

It was shown previously that endotubin, an integral membrane protein of endosomes, regulates the trafficking of tight junction proteins between endosomes and the tight junctions. Here it is shown that endotubin regulates YAP localization on endosomes through its interaction with AMOT and thus may play a role in contact inhibition.
Abstract
Contact-mediated inhibition of cell proliferation is an essential part of organ growth control; the transcription coactivator Yes-associated protein (YAP) plays a pivotal role in this process. In addition to phosphorylation-dependent regulation of YAP, the integral membrane protein angiomotin (AMOT) and AMOT family members control YAP through direct binding. Here we report that regulation of YAP activity occurs at the endosomal membrane through a dynamic interaction of AMOT with an endosomal integral membrane protein, endotubin (EDTB). EDTB interacts with both AMOT and occludin and preferentially associates with occludin in confluent cells but with AMOT family members in subconfluent cells. EDTB competes with YAP for binding to AMOT proteins in subconfluent cells. Overexpression of the cytoplasmic domain or full-length EDTB induces translocation of YAP to the nucleus, an overgrowth phenotype, and growth in soft agar. This increase in proliferation is dependent upon YAP activity and is complemented by overexpression of p130-AMOT. Furthermore, overexpression of EDTB inhibits the AMOT:YAP interaction. EDTB and AMOT have a greater association in subconfluent cells compared with confluent cells, and this association is regulated at the endosomal membrane. These data provide a link between the trafficking of tight junction proteins through endosomes and contact-inhibition-regulated cell growth.
INTRODUCTION
Regulation of signals that govern cell proliferation is required for the maintenance of tissue homeostasis. Proliferation may be controlled by several parameters, including growth factors, tissue architecture, and cell–cell contact (Polyak et al., 1994; Faust et al., 2005; McClatchey and Yap, 2012; Alkasalias et al., 2014). Contact inhibition of the cell cycle has been recognized for decades (Eagle and Levine, 1967), and loss of contact-mediated growth control is one of the hallmarks of cancer (Hanahan and Weinberg, 2011). Physical contact between cells has been shown to mediate contact inhibition, and adhesion junctions and proteins required for establishing polarity all regulate contact-mediated cell proliferation (Li and Mrsny, 2000; Perrais et al., 2007; Chen et al., 2010; Kim et al., 2011; Archibald et al., 2014). However, our understanding of how signals generated at the plasma membrane are communicated to the nucleus to limit proliferation remains incomplete.
The Hippo pathway is conserved from Drosophila to mammals and regulates organ size, tissue homeostasis, and contact-mediated cell growth (Camargo et al., 2007; Dong et al., 2007; Zhao et al., 2007; Gumbiner and Kim, 2014). The Hippo pathway was originally identified as a kinase cascade with the downstream effector the oncoprotein Yki/YAP (Drosophila/mammals; Justice et al., 1995; Harvey et al., 2003; Huang et al., 2005; Lai et al., 2005; Callus et al., 2006; Zhang et al., 2008; Boggiano and Fehon, 2012). Nonphosphorylated YAP translocates to the nucleus and acts as a transcriptional cofactor (Dong et al., 2007; Wu et al., 2008; Zhao et al., 2008); this transcriptional complex in turn regulates expression of genes involved in cell proliferation and antiapoptosis (Basu et al., 2003; Huang et al., 2005; Overholtzer et al., 2006). Polarity proteins such as Crumbs, adherens proteins such as E-cadherin, mechanotransduction, and growth factors, all through upstream regulation of the kinase cascade, regulate the Hippo pathway (Faust et al., 2005; Grzeschik et al., 2010; Ling et al., 2010; Dupont et al., 2011; Kim et al., 2011; Halder et al., 2012). However, an additional mode of regulation of YAP is through angiomotin (AMOT) family members (Chan et al., 2011; Wang et al., 2011; Zhao et al., 2011). AMOT regulation is phosphorylation independent, in that YAP associates with AMOT at the membrane, regulating its availability to translocate to the nucleus (Chan et al., 2011; Wang et al., 2011; Zhao et al., 2011). AMOT has been localized to the tight junction (TJ) and endosomes, and overexpression of AMOT rescues loss of contact inhibition induced by YAP activation (Bratt et al., 2005; Heller et al., 2010; Zhao et al., 2011; Mana-Capelli et al., 2014). However, how contact inhibition regulates this interaction and YAP activity is unknown.
Endocytic membrane trafficking is essential for maintenance of TJ integrity (Ivanov et al., 2005; Morimoto et al., 2005; Terai et al., 2006; Yamamura et al., 2008; Marchiando et al., 2010) and epithelial polarity, and disruption of membrane trafficking induces a loss of polarity and an overgrowth phenotype (Lu and Bilder, 2005). Endotubin (EDTB; also known as MAMDC4, AEGP) is an integral membrane protein that localizes to specialized apical endosomes (Wilson et al., 1987, 2000; Wilson and Colton, 1997; Gokay and Wilson, 2000). Although EDTB does not localize to tight junctions at steady state, it regulates their assembly and maintenance (Wilson et al., 1987; McCarter et al., 2010). Here we report that EDTB regulates YAP through modulation of the AMOT:YAP interaction at endosomal membranes, providing a mechanism for contact-mediated inhibition through trafficking between endosomes and tight junctions.
RESULTS
YAP localizes to intracellular puncta and colocalizes with a specialized endosomal marker
Contact inhibition requires communication between the plasma membrane/junctions and the nucleus, and YAP has been localized to lateral membranes, the nucleus, and intracellular puncta (Gilbert et al., 2011; Mana-Capelli et al., 2014). EDTB affects tight junction assembly but does not localize to lateral membranes at steady state (McCarter et al. 2010); it resides on specialized apical endosomes and does not colocalize with classical early endosomal markers such as EEA1 (Wilson et al., 1987, 2000). In addition, EDTB does not colocalize with transferrin receptor or Rab11 but does label with internalized tracer and colocalizes with Rab14 (Wilson and Colton, 1997; Gokay and Wilson, 2000; Kitt et al., 2008). To determine whether YAP is present on EDTB endosomes, we colabeled for endogenous EDTB and YAP. We find that there is colocalization of these markers on some intracellular puncta with a Pearson's r = 0.31. Of interest, in contrast to the findings of Gilbert et al. (2011), labeling of EEA1 and YAP results in very little colocalization (Figure 1A, Pearson's r = 0.18), suggesting that, in Madin–Darby canine kidney (MDCK) cells, YAP resides on the specialized endosomal compartment marked by EDTB.
