 6 h of incubation.13 Cells, resuspended in lysis buffer (50 mM Tris–HCl, 5 mM benzamidine, 2 mM ethylenediaminetetraacetic acid (EDTA) at pH 7.5), were disrupted by sonication, centrifuged to remove cell debris (43,000g), incubated for 5 min at 80°C then separated by centrifugation (17,000g). The heat treatment causes mesophilic native E. coli proteins to precipitate while the thermophilic NADH oxidase remains in solution.29 The sample was further purified using DEAE anion-exchange chromatography, in which impurities, but not NOX, bound to the column. The protein solution was further purified by adding 50 mM acetate buffer, reducing the pH to 5 loading on an SP–sepharose cation-exchange column and applying a 0–2M NaCl gradient, with elution occurring at 50 mM NaCl. Size-exclusion chromatography was performed using a HiLoad 16/60 Superdex 75 FPLC (GE Healthcare, Little Chalfont, UK) column as a final purification step. The sample was dialyzed into a 50 mM potassium phosphate, pH 7.2, consistent with sample conditions used previously for enzymatic activity assays.11 The NMR samples contained 50 mM potassium phosphate pH 7.2, between 0.8 and 1 mM NOX (monomer concentration) with 2.4 mM riboflavin 5′-monophosphate sodium salt (FMN, Sigma-Aldrich, St. Louis MO, USA) unless otherwise specified, 5% D2O and 10 μM 2,2-dimethyl-2-silapentane-5-sulfonate (DSS, Sigma-Aldrich, St. Louis, MO).
NMR spectroscopy
All NMR experiments were recorded at 50°C, except for the temperature titration and low-temperature urea titration, using Varian INOVA spectrometers equipped with high-sensitivity cold probes operating at 1H Larmor frequencies of 800 MHz (18.8 T) and 500 MHz (11.7 T). This temperature was found to be optimal for sample stability and spectral quality. The temperature settings of both instruments were calibrated using a methanol reference.61 The data were processed and analyzed using the NMRPipe/NMRDraw suite of programs.62
NMR titrations
15N/1H HSQC correlation spectra63 were collected at 18.8 T with 102 increments in the indirect dimension, 8 scans and 64 steady-state scans, for a total acquisition time of 37 min per spectrum. The urea titrations consisted of spectra collected for FMN-saturated NOX in the presence of 0–2M urea in 0.2M increments repeated at 20 and 50°C (Supporting Information Fig. S1). For the temperature titration, a series of spectra was collected from 25 to 50°C in 5°C intervals with 0M urea (Supporting Information Fig. S2). Amide proton and nitrogen temperature coefficients of each residue, indicative of hydrogen bonding and flexibility patterns, were derived from the slope (ppb/K) of chemical shift changes with increasing temperature.64 The FMN and FAD titrations consisted of NOX spectra collected with 0.00, 0.05, 0.15, 0.20, 0.25, 0.30, 0.50, 1.00, 2.00, 2.40, 3.00, and 5.00 mM FMN and 0.0, 0.2, 0.4, 1.0, 2.0, 2.5, 3.0, 3.5, 14.9, and 39.9 mM FAD, respectively (Supporting Information Fig. S3). Titrant stock solutions were prepared in NMR protein buffer so that the final concentrations of all buffer components were identical to those of the protein NMR sample. Titrant was added by removing the protein sample from the NMR tube using a Pasteur pipette and placing it in an 1.5 mL disposable test tube, adding the required amount of the stock solution, mixing thoroughly, and replacing it in the NMR tube, using the same Pasteur pipette and test tube for each addition. The maximum changes in pH due to titrant addition (2M urea, 5.0 mM FMN, 39.9 mM FAD) were measured to be negligible (less than 0.1 pH units) All 1H and 15N chemical shifts were referenced using the 1H methyl signal of DSS for all temperature and cosolute titrations.65
The shifts in 15N and 1H resonance frequencies were analyzed as a function of FAD concentration according to:30
| 2 |
where
[L]0 and [P]0 are the total concentrations of FAD and NOX (monomer), δ is the observed chemical shift, δP and δC are the chemical shifts of the free protein and the complex, respectively, and Δδ = δP − δC. The titration data for 20 residues were analyzed simultaneously by nonlinear least-squares optimization using inhouse MATLAB scripts to yield a global equilibrium dissociation constant, KD, and an individual value of Δδ for each set of 15N and 1H chemical shifts used. Uncertainties in the global KD were estimated by a bootstrap method66 where the fitting was repeated 100 times on Monte Carlo datasets comprising data from randomly-selected sets of residues. The error in the global KD value was calculated as the standard deviation of the Monte Carlo KD estimates. The FMN titration was not analyzed in this manner as the affinity of the interaction is too great to yield a reliable KD value by this method.
