

Abstract
A number of studies have previously proposed the existence of glucocorticoid receptors on the plasma membrane of many cell types, including skeletal muscle fibres. However, their exact localisation and the cellular signalling pathway(s) they utilise to communicate with the rest of the cell are still poorly understood. In this study, we investigated the localisation and the mechanism(s) underlying the non-genomic physiological functions of these receptors in mouse skeletal muscle cells. The results show that the receptors were localised in the cytoplasm in myoblasts, in the nucleus in myotubes, in the extracellular matrix, in satellite cells and in the proximity of mitochondria in adult muscle fibres. Also, they bound laminin in a glucocorticoid-dependent manner. Treating small skeletal muscle fibre bundles with the synthetic glucocorticoid beclomethasone dipropionate increased the phosphorylation (= activation) of extracellular signal-regulated kinases 1 and 2, c-Jun N-terminal kinase and p38 mitogen-activated protein kinase. This occurred within 5 min and depended on the fibre type and the duration of the treatment. It was also abolished by the glucocorticoid receptor inhibitor, mifepristone, and a monoclonal antibody against the receptor. From these results we conclude that the non-genomic/non-canonical physiological functions of glucocorticoids, in adult skeletal muscle fibres, are mediated by a glucocorticoid receptor localised in the extracellular matrix, in satellite cells and close to mitochondria, and involve activation of the mitogen-activated protein kinase pathway.
Key points
Many studies have previously suggested the existence of stress hormone receptors on the cell membrane of many cell types, including skeletal muscle fibres; however, the exact localisation of these receptors and how they signal to the rest of the cell is poorly understood.
In this study, we investigated the localisation and the mechanism(s) underlying the physiological functions of these receptors in mouse skeletal muscle cells.
We found that the receptors were present throughout muscle development and that, in adult muscle fibres, they were localised in the extracellular matrix, satellite cells (muscle stem cells) and close to mitochondria.
We also found that they signalled to the rest of the cell by activating enzymes called mitogen-activated protein kinases.
From these results we suggest that, at physiological concentrations, stress hormones may be important in skeletal muscle differentiation, repair and regeneration.
Introduction
Glucocorticoids (GCs, steroids) are the main stress hormones in the body. They are synthesised from cholesterol and released from cells in the zona fasciculata of the adrenal cortex. In healthy individuals, they are released in short irregular bursts that peak ∼2 h before the start of the day and their plasma concentration rarely exceeds 250 nm except during stress when it can be as high as 700 nm (Krieger et al. 1971). GCs are essential for life and affect most tissues in the body including skeletal muscle. Indeed, skeletal muscle plays a key role in the stress response because it enables individuals to evade or to fight the stressor. Despite this, little is known about the physiological functions of GCs in skeletal muscle especially at the low concentrations normally found in plasma. Instead, what has been extensively studied and reported are the effects of treating laboratory animals for long periods with high doses of synthetic GCs (especially dexamethasone) on skeletal muscle function (Nava et al. 1996; van Balkom et al. 1996; Ma et al. 2003) and protein metabolism (Goldberg, 1969; Rannels & Jefferson, 1980; Wing & Goldberg, 1993). Much is also known about the effects of lack of (Addison’s disease) and excess of (Cushing’s syndrome) GCs on skeletal muscle mass and function.
GCs exert most of their physiological and phar-macological effects through an intracellular glucocorticoid receptor (GR, GCR, NR3C1). The GR belongs to the super family of ligand-activated nuclear transcription factors that include the steroid, thyroid and retinoic acid receptors (Kadmiel & Cidlowski, 2013). It is coded for by a single gene (the NR3C1 gene) that is spliced into two main isoforms, GRα (which is the active form) and GRβ (which is inactive and resides in the nucleus where it acts as a dominant negative inhibitor of GRα) (Kadmiel & Cidlowski, 2013). GRα (henceforth referred to as GR) is expressed in most cells, including muscle fibres (= cells), where the inactive receptor is thought to reside in the cytosol (hence cytosolic GR). In this state, the receptor is maintained in its high affinity conformation and is protected from inactivation by chaperone molecules such as heat shock proteins (HSPs), e.g. HSP 90, and immunophilins, e.g. FK506 binding protein (FKBP) 51 (Pratt & Toft, 2003). However, in a recent study we showed that, in adult mammalian skeletal muscle fibres, GRs were localised close to the surface of muscle fibres where they co-localised with laminin. We were also able to inhibit their effects on force using a monoclonal antibody against the receptor suggesting that they were easily accessible from the cell surface (Pérez et al. 2013). Although a number of studies have previously alluded to the existence of membrane glucocorticoid receptors in other cell types, including human lymphoma cells (Gametchu, 1993), peripheral blood mononuclear cells (Bartholome et al. 2004) and neuronal cells (Orchinik et al. 1991), their precise location is still unknown. It is also uncertain whether in skeletal muscle the GRs are localised close to the cell surface throughout myogenesis or just in adult muscle fibres.
In addition to their genomic actions, GCs also exert actions that occur within minutes and are relatively insensitive to inhibitors of transcription and translation (Stahn & Buttgereit, 2008; Pérez et al. 2013). These non-canonical/non-genomic actions have been described in many cell types including HeLa cells (Lasa et al. 2002; Bruna et al. 2003), guinea pig neurones (Hua & Chen, 1989), human endometrial cells (Hafezi-Moghadam et al. 2002), human adenocarcinoma cells (Croxtall et al. 2000), rat thymocytes (Buttgereit et al. 1997), airway vasculature (Alangari, 2010) and skeletal muscle (Pérez et al. 2013). However, the cellular signal transduction events mediating them are still poorly understood. The receptor/mechanism underlying their actions is also controversial.
The primary aims of this study were twofold: (1) to investigate the localisation and physiological functions of the GR in mouse skeletal muscle cells and (2) to determine the cellular signal transduction events mediating their non-genomic physiological actions in adult mammalian skeletal muscle fibres.
Methods
Small intact skeletal muscle fibre bundles
The experiments reported here were performed at room temperature (∼20°C) using small skeletal muscle fibre bundles (∼540 ± 26 μm in diameter) isolated from the extensor digitorum longus (EDL; a fast-twitch) or the soleus (slow-twitch) muscles of adult male C57BL/6 mice. The mice were killed by cervical dislocation according to UK legislation (for a summary of the legislation see Drummond, 2009) and all the experiments conformed to the University of East Anglia animal welfare committee guidelines. Throughout the experiments, the muscles and muscle fibre bundles were bathed in mammalian Ringer solution with the following composition (in mm): 109 NaCl, 5 KCl, 1 MgCl2, 4 CaCl2, 24 NaHCO3, 1 NaHPO4, 10 sodium pyruvate plus 400 mg l−1 fetal bovine serum (FBS). The pH of the Ringer solution was maintained at ∼7.42 by continuously bubbling it with 95% O2 and 5% CO2. To avoid any confounding influence of the GCs present in the FBS, serum from the same batch was used in all the experiments.
Cell culture
C2C12 cells (an immortalised mouse skeletal muscle cell line) were grown on coverslips in growth media (Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% l-glutamine plus10% FBS and 1% penicillin and streptomycin (all purchased from Sigma-Aldrich, Poole, UK) for 3–4 days or until 90% confluence. Once 90% confluent, they were induced to differentiate by replacing the growth media with differentiation media, i.e. growth media containing 2% horse serum instead of FBS. At the end of the experiment the cells were washed in phosphate-buffered saline (PBS), air dried and permeabilised for 15 min using Tris-buffered saline with Tween 20 (TBST). They were blocked for non-specific antibody binding for 10 min using 1% bovine serum albumin (BSA) in PBS. Finally, they were immunoblotted for the expression of the GR, laminin and double stranded DNA as described below.