FIGURE 1:

YAP and EDTB colocalize on endosomes. (A) MDCK cells grown on coverslips were labeled for endogenous EDTB, YAP, and EEA1. YAP colocalizes with EDTB (arrowheads) on intracellular puncta with a Pearson r of 0.31, whereas the Pearson r of YAP with EEA1 is 0.18. Scale bars, 10 μm (top), 2.5 μm (inset). (B–D) EDTB overexpression induces enrichment of YAP in the nucleus. MDCK cells expressing full-length EDTB (B, ET-FL) or control plasmid were grown on coverslips to confluence, and YAP localization was assessed using immunofluorescence. DAPI staining was used to determine total cell number. Representative images. (C) Quantification of B. ImageJ was used for quantification of nuclear enrichment of YAP protein. An identical threshold was set for all images, and YAP-positive nuclei were counted using the analyze particles function of ImageJ. Error bars, SEM of percentage of cells with nuclear enrichment of YAP; *p < 0.05. (D) GFP-CD and control cells grown to confluence and labeled with YAP. Expression of GFP-CD results in increased nuclear YAP (arrows).
Expression of EDTB full-length or cytoplasmic domain results in translocation of YAP to the nucleus and loss of growth control
It is possible that cycling of junctional proteins or YAP regulators to endosomes could regulate YAP activity. We generated stable MDCK cell lines expressing full-length endotubin (ET-FL), a green fluorescent protein (GFP) fusion protein containing only the cytoplasmic domain of EDTB (GFP-CD; Supplemental Figure S1A; McCarter et al., 2010) or GFP. These constructs are expressed at 1.2–2 times the level of endogenous EDTB (Supplemental Figure S1, B–E). At steady state, expression of ET-FL or GFP-CD does not affect the distribution of tight junction proteins (McCarter et al., 2010). Labeling of confluent cells expressing full-length endotubin (Figure 1, B and C) or GFP-CD (Figure 1D) showed extensive localization of YAP in the nucleus. However, although there is no change in the protein or phosphorylation level of the Hippo pathway kinase Mst1/2 (Supplemental Figure S1F), there is an increase in YAP protein levels (Supplemental Figure S1G) and YAP phosphorylation (Supplemental Figure S1H) in cells expressing both ET-FL and GFP-CD. However, because the ratio of total and phosphorylated YAP is unchanged, there is more unphosphorylated (potentially active) YAP in the cells overexpressing ET-FL or GFP-CD.
We next tested whether the increased nuclear YAP resulted in increased cell proliferation. Phospho–histone H3 (PHH3) labeling shows that proliferation is increased twofold in cells expressing ET-FL or GFP-CD (Figure 2A). Furthermore, when these cells are grown to confluence, expression of GFP-CD results in the formation of many small, multilayered foci (Figure 2B). Transmission electron microscopy of the GFP-CD foci indicates that the cells retain an epithelial morphology, including apical microvilli (Figure 2B), and quantification shows that foci formation increases more than fivefold (Figure 2C). Formation of foci could indicate a loss of epithelial polarity. To assess polarity, we labeled control and GFP-CD cells grown on permeable supports with antibodies against the apical marker gp135/podocalyxin and the basolateral marker E-cadherin. As shown in Figure 2D, GFP-CD cells exhibit apical gp135 and basolateral E-cadherin expression. In addition, foci are also polarized and sometimes form polyp-like structures that retain polarity (Supplemental Figure S2E).
FIGURE 2:

EDTB overexpression results in loss of growth control. (A) Representative cells expressing ET-FL or GFP-CD labeled with PHH3. Proliferation is increased with overexpression of GFP-CD or ET-FL. Error bars, SEM of percentage of cells that are PHH3 positive; ***p < 0.005, **p < 0.01. (B) GFP-CD cells form multiple small, multilayered foci (arrows) when grown on Transwell filters, whereas control cells remain as a monolayer with limited number of foci. Transmission electron microscopy (TEM) of control and GFP-CD cells grown on filters shows that control cells grow as a monolayer but GFP-CD cells form multicellular foci. Foci of GFP-CD–expressing cells maintain epithelial characteristics, such as apical microvilli (arrowheads). Scale bars, 100 μm (phase), 1 μm (TEM). (C) Quantification of the multilayer area shows an increase in foci formation in cells expressing GFP-CD. Error bars, SEM; ***p < 0.005. (D) Immunofluorescence labeling of apical (gp135, podocalyxin) and basolateral (E-cadherin) markers shows that the polarized distribution of these proteins is maintained with expression of GFP-CD. The foci (arrowheads) also maintain this polarized expression pattern. Scale bar, 20 μm.
Because results obtained using GFP-CD were often more pronounced than those obtained using full-length endotubin, most experiments used overexpression of GFP-CD, as it provides greater mechanistic insight into the domains important for the observed effects. In fact, this approach has been used successfully to define the function of other integral membrane proteins (Wodarz et al., 1993; Delmas et al., 1999). Specifically, overexpression of the cytoplasmic domain of Crumbs defined its role in epithelial polarity and establishment of apical membranes (Wodarz et al., 1993, 1995), and expression of the Crumbs cytoplasmic domain drives proliferation through activation of Yorkie (Ling et al., 2010; Robinson et al., 2010).
