

Abstract
Site-directed mutagenesis identified residues in the substrate channel of LodA that play multiple roles in regulating Km values of substrates, kcat and the extent of biosynthesis of the protein-derived cysteine tryptophylquinone (CTQ) cofactor. Mutations of Cys448 increase Km values for lysine and O2, with the larger effect on Klysine. Tyr211 resides within a mobile loop and is seen in the crystal structure of LodA to form a hydrogen bond with Lys530 that appears to stabilize its position in the channel. Y211F LodA had reduced levels of CTQ but near normal levels of kcat. K530A and K530R variants exhibited diminished levels of CTQ but significantly increased kcat. The Y211F, K530A and K530R mutations each caused large increases in the Km values for lysine and O2. These effects of the mutations of Tyr211 and Lys530 suggest that when these residues are hydrogen-bonded they may form a gate that controls entry and exit of substrates and products from the active site. Y211A and Y211E variants had the highest level of CTQ but exhibited no activity. These results highlight the different evolutionary factors that must be considered for enzymes which possess protein-derived cofactors, in which the catalytic cofactor must be generated by posttranslational modifications.
Graphical abstract
Introduction
LodA (previously called marinocine) is a ε-lysine oxidase that was first discovered in Marinomonas mediterranea, a melanogenic marine bacterium [1–3]. It catalyzes the oxidative deamination of lysine by removing the ε-amino group and releasing the aldehyde product plus hydrogen peroxide (eq 1). When secreted out into the biofilm in which the bacterium resides, LodA exerts antimicrobial activity as a consequence of the production of H2O2. This is beneficial because causing death of a subpopulation of cells facilitates biofilm differentiation and dispersal of surviving cells with phenotypic variation among dispersed cells [4]. Wild type (WT) LodA has been recombinantly expressed in E. coli [5] and isolated. LodA contains a protein-derived cofactor [6] which is generated by posttranslational modifications. The cofactor is cysteine tryptophylquinone (CTQ) which is formed by the di-oxygenation of Trp581 and the crosslinking of this residue with Cys516 (Figure 1). Expression of active LodA with correctly synthesized CTQ requires the co-expression of the lodB gene [5], which encodes a flavoprotein that is required for the posttranslational modifications that generate CTQ.
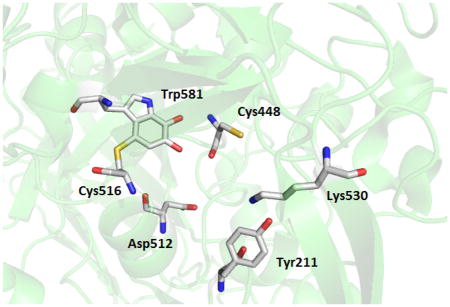
Figure 1.

Residues of interest in the active site of LodA. A portion of the structure of LodA (PDB entry 2YMW) is shown with the residues that form CTQ and other residues of interest in the active site shown as sticks.
| [1] |
LodA is the first enzyme that was shown to use a tryptophylquinone cofactor that functions as an oxidase. All previously described enzymes that use CTQ or tryptophan tryptophylquinone (TTQ) cofactors are dehydrogenases that use other redox proteins or small molecules other than NAD(P)+ as electron acceptors [7]. LodA is distinct from the other known tryptophylquinone enzymes in using an amino acid as a substrate. LodA is also unusual among amino acid oxidases since the other enzymes previously described in this group utilize a flavin cofactor. Three crystal structures of LodA have been determined, LodA isolated from M. mediterranea alone (PDB 3WEU) and with the lysine substrate covalently-bound to CTQ (PDB 3WEV) [8], and recombinant LodA isolated from E. coli (PDB 2YMW). Inspection of these structures identified the residues which are modified to form CTQ, Trp581 and Cys516. It was subsequently shown that mutation of either of these two residues to Ala resulted in no CTQ biosynthesis [9]. It was also shown that conversion of Asp512, which is in close proximity to CTQ, to Ala led to expression of a precursor of LodA without CTQ [9]. A similar effect was reported when the structurally analogous Asp was mutated in methylamine dehydrogenase (MADH). That led to expression of a precursor of MADH without TTQ [10].