15N spin relaxation experiments
15N R1, R1ρ,67 relaxation rates and 1H/15N steady-state NOE (ssNOE) values were recorded for FMN-saturated NOX in the presence of 1.2M urea. The R1 experiments employed relaxation delays of 0.0111, 0.0999, 0.200, 0.322, 0.433, 0.566, 0.699, 0.855, 1.03, 1.22, 1.42, 1.67 s (11.7 T) and 0.0109, 0.0978, 0.196, 0.315, 0.424, 0.554, 0.685, 0.837, 1.01, 1.20, 1.39, 1.63 s (18.8 T). The R1ρ experiments employed relaxation delays of 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1 s and spin–lock field strengths of 1.5 kHz at both 11.7 and 18.8 T. NOE experiments were performed with 5 s of proton presaturation and a 7 s interscan delay, and with no 1H saturation and 12 s interscan delay at 18.8 T. ssNOEs were calculated as the ratios of peak intensities in the saturated and unsaturated spectra while uncertainties were obtained from noise level estimates in NMRDraw. R1 and R1ρ relaxation rates were determined by nonlinear least-squares fitting of the peak intensities, I(t), to a monoexponential decay function as a function of the relaxation delay, t:
| 4 |
Error estimates were obtained from the deviations between experimental peak intensities and their back-calculated values. R2 relaxation rates were calculated according to:
| 5 |
where ν1 is the strength of the spin–lock pulse (1.5 kHz) and Δν is the offset of 15N resonance frequency from the carrier frequency. The R1, R2, and ssNOE values were analyzed according to the Lipari–Szabo model-free formalism42 as described previously.6 Our approach simultaneously optimizes the order parameters, S2, and global tumbling parameters.68 In this case, an axially symmetric model of rotational diffusion in solution with D|| = 1.04 × 107 s−1 and D⊥ = 1.2 × 107 s−1 gave significantly better agreement with the relaxation data than an isotropic tumbling model. A fully anisotropic tumbling model did not significantly improve the fit and the axially symmetric rotational diffusion model was used. All calculations were performed using inhouse MATLAB programs.
Isothermal titration calorimetry (ITC)
ITC experiments were performed on a MicroCal VP-ITC instrument (MicroCal, North Hampton, MA) at 50°C using buffer conditions identical to the NMR experiments. Three replicates of each titration were collected as well as three blank titrations of FAD into buffer. For FMN experiments, the syringe contained 122 µM FMN and the sample cell contained 8.7 µM NOX. Most titrations employed an initial delay of 60 s, a 2 μL initial injection followed by a 150 s delay and 27 × 10 μL injections each followed by a 300 s delay. Two replicates of the FMN experiments used different settings: an initial delay of 120 s, a 2 μL initial injection volume followed by a 150 s delay, 11 × 5 μL injections followed by 180 s delays and 44 × 10 μL injections followed by 240 s delays. For FAD experiments, the syringe contained 10 mM FAD and the sample cell contained 104 µM NOX or buffer alone. Titrations employed an initial delay of 60 s, a 1 μL initial injection volume followed by a 300 s delay and 10 × 5.6 μL injections followed by a 300 s delay. The FAD blank titration injection heats were subtracted from those of the NOX titration prior to analysis.