Determination of the effects of beclomethasone dipropionate on the MAPK pathway
This experiment was performed as previously described in Hamdi & Mutungi (2010, 2011). Briefly, the muscle fibre bundles were mounted horizontally between two stainless steel hooks in a Perspex muscle chamber (total volume ∼40 ml) with a glass bottom. Half of the bundles (controls) were treated for 5, 10, 30 and 60 min with the standard Ringer solution plus 34 nm ethanol (the vehicle used to dissolve beclomethasone (beclometasone) dipropionate (BDP)). The other half (experimental) was treated for the same period of time with the Ringer solution containing 250 nm BDP. At the end of the experiment, the fibre bundles were snap-frozen in liquid nitrogen and cytosolic proteins were extracted using NP40 lysis buffer. The proteins were immunoblotted for the activation/phosphorylation of c-Jun N-terminal kinase (JNK; also known as stress-activated protein kinase (SAPK)), extracellular signal-regulated kinases 1 and 2 (ERK1&2) and p38 mitogen-activated protein kinase (MAPK) as described below.
Determination of the receptor/mechanism mediating the effects of BDP in mammalian skeletal muscle fibres
To determine whether the effects of BDP on the MAPK pathway were mediated through the GR and whether the receptor was localised close to the fibre surface, another set of fast-twitch and slow-twitch bundles was divided into three groups. Group 1 (controls) was treated with the standard Ringer solution plus the vehicle used to dissolve the GC (ethanol) and mifepristone (DMSO). Group 2 was pre-incubated for 10 min in Ringer solution containing 10 μm mifepristone (RU486; a GR inhibitor) (Sigma-Aldrich, Gillingham, UK), whereas Group 3 was pre-incubated for the same period in a 1:5000 dilution of a monoclonal anti-GR (ab109022; Abcam, Cambridge, UK). The fibres in Group 2 were then treated for a further 30 min with the Ringer solution containing 10 μm mifepristone alone or mifepristone plus 250 nm BDP, whereas those in Group 3 were treated for 5 min with the Ringer solution plus 1:5000 dilution of the anti-GR alone or 1:5000 dilution of the anti-GR plus 250 nm BDP. To determine whether the effects of BDP were mediated via focal adhesion kinase (FAK), another set of fibres was pre-treated with 10 μm FAK inhibitor (FAKi) 14 (Abcam). They were then incubated for a further 10 min in Ringer solution containing the inhibitor alone or the inhibitor plus 250 nm BDP. At the end of these experiments, the fibre bundles were frozen and cytosolic proteins were extracted using NP40 lysis buffer. They were then immunoblotted for the activation of ERK1&2, JNK and p38 MAPK as described below.
Immunoblotting
To determine the effects of BDP on the activa-tion/phosphorylation of ERK1&2, JNK and p38 MAPK, equal amounts of the proteins were immunoblotted as previously described in Pérez et al. (2013). Briefly, 10 μg of the proteins were separated by standard gel-electrophoresis and transferred onto nitrocellulose membranes. The membranes were blocked for non-specific antibody binding using 5% milk. They were then immunoblotted for the expression of phosphorylated (p) ERK1&2, pJNK and pp38 MAPK using a 1:500 dilution of rabbit monoclonal antibodies (4377, 4671 and 9215, respectively) from Cell Signaling Technology (New England Biolabs, Hitchin, UK) and the expression of the GR using a 1:500 dilution of the monoclonal anti-GR from Abcam used in the experiment above (ab109022; Cambridge, UK). Finally, they were visualised using SuperSignal WestPico chemiluminescence substrate (Perbio Science UK Ltd, Cramlington, UK) and exposure to film.
The following day, the membranes were stripped and re-probed with a pan-actin antibody (ab3280, Abcam, Cambridge, UK) to check for loading. In some experiments, the fibre bundles were fibre typed using monoclonal antibodies against fast and slow MyHCs (Sigma-Aldrich, Gillingham, UK). All the blots were run in duplicate and each experiment was repeated at least twice.
Immunocytochemistry
The expression and localisation of the GR in myoblasts, myotubes and adult mammalian skeletal muscles fibre bundles was determined as previously described in Pérez et al. (2013). Briefly, the EDL and soleus muscles isolated from adult male C57BL/6 mice were mounted perpendicularly on cryostat chucks using tissue Tek OCT compound (Sakura Finetek UK Ltd, Thatcham, UK). They were snap frozen in liquid nitrogen-cooled isopentane and 10 μm thick serial sections were cut. Two of the sections and the two coverslips of myoblasts and myotubes (grown as described above) were blocked for non-specific antibody labelling using 1% BSA dissolved in PBS. They were incubated at room temperature, for at least 1 h, with a 1:200 dilution of a monoclonal rat anti-laminin (MAB1905; Millipore Corporation, Billerica, MA, USA) and a 1:100 dilution of the monoclonal rabbit anti-GR (ab109022; Abcam, Cambridge, UK). They were then washed 5 times in PBS and incubated in 1:500 dilution of species-specific secondary antibodies conjugated to Alexa fluorophores for 1 h. Finally, Hoechest 33342 (a double stranded DNA-staining dye; Life Technologies, Paisley, UK) was added 5 min before the end of this incubation. Two more sections and coverslips (controls) were incubated with the primary or the secondary antibodies only. At the end of the incubation period, the sections and coverslips were washed 4 times in TBST, dried and mounted for viewing. Finally, they were visualised and photographed using an Axioplan 2 Imaging microscope and AxioVision Release 4.8 software (Carl Zeiss, Cambridge, UK). Like all the immunoblotting experiments, the immunocytohistochemistry was performed in duplicate and each experiment was repeated at least twice.
Pull-down assay
To determine whether the GR was able to bind laminin, a modification of the pull-down assay described in De Beer et al. (1981) was used. Briefly, 250 μg recombinant human merosin (laminin 2; laminin 211; Millipore (UK) Ltd, Watford, UK), the main laminin in skeletal muscle, or 100 μg recombinant mouse integrin α7β1 (the main integrins in mammalian skeletal muscle; R&D Systems, Abingdon, UK) were coupled to cyanogen bromide (CNBr)-activated sepharose beads (Sigma-Aldrich), according to the manufacturer’s instructions. The excess ligand was washed off with coupling buffer and all the unbound active groups were blocked using 0.2 m glycine. A 300 μl volume of slow-twitch muscle lysate was added to the beads alone, the beads plus laminin, or the beads plus integrin, and left overnight at 4°C on a rotator. Slow-twitch muscle lysate was used because slow muscle expresses 4–5 times more GR than fast-twitch muscle (Pérez et al. 2013). The following morning the beads were washed 2 times with TBST and once in PBS. They were then mixed with 30–40 μl elution buffer (Laemmli sample loading buffer) and left on a rotator for 1 h. Finally, they were centrifuged at 2000 g for 20 s and the supernatant was collected. A 10 μl volume of each supernatant was separated using standard gel-electrophoresis, transferred onto nitrocellulose membrane and finally immunoblotted for the GR using a 1:500 dilution of the rabbit monoclonal anti-GR antibody from Abcam as described above.