A steady-state kinetic study showed that LodA follows a ping-pong mechanism in which it first interacts with lysine forming a Schiff base intermediate with CTQ, then releases the aldehyde product, and then binds O2 to finally release NH3 and H2O2 [11] (Figure 2). From the structure of LodA it was also possible to identify other residues of interest in the active site of LodA which could participate in binding of one or both substrates, or catalysis, as well as CTQ biosynthesis (Figure 1). These residues are the topic of this study. Cys 448 is located in close proximity to the quinone oxygens of CTQ. Two other quinoproteins, the CTQ-containing quinohemoprotein amine dehydrogenase (QHNDH) [12] and the TTQ-containing MADH [13], have a structurally conserved Asp residue in the position corresponding to Cys448 in the LodA structure in the active site. Mutation of the corresponding Asp in MADH affected the efficiency of MauG-dependent TTQ biosynthesis but had no effect on the catalytic activity of the population of the isolated MADH with fully formed TTQ [10]. As such, Cys448 in LodA was converted to Asp, as well as Ala, to ascertain its role in CTQ biosynthesis or catalysis or both. Tyr211 resides in a loop comprised of residues 206 to 215, and it is flanked by one and three Gly residues on either side. This loop which is located close to the entrance of the active site was not visible in the crystals structures of the free and substrate bound native enzymes. In the structure of recombinant LodA, this loop was defined in the structure of one of the two molecules in the asymmetric unit but not in the other. These observations are consistent with mobility of this loop and a possible role in controlling substrate binding or product release, or both, from the active site. To probe the role of Tyr211 it was converted to Phe, Ala and Glu. Lys530 is of interest because it appears to form a hydrogen bond (2.7 Å) between its ε-amino N and the phenolic O of Tyr211. It was converted to Ala and Arg. The results of this study demonstrate that these residues in the active site of LodA play multiple roles in binding of lysine and O2, catalysis and CTQ biosynthesis. Analysis of the Integrated Microbial Genomes database of genome sequences identified 168 genes from 144 different bacterial sequences that encoded LodA-like proteins [14]. LodA from M. mediterranea was classified as a member of Group IA LodA-like proteins. The residues which were mutated in this study were highly conserved in these sequences. These results are discussed in the context of the different evolutionary factors that must be considered for enzymes which possess protein-derived cofactors, in which the catalytic cofactor must be generated by posttranslational modifications.
Figure 2.

Reaction mechanism for the LodA-catalyzed oxidative deamination of lysine. B represents an active-site residue which participates in acid–base chemistry.
Experimental
Construction, expression and purification of recombinant proteins
Wild-type (WT) LodA and LodA variants that were generated by site-directed mutagenesis were expressed in E. coli Rosetta cells as described previously with the lodB gene co-expressed [5, 11]. The recombinant LodA possessed a 6XHis tag to facilitate purification as previously described [11]. The following LodA variants were generated by site directed mutagenesis: C448A, C448D, Y211A, Y211E, Y211F, K530A and K530R. The forward and reverse primers that were used are shown in Table 1. Most of the mutations were made using the QuikChange Lightning kit. The Y211A and C448A mutations were constructed using the overlapping extension method (Ho et al., 1989). In the latter case, pETLODAB15 was used as a template for the mutagenesis PCR [9] using Pfu DNA polymerase (Promega). The outer primers used to perform the last PCR step containing the mutations were MARDIRsac1 (5′-CTCTGGTGAGCTCCTACAG-3) and MAREVeco2 (5′-GTGCTTGGGAGAATTCGCCTC-3′). After PCR amplification, fragments were cloned using the SacI-SacII sites in pETLODAB15, substituting the wild type copy of lodA. All mutations were confirmed by automated DNA sequencing. All cloning steps were performed using E. coli DH5α and the plasmids obtained were electroporated or transformed into E. coli Rosetta just before induction.
Table 1.
Primers used to generate site-directed mutations
| Mutation | Primers |
|---|---|
| C488A | Forward 5′-GTTGGATTGCCTATGGCTCCAGGAATAGAAATG-3′ Reverse 5′-CATTTCTATTCCTGGAGCCATAGGCAATCCACC-3′ |
| C448D | forward: 5′ GATTGCCTATGGATCCAGGAAT 3′ reverse: 5′ ATTCCTGGATCCATAGGCAATC 3′ |
| Y211A | Forward 5′-CAGATCTCAGCGGTGCTGGTGGTGGAGATGA-3′ Reverse 5′-TCATCTCCACCACCAGCACCGCTGAGATCTG-3′ |
| Y211E | forward: 5′ GATCTCAGCGGTGAAGGTGGT 3′ reverse: 5′ ACCACCTTCACCGCTGAGATC 3′ |
| Y211F | forward: 5′ GATCTCAGCGGTTTTGGTGGT 3′ reverse: 5′ ACCACCAAAACCGCTGAGATC 3′ |
| K530A | forward: 5′ AGCGTCAATGCAGCAAGTCAG 3′ reverse: 5′ CTGACTTGCTGCATTGACGCT 3′ |
| K530R | forward: 5′ AGCGTCAATCGAGCAAGTCAG 3′ reverse: 5′ CTGACTTGCTCGATTGACGCT 3′ |
The concentration of purified LodA was determined using an ε280= 125,180 M−1cm−1 which was calculated from its amino acid sequence [15].