Relative reagent accessible surface area calculations
All surface area calculations were performed using UCSF Chimera and the 1NOX PDB atomic coordinates.69 The relative reagent accessible surface areas (column 6 in Table2) were calculated as the sum of the nitrogen and hydrogen solvent accessible surface areas (SASA) for each residue, normalized by the sum of the nitrogen and hydrogen surface areas for the second glycine residue in an extended Gly-Gly-Gly tripeptide. For the side-chain NH group of W47, the normalization was taken relative to the sum of exposed nitrogen and hydrogen surface areas in an individual tryptophan residue. All SASA calculations were performed using a 2.5 Å probe. This is slightly smaller than the Van der Waals radius of a urea molecule relative to its geometric centre, rVDW = 3.6 Å.70
Acknowledgments
The authors declare that they have no conflicting interests. NOX expression plasmid was a generous gift from Prof. Mathias Sprinzl (U. Bayreuth). We would also like to thank Dr. Sameer Al-Abdul-Wahid for invaluable discussions and technical guidance, and Prof. Wulf Blankenfeldt and Dr. Joachim Reichelt for generously providing the atomic coordinates for FAD-bound NOX.
Glossary
- 2D
two-dimensional
- Δδ
chemical shift displacement
- DEAE
diethylaminoethyl
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- FPLC
fast protein liquid chromatography
- HSQC
heteronuclear single quantum correlation
- IPTG
isopropyl β-d-thiogalactoside
- ITC
isothermal titration calorimetry
- KD
dissociation constant
- MD
molecular dynamic
- 15NH4Cl
ammonium chloride
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOX
NADH oxidase
- OD600
optical density 600
- PCR
polymerase chain reaction
- RMSD
root-mean-square deviation
- RT
room temperature
- S2
order parameter.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Supporting Information
References
- Gianfreda L, Rao MA. Potential of extra cellular enzymes in remediation of polluted soils: a review. Enzyme Microb Technol. 2004;35:339–354. [Google Scholar]
- Panke S, Held M, Wubbolts M. Trends and innovations in industrial biocatalysis for the production of fine chemicals. Curr Opin Biotechnol. 2004;15:272–279. doi: 10.1016/j.copbio.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Bistolas N, Wollenberger U, Jung C, Scheller FW. Cytochrome P450 biosensors—a review. Biosens Bioelectron. 2005;20:2408–2423. doi: 10.1016/j.bios.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Amine A, Mohammadi H, Bourais I, Palleschi G. Enzyme inhibition-based biosensors for food safety and environmental monitoring. Biosens Bioelectron. 2006;21:1405–1423. doi: 10.1016/j.bios.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Ahuja T, Mir IA, Kumar D, Rajesh Biomolecular immobilization on conducting polymers for biosensing applications. Biomaterials. 2007;28:791–805. doi: 10.1016/j.biomaterials.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Engineering the third wave of biocatalysis. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- Fan YX, Ju M, Zhou JM, Tsou CL. Activation of chicken liver dihydrofolate reductase by urea and guanidine hydrochloride is accompanied by conformational change at the active site. Biochem J. 1996;315:97–102. doi: 10.1042/bj3150097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HJ, Sheng XR, Pan XM, Zhou JM. Activation of adenylate kinase by denaturants is due to the increasing conformational flexibility at its active sites. Biochem Biophys Res Commun. 1997;238:382–386. doi: 10.1006/bbrc.1997.7301. [DOI] [PubMed] [Google Scholar]
- Inui T, Ohkubo T, Urade Y, Hayaishi O. Enhancement of lipocalin-type prostaglandin D synthase enzyme activity by guanidine hydrochloride. Biochem Biophys Res Commun. 1999;266:641–646. doi: 10.1006/bbrc.1999.1881. [DOI] [PubMed] [Google Scholar]