Immunofluorescence confocal microscopy
Soleus muscles from 6-month-old C57BL/6 mice were fixed in 2% paraformaldehyde in phosphate-buffered saline (PBS) for 2 h at room temperature. Small bundles were washed twice with PBS and blocked for 1 h in PBS containing 1% BSA, 10% goat serum and 0.5% Triton X-100 (added to permeabilise the membrane). They were incubated overnight at 4°C in primary antibody and the following morning they were washed 3 times in PBS. They were incubated with the secondary antibody for 1 h at room temperature before being mounted on coverslips with anti-bleach media (Slowfade Gold antifade reagent; Invitrogen (Molecular Probes) Eugene, Oregon, USA). Primary antibodies were: anti-RYR1 (Ryanodine receptor type 1), 34C (dilution 1:30; Developmental Studies Hybridoma bank, University of Iowa); rabbit monoclonal anti-GR (dilution 1:100; Abcam). Secondary antibodies were: Cy3-labelled goat anti-rabbit IgG (dilution 1:300) for single GR labelling; Cy5-labelled goat anti-mouse IgG (dilution 1:200) and Cy3-labelled goat anti-rabbit IgG for double labelling (dilution 1:300). All secondary antibodies were from Jackson ImmunoResearch Laboratories, Lexington, KY. Specimens were finally viewed using a scanning laser confocal microscope (LS510 META; Carl Zeiss, Jena, Germany).
Immunogold labelling electron microscopy
Soleus muscles were fixed for 20 min at room temperature in a fixative mixture containing 2% paraformaldehyde and 0.5% glutaraldehyde (in PBS buffer). Small bundles were permeabilised and blocked as described above. After incubation with the rabbit monoclonal anti-GR from Abcam, secondary antibodies conjugated with Nanogold particles were applied for 2 h at 4°C (dilution 1:100). Samples were then post-fixed with 1% glutaraldehyde (in PBS buffer), at room temperature, and incubated with reagents to enhance the signal (Goldenhance EM Formulation; Nanoprobes, Yaphank, NY, USA) for 5 min. Fibre bundles were finally embedded and sectioned using standard electron microscopy protocols (Boncompagni et al. 2009). Ultrathin sections of ∼40 nm were cut using a Leica Ultracut R microtome (Leica Microsystems, Vienna, Austria) with a Diatome diamond knife (Diatome Ltd, CH-2501 Biel, Switzerland) and double-stained with uranyl acetate and lead citrate. Sections were viewed using a FP 505 Morgagni Series 268D electron microscope (FEI Company, Brno, Czech Republic), equipped with Megaview III digital camera and Soft Imaging System (Münster, Germany).
As the monoclonal antibody used in the immunocytohistochemistry, immunofluorescence confocal microscopy and immunogold-labelling electron microscopy has never been tested before for use in these techniques, the findings were confirmed using a polyclonal rabbit anti-GR (Abcam ab35780) that is recommended for use in immunocytochemistry.
Statistical analysis
As mentioned above, all the immunoblotting experiments were run in duplicate and each experiment was repeated at least twice. To determine the expression of GRs in the various muscles, the Western blots were digitised and the intensity of the various protein bands were analysed using Scion Image from NIH and normalised to that of the loading control (actin). Statistical analysis of the data was performed using SigmaPlot 11.2 (Systat Software Inc., London, UK). The data obtained from each fibre type under control and from the various experimental conditions were compared using a two-way ANOVA with Tukey’s post hoc test and a P < 0.05 was considered to be statistically significant.
Results
The results in Fig.1 show the expression and localisation of GRs in myoblasts (Fig.1A and B), myotubes (Fig.1C and D) and adult muscle fibres (Fig.1E and F). They confirm our previous findings in adult skeletal muscle fibres (Fig.1E and F) but also show that the receptors were expressed throughout myogenesis and that their localisation, within the cell, changed with differentiation. Thus, they were localised in the cytoplasm in myoblasts (Fig.1A and B), in the nucleus in myotubes (Fig.1C and D) and around the periphery of the muscle fibres, where they co-localised with laminin, in adult skeletal muscle fibres (Fig.1E and F). Moreover, this shift in the localisation of GRs was accompanied by an increase in the expression of laminin (green staining in Fig.1D and F).
Figure 1.
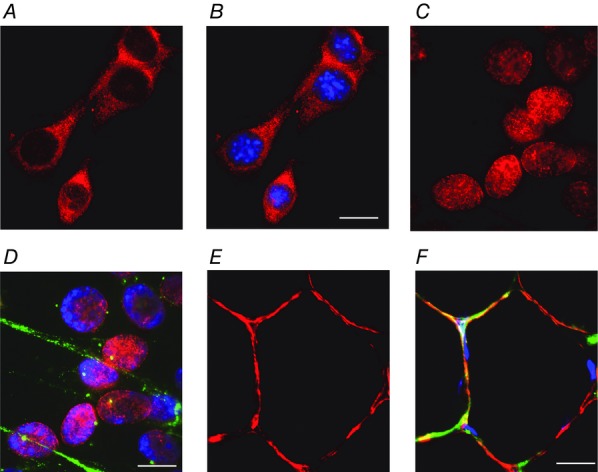
The localisation of GRs changes with muscle differentiation
Photographs showing the expression and localisation of GRs (red), laminin (green) and double stranded DNA (nucleus; blue) in myoblasts (A and B), myotubes (C and D) and muscle fibres (E and F). Note that the GRs are expressed mainly in the cytoplasm in myoblasts, in the nucleus in myotubes, and in a narrow band along the cell surface in adult muscle fibres. Scale bars: A and B, 5 μm; C and D, 2.5 μm; E and F, 15 μm.
To visualise the precise localisation of the receptors, in adult mammalian skeletal muscle fibres, a combination of immunofluorescence confocal microscopy (CM) and immunogold-labelling electron microscopy was used. Since we have previously shown that the GR is expressed mainly in oxidative fibres (Pérez et al. 2013), the soleus muscle, which contains mostly oxidative fibres (i.e. type I and IIA fibres), was used in this experiment. The immunofluorescence CM confirmed some of our previous findings. For example, it showed that most receptors were localised in clusters on the fibre surface where they generated a narrow, but discontinuous band around the fibres (Fig.2A; white dashed square). In addition, it provided new evidence showing that the receptors were also localised in large quantities in satellite cells (white arrow, Fig.2B) and to a lesser extent inside the muscle fibres where they formed a striated pattern. The immunogold labelling confirmed the CM data (Fig.2C, D and E), and clearly demonstrated that the GR was quite abundant in the extracellular matrix (Fig.2C) and in satellite cells (Fig.2E). Gold particles were also found inside the fibres, preferentially, but not exclusively, in the vicinity of mitochondria (Fig.2F), that we know are specially localised at I-bands (on both sides of Z lines) in adult fibres (Boncompagni et al. 2009). To determine the exact location of the GR signal inside the fibres, some of the fibres were double stained with an anti-GR and an antibody (anti-RYR1, 34C) that marks the position of Ca2+ release units (CRUs), or triads. In adult fibres, CRUs are specifically placed at the transition between A- and I-bands, where they form a very peculiar transverse double-cross striation on either side of the I-band (Boncompagni et al. 2009a). They also contain the sarcoplasmic reticulum Ca2+ release channels, type-1 ryanodine receptors (RYR1s). Double labelling for GRs and RYR1s (red and green, respectively, in Fig.2G) indicated that the GR staining inside the fibres was exclusively localised at the I-band, flanked on either side by the two green lines marking the position of the triads. These findings also suggested that some of the gold particles that were not at the I-band may have been non-specific (Fig.2C and D). As the I-band region of adult fibres is rich in mitochondria (Boncompagni et al. 2009), the results in Fig.2G agree with those in Fig.2E and F showing gold particles in the proximity of mitochondria (the dark and branched organelles).
Figure 2.