Changes induced in cells expressing GFP-CD are not the result of epithelial-to-mesenchymal transition
Migration assays indicate that GFP-CD cells have increased rates of migration (Supplemental Figure S2A), and GFP-CD cells plated on type II collagen grow as spindle-shaped cells when subconfluent (Supplemental Figure S2B). However, they never invade the collagen matrix. Changes in morphology and migration of epithelial cells can be the result of reprogramming to a mesenchymal phenotype, with loss of E-cadherin and up-regulation of the intermediate filament protein vimentin and epithelial-to-mesenchymal transition (EMT) transcription factors (Thiery et al., 2009), and forced expression of YAP results in EMT in the epithelial cell line MCF10A (Overholtzer et al., 2006). However, analysis of protein levels of EMT markers shows that E-cadherin levels are maintained and there is no increase in vimentin (Supplemental Figure S2C). Furthermore, quantitative PCR shows that transcription factors and markers associated with EMT are decreased (Supplemental Figure S2D). Surprisingly, the levels of the transcription factor Snail are increased without a significant reduction in the proteins levels of E-cadherin and only a small decrease in E-cadherin message or an increase in Zeb transcription factors, all direct targets of Snail (Dave et al., 2011). We interpret this to mean that the expression of GFP-CD is not causing EMT.
To examine other proliferation pathways, such as mitogen-activated protein kinase (MAPK) and AKT, that could drive increased proliferation seen with overexpression of ET-FL or GFP-CD, we analyzed their activation. As shown in Supplemental Figure S3, A and B, neither MAPK nor AKT signaling is activated after overexpression of the full-length or cytoplasmic domain of EDTB. In contrast, there is an increase in the cell cycle regulatory protein cyclin D1 when either full-length or cytoplasmic domain of EDTB is overexpressed (Supplemental Figure S3, A and B), consistent with increased activity of YAP (Camargo et al., 2007; Cao et al., 2008).
AMOT and EDTB colocalize on endosomes
An additional mechanism of control of YAP activation and nuclear localization is YAP association with the scaffold proteins p130-AMOT and p100-AMOTL2 (Chan et al., 2011; Wang et al., 2011; Zhao et al., 2011). AMOT isoforms localize to junctions and intracellular puncta that have been identified as endosomal membranes (Bratt et al., 2005; Wells et al., 2006; Heller et al., 2010). As with YAP, immunofluorescence of endogenous EDTB and AMOT or AMOTL2 shows extensive colocalization of these proteins on intracellular puncta (AMOT:EDTB, Pearson's r = 0.65 and AMOTL2:EDTB, Pearson's r = 0.79), suggesting an association on specialized early endosomes. Furthermore, labeling for endogenous AMOT and YAP also shows colocalization on intracellular puncta (Pearson's r = 0.56; Figure 3A). However, as with YAP:EEA1, there is limited colocalization of AMOT with EEA1 (Pearson's r = 0.18).
FIGURE 3:

EDTB, AMOT, and YAP all localize to endosomes, and EDTB regulates AMOT interaction with YAP. (A) Localization of endogenous AMOT, AMOTL2, YAP, EDTB, and EEA1 in MDCK cells grown on coverslips. EDTB colocalizes with AMOT (Pearson's r = 0.65) and AMOTL2 (Pearson's r = 0.79) on intracellular puncta (arrows), but there is limited colocalization with EEA1 (Pearson's r for AMOT/EEA1 is 0.18). AMOT and YAP also colocalize on intracellular puncta (Pearson's r = 0.56). Scale bars, 5 μm (top), 1 μm (insets). (B) AMOT and AMOTL2 domain structure. p80-AMOT and p60-AMOTL2 isoforms result from an internal start site. The amino-terminal domains of p130-AMOT and p100-AMOTL2 interact with YAP through the PPxY motif. (C) MDCK lysates collected at 90% confluence were used to assess the physical interaction of EDTB and AMOT/AMOTL2. Pull-down using the cytoplasmic domain of EDTB fused to GST (GST-CD) analyzed by Western blot shows interaction with AMOT and AMOTL2 relative to the GST-only control. AMOT input is sometimes not detected, indicating concentration by pull down. (D) Coimmunoprecipitation of ET-FL and HA-p130-AMOT. HEK293 cells were transfected with ET-FL and HA-p130AMOT. Lysates were immunoprecipitated with EDTB or HA antibodies. Western blot analysis shows coimmunoprecipitation of EDTB and AMOT. (E) Competition of EDTB with YAP for binding to AMOT and AMOTL2. MDCK cells expressing GFP-CD or control plasmid were immunoprecipitated with antibody against YAP and analyzed by Western blot for AMOT or AMOTL2. In the presence of GFP-CD, there is a significant decrease in the amount of AMOT and AMOTL2 associated with YAP compared with control. (F) Quantification of E. Error bars, SEM from three independent experiments. **p < 0.01, ***p < 0.005.
EDTB modulates the interaction between AMOT family members and YAP
AMOT is synthesized as several isoforms due to different translational start sites (Figure 3B), in addition to being a member of a multigene family (Bratt et al., 2002). Interaction between YAP and AMOT occurs with only the p130 form of AMOT, as well as the long form of AMOTL1 and AMOTL2 (Chan et al., 2011; Wang et al., 2011; Zhao et al., 2011). To test for an interaction between EDTB and AMOT, we used the EDTB cytoplasmic domain fused to glutathione S-transferase (GST). We find that this domain of EDTB brings down p130-AMOT (Figure 3C). In addition, GST-CD also interacts with the highly related AMOTL2 protein (Figure 3C). Consistent with the GST pull-down results, coimmunoprecipitation from cells expressing ET-FL and HA-p130AMOT confirms the interaction of EDTB and AMOT (Figure 3D). Of importance, EDTB competes with YAP for binding to both AMOT and AMOTL2, as expression of GFP-CD results in decreased AMOT:YAP and AMOTL2:YAP association (Figure 3, E and F).