- Deshpande RA, Kumar AR, Khan MI, Shankar V. Ribonuclease Rs from Rhizopus stolonifer: lowering of optimum temperature in the presence of urea. Biochim Biophys Acta Protein Struct Mol Enzymol. 2001;1545:13–19. doi: 10.1016/s0167-4838(00)00256-9. [DOI] [PubMed] [Google Scholar]
- Zoldak G, Sut'ak R, Antalik M, Sprinzl M, Sedlak E. Role of conformational flexibility for enzymatic activity in NADH oxidase from Thermus thermophilus. Eur J Biochem. 2003;270:4887–4897. doi: 10.1046/j.1432-1033.2003.03889.x. [DOI] [PubMed] [Google Scholar]
- Zavodszky P, Kardos J, Svingor A, Petsko GA. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc Natl Acad Sci USA. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruins ME, Janssen AEM, Boom RM. Thermozymes and their applications—a review of recent literature and patents. Appl Biochem Biotechnol. 2001;90:155–186. doi: 10.1385/abab:90:2:155. [DOI] [PubMed] [Google Scholar]
- Li WF, Zhou XX, Lu P. Structural features of thermozymes. Biotechnol Adv. 2005;23:271–281. doi: 10.1016/j.biotechadv.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Feng J-Y, Liu J-Z, Ji L-N. Thermostability, solvent tolerance, catalytic activity and conformation of cofactor modified horseradish peroxidase. Biochimie. 2008;90:1337–1346. doi: 10.1016/j.biochi.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Machielsen R, Looger LL, Raedts J, Dijkhuizen S, Hummel W, Hennemann H-G, Daussmann T, Van der Oost J. Cofactor engineering of Lactobacillus brevis alcohol dehydrogenase by computational design. Eng Life Sci. 2009;9:38–44. [Google Scholar]
- Brustad EM, Arnold FH. Optimizing non-natural protein function with directed evolution. Curr Opin Chem Biol. 2011;15:201–210. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann H, Hecht HJ, Park HJ, Sprinzl M, Schomburg D, Schmid RD. Crystallization and preliminary X-ray diffraction studies of a NADH oxidase from Thermus thermophilus HB8. J Mol Biol. 1993;230:1086–1088. doi: 10.1006/jmbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- Zoldak G, Sprinzl M, Sedlak E. Modulation of activity of NADH oxidase from Thermus thermophilus through change in flexibility in the enzyme active site induced by Hofmeister series anions. Eur J Biochem. 2004;271:48–57. doi: 10.1046/j.1432-1033.2003.03900.x. [DOI] [PubMed] [Google Scholar]
- Zoldak G, Musatov A, Stupak M, Sprinzl M, Sedlak E. pH-induced changes in activity and conformation of NADH oxidase from Thermus thermophilus. Gen Physiol Biophys. 2005;24:279–298. [PubMed] [Google Scholar]
- Hecht HJ, Erdmann H, Park HJ, Sprinzl M, Schmid RD. Crystal structure of NADH oxidase from Thermus thermophilus. Nat Struct Biol. 1995;2:1109–1114. doi: 10.1038/nsb1295-1109. [DOI] [PubMed] [Google Scholar]
- Palmer DH, Chen MJ, Searle PF, Kerr DJ, Young LS. Inhibition of NF-kappa B enhances the cytotoxicity of virus-directed enzyme prodrug therapy and oncolytic adenovirus cancer gene therapy. Gene Ther. 2005;12:1187–1197. doi: 10.1038/sj.gt.3302510. [DOI] [PubMed] [Google Scholar]
- Hritz J, Zoldak G, Sedlak E. Cofactor assisted gating mechanism in the active site of NADH oxidase from Thermus thermophilus. Proteins. 2006;64:465–476. doi: 10.1002/prot.20990. [DOI] [PubMed] [Google Scholar]