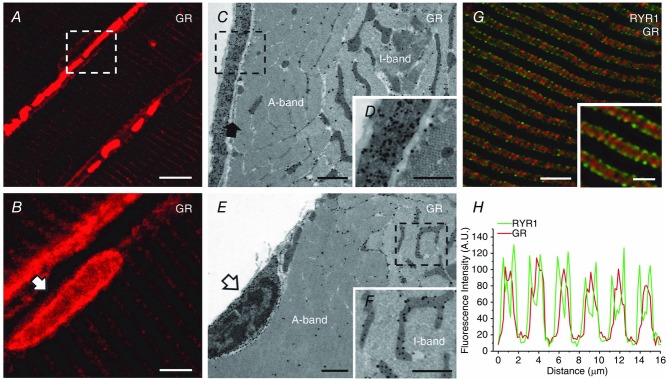
GRs are localised in the extracellular matrix and around satellite cells in adult mouse muscle fibres
A and B, immunofluorescence confocal microscopy images showing the localisation of GRs in mouse soleus. Note that the GRs are localised either in the proximity of the surface membrane, where they appear to form a narrow and discontinuous band (A; white dashed square) and in satellite cells (B, white arrow) or in the fibre interior where the signal is mostly localised in the I-band. C, D, E and F, immunogold-labelling electron microscopy photographs showing the localisation of GRs in mouse slow-twitch fibres. Note that the GRs (dark dots, i.e. gold particles) are abundant in the extracellular matrix (C and enlargement of the dotted box in D) outside the sarcolemma (C, filled arrow) and in satellite cells (E, open arrow). GRs are also present within the fibre interior almost exclusively at the I-band, and mostly in the proximity of mitochondria (C, E and F). G, a confocal micrograph showing a muscle section immunoblotted for both GRs and RYR1s. Note that the GRs (red) are mostly spread throughout the I-band inside the double green lines generated by the staining of RYR1s, which marks the position of triads. H, graph representing the fluorescence intensity profile calculated from images obtained from samples co-immunostained for RYR1s and GRs. Scale bars: A, 5 μm; B, 10 μm; C and E, 5 μm; D and F, 0.5 μm; G and inset, 10 μm and 1 μm, respectively.
To determine what tethers the GR in the extracellular matrix, laminin 2 (merosin, laminin 211) or integrins of the α7β1 type coupled to cyanogen bromide-activated sepharose beads were used to pull down the GR from slow muscle lysates. As the results displayed in Fig.3A show, only laminin-coupled beads were able to bind and pull-down the GR. Moreover, their ability to bind the receptor depended on the presence or absence of the ligand (GC). Thus, adding 500 nm BDP to the muscle lysate significantly reduced the amount of GR pulled down by the beads (Fig.3B) suggesting that the GC was able to dissociate the GR from laminin.
Figure 3.
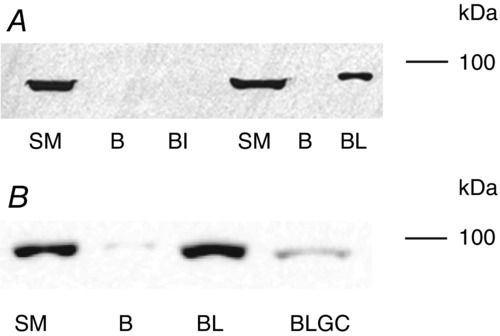
GRs bind laminin in a GC-dependent manner
A and B, Western blots showing the expression of GRs in a slow-twitch muscle (SM; control), in loading buffer used to elute GRs from CNBr-activated sepharose beads alone (B), or with the beads cross-linked with laminin 211 (BL) or with integrin α7β1 (BI). In B the lysate used in the pull-down assay of the sample labelled (BLGC) was pre-treated with 500 nm BDP for 10 min before it was used in the assay. Note that only the beads coupled with laminin are able to bind and pull down the GR and treating the lysate with GC abolishes this interaction.
To determine the cellular signalling pathways activated by physiological concentrations of GCs in adult skeletal muscle, small muscle fibre bundles isolated from the EDL (a fast-twitch muscle) and soleus (a slow-twitch muscle) were treated with 250 nm BDP (a synthetic GC widely used in the management of asthma). The phosphorylation (= activation) of JNK, ERK1&2 and p38 MAPK was then determined. As the results displayed in Figs4 and 5 show, the effects of BDP on the phosphorylation of these kinases was complex and depended on the fibre type and the duration of the treatment. For example, treating the muscle fibre bundles with BDP for 5 min increased the phosphorylation of p38 MAPK and ERK1&2 in the fast-twitch muscle fibres (Figs4A and C, and 5A, D and E) and that of JNK in the slow-twitch fibres (Figs4B, and 5B and C). Increasing the treatment time to 30 min further enhanced the activation of p38 MAPK in the fast-twitch fibres (Figs4A and 5A) and that of ERK1&2 (Figs4C, and 5D and E). However, it led to the activation of JNK in both fibre types (see Figs4B and C, and 5B and C). In contrast, prolonging the treatment duration to 60 min led to a slight decrease in the phosphorylation of p38 MAPK and a marked decline in that of JNK in the fast-twitch fibres (Fig.4A and B, and 5A, B and C) but further enhanced that of JNK in the slow-twitch fibres (Fig.4B, and 5B and C). On the other hand, the phosphorylation of ERK1&2 in the fast-twitch fibres remained elevated and was similar to that seen after the 30 min treatment (Figs4C, and 5D and E), whereas in the slow-twitch fibres it was slightly reduced (Fig.5D and E).
Figure 4.
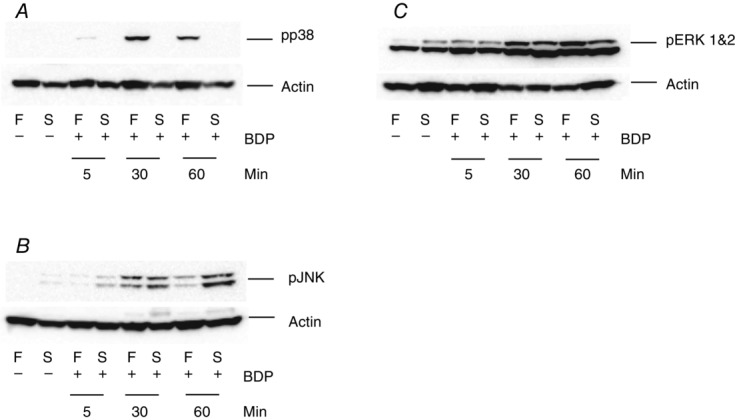
The activation of MAPKs by BDP in mammalian skeletal muscle fibres depends on the fibre type and the duration of the treatment
Western blots showing the effects of treating fast-twitch (F) and slow-twitch (S) muscle fibre bundles with the Ringer solution only (–) or the Ringer solution plus 250 nm BDP (+), for 5, 30 and 60 min, on the phosphorylation of p38 MAPK (A), JNK (B) and ERK1&2 (C). Note that the effects of BDP depend on the fibre type and the duration of the treatment.
Figure 5.
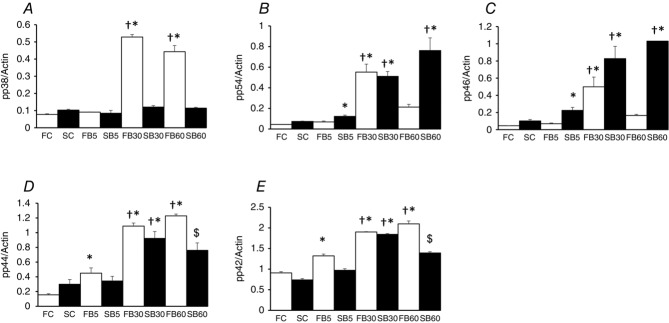
BDP activates MAPKs in mouse skeletal muscle fibres in a fibre-type and duration-dependent manner
Summary data showing the effects of treating fast-twitch (F) and slow-twitch (S) muscle fibre bundles with the Ringer solution only (FC; SC) or the Ringer solution plus 250 nm BDP for 5 (FB5; SB5), 30 (FB30; SB30) and 60 min (FB60; SB60) on the phosphorylation of p38 MAPK (A), the 54 kDa isoform of JNK (B; p54); the 46 kDa isoform of JNK (C; p46); ERK1 (D; p44) and ERK2 (D; p42). Note that the effects of BDP depend on the fibre type and the duration of the treatment. *P < 0.05 when the data indicated are compared to those of the corresponding untreated (control) fibres. †P < 0.05 when the data indicated are compared to those of the corresponding fibres treated with BDP for 5 min. $P < 0.05 when the data indicated are compared to those of the corresponding fibres from the other treatments.