Steady-state kinetic analysis
The steady-state activity of ε-lysine oxidase was monitored using a coupled assay that included horseradish peroxidase (HRP), which uses the H2O2 product of the reaction to oxidize Amplex Red to resorufin, which has an extinction coefficient of 54000 M−1cm−1 at 570 nm [11]. The assay mixture contained 0.05 mM Amplex Red, 0.1 U/ml of HRP and 0.42 μM LodA (WT or variants) in 50 mM potassium phosphate buffer, pH 7.5, at 25 °C with the indicated amounts of lysine and O2 included. To vary [O2] the buffer was made anaerobic by repeated cycles of vacuum and purging argon and mixed with an appropriate amount of air saturated ([O2]=252 μM) or 100% O2-saturated ([O2]= 1150 μM) buffer [16]. In order to investigate the possibility that any of the mutations had converted LodA from an oxidase to a dehydrogenase, reactions were also performed under anaerobic conditions using artificial electron acceptors as described previously [11]. In these cases the reaction was initiated by addition of lysine and monitored by the spectral change associated each potential electron acceptor, phenezine ethosulfate (PES) plus 2,6-dichloroinophenol (DCIP) (ε600=21,500 M−1cm−1), ferricyanide (ε420=1,040 M−1cm−1), or NAD+ (ε340=6,220 M−1cm−1). The data were fit by
| [2] |
Estimation of the extent of CTQ biosynthesis
The absorption spectrum of the purified oxidized form of active LodA exhibits characteristic features in the 350–450 nm range (Figure 3A). Inspection of the absorption spectra of the purified variant enzymes revealed the ratio of absorbance at 400 nm to absorbance at 280 nm (A400/A280) was affected by some of the mutations, despite the fact that proteins were pure as judged by SDS-PAGE. The features of the absorption spectra of the variants are essentially identical to that of WT LodA except for the intensity (Figure 3B). The most likely explanation for this is that some mutations affect the extent to which CTQ biosynthesis has occurred, yielding a mixed population of active LodA with mature CTQ and inactive preLodA with incompletely synthesized CTQ. Unfortunately, it was not possible to verify this by determining a molar extinction coefficient for CTQ in LodA by performing a spectrochemical quantitative redox titration because of poor reactivity of the protein-bound CTQ towards dithionite and other reductants. For ease of comparison, the ratio of A400/A280 for each variant is presented, normalized with the value for WT LodA set as 1.0 (Table 2). To be sure that the A400 was not varying due to changes in the active-site environment that were caused by mutations, the ratio of A400/A280 was also determined for proteins after incubation in 6 M guanidine HCl. The values for these unfolded proteins were the same as for the native proteins indicating that the ratio of A400/A280 was a valid indicator of the extent of CTQ biosynthesis. For determination of kcat values, the concentration of enzyme was determined from A280. As such one may wish to adjust the kcat values for variants with normalized A400/A280 values greater or less than 1.0. The Km values will not be affected by the variation in A400/A280 since the experimentally determined Km is not dependent on the concentration of the enzyme.
Figure 3.

The absorbance spectra of WT LodA and LodA variants. A. The absorbance spectrum of WT LodA with the visible absorbance features of CTQ magnified in the inset. B. The visible absorbance spectra of LodA variants in this study. The 280 nm absorbance in each of these spectra is identical so the observed variations in the intensity of the absorbance reflect the variation in A400/A280 nm of these variant proteins. The spectra from top to bottom are C448D, Y211A, Y211E, WT, C448A, Y211F, K530A and K530R LodA.
Table 2.