To assess the requirement for YAP in GFP-CD-induced proliferation, we knocked down YAP in MDCK cells expressing GFP-CD. In these cells, YAP protein levels are reduced >90%, and the proliferation of cells expressing GFP-CD is greatly reduced (Figure 4, A and B), suggesting that GFP-CD–induced proliferation is mediated through the YAP pathway. Control cells also had slightly decreased proliferation (0.74-fold) as measured by PHH3 labeling (p < 0.1). YAP knockdown decreases cell growth of several carcinoma and transformed cell lines (Zhao et al., 2008; Zhi et al., 2012; Huang et al., 2013; Xie et al., 2013) when measured over several days, and it may be that these cells have greater sensitivity to YAP activity at steady state. We next reasoned that, if EDTB competes with YAP for binding to AMOT, overexpression of p130-AMOT would rescue the GFP-CD-induced increase in proliferation. As shown in Figure 4C, p130-AMOT overexpression in GFP-CD cells complements the increased proliferation phenotype. Conversely, knockdown of EDTB should result in decreased proliferation and a corresponding increase in YAP:AMOT localization in MDCK cells due to the greater availability of AMOT to bind YAP. In fact, when EDTB expression is reduced using short hairpin RNA (shRNA), there is both a reduction in the proliferation rate (0.68-fold, p < 0.05) and an increase in the colocalization of YAP and AMOTL2 (Figure 4, D–F).
FIGURE 4:

YAP activity is required for GFP-CD–induced increase in proliferation. (A) MDCK cells expressing GFP or GFP-CD were infected with YAP shRNA lentivirus. Lysates were analyzed by Western blot for expression of YAP. YAP protein levels are decreased >90% in both the control and GFP-CD–expressing cells. (B) Changes in proliferation were determined by PHH3 labeling. Proliferation of GFP-CD cells is significantly decreased with YAP knockdown. Error bars, SEM; ***p < 0.005. YAP knockdown in control cells also resulted in decreased proliferation (0.74-fold of that of parent lines). (C) MDCK cells expressing GFP-CD were transfected with p130AMOT. Overexpression of p130-AMOT complements the GFP-CD increased-proliferation phenotype. Error bars, SEM; *p < 0.05. (D) Endogenous EDTB was knocked down in MDCK cells using two independent shRNAs, individually or together. Lysates were analyzed by Western blot to verify the reduction in EDTB expression. (E) Proliferation is decreased when EDTB expression is reduced. Error bars, SEM; *p < 0.05. (F) Pearson r of AMOTL2:YAP colocalization in EDTB knockdown cells. Knockdown of endotubin results in a significant increase in AMOTL2:YAP colocalization. Four to six images were quantified for each condition. Error bars, SEM; ***p < 0.005.
Cell confluence affects the EDTB:AMOT association
To test whether EDTB could regulate YAP via the AMOT pathway, we examined AMOT:EDTB association in subconfluent and confluent cultures of MDCK cells. If EDTB regulates the AMOT:YAP interaction during contact inhibition, there should be greater EDTB:AMOT interaction in subconfluent cells, freeing YAP to translocate to the nucleus. There is no change in the amount of EDTB, YAP, AMOT, or AMOTL2 in cells, regardless of confluence (Figure 5A). This contrasts with previous reports, in which levels of the YAP were found to be either up or down under confluent conditions (Zhao et al., 2007, 2010). This may relate to cell-type differences, as experiments in these studies were performed in fibroblasts. After coimmunoprecipitation, we find increased EDTB:AMOT and EDTB:AMOTL2 complexes in subconfluent cells (Figure 5B). In addition, there is an increase in AMOT:YAP association in confluent cultures (Figure 5B), consistent with AMOT sequestering YAP to prevent proliferation. Analysis of the localization of EDTB and AMOT or AMOTL2 on endosomes in subconfluent and confluent cultures shows decreased colocalization of EDTB:AMOT and EDTB:AMOTL2 in confluent cultures (Figure 5, C and D).
FIGURE 5:

Interaction between EDTB and AMOT is regulated by cell density. (A) The expression levels of EDTB, AMOT, AMOTL2, YAP, and occludin in subconfluent and confluent MDCK cultures were analyzed by Western blot analysis. (B) Top, immunoprecipitation of MDCK subconfluent and confluent cultures with EDTB was analyzed by Western blot for AMOTL2. Middle, immunoprecipitation of MDCK subconfluent and confluent cultures with AMOT was analyzed by Western blot for EDTB. Bottom, immunoprecipitation of MDCK subconfluent and confluent cultures with YAP was analyzed by Western blot for AMOT. (C) MDCK cells plated on coverslips were immunolabeled for EDTB and AMOT or AMOTL2. (D) The amount of colocalization between AMOT/EDTB and AMOTL2/EDTB was determined by calculating the Pearson r. The colocalization of AMOT/EDTB and AMOTL2/EDTB is significantly reduced in confluent cultures. Average endosome number per image is 475, and eight images were quantified per condition. Error bars, SEM; ***p < 0.005. (E) Immunoprecipitation of MDCK subconfluent and confluent cultures with occludin was analyzed by Western blot for EDTB. The image is spliced from the same gel.
To further characterize the relationship between contact inhibition of proliferation and junction assembly, we next tested whether EDTB interacts with the tight junction protein occludin and whether that interaction depends on cell confluence. To test this, we performed coimmunoprecipitation experiments with occludin, followed by immunoblotting for EDTB in subconfluent and confluent cells. Immunoblotting analysis shows that EDTB associates with occludin in confluent, but not subconfluent, cells (Figure 5E).
Disruption of EDTB results in growth in soft agar
Growth in soft agar is a hallmark of transformation and loss of growth control. When cells expressing GFP or GFP-CD are plated in soft agar, control cells form occasional small, irregular colonies (Figure 6A). However, cells expressing GFP-CD form large, branching structures and generate more colonies than control cells (Figure 6A). In addition, in GFP-CD cultures, after growth in soft agar for 3 wk, we observe cells dispersing from the original colony and the formation of additional foci. This was never observed in control cultures.