The findings in Figs4 and 5 suggested that the activation of MAPKs by GCs in mammalian skeletal muscles was non-genomic (i.e. it occurred within 5 min). However, they did not tell us the receptor/mechanism underlying this effect. As the mechanisms underlying the non-genomic effects of GCs are still controversial (Lipworth, 2000; Stahn & Buttgereit, 2008; Pérez et al. 2013), we investigated whether the activation of the var-ious MAPKs by BDP were mediated through the GR or through another receptor/mechanism. As the results displayed in Fig.6A and B show, pre-treating the muscle fibre bundles with 10 μm mifepristone (RU486, a GR inhibitor), for 10 min, completely abolished or significantly reduced the 30 min BDP-induced activation of p38 MAPK and JNK suggesting that they were mediated by a GR. To determine whether they were mediated by a cytosolic GR or a membrane GR, the experiment above was repeated using the rabbit monoclonal anti-GR used in the immunocytochemistry and Western blotting experiment. The rationale behind this experiment was that the antibody was too large to cross the cell membrane and that it could only bind the GR and inhibit its effects if the receptor was easily accessible, i.e. located close to the cell surface. As the results presented in Fig.6C and D show, pre-treating the muscle fibre bundles with the antibody for 10 min completely abolished the BDP-induced activation of JNK and p38 MAPK in both fibre types suggesting that they were mediated by a GR that was easily accessible to the antibody (i.e. a cell surface GR). They also suggested that the antibody bound the GR close to the ligand binding site or caused a conformational change to the receptor that inhibited its activation of the MAPK pathway.
Figure 6.
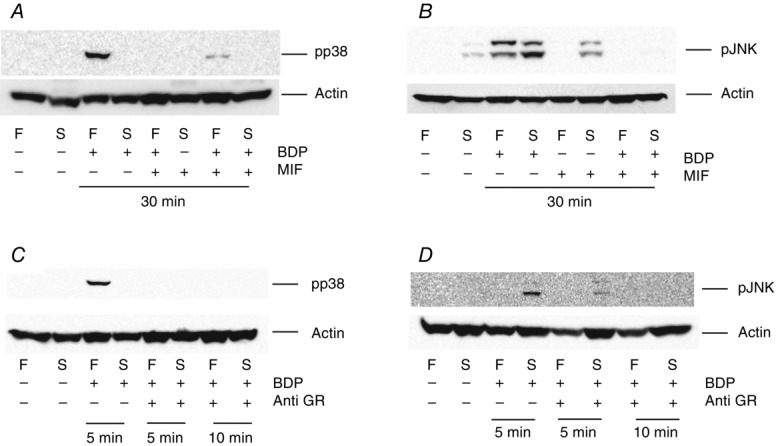
The effects of BDP are mediated through cell surface (membrane) GRs
Western blots showing the effects of pre-treating fast-twitch (F) and slow-twitch (S) muscle fibre bundles with10 μm mifepristone (MIF, a GR inhibitor; A and B) or a 1:5000 dilution of an anti-GR (C and D), for 10 min, on the pho-sphorylation of p38 MAPK (A and C) and JNK (B and D). The muscle fibre bundles were then treated with the compounds for the durations shown below each blot. Note that both mifepristone and the antibody abolish the BDP-induced increase in the phosphorylation of both MAPKs.
The results presented so far suggested that the effects of BDP on the activation of the MAPK pathway were non-genomic and were mediated by a GR located close to the cell surface. They also suggested that it may be bound to laminin. However, they did not tell us how the GC was able to activate the MAPKs. In mammalian skeletal muscle fibres, laminin is localised in the basal laminar (one of the two layers that form the basement membrane of skeletal muscles) and is thought to communicate with the rest of the cell through integrins (mainly integrin α7β1 in skeletal muscle) and dystroglycan (Gawlik & Durbeej, 2011). This led us to hypothesise that the GR signalled to the MAPK through the activation of focal adhesion kinase (FAK) by integrins (see Fig.9). To test this hypothesis, we pre-treated another set of muscle fibre bundles with the FAK specific inhibitor FAK inhibitor 14. As the results displayed in Figs7 and 8 show, pre-treatment of the fibre bundles with the inhibitor completely abolished the GC-induced activation of p38 MAPK (Figs7A and 8A) and ERK1 (p44; Figs7C, and 8D and E). However, pre-treatment of the muscle bundles with the inhibitor alone augmented the activation of JNK (especially the 46 kDa isoform) in the slow twitch fibres and returned to the level observed in the BDP-treated fibres when the bundles were treated with both the inhibitor and the GC (Figs7B, and 8B and C).
Figure 9.
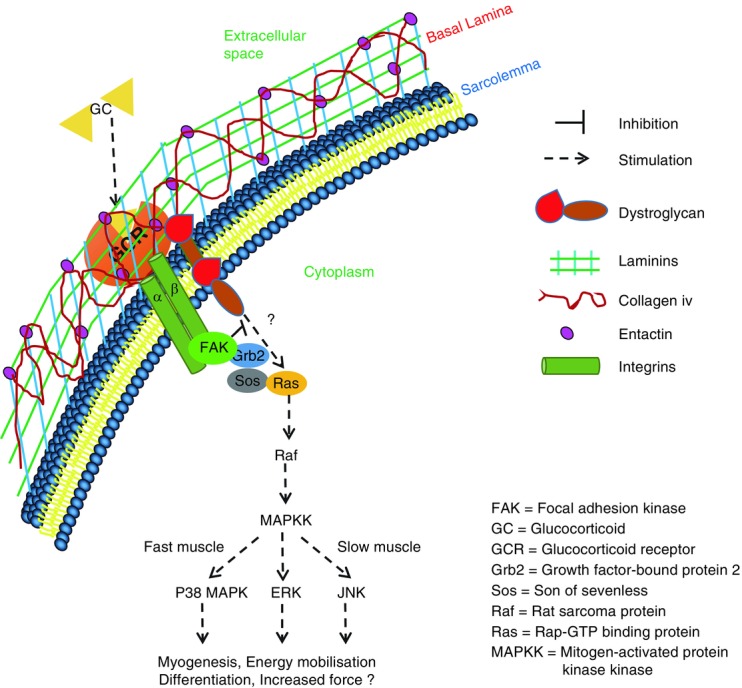
A schematic diagram summarising the mechanism we propose underlies the non-genomic effects of BDP in mammalian muscle fibres
Our hypothesis is that some of the GRs (‘GCR’ in the figure) are localised in the basement membrane where they are bound by laminin and that their stimulation by GCs leads to the activation of either integrins (in fast-twitch fibres) or dystroglycan (in slow-twitch fibres). We also think that integrins recruit and activate FAK leading eventually to the phosphorylation of p38 MAPK and ERK1&2, whereas dystroglycans act lower down the pathway and lead to the activation of JNK. We also think that FAK has inhibitory effects on the dystroglycan pathway.
Figure 7.