Steady-state kinetic parameters for LodA variants
| Normalized value of A400/A280a | kcat (s−1)b | Km lysine (μM) | Km O2 (μM) | |
|---|---|---|---|---|
| WT LodA | 1.0 | 0.34 ± 0.01 | 3.9 ± 0.2 | 199 ± 20 |
| C448A LodA | 1.0 | 0.39 ± 0.02 | 48 ± 4 | 543 ± 94 |
| C448D LodA | 1.5 | 0.29 ± 0.03 (0.19) | 142 ± 33 | 386 ± 70 |
| Y211F LodA | 0.8 | 0.19 ± 0.02 (0.23) | 129 ± 22 | 522 ± 162 |
| Y211A LodA | 1.5 | No reaction | No reaction | No reaction |
| Y211E LodA | 1.5 | No reaction | No reaction | No reaction |
| K530A LodA | 0.7 | 1.2 ± 0.1 (1.7) | 52 ± 5 | 1043 ± 102 |
| K530R LodA | 0.3 | 0.68 ± 0.04 (2.3) | 113 ± 13 | 706 ± 95 |
For comparison, this data is presented with the ratio of A400/A280 for each variant normalized with the value for WT LodA set as 1.0.
For determination of kcat the concentration of enzyme was that which was determined from A280. Values in parentheses are adjusted for variants with normalized A400/A280 values greater or less than 1.0 to reflect what the kcat values would be if these variants had the same level of CTQ biosynthesis as the WT LodA.
Sequence alignments of LodA-like proteins
LodA-like proteins belonging to group IA, as defined previously [15] includes LodA and the most similar proteins. The proteins in this group were aligned using the program clustal omega [17] available at http://www.ebi.ac.uk/Tools/msa/clustalo/.
Results
Modification of the steady-state assay conditions to allow comparison of LodA variants
In the previous steady-state kinetic study of LodA, the maximum concentrations of lysine and O2 that were used were 30 μM and 252 μM, respectively [11]. Those values were well above the Km values that were determined for each substrate. Preliminary examination of the LodA variants which are the topic of the current study revealed that some of them exhibited significantly greater Km values for these substrates. In order to obtain reasonable estimates for the Km and kcat values for these variants, it was necessary to extend the range of substrate concentrations used to 1 mM for lysine and 1150 μM for O2 (100% O2 saturation). In the presence of 1150 μM for O2, the reaction with WT LodA exhibited values of kcat of 0.34 ± 0.01 s−1, Km for lysine of 3.9 ± 0.2 μM. These values of kcat and Km for lysine are similar to the previous reported values of 0.22 ± 0.04 s−1 and 3.2 ± 0.2 μM, respectively [11]. However in the presence of 1 mM for lysine, a Km for O2 of 199 ± 20 μM was obtained which is larger than the previously reported value of 37 ± 6 μM. The reason for this discrepancy is unclear. It may be an artifact caused by the very high lysine concentration which is required to study the variants. It is possible that lysine is binding non-specifically to the enzyme, or perhaps competing with the other substrate (O2) in the ping-pong mechanism. Despite this peculiarity, in this study all LodA variants and WT LodA were analyzed at the higher concentration ranges so that the values of the kinetic parameters for each enzyme could be compared under the same conditions. Furthermore, as a consequence of some of the mutations, the apparent Km for O2 approached the [O2] in 100% O2-satuated buffer. As, such it was not possible to perform experiments in which [O2] could be varied over an appropriate range so as to obtain true kinetic parameters. As a consequence of this, the Km values that are reported here should be considered apparent Km values. However, as can be seen in the results which follow (Table 2), the effects of these mutations on kinetic parameters are quite evident and so the conclusions that can be drawn from these results are not compromised by these limitations imposed by the experimental system.
Effects of Cys448 mutations
Cys448 is present at the active site of LodA in close proximity (5.3 Å) to the CTQ cofactor. This residue was of particular interest because in the TTQ-dependent MADH, mutation of an Asp residue which occupies the analogous position near TTQ in the active site, to Asn caused decreased efficiency of TTQ biosynthesis [10]. C448A and C448D LodA mutations were made and each of these variant proteins was expressed and isolated at yields comparable to that of the WT LodA. The C448D LodA mutation actually increased the A400/A280 ratio of the purified protein. This suggests that WT LodA contains a subpopulation with incompletely biosynthesized CTQ and that introducing Asp at this position in LodA has enhanced the efficiency of CTQ biosynthesis relative to WT LodA.
The C448A and C448D LodA variants each exhibited lysine oxidase activity (Figure 4, Table 2). The kcat values of 0.39 ± 0.02 s−1 for C448A LodA and 0.29 ± 0.03 s−1 for C448D LodA are similar to that of WT LodA of 0.34 ± 0.01 s−1. Previous studies of LodA have indicated that an active site base is needed to deprotonate the lysine-CTQ adduct during catalysis [8, 11]. As the Cys448 mutations do not significantly decrease kcat, this residue does not appear to function as an active-site base or play any direct role in the catalytic reaction steps. However, the Km values for both substrates were increased. The Km for O2 of 544 ± 94 μM for C448A LodA is about 3-fold greater than that for WT LodA. For C448D LodA the Km of 386 ± 70 μM is about 2-fold greater. The Km for lysine of 48 ± 4 μM for C448A LodA is 12-fold greater than that for WT LodA and for C448D LodA the Km of 142 ± 33 s−1 is 36-fold greater. These data suggest an important role for Cys448 in lysine binding, or more specifically, facilitating the formation of the lysine-CTQ adduct.