- Merkley ED, Parson WW, Daggett V. Temperature dependence of the flexibility of thermophilic and mesophilic flavoenzymes of the nitroreductase fold. Protein Eng Des Sel. 2010;23:327–336. doi: 10.1093/protein/gzp090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkley ED, Daggett V, Parson WW. A temperature-dependent conformational change of NADH oxidase from Thermus thermophilus HB8. Proteins. 2011;80:546–555. doi: 10.1002/prot.23219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miletti T, Farber PJ, Mittermaier A. Active site dynamics in NADH oxidase from Thermus thermophilus studied by NMR spin relaxation. J Biomol NMR. 2011;51:71–82. doi: 10.1007/s10858-011-9542-0. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. 2004. pp. 3607–3362. ) Unfolded proteins and protein folding studied by NMR. Chem Rev 104: [DOI] [PubMed]
- Merkley ED, Daggett V, Parson WW. A temperature-dependent conformational change of NADH oxidase from Thermus thermophilus HB8. Proteins. 2012;80:546–555. doi: 10.1002/prot.23219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Reiser CO, Kondruweit S, Erdmann H, Schmid RD, Sprinzl M. Purification and characterization of a NADH oxidase from the thermophile Thermus thermophilus HB8. Eur J Biochem. 1992;205:881–885. doi: 10.1111/j.1432-1033.1992.tb16853.x. [DOI] [PubMed] [Google Scholar]
- Macomber RS. An Introduction to NMR titration for studying rapid reversible complexation. J Chem Educ. 1992;69:375–378. [Google Scholar]
- Bai YW, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen-exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly GP, Bai YW, Jeng MF, Englander SW. Isotope effects in peptide group hydrogen-exchange. Proteins. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- Radford SE, Buck M, Topping KD, Dobson CM, Evans PA. Hydrogen-exchange in native and denatured states of hen egg-white lysozyme. Proteins. 1992;14:237–248. doi: 10.1002/prot.340140210. [DOI] [PubMed] [Google Scholar]
- Crowhurst KA, Tollinger M, Forman-Kay JD. Cooperative interactions and a non-native buried Trp in the unfolded state of an SH3 domain. J Mol Biol. 2002;322:163–178. doi: 10.1016/s0022-2836(02)00741-6. [DOI] [PubMed] [Google Scholar]
- Perkins SJ, Dwek RA. Comparisons of ring-current shifts calculated from the crystal-structure of egg-white lysozyme of hen with the proton nuclear magnetic-resonance spectrum of lysozyme in solution. Biochemistry. 1980;19:245–258. doi: 10.1021/bi00543a001. [DOI] [PubMed] [Google Scholar]
- Baxter NJ, Williamson MP. Temperature dependence of 1H chemical shifts in proteins. J Biomol NMR. 1997;9:359–369. doi: 10.1023/a:1018334207887. [DOI] [PubMed] [Google Scholar]
- Cordier F, Grzesiek S. Temperature-dependence properties as studied by of protein hydrogen bond high-resolution NMR. J Mol Biol. 2002;317:739–752. doi: 10.1006/jmbi.2002.5446. [DOI] [PubMed] [Google Scholar]
- Holtzer ME, Lovett EG, dAvignon DA, Holtzer A. Thermal unfolding in a GCN4-like leucine zipper: C-13(alpha) NMR chemical shifts and local unfolding curves. Biophys J. 1997;73:1031–1041. doi: 10.1016/S0006-3495(97)78136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Wu L, McElheny D, Bour P, Roy A, Keiderling TA. Cross-strand coupling and site-specific unfolding thermodynamics of a Trpzip beta-hairpin peptide using C-13 isotopic labeling and IR spectroscopy. J Phys Chem B. 2009;113:5661–5674. doi: 10.1021/jp9014299. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Tanford C. Viscosity and Density of Aqueous Solutions of Urea and Guanidine Hydrochloride. J Biol Chem. 1966;241:3228–3232. [PubMed] [Google Scholar]
- Hamada H, Arakawa T, Shiraki K. Effect of additives on protein aggregation. Curr Pharm Biotechnol. 2009;10:400–407. doi: 10.2174/138920109788488941. [DOI] [PubMed] [Google Scholar]
- Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- Doucet N, Watt ED, Loria JP. The flexibility of a distant loop modulates active site motion and product release in ribonuclease A. Biochemistry. 2009;48:7160–7168. doi: 10.1021/bi900830g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittung-Stafshede P. Role of cofactors in protein folding. Acc Chem Res. 2002;35:201–208. doi: 10.1021/ar010106e. [DOI] [PubMed] [Google Scholar]