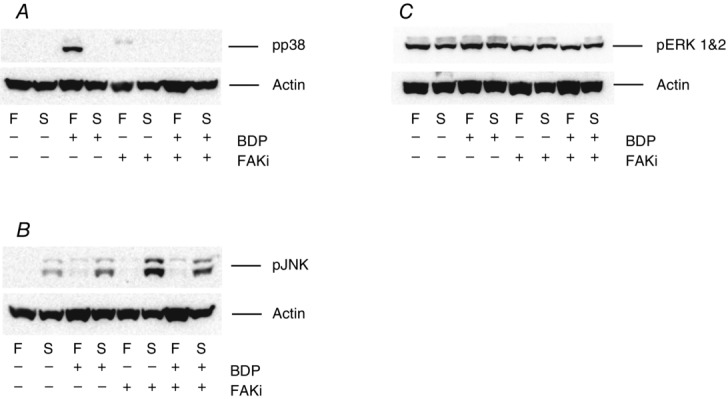
The BDP-induced activation of p38 MAPK is mediated through focal adhesion kinase (FAK)
Western blots showing the effects of pre-treating fast-twitch (F) and slow-twitch (S) muscle fibre bundles with 10 μm FAK inhibitor (FAKi) 14, for 10 min, on the phosphorylation of p38 MAPK (A), JNK (B) and ERK1&2 (C). The muscle fibre bundles were then treated with the Ringer solution containing BDP alone or BDP plus the inhibitor as shown below each Western blot for a further 10 min. Note that pre-treating the fibre bundles with FAKi 14 abolishes the BDP-induced increase in the phosphorylation of p38 MAPK (A) and ERK1&2 (C) but not that of JNK (B).
Figure 8.
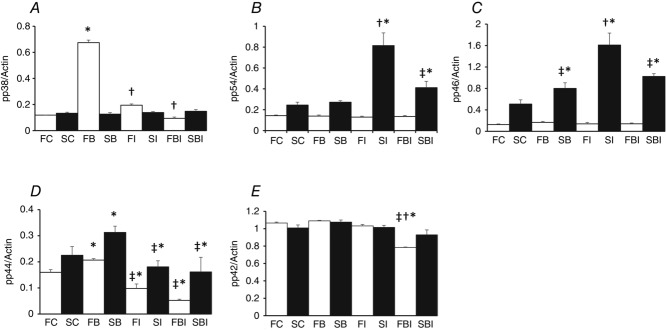
FAKi 14 inhibits the BDP-induced increase in phosphorylation of p38 MAPK and ERK1 (p44)
Summary data showing the effects of pre-treating fast-twitch (F) and slow-twitch (S) muscle fibre bundles with the Ringer solution only (FC; SC), the Ringer solution plus 10 μm FAKi (FI; SI) or the Ringer solution plus the inhibitor as well as the GC (FBI; SBI) for 10 min, on the phosphorylation of p38 MAPK (A), the 54 kDa isoform of JNK (B; p54); the 46 kDa isoform of JNK (C; p46); ERK1 (D; p44) and ERK2 (E; p42). Note that pre-treatment with FAKi inhibits the BDP-induced increase in the phosphorylation of p38 MAPK and ERK1 but not that of the two JNK isoforms. *P < 0.05 when the data indicated are compared to those of the corresponding untreated (control) fibres. †P < 0.05 when the data indicated are compared to those of the corresponding fibres treated with BDP. ‡P < 0.05 when the data indicated are compared to those of the fibres treated with the inhibitor only.
Discussion
Localisation of the GR in mouse skeletal muscle cells
The results we report here show for the first time that GRs are expressed at all stages of myogenesis. However, their localisation changes with differentiation (Fig.1). They also revealed that, in adult mammalian skeletal muscle fibres, they were localised mostly in the extracellular matrix (Fig.2A and C), in satellite cells (Fig.2B and D) and in the I-band region where they were closely associated with mitochondria. Although a number of studies have previously suggested that a membrane glucocorticoid receptor exists in many cell types (Orchinik et al. 1991; Gametchu, 1993; Bartholome et al. 2004; Pérez et al. 2013), this is the first study to show that a GR localised close to the cell membrane exists. However, as our results show, it is localised in the extracellular matrix and in satellite cells and not in the plasma membrane as previous studies have suggested.
Adult mammalian skeletal muscle fibres, unlike most other cell types, are long, cylindrical, multinucleated cells whose nuclei are located close to the fibre surface. Their cytoplasm (= sarcoplasm) is also packed with contractile proteins that are organised into striations consisting of highly regular repeats of thin (actin) and thick (myosin) filaments. Slow-twitch fibres also contain large quantities of mitochondria that are mostly found at the I-band region (Boncompagni et al. 2009) and under the sarcolemma (mostly in the proximity of capillaries). Here, we propose that this unique structural organisation of mammalian skeletal muscle fibres is responsible for the distinctive distribution and localisation of GRs in adult mammalian skeletal muscle fibres. We also hypothesise that, as myoblasts differentiate into myotubes, GRs migrate to the nucleus and as contractile proteins are laid down, the nucleus, the GRs and most other cell organelles are pushed to the periphery of the muscle fibres. However, how the GRs get out of the muscle fibres and localise in the extracellular matrix is uncertain. Also, why and how the receptor localises in the proximity of mitochondria is also uncertain and further studies to investigate these uncertainties are necessary.
Another possibility is that GRs are bound to or are closely associated with cytoskeletal proteins such as dystrophins, dystroglycans, utrophins and laminins. In adult mammalian skeletal muscle fibres, these proteins are important in the maintenance of cell shape, structure and function, and are closely associated with the cell membrane (Gawlik & Durbeej, 2011). Indeed, GCs have been shown to increase the content of dystrophin and utrophin in both normal and dystrophic differentiating human skeletal muscle satellite cells (Sklar et al. 1991; Pasquini et al. 1995). In the present study, GRs were also found to be able to bind laminin (Fig.3). The reason why GC treatment increases the accumulation of these proteins is uncertain. Here we postulate that, in mammalian skeletal muscles, GCs, and hence GRs, may be important for the synthesis, recruitment and/or organisation of these extracellular matrix proteins. Indeed, the present findings show that myoblasts expressed little or no laminin (Fig.1A and B) and laminin only started to appear in myotubes when the GRs translocated into the nucleus (Fig.1C and D).
Effects of GCs on the MAPK pathway
Another novel finding in the present study is the observation that treating mammalian skeletal muscle fibre bundles with physiological concentrations of BDP increases the phosphorylation/activation of ERK1&2, JNK and p38 MAPK in a fibre type- and duration-dependent manner (Figs 4 and 5). MAPKs are also activated in brain tissues of rats subjected to short periods of psychological stress (Gutièrrez-Mecinas et al. 2011) as well as in hippocampal and PC12 (rat phaeochromocytoma) cells treated with low doses of corticosterone (Li et al. 2001; Qi et al. 2005). However, treatment of most other cell types, including HeLa cells, vascular endothelial cells, macrophages, mast cells and monocytes, with low doses of GCs has been shown to have the opposite effect, i.e. it decreases MAPK activation (Franklin et al. 1997; Caelles et al. 1997; González et al. 2000; Kassel et al. 2001; Imasato et al. 2002; Lasa et al. 2002; Bruna et al. 2003). Together these findings suggest that the effects of GCs on the MAPK pathway depend on the tissue that the cells are derived from and the physiological role of stress and MAPKs in that tissue. For example, in tissues such as the brain and skeletal muscle that are directly involved in the stress response and where activation of MAPKs by short term stress is beneficial (Gutièrrez et al. 2011; Pérez et al. 2013; present study), physiological concentrations of GCs increase the activation of MAPKs. Conversely, in tissues such as the immune system where activation of MAPKs by stress is detrimental to the long term survival of the organism, GCs inhibit their activation (Franklin et al. 1997; Caelles et al. 1997; González et al. 2000; Bruna et al. 2003).