Figure 4.
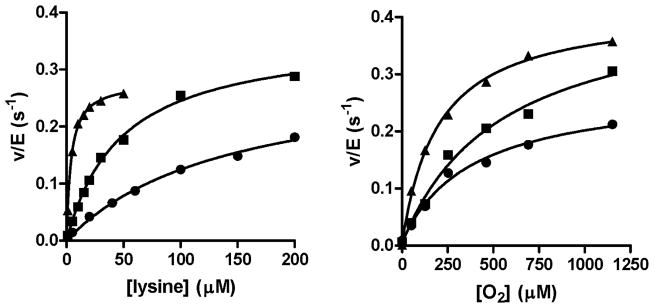
Effects of the C448A and C448D mutations on the steady-state activity of LodA. A. Assays were performed in the presence of a fixed concentration of 1150 O2. B. Assays were performed in the presence of a fixed concentration of 1 mM lysine. The symbols represent WT LodA (▲), C448A LodA (■) and C448D LodA (●). The lines are fits of each data set to eq 1.
Effects of Tyr211 mutations
Tyrosine 211 is situated on a mobile loop in proximity to the active site of LodA (discussed earlier). Tyr211 was mutated to Ala, Glu and Phe. In each case, the amount of protein expressed and isolated was similar to that of WT LodA. However, only the Y211F LodA exhibited activity. Interestingly the A400/A280 for the purified Y211A LodA and Y211E LodA were each greater than that of WT LodA and equal to that of C448D LodA which is the highest value observed in this study. This indicates that the lack of enzymatic activity of Y211A and Y211E LodA is not due to lack of CTQ biosynthesis which was not compromised, but apparently optimized by these mutations. In contrast to those mutations, Y211F LodA exhibited an A400/A280 less than that of WT LodA suggesting that the biosynthesis of CTQ is a bit less efficient in this variant. However, Y211F LodA was active (Figure 5, Table 2). The kcat value of 0.19 ± 0.02 s−1 for Y211F is similar to that of WT LodA of 0.34 ± 0.01 s−1, particularly when one takes into consideration the lower A400/A280 for Y211F LodA. The Km for lysine is 129 ± 22 μM which is 30-fold greater than for WT LodA. The Km of O2 is 522 ± 162 μM which is 2.7-fold greater than for WT LodA. These results highlight the importance of an aromatic residue at position 211 for substrate binding. While Y211F LodA has a near normal kcat, the affinity for each substrate is significantly compromised. Y211A and Y211E LodA are completely inactive, despite the presence of CTQ, suggesting that binding of substrates is further compromised in these variants to the extent that activity is undetectable.
Figure 5.
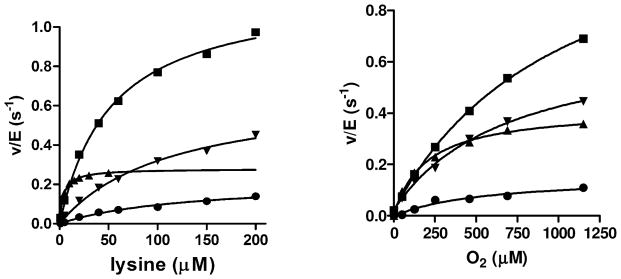
Effects of the Y211A, K530R and K530A mutations on the steady-state activity of LodA. A. Assays were performed in the presence of a fixed concentration of 1150 O2. B. Assays were performed in the presence of a fixed concentration of 1 mM lysine. The symbols represent WT LodA (▲), K530R (▼), K530A LodA (■) and Y211F LodA (●). The lines are fits of each data set to eq 1.
Effects of Lys530 mutations
Inspection of the one crystal structure in which Tyr211 is visible (PDB 3WEV) reveals that it forms a hydrogen bond with Lys530 (Figure 1). As such, this residue was mutated to Ala and Arg. The protein was expressed and isolated at levels comparable to WT LodA. However, the A400/A280 was significantly decreased indicating that the presence or absence of Lys530 does affect the extent of CTQ biosynthesis. While this residue is not that close to CTQ, it is situated on the outer side of the active site and hence could be involved in CTQ biosynthesis by a direct or indirect involvement with the modifying enzyme LodB.