FIGURE 6:

Increased expression of EDTB results in growth in soft agar and is associated with early-stage LIHC samples. (A) MDCK cells expressing GFP (control) or GFP-CD were grown in soft agar for 3 wk. GFP-CD cells form long, branching structures (arrows) that produce satellite colonies (arrowheads) after 3 wk in culture. Scale bars, 100 μm (left), 20 μm (right). Right, quantification of colony formation after 14 d in cells expressing GFP-CD. Error bars, SEM; **p < 0.01. (B) Using the Cancer Genome Database, we collected the rsem expression values for EDTB in 105 liver hepatocellular carcinomas. The tumors were binned into three equal groups based on EDTB expression levels. Separating these groups by tumor stage shows that 44% of stage I tumors express EDTB at high levels.
EDTB is overexpressed in early-stage liver cancer
In addition to its known role in the control of organ size and contact inhibition, YAP expression is increased in the majority of liver hepatocellular carcinomas (LIHCs; Bai et al., 2012). To determine whether EDTB expression is altered in these cancers, we examined the levels of EDTB mRNA expression (RNASeqV2 rsem values) using data available from the Cancer Genome Database (tcga-data.nci.nih.gov). Using 48 matched tumor and normal pairs, we found a twofold or greater increase in EDTB expression in 17% of patients (Supplemental Table S1). Of interest, within this group, 75% are stage I tumors. The rsem expression values for EDTB in these tumor samples (N = 105) range from 7 to 6579. Binning these samples into three groups based on these expression values shows a trend in which the majority of stage I tumors are medium to high expressing, and the percentage decreases with increasing stage (Figure 6B). Examination of only EDTB high-expressing tumors (rsem >450) indicates that 44% of stage I tumors are high expressing, and 54% of all high-expressing tumors are stage I (Supplemental Table S2). This may indicate that dysregulation of EDTB promotes early events in the development of hepatic cancer.
DISCUSSION
Contact inhibition of cell proliferation is essential for regulation of organ size and to prevent the overproliferation that characterizes cancer (Hanahan and Weinberg, 2011). In transformed cells, overexpression of occludin restores assembly of the tight junction and cell–cell contact–mediated growth control, suggesting that the formation and maintenance of tight junctions are important components of contact inhibition (Chen et al., 2000; Li and Mrsny, 2000). Adherens junctions are also an important component of contact inhibition, and E-cadherin regulates the YAP pathway through the regulation of phosphorylation of Mst/Lats (Kim et al., 2011). We do not see any change in the upstream phosphorylation of YAP regulators with EDTB overexpression, suggesting that EDTB regulates YAP through modulation of the interaction of YAP with AMOT at the endosomes.
EDTB is a member of the MAMDC family. The extracellular domain of EDTB contains several meprin-A5 protein–protein tyrosine phosphatase μ (MAM) domains (Speelman et al., 1995), which are believed to mediate protein interactions and adhesion (Zondag et al., 1995; Gao and Garbers, 1998), whereas the short (40 amino acids) cytoplasmic tail contains apical endosomal sorting motifs (Gokay et al., 2001). Diet1, a MAMDC protein related to EDTB, affects bile acid synthesis through regulation of fibroblast growth factor (FGF) trafficking. Of interest, Diet1, like EDTB, is localized to a specialized endosomal compartment, where it colocalizes with FGF15/19 (Vergnes et al., 2013). This interaction is critical in maintaining signaling from the small intestine to the liver (Vergnes et al. 2013). Based on this together with the results reported here, it appears that the MAMDC family proteins play a role in signal transduction through modulation of protein trafficking.
We find that the endosomal protein EDTB both colocalizes and interacts with AMOT family members. EDTB localizes to EEA1-negative early endosomes in a variety of cell types, whereas AMOT and AMOTL2 localize to both the tight junction and endosomes (Wilson and Colton, 1997; Wilson et al., 2000; Wells et al., 2006; Heller et al., 2010; Zhao et al., 2011; Mana-Capelli et al., 2014). Reduced expression of AMOT induces loss of contact inhibition and the formation of foci (Zhao et al., 2011), and AMOT regulates YAP activity by sequestering YAP in the cytoplasm (Chan et al., 2011; Zhao et al., 2011; Mana-Capelli et al., 2014). Although the AMOT:YAP association has traditionally been presumed to occur at the plasma membrane, overexpression of AMOT localizes YAP to the cytosol (Wang et al., 2011). Our results show that AMOT, YAP, and EDTB are located on endosomes, and EDTB competes with YAP for binding to AMOT, likely at the endosomal membrane. Under conditions of increased EDTB, YAP is displaced from p130-AMOT and/or AMOTL2 and translocates to the nucleus. Our findings of increased colocalization and greater association of EDTB with AMOT and AMOTL2 in subconfluent cells, and conversely an increased interaction of EDTB and occludin in confluent cells, further support the model that endosomes play a role in mediating signaling from this pathway. Although we cannot exclude that this interaction occurs at the junction, it is important to note that EDTB overexpression does not disrupt tight junctions at steady state, and endotubin is not localized to junctions at steady state (Wilson et al., 1987; McCarter et al., 2010).
We find that EDTB message levels are increased in many cases of early-stage liver cancer. This suggests that EDTB may be a predisposing factor in the development of hepatic cancer. This is consistent with another report that identified mutations of EDTB in early events in the development of breast cancer (Sjoblom et al., 2006). Although recent data in transgenic mice argued against the role of AMOT as a suppressor of YAP activity (Yi et al., 2013), contrasting data indicated that AMOT can sequester YAP and prevent its translocation to the nucleus. There are multiple pathways that lead to the activation of YAP, and it is plausible that this activation is context dependent and the regulation of AMOT:YAP interaction via EDTB is important for a subset of cancers.
Recent evidence shows that F-actin competes with YAP for AMOT binding, and disruption of the actin-binding domain on AMOT results in colocalization of AMOT and YAP on endosomes (Mana-Capelli et al. 2014). On the basis of this and the results reported here, we propose a model in which the AMOT:YAP interaction may occur predominantly on endosomes and the availability of EDTB to interact with AMOT is regulated by the presence of tight junction proteins in the endosomes (Figure 7). Thus, in confluent cells, tight junction proteins bind to EDTB, preventing the EDTB:AMOT interaction and allowing AMOT to sequester YAP to prevent nuclear translocation and proliferation.