Physiological functions of GCs
In mammalian skeletal muscle cells, all three MAPKs (p38 MAPK, JNK and ERK) have been implicated in the regulation of myogenesis and the expression of skeletal muscle-specific genes (Bennett & Tonks, 1997; Cuenda et al. 1999; Weston et al. 2003). Additionally, intense exercise (Widegren et al. 2000) and marathon running (Yu et al. 2001), both of which lead to fibre type switch, have been shown to induce the release of GCs and the activation of p38 and ERK1&2 in the vastus lateralis portion of the quadriceps femoris muscle (note that the vastus lateralis consists mostly of fast-twitch muscle fibres). p38 MAPKs have also been shown to act as molecular switches in the activation of quiescent satellite cells (Jones et al. 2005). Myogenesis is important in skeletal muscle development, whereas satellite cell activation is essential for skeletal muscle repair/regeneration. The fact that GC treatment increases the activation of all three MAPKs in mouse skeletal muscle fibres (present study) and strenuous exercise activates p38 and ERK1&2 in fast-twitch fibres suggests that GCs may play an important role in both myogenesis and skeletal muscle repair/regeneration. However, the exact role of GCs in myogenesis and satellite cell activation is uncertain and further studies to confirm this are necessary.
The GC-induced activation of ERK1&2 we report here may also be responsible for the GC-induced increase in maximum isometric force (Po) in slow-twitch muscle fibres previously reported by Pérez and her colleagues (Pérez et al. 2013). Here we speculate that the GC-induced activation of ERK1&2 leads to the phosphorylation of the regulatory myosin light chain (RMLC) and the potentiation of Po as previously shown in Hamdi & Mutungi (2010). Fast-twitch and slow-twitch muscle fibres express different isoforms of RMLCs that are phosphorylated at low stimulation frequencies in fast-twitch fibres and at high frequencies in slow-twitch fibres (Moor & Stull, 1984). The phosphorylation of RMLCs in fast-twitch fibres increases sub-maximal force (including twitch contraction), whereas their function in slow-twitch muscles is unknown as their phosphorylation does not seem to affect twitch amplitude (Moore & Stull, 1984). Here we speculate that the phosphorylation of RMLCs in slow-twitch fibres leads to the potentiation of maximum isometric force. However, the exact mechanism underlying the GC-induced increase in force is uncertain and further studies to confirm this are needed.
Mechanism underlying the effects of GCs in skeletal muscle
As mentioned above, in most cells, GCs inhibit the MAPK pathway. They do this either by up-regulat-ing the synthesis and activation of proteins such as glucocorticoid-induced-leucine zipper, MAPK phosphatase-1 and annexin-1 (González et al. 2000; Lasa et al. 2002) or by directly interacting with components of the MAPK pathway (Caelles et al. 1997; Bruna, 2003). The results reported here suggest that the effects of GCs in mammalian skeletal muscle fibres are mediated through GRs localised close to the cell surface (Fig.6) and that their stimulation leads to the activation of focal adhesion kinase (FAK, Fig.9). Here we hypothesise that in mammalian skeletal muscle fibres, membrane GRs are bound to laminin and that their stimulation by GCs leads to a conformational change in laminin and the activation of FAK by integrins (Fig.9). In mammalian skeletal muscles, laminin binds to and signals through both integrin α7β1 and dystroglycan (Gawlik & Durbeej, 2011). Therefore, we think that the conformational change in laminin activates integrin α7β1 in fast-twitch muscle fibres and dystroglycan in slow-twitch fibres (Fig.9). We also think that integrins recruit and activate FAK leading eventually to the phosphorylation of p38 MAPK and ERK1&2, whereas dystroglycans act slightly lower down the pathway and their stimulation leads to the activation of JNK. We also think that FAK has inhibitory effects on the dystroglycan pathway (Fig.9). However, the exact mechanism mediating the GC-induced increase in the activation of MAPKs in mammalian skeletal muscle fibres is uncertain and further studies to determine this are necessary.
Glossary
- BDP
beclomethasone dipropionate
- EDL
extensor digitorum longus
- ERK1&2
extracellular signal-regulated kinases 1 and 2
- FAK
focal adhesion kinase
- FAKi-14
focal adhesion kinase inhibitor 14
- GC
glucocorticoid
- GR
glucocorticoid receptor
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- RYR1
type-1 ryanodine receptor
Additional information
Competing interests
None declared.
Author contributions
G.M. was responsible for the conception and design of the experiments. He dissected the small muscle fibre bundles and helped with the various treatments. He also did the immunocytochemistry experiments and wrote the manuscript. S.B. and F.P. did all the confocal and electron microscopy experiments including the immunogold labelling. L.A. did most of the Western blotting under the supervision of G.M. He also helped with the treatment of the muscle fibre bundles with the various compounds. E.A. and L.A. did the pull-down assays with the help of D.S. T.P. performed all the C2C12 cell experiments. All authors approved the final version of the manuscript. All the immunocytohistochemistry and cell signalling experiments were performed in the BioMedical Research Centre, University of East Anglia; whereas, the immunofluorescence confocal microscopy and immunogold labelling electron microscopy experiments were performed in the Department of Neuroscience, Imaging and Clinical Sciences, University G. d’ Annunzio.
Funding
This study was funded by BBSRC grant BB/J01754X/1 to G.M. L.A. and E.A. were Wellcome Trust and Society of Biology vacation students, respectively. This study was also supported by the following grants: Italian Telethon ONLUS Foundation GGP13213 (to F.P.) and RBFR13A20K Future in Research 2013 MIUR (Italian Ministry of Education, Universities and Research) (to S.B.).