Interestingly, the mutations of Lys530 increased kcat (Figure 5). For the reaction catalyzed by K530A LodA the kcat is 1.2 ± 0.1 s−1 which is around 3.5-fold greater than for WT LodA and that for K530R is 0.68 ± 0.04 s−1 which is double that of WT LodA. Considering the lower A400/A280 for K530A and K530R LodA, the true kcat may be even greater if one considers the lower level of CTQ that is present. While the kcat has been significantly enhanced by these mutations, the affinity for each substrate was greatly decreased. For K530A, the Km value for lysine is 52 ± 5 μM and that for O2 is 1043 ± 102 μM. For K530R, the Km value for lysine is 112.5 ± 12.9 μM and that for O2 is 706.1 ± 94.8 μM. While the affinity for O2 is significantly decreased, because of the increase in kcat, the kcat/Km (O2) values for K530 and K530R LodA of 1150 s−1M−1 and 960 s−1M−1 and not that much lower than the value for WT LodA of 1710 s−1M−1.
Conservation of Cys448, Tyr211 and Lys530 in sequences of proteins encoded by LodA-like genes
Analysis of the Integrated Microbial Genomes database of genome sequences identified 168 genes from 144 different bacterial genomes that encoded LodA-like proteins [14]. LodA from M. mediterranea was classified as a member of Group IA LodA-like proteins. Alignment of the sequences of the 20 members of the Group IA LodA-like proteins reveals that the residues which were mutated in this study are highly conserved in these sequences. Alignments of the regions of the sequence in which each of these three residues reside revealed that Cys448 (Supplemental Table S1) and Lys530 (Supplemental Table S2) are each conserved in each sequence. Tyr211 is conserved in 15 sequences and the other five have a Phe in this position (Supplemental Table S3).
Discussion
Site-directed mutagenesis of residues in the substrate channel of LodA have demonstrated that Cys448, Tyr211 and Lys530 play multiple roles in regulating Km values of substrates, kcat and the extent of biosynthesis of the protein-derived CTQ cofactor. It may seem intuitively odd that the WT enzyme would have less CTQ formed than some LodA variants. However, the fact that certain mutations either decrease or increase the efficiency of LodB-dependent CTQ biosynthesis is significant. This means that, in addition to furthering our understanding of the functional roles of these residues in the activity of LodA, these results highlight the point that different evolutionary factors must be considered for enzymes which possess protein-derived cofactors. Residues present in the active site that may be optimal for catalytic efficiency may not be the best to participate in CTQ biosynthesis, and vice versa. While CTQ biosynthesis is catalyzed by another enzyme, the residues surrounding the nascent CTQ may play important roles in properly positioning the Cys and Trp residues for modification or substrate oxygen for insertion, or both. In this regard, it has been shown that the first modification in the biosynthesis of CTQ takes place auto-catalytically in the absence of LodB, and that some LodA residues are involved in that process [9].
The residue which corresponds in structure to Cys448 in two other quinoproteins, the CTQ-containing QHNDH [12] and the TTQ-containing MADH [13], is a structurally conserved Asp residue. Mutation of this corresponding Asp in MADH affected the efficiency of MauG-dependent TTQ biosynthesis but had no effect on the catalytic activity of the population of the isolated MADH with fully formed TTQ [10]. Consistent with the results of the MADH study, it can be concluded that the residue at this position, Cys448 in LodA, is not the active-site base that is involved in deprotonation of the lysine-CTQ adduct during catalysis as the C448A mutation has no significant effect on kcat. However, in contrast to the results of the MADH, mutation of Cys448 in LodA to Ala did not result in decreased levels of CTQ biosynthesis. Furthermore, the C448D LodA mutation appears to have enhanced the efficiency of CTQ biosynthesis. This suggests that the native LodA is a mixed population in which some of the protein has incompletely synthesized CTQ and is inactive. Together with the previous study that showed a role for the corresponding Asp in TTQ biosynthesis in MADH, it appears that an Asp residue in this position is preferred for tryptophylquinone cofactor biosynthesis. However, for LodA the presence of Asp at this position is not optimal for catalysis. In particular, this mutation increases the Km for lysine 35-fold. Thus, the sub-optimal role in CTQ biosynthesis of Cys448 is more than compensated for by the greater catalytic efficiency of the enzyme.