FIGURE 7:
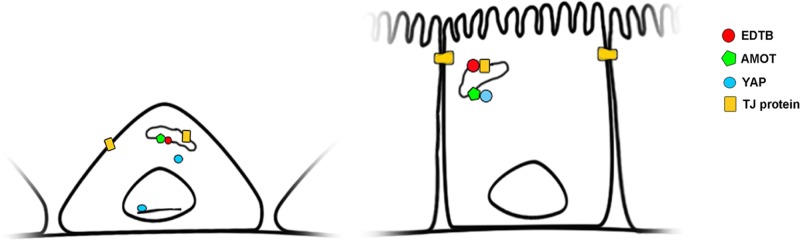
Model for endosomal control of contact inhibition through YAP:AMOT. In subconfluent cells, AMOT associates with EDTB on endosomes, allowing YAP to localize to the nucleus. When cells reach confluence, tight junction proteins cycling through endosomes associate with EDTB, allowing AMOT to sequester YAP at the endosomal membrane.
MATERIALS AND METHODS
Reagents
Primary antibodies used for this study include rabbit anti-AMOT (H-66), goat anti-AMOTL2 (N-14), goat anti–β-catenin, rabbit anti–cyclin D1 (H-295), and rabbit anti-YAP (H-125), purchased from Santa Cruz Biotechnology (Dallas, TX). Rabbit anti–phospho-Akt (473), rabbit anti–phospho-p44/42 MAPK, rabbit anti–p44/42 MAPK, rabbit anti-YAP, rabbit anti–phospho-YAP (Ser-127), rabbit anti-Mst1/2 (3682), and phospho-Mst1 (3681) were purchased from Cell Signaling Technology (Beverly, MA). Mouse anti–β-actin, rat anti–E-cadherin (clone DECMA-1), and mouse anti–phospho-histone H3 were purchased from Sigma-Aldrich (St. Louis, MO). Mouse anti–E-cadherin was purchased from BD Biosciences (San Jose, CA), mouse anti-hemagglutinin (HA) from Covance (San Diego, CA), rabbit anti-AMOT from Antibody Verify, and rabbit anti-AKT from BD Biosciences. Secondary antibodies used with the LI-COR Odyssey infrared imager include anti-rabbit, -goat, and -mouse purchased from LI-COR (Lincoln, NE). Polyethyleneimine (PEI; Polysciences, Warrington, PA) was prepared as a 1 μg/ul solution in water and sterilized by filtration.
Cell culture
MDCKII cells were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, 1000 U of penicillin, 1 mg/ml streptomycin, 20 mM l-glutamine (Sigma-Aldrich), and nonessential amino acids (Mediatech, Manassas, VA). Generation of cell lines expressing the EDTB and EDTB cytoplasmic domain (GFP-CD) has been described previously (Gokay and Wilson, 2000; McCarter et al., 2010). The GFP-CD cell lines were maintained in DMEM supplemented with G418 (400 μg/ml). Both the EDTB knockdown (ETKD) and the YAP knockdown (YAPKD) cell lines were generated using lentiviral constructs.
ET 1014 shRNA:
Forward primer
ccggaactgctcgctcgcttctatctcgagatagaagacgagggagcagtttttttg
Reverse primer
aattcaaaaaaactgctcgctcgtcttctatctcgagatagaagacgagcgagcagtt
ET 3603 shRNA:
Forward primer
ccggaacatcgtcttcaatgcggatctcgagatccgcattgaagacgatgtttttttg
Reverse primer
aattcaaaaaaacatcgtcttcaatgcggatctcgagatccgcattgaagacgatgtt
YAPKD:
Forward primer
ccggaagctctgagctctgacatccctcgagggatgtcagagctcagagctttttttg
Reverse primer
aattcaaaaaaagctctgagctctgacatccctcgagggatgtcagagctcagagctt
All cultures were selected using puromycin (2 μg/ml) after lentiviral transduction. For controls, we used the pLK0.1 puro construct (Addgene; Stewart et al., 2003).
Transwell filters
Twelve-well Transwell filters (Corning, Corning, NY) were seeded with 250,000 cells/well. Fresh medium was added daily and cells incubated for an additional 4–5 d. For immunolabeling, filters were processed as described later. For multilayer analysis, images were obtained using an Olympus IMT-2 microscope equipped with Hoffman modulation contrast filters, and multilayer area was calculated using ImageJ (National Institutes of Health, Bethesda, MD).
Immunofluorescence labeling and confocal microscopy
For immunofluorescence labeling, cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% saponin/phosphate-buffered saline, and incubated in primary antibodies for 2 h at room temperature, followed by incubation with fluorescent secondary antibodies (Molecular Probes, Grand Island, NY; Jackson Labs, Bar Harbor, ME). Images were acquired using an Olympus FluoView 1100 or 1200 confocal microscope with a 60× (numerical aperture 1.4) oil immersion objective. Excitation wavelengths of 405, 488, 568, and 594 nm were used for simultaneous two-or three-channel recording. Images were processed and merged using Photoshop software (Adobe Systems, San Jose, CA) or ImageJ. For comparison between control and knockdown cells, identical imaging and processing parameters were used. Pearson's r was calculated from 60× images generated using the Olympus FluoView confocal microscope and software. The average Pearson's r was derived from seven images.
Transient transfection
Cells were plated at ∼60% confluence 24 h before transfection. PEI was used for transient transfections. The PEI:DNA ratio for the transfections was 3:1, with 4.5 μg of PEI used for transfections in a six-well plate. Cells were incubated with the PEI:DNA solution for 16 h, and cells were fixed or lysed 48 h later.