References
- Alangari AA. Genomic and non-genomic actions of glucocorticoids in asthma. Ann Thorac Med. 2010;5:133–139. doi: 10.4103/1817-1737.65040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, Bienert M, Radbruch A, Burmester GR, Lauster R, Scheffold A. Buttgereit F. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J. 2004;18:70–80. doi: 10.1096/fj.03-0328com. [DOI] [PubMed] [Google Scholar]
- Bennett AM. Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT. Protasi F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell. 2009;20:1058–1067. doi: 10.1091/mbc.E08-07-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A, Nicolas M, Munoz A, Kyriakis JM. Caelles C. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 2003;22:6035–6044. doi: 10.1093/emboj/cdg590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit F, Krauss S. Brand MD. Methylprednisolone inhibits uptake of Ca2+ and Na+ ions into concanavalin A-stimulated thymocytes. Biochem J. 1997;326:329–332. doi: 10.1042/bj3260329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caelles C, González-Sancho JM. Muñoz A. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 1997;11:3351–3364. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxtall JD, Choudhury Q. Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br J Pharmacol. 2000;130:289–298. doi: 10.1038/sj.bjp.0703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A. Cohen P. Stress-activated protein kinase-2 p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J Biol Chem. 1999;274:4341–4346. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- De Beer FC, Baltz ML, Holford S, Feinstein A. Pepys MB. Fibronectin and C4-binding protein are selectively bound by aggregated amyloid P component. J Exp Med. 1981;154:1134–1149. doi: 10.1084/jem.154.4.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin CC. Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem. 1997;272:16917–16923. doi: 10.1074/jbc.272.27.16917. [DOI] [PubMed] [Google Scholar]
- Gametchu B, Watson CS. Wu SH. Use of receptor antibodies to demonstrate membrane glucocorticoid receptor in cells from human leukemic patients. FASEB J. 1993;7:1283–1292. doi: 10.1096/fasebj.7.13.8405814. [DOI] [PubMed] [Google Scholar]
- Gawlik KI. Durbeej M. Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle. 2011;1:9. doi: 10.1186/2044-5040-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Protein turnover in skeletal muscle. 2. Effects of denervation and cortisone on protein catabolism in skeletal muscle. J Biol Chem. 1969;244:3223–3229. [PubMed] [Google Scholar]
- González MV, Jiménez B, Berciano MT, González-Sancho JM, Caelles C, Lafarga M. Muñoz A. Glucocorticoids antagonize AP-1 by inhibiting the activation/phosphorylation of JNK without affecting its subcellular distribution. J Cell Biol. 2000;150:1199–1207. doi: 10.1083/jcb.150.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutièrrez-Mecinas M, Trollope AF, Collins A, Morfett H, Hesketh SA, Kersanté F. Reul JM. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc Natl Acad Sci U S A. 2011;108:13806–13811. doi: 10.1073/pnas.1104383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Simoncini T, Yang ZQ, Limbourg FP, Plumier JC, Rebsamen MC, Hsieh CM, Chui DS, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K. Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi M. Mutungi G. Dihydrotestosterone activates the MAPK pathway and modulates maximum isometric force through the EGF receptor in isolated intact mouse skeletal muscle fibres. J Physiol. 2010;588:511–525. doi: 10.1113/jphysiol.2009.182162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi M. Mutungi G. Dihydrotestosterone stimulates amino acid uptake and the expression of LAT2 in mouse skeletal muscle fibres through an ERK1/2-dependent mechanism. J Physiol. 2011;589:3623–3640. doi: 10.1113/jphysiol.2011.207175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua SY. Chen YZ. Membrane receptor-mediated electrophysiological effects of glucocorticoid on mammalian neurons. Endocrinology. 1989;124:687–691. doi: 10.1210/endo-124-2-687. [DOI] [PubMed] [Google Scholar]
- Imasato A, Desbois-Mouthon C, Han JH, Kai H, Cato ACB, Akira S. Li JD. Inhibition of p38 MAPK by glucocorticoids via induction of MAPK phosphatase-1 enhances nontypeable Haemophilus influenzae-induced expression of toll-like receptor 2. J Biol Chem. 2002;277:47444–47450. doi: 10.1074/jbc.M208140200. [DOI] [PubMed] [Google Scholar]
- Jones NC, Tyner KJ, Nibarger L, Stanley HM, Cornelison DDW, Fedorov YV. Olwin BB. The p38α/β MAPK functions as a molecular switch to activate the quiescent satellite cell. J Cell Biol. 2005;169:105–116. doi: 10.1083/jcb.200408066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmiel M. Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34:518–530. doi: 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M. Cato ACB. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001;20:7108–7116. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger DT, Allen W, Rizzo F. Krieger HP. Characterization of the normal temporal pattern of plasma corticosteroid levels. J Clin Endocrinol Metab. 1971;32:266–284. doi: 10.1210/jcem-32-2-266. [DOI] [PubMed] [Google Scholar]
- Lasa M, Abraham SM, Boucheron C, Saklatvala J. Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22:7802–7811. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Qiu J, Wang J, Zhong Y, Zhu J. Chen Y. Corticosterone-induced rapid phosphorylation of p38 and JNK mitogen-activated protein kinases in PC12 cells. FEBS Lett. 2001;492:210–214. doi: 10.1016/s0014-5793(01)02254-2. [DOI] [PubMed] [Google Scholar]
- Lipworth BJ. Therapeutic implications of non-genomic glucocorticoid activity. Lancet. 2000;356:87–89. doi: 10.1016/S0140-6736(00)02463-6. [DOI] [PubMed] [Google Scholar]
- Ma K, Mallidis C, Bhasin S, Mahabadi V, Artaza J, Gonzalez-Cadavid N, Arias J. Salehian B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab. 2003;285:E363–E371. doi: 10.1152/ajpendo.00487.2002. [DOI] [PubMed] [Google Scholar]
- Moore RL. Stull JT. Myosin light chain phosphorylation in fast and slow skeletal-muscles in situ. Am J Physiol. 1984;247:C462–C471. doi: 10.1152/ajpcell.1984.247.5.C462. [DOI] [PubMed] [Google Scholar]
- Nava S, Gayan-Ramirez G, Rollier H, Bisschop A, Dom R, de Bock V. Decramer M. Effects of acute steroid administration on ventilatory and peripheral muscles in rats. Am J Respir Crit Care Med. 1996;153:1888–1896. doi: 10.1164/ajrccm.153.6.8665051. [DOI] [PubMed] [Google Scholar]
- Orchinik M, Murray TF. Moore FL. A corticosteroid receptor in neuronal membranes. Science. 1991;252:1848–1851. doi: 10.1126/science.2063198. [DOI] [PubMed] [Google Scholar]
- Pasquini F, Guerin C, Blake D, Davies K, Karpati G. Holland P. The effect of glucocorticoids on the accumulation of utrophin by cultured normal and dystrophic human skeletal-muscle satellite cells. Neuromuscul Disord. 1995;5:105–114. doi: 10.1016/0960-8966(94)00042-8. [DOI] [PubMed] [Google Scholar]
- Pérez MH-A, Cormack J, Mallinson D. Mutungi G. A membrane glucocorticoid receptor mediates the rapid/non-genomic actions of glucocorticoids in mammalian skeletal muscle fibres. J Physiol. 2013;591:5171–5185. doi: 10.1113/jphysiol.2013.256586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WB. Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- Qi AQ, Qiu J, Xiao L. Chen YZ. Rapid activation of JNK and p38 by glucocorticoids in primary cultured hippocampal cells. J Neurosci Res. 2005;80:510–517. doi: 10.1002/jnr.20491. [DOI] [PubMed] [Google Scholar]
- Rannels SR. Jefferson LS. Effects of glucocorticoids on muscle protein-turnover in perfused rat hemicorpus. Am J Physiol. 1980;238:E564–E572. doi: 10.1152/ajpendo.1980.238.6.E564. [DOI] [PubMed] [Google Scholar]
- Sklar RM. Brown RH. Methylprednisolone increases dystrophin levels by inhibiting myotube death during myogenesis of normal human muscle in vitro. J Neurol Sci. 1991;101:73–81. doi: 10.1016/0022-510x(91)90019-4. [DOI] [PubMed] [Google Scholar]
- Stahn C. Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol. 2008;4:525–533. doi: 10.1038/ncprheum0898. [DOI] [PubMed] [Google Scholar]
- van Balkom RHH, van der Heijden HFM, van Moerkerk HTB, Veerkamp JH, Fransen JAM, Ginsel LA, Folgering HTM, van Herwaarden CLA. Dekhuijzen PNR. Effects of different treatment regimens of methylprednisolone on rat diaphragm contractility, immunohistochemistry and biochemistry. Eur Respir J. 1996;9:1217–1223. doi: 10.1183/09031936.96.09061217. [DOI] [PubMed] [Google Scholar]
- Weston AD, Sampaio AV, Ridgeway AG. Underhill TM. Inhibition of p38 MAPK signaling promotes late stages of myogenesis. J Cell Sci. 2003;116:2885–2893. doi: 10.1242/jcs.00525. [DOI] [PubMed] [Google Scholar]
- Widegren U, Wretman C, Lionikas A, Hedin G. Henriksson J. Influence of exercise intensity on ERK/MAP kinase signalling in human skeletal muscle. Pflugers Arch. 2000;441:317–322. doi: 10.1007/s004240000417. [DOI] [PubMed] [Google Scholar]
- Wing SS. Goldberg AL. Glucocorticoids activate the ATP-ubiquitin-dependent proteolytic system in skeletal-muscle during fasting. Am J Physiol. 1993;264:E668–E676. doi: 10.1152/ajpendo.1993.264.4.E668. [DOI] [PubMed] [Google Scholar]
- Yu M, Blomstrand E, Chibalin AV, Krook A. Zierath JR. Marathon running increases ERK1/2 and p38 MAP kinase signalling to downstream targets in human skeletal muscle. J Physiol. 2001;536:273–282. doi: 10.1111/j.1469-7793.2001.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]