Mutation of Tyr211 and its H-bonding partner Lys530 had significant effects on both kcat and Km values. As discussed earlier, Tyr211 is present on a mobile loop and it is possible that the H-bond to Lys530 could anchor it in place at certain times during the catalytic cycle. The aromatic character of residue 211 seems critical for activity since the Y211F LodA retains function, whereas Y211A LodA and Y211E LodA are inactive. The K530A and K530R mutations significantly increased the Km values for the substrates, particularly O2. For lysine is increased 13- and 29-fold, respectively while for O2 it increased 5- and 3,5-fold respectively. Since the loop containing Tyr211 was not observed in the structure of the lysine-CTQ adduct of LodA [8] it may not be important in stabilizing that intermediate. It is possible that Tyr211 in concert with Lys530 could aid in O2 binding, or retention of O2 in the active site to poise it for action. This would be consistent with the hydrophobic nature of residue 211 being important. Interestingly, the K530A mutation significantly increased kcat. Since the rate-limiting step for the overall reaction catalyzed by LodA is not known, it is difficult to interpret this effect. Given the position of Lys530 in the active site, and the fact that mutations to Ala and Arg each enhance rather than eliminate activity, these results suggest that Lys530 is not playing a direct role in acid-base chemistry or nucleophilic attack during catalysis. However, it is possible that that it could control the rate of binding of one or both of the substrates, or release of one or more of the products. If the hydrogen-bond between Lys530 and Tyr211 stabilizes the positions of these residues such that they impede the entry into or exit from the active site, then the removal of this blockage of the active site channel caused by the K530A mutation could possibly accelerate a rate-limiting product release step which could account for the increase in kcat. This would also be consistent with the large increases in the Km values that are caused by mutation of Lys530. In this sense the mobile loop which can be anchored by a hydrogen-bond between Tyr211 and Lys530 could act as a switch that regulates entry of substrates and exit of products to and from the active site.
A couple of points require consideration regarding the kinetic analysis of the LodA variants. As stated earlier, it was not possible to perform a detailed steady-state analysis in which both substrates were varied over an appropriate range of concentrations of each substrate. As such, it is an assumption that each variant obeys a ping-pong mechanism as does WT LodA. For example, with some flavoprotein oxidases it may be possible for reoxidation of the cofactor to precede or follow the release of the oxidized product. However, in the case of LodA, the substrate forms a covalent adduct with CTQ (Figure 2) and it is difficult to imagine how these mutations could alter the kinetic mechanism to allow O2 to reoxidize the cofactor prior to release of the aldehyde product. It should also be noted that the mutations in this study involve residues with ionizable groups. It was previously shown [11] that kcat for LodA exhibits a bell-shaped pH profile with a maximum at pH 7.5, implicating the involvement of at least two ionizable groups, and that the Km for lysine also varied in this range. As such, it was not possible to compare kinetic parameters in a pH-independent region.
These results reflect the different evolutionary factors that must be considered for enzymes which possess protein-derived cofactors. Since the catalytic cofactor must be generated by posttranslational modifications, this suggests that the protein must have originally evolved for some other purpose. The modifying enzyme likely also originally evolved for a different purpose. The residues in the active site could not have evolved to optimize function as a lysine oxidase until after the evolution of the LodA that led to its specific interaction with the modifying enzyme LodB and the ability to catalyze CTQ biosynthesis. This is well-described by the observation that some Y211A and Y211E mutations maximize levels of CTQ biosynthesis but are inactive in catalysis. Also consistent with this argument is the observation that Lys530 mutations lead to decreased CTQ biosynthesis but increased kcat. The fact that each of the mutations in this study increased Km values for lysine and O2 further indicate that the combination of residues throughout the active site channel together participate in the specificity and catalytic efficiency of the enzyme.
Supplementary Material
Highlights.
Active site residues of LodA play multiple roles in catalysis and CTQ biogenesis
A hydrogen bond between Tyr211 and Lys530 may control access to the active site
Cys448 is a strong determinant of substrate specificity for lysine
Acknowledgments
We acknowledge the collaboration of Patricia Lucas-Elío in the construction of some of the mutants used in this study, Yu Tang for technical assistance and Heather Williamson for helpful discussions. This research was supported by internal funds from the Burnett School of Biomedical Sciences and in part by the National Institute of General Medical Sciences of the National Institutes of Health under award number R37GM41574 (V.L.D.)