Immunoblot analysis
For quantification of signaling pathways, lysates were collected at ∼90% confluence. Cells were lysed with RIPA (50 mM Tris, 100 mM NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% SDS) containing protease inhibitors (Complete Mini; Roche Diagnostics, Indianapolis, IN) supplemented with calyculin A (Sigma-Aldrich) and sodium orthovanadate (Sigma-Aldrich) for 5 min on ice. Cells were scraped, passed through a 27-gauge needle, vortexed, and incubated on ice for 30 min. The insoluble materials were removed by centrifugation, and supernatant was retained for analysis. For immunoblotting, 12–25 μg of protein was placed in LI-COR SDS sample buffer containing 100 μM dithiothreitol and separated on a 7.5% SDS–PAGE gel. Protein was transferred to nitrocellulose, blocked using 5% milk, and incubated with primary antibodies in Tris-buffered saline–Tween/5% milk for 2 h at RT or overnight at 4°C. For phosphospecific antibodies, 5% bovine serum albumin was used as the blocking solution. For all experiments, samples were run in triplicate or quadruplicate, and each experiment was run at least three independent times. Blots were imaged and quantified using the LI-COR Odyssey system.
Immunoprecipitation of subconfluent and confluent cultures
For analysis of protein expression and protein–protein interaction of subconfluent and confluent cultures, cells were grown in 10-cm dishes. For subconfluent cells, the cultures were lysed at 50–60% confluence. For confluent cultures, cells were grown to 100% confluence, fresh medium was added, and cells were incubated for an additional 24 h. For immunoprecipitation, cells were lysed in an NP-40 buffer (1% NP-40, 25 mM Tris, pH 7.2, 60 mM NaCl, 5 mM MgCl2, 5% glycerol). Lysates (500 μg of protein) were precleared for 2 h at 4°C using protein G Dynabeads (Life Technologies, Foster City, CA) and incubated with 2 μg of primary antibody overnight at 4°C, followed by 2-h incubation with protein G Dynabeads at 4°C. Beads were washed with lysis buffer, resuspended in SDS loading buffer, and separated by electrophoresis. As described earlier, experiments were performed in triplicate or quadruplicate and repeated at least three times.
GST pull down
EDTB-CD GST proteins were purified by sonication of bacterial lysates in ice-cold STE buffer (0.1 M NaCl, 10 mM Tris-HCl, pH 8, 1 mM EDTA) supplemented with phenylmethylsulfonyl fluoride/leupeptin and lysozyme and incubated with Glutathione Sepharose 4 Fast Flow beads (GE Healthcare, Piscataway, NJ) for 2 h at 4°C. Lysates from MDCK were prepared using PLC lysis buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 150 mM NaCl, 5% glycerol, 0.5% Triton X-100, 1.5 mM MgCl2, 1 mM ethylene glycol tetraacetic acid, pH 8.0) supplemented with protease inhibitor cocktail and calyculin A. Lysates were incubated with GST beads for 3 h at 4°C. The GST beads were washed four times with PLC lysis buffer, and proteins were eluted from beads by incubation at 95°C for 5 min in SDS–PAGE loading buffer.
Proliferation assay
We plated 5 × 104 cells on coverslips and incubated them for 24 h before fixation and labeling with anti–phospho-histone H3. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and the percentage of phospho-histone H3–labeled cells was determined though direct counting of blinded samples. For the p130-AMOT rescue, 2.5 × 104 cells were plated and transfected with p130 AMOT after 24 h. Cells were incubated for an additional 24 h before fixation and labeling. For all proliferation experiments, three images at 40× magnification were collected from each coverslip, and three coverslips were used for each experimental and control condition. All cells in the field of view were counted, and the total number of cells ranged from 100 to 300 for each image (n = 500–1000 cells/condition).
AMOT rescue
MDCK cells expressing GFP-CD were transfected with HA p130-AMOT (Addgene, Cambridge, MA) (Zhao et al., 2011) using PEI. After 48 h, the cells were assessed for phospho-histone H3–positive nuclei as described. For controls, pCDNA3 was transfected into GFP-CD–expressing cells.
Soft agar colony-forming assay
Anchorage-independent growth was determined using a soft agar assay. MDCK control and GFP-CD cells were plated in 0.3% agar over a base of 0.5% agar prepared in DMEM. One thousand cells were seeded into six-well plates, and colony formation was assessed after 14–21 d of culture. Colonies were counted after 14 d in culture from three independent wells, and the experiment was repeated at least five times.
Electron microscopy
MDCK cells stably expressing control or GFP-CD constructs were fixed in 3% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2, and embedded in Spurrs resin (Electron Microscopy Sciences, Hatfield, PA). Sections were examined using a Philips 410STEM at 80 kV. Images were acquired using an AMT-XR40 (Advanced Microscopy Techniques, Woburn, MA) digital camera.
Migration assay
Cells were grown to confluence in a six-well plate (Corning). Confluent monolayers were wounded using a pipette tip to create a small circular area devoid of cells. The plate was marked to allow tracking of individual wounds. The wounds were imaged using an Olympus IMT-2 microscope equipped with Hoffman modulation contrast filters at hourly intervals for 3 h, and wound area was quantified using ImageJ. Wounds to the monolayer were made in separate wells of the six-well plate. Quantification of four wounds for both the control and GFP-CD– overexpressing cells were measured. The experiment was repeated tree times.
Quantitative real-time PCR
Total RNA was extracted from GFP-CD or control cells cultured for 7 d on Transwell filters using TRIZOL reagent (Life Technologies) and DNase treated using the TURBO-DNA Free kit (Ambion, Grand Island, NY). cDNA was transcribed using the iScript CDNA synthesis kit (Bio-Rad). Primer sequences were from Wyatt et al. (2007). Data were normalized against the housekeeping gene actin. cDNA was measured using a fluorometer (Turner Biosystems, Madison, WI) with the Quanti-iT Oligreen ssDNA reagent and kit (Molecular Probes). Equal aliquots were pipetted into each reaction tube. Real-time PCR was performed using the Platinum SYBR Green qPCR Supermix UDG (Invitrogen, Grand Island, NY). In each experiment, triplicate reactions were averaged for each gene analyzed.
Statistical analysis
Statistical comparisons were made using Excel (Microsoft) and a Student's t test. Data are expressed as the mean ± SEM.