- Fisher WR, Taniuchi H, Anfinsen CB. Role of heme in formation of structure of cytochrome c. J Biol Chem. 1973;248:3188–3195. [PubMed] [Google Scholar]
- Pagani S, Vecchio G, Iametti S, Bianchi R, Bonomi F. On the role of the 2Fe–2S cluster in the formation of the structure of spinach ferredoxin. Biochim Biophys Acta. 1986;870:538–544. [Google Scholar]
- Freiburger LA, Baettig OM, Sprules T, Berghuis AM, Auclair K, Mittermaier AK. Competing allosteric mechanisms modulate substrate binding in a dimeric enzyme. Nat Struct Biol. 2011;18:288–U270. doi: 10.1038/nsmb.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittung-Stafshede P. A stable, molten-globule-like cytochrome c. Biochim Biophys Acta Protein Struct Mol Enzymol. 1998;1382:324–332. doi: 10.1016/s0167-4838(97)00176-3. [DOI] [PubMed] [Google Scholar]
- Wolf-Watz M, Thai V, Henzler-Wildman K, Hadjipavlou G, Eisenmesser EZ, Kern D. Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat Struct Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Xu XP, Case DA. Probing multiple effects on N-15, C-13 alpha, C-13 beta, and C-13' chemical shifts in peptides using density functional theory. Biopolymers. 2002;65:408–423. doi: 10.1002/bip.10276. [DOI] [PubMed] [Google Scholar]
- Parker LL, Houk AR, Jensen JH. Cooperative hydrogen bonding effects are key determinants of backbone amide proton chemical shifts in proteins. J Am Chem Soc. 2006;128:9863–9872. doi: 10.1021/ja0617901. [DOI] [PubMed] [Google Scholar]
- Williamson MP. Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- Onuchic JN, LutheySchulten Z, Wolynes PG. Theory of protein folding: the energy landscape perspective. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- Watt ED, Shimada H, Kovrigin EL, Loria JP. The mechanism of rate-limiting motions in enzyme function. Proc Natl Acad Sci USA. 2007;104:11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462:669–U149. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Kreutzer R, Reiser CO, Sprinzl M. Molecular cloning and nucleotide sequence of the gene encoding a H2O2-forming NADH oxidase from the extreme thermophilic Thermus thermophilus HB8 and its expression in Escherichia coli. Eur J Biochem. 1992;205:875–879. doi: 10.1111/j.1432-1033.1992.tb16852.x. [DOI] [PubMed] [Google Scholar]
- Amman C, Meier P, Merbach AE. A simple multinuclear NMR thermometer. J Magn Reson. 1982;46:319–321. [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Kay LE, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- Baxter NJ, Williamson MP. Temperature dependence of H-1 chemical shifts in proteins. J Biomol NMR. 1997;9:359–369. doi: 10.1023/a:1018334207887. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat Sci. 1986;1:54–75. [Google Scholar]
- Dmitry MK, Nikolai RS, Oscar M, Dennis AT, Lewis EK. An NMR experiment for the accurate measurement of heteronuclear spin–lock relaxation rates. J Am Chem Soc. 2002;124:10743–10753. doi: 10.1021/ja0204776. [DOI] [PubMed] [Google Scholar]
- Bruschweiler R, Liao XB, Wright PE. Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. Science. 1995;268:886–889. doi: 10.1126/science.7754375. [DOI] [PubMed] [Google Scholar]
- Pettersen E, Goddard T, Huang C, Couch G, Greenblatt D, Meng E, Ferrin T. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Krieger E, Vriend G. YASARA View-molecular graphics for all devices-from smartphones to workstations. Bioinformatics. 2014;30:2981–2982. doi: 10.1093/bioinformatics/btu426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information