Abbreviations
- CTQ
cysteine tryptophylquinone
- TTQ
tryptophan tryptophylquinone
- WT
wild-type
- MADH
methylamine dehydrogenase
- QHNDH
quinohemoprotein amine dehydrogenase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gomez D, Lucas-Elio P, Sanchez-Amat A, Solano F. A novel type of lysine oxidase: L-lysine-epsilon-oxidase. Biochim Biophys Acta. 2006;1764:1577–1585. doi: 10.1016/j.bbapap.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 2.Lucas-Elio P, Gomez D, Solano F, Sanchez-Amat A. The antimicrobial activity of marinocine, synthesized by Marinomonas mediterranea, is due to hydrogen peroxide generated by its lysine oxidase activity. J Bacteriol. 2006;188:2493–2501. doi: 10.1128/JB.188.7.2493-2501.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas-Elio P, Hernandez P, Sanchez-Amat A, Solano F. Purification and partial characterization of marinocine, a new broad-spectrum antibacterial protein produced by Marinomonas mediterranea. Biochim Biophys Acta. 2005;1721:193–203. doi: 10.1016/j.bbagen.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Mai-Prochnow A, Lucas-Elio P, Egan S, Thomas T, Webb JS, Sanchez-Amat A, Kjelleberg S. Hydrogen peroxide linked to lysine oxidase activity facilitates biofilm differentiation and dispersal in several gram-negative bacteria. J Bacteriol. 2008;190:5493–5501. doi: 10.1128/JB.00549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chacon-Verdu MD, Gomez D, Solano F, Lucas-Elio P, Sanchez-Amat A. LodB is required for the recombinant synthesis of the quinoprotein L -lysine-epsilon-oxidase from Marinomonas mediterranea. Appl Microbiol Biot. 2014;98:2981–2989. doi: 10.1007/s00253-013-5168-3. [DOI] [PubMed] [Google Scholar]
- 6.Davidson VL. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry. 2007;46:5283–5292. doi: 10.1021/bi700468t. [DOI] [PubMed] [Google Scholar]
- 7.Davidson VL. Structure and mechanism of tryptophylquinone enzymes. Bioorg Chem. 2005;33:159–170. doi: 10.1016/j.bioorg.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Okazaki S, Nakano S, Matsui D, Akaji S, Inagaki K, Asano Y. X-Ray crystallographic evidence for the presence of the cysteine tryptophylquinone cofactor in L-lysine epsilon-oxidase from Marinomonas mediterranea. J Biochem. 2013;154:233–236. doi: 10.1093/jb/mvt070. [DOI] [PubMed] [Google Scholar]
- 9.Chacon-Verdu MD, Campillo-Brocal JC, Lucas-Elio P, Davidson VL, Sanchez-Amat A. Characterization of recombinant biosynthetic precursors of the cysteine tryptophylquinone cofactors of l-lysine-epsilon-oxidase and glycine oxidase from Marinomonas mediterranea. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbapap.2014.1012.1018. [DOI] [PubMed] [Google Scholar]
- 10.Jones LH, Pearson AR, Tang Y, Wilmot CM, Davidson VL. Active site aspartate residues are critical for tryptophan tryptophylquinone biogenesis in methylamine dehydrogenase. J Biol Chem. 2005;280:17392–17396. doi: 10.1074/jbc.M500943200. [DOI] [PubMed] [Google Scholar]
- 11.Sehanobish E, Shin S, Sanchez-Amat A, Davidson VL. Steady-state kinetic mechanism of LodA, a novel cysteine tryptophylquinone-dependent oxidase. Febs Lett. 2014;588:752–756. doi: 10.1016/j.febslet.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, Mathews FS. Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking. Proc Natl Acad Sci USA. 2001;98:14268–14273. doi: 10.1073/pnas.241429098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen LY, Doi N, Durley RCE, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. Refined crystal structure of methylamine dehydrogenase from Paracoccus denitrificans at 1.75 angstrom resolution. J Mol Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 14.Campillo-Brocal JC, Chacon-Verdu MD, Lucas-Elio P, Sanchez-Amat A. Distribution in microbial genomes of genes similar to lodA and goxA which encode a novel family of quinoproteins with amino acid oxidase activity. BMC Genomics. 2015;16:231. doi: 10.1186/s12864-015-1455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 16.Grzyska PK, Ryle MJ, Monterosso GR, Liu J, Ballou DP, Hausinger RP. Steady-state and transient kinetic analyses of taurine/alpha-ketoglutarate dioxygenase: effects of oxygen concentration, alternative sulfonates, and active-site variants on the FeIV-oxo intermediate. Biochemistry. 2005;44:3845–3855. doi: 10.1021/bi048746n. [DOI] [PubMed] [Google Scholar]
- 17.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.