Supplementary Material
Acknowledgments
We thank members of the Wilson and Ghosh laboratories for helpful discussions and Patrick Mantyh for access to the Olympus FluoView confocal microscope. In addition, we thank James Casanova and Ghassan Mouneimne for critical reading of the manuscript and help with construction of the model. This work was supported by grants from the National Institutes of Health (RO1 DK84047, J.M.W.; RO1 CA95060, S.G.).
Abbreviations used:
- AMOT
angiomotin
- AMOTL2
angiomotin-like 2
- EDTB
endotubin
- EEA1
early endosomal antigen 1
- EMT
epithelial-to-mesenchymal transition
- ET-FL
full-length endotubin
- GFP-CD
endotubin cytoplasmic domain
- GST
glutathione S-transferase
- GST-CD
GST fused to endotubin cytoplasmic domain
- MDCK
Madin–Darby canine kidney epithelial cells
- PHH3
phospho-histone H3
- YAP
YES-associated protein
- Yki
Yorkie.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-04-0224) on May 20, 2015.
REFERENCES
- Alkasalias T, Flaberg E, Kashuba V, Alexeyenko A, Pavlova T, Savchenko A, Szekely L, Klein G, Guven H. Inhibition of tumor cell proliferation and motility by fibroblasts is both contact and soluble factor dependent. Proc Natl Acad Sci USA. 2014;111:17188–17193. doi: 10.1073/pnas.1419554111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archibald A, Mihai C, Macara IG, McCaffrey L. Oncogenic suppression of apoptosis uncovers a Rac1/JNK proliferation pathway activated by loss of Par3. Oncogene. 2014:doi:10.1038/onc.2014.242. doi: 10.1038/onc.2014.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Gayyed MF, Lam-Himlin DM, Klein AP, Nayar SK, Xu Y, Khan M, Argani P, Pan D, Anders RA. Expression of Yes-associated protein modulates Survivin expression in primary liver malignancies. Hum Pathol. 2012;43:1376–1385. doi: 10.1016/j.humpath.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11:11–23. doi: 10.1016/s1097-2765(02)00776-1. [DOI] [PubMed] [Google Scholar]
- Boggiano JC, Fehon RG. Growth control by committee: intercellular junctions, cell polarity, and the cytoskeleton regulate Hippo signaling. Dev Cell. 2012;22:695–702. doi: 10.1016/j.devcel.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratt A, Birot O, Sinha I, Veitonmaki N, Aase K, Ernkvist M, Holmgren L. Angiomotin regulates endothelial cell-cell junctions and cell motility. J Biol Chem. 2005;280:34859–34869. doi: 10.1074/jbc.M503915200. [DOI] [PubMed] [Google Scholar]
- Bratt A, Wilson WJ, Troyanovsky B, Aase K, Kessler R, Van Meir EG, Holmgren L. Angiomotin belongs to a novel protein family with conserved coiled-coil and PDZ binding domains. Gene. 2002;298:69–77. doi: 10.1016/s0378-1119(02)00928-9. [DOI] [PubMed] [Google Scholar]
- Callus BA, Verhagen AM, Vaux DL. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J. 2006;273:4264–4276. doi: 10.1111/j.1742-4658.2006.05427.x. [DOI] [PubMed] [Google Scholar]
- Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–2060. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22:3320–3334. doi: 10.1101/gad.1726608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J Biol Chem. 2011;286:7018–7026. doi: 10.1074/jbc.C110.212621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, Tao C, Halder G. The apical-basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc Natl Acad Sci USA. 2010;107:15810–15815. doi: 10.1073/pnas.1004060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Lu Q, Schneeberger EE, Goodenough DA. Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Mol Biol Cell. 2000;11:849–862. doi: 10.1091/mbc.11.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, de Herreros AG. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011;286:12024–12032. doi: 10.1074/jbc.M110.168625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas V, Pla P, Feracci H, Thiery JP, Kemler R, Larue L. Expression of the cytoplasmic domain of E-cadherin induces precocious mammary epithelial alveolar formation and affects cell polarity and cell-matrix integrity. Dev Biol. 1999;216:491–506. doi: 10.1006/dbio.1999.9517. [DOI] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Eagle H, Levine EM. Growth regulatory effects of cellular interaction. Nature. 1967;213:1102–1106. doi: 10.1038/2131102a0. [DOI] [PubMed] [Google Scholar]
- Faust D, Dolado I, Cuadrado A, Oesch F, Weiss C, Nebreda AR, Dietrich C. p38alpha MAPK is required for contact inhibition. Oncogene. 2005;24:7941–7945. doi: 10.1038/sj.onc.1208948. [DOI] [PubMed] [Google Scholar]
- Gao Z, Garbers DL. Species diversity in the structure of zonadhesin, a sperm-specific membrane protein containing multiple cell adhesion molecule-like domains. J Biol Chem. 1998;273:3415–3421. doi: 10.1074/jbc.273.6.3415. [DOI] [PubMed] [Google Scholar]
- Gilbert MM, Tipping M, Veraksa A, Moberg KH. A screen for conditional growth suppressor genes identifies the Drosophila homolog of HD-PTP as a regulator of the oncoprotein Yorkie. Dev Cell. 2011;20:700–712. doi: 10.1016/j.devcel.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokay KE, Wilson JM. Targeting of an apical endosomal protein to endosomes in Madin-Darby canine kidney cells requires two sorting motifs. Traffic. 2000;1:354–365. doi: 10.1034/j.1600-0854.2000.010408.x. [DOI] [PubMed] [Google Scholar]
- Gokay KE, Young RS, Wilson JM. Cytoplasmic signals mediate apical early endosomal targeting of endotubin in MDCK cells. Traffic. 2001;2:487–500. doi: 10.1034/j.1600-0854.2001.20706.x. [DOI] [PubMed] [Google Scholar]
- Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE. Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr Biol. 2010;20:573–581. doi: 10.1016/j.cub.2010.01.055. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM, Kim NG. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci. 2014;127:709–717. doi: 10.1242/jcs.140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13:591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114:457–467. doi: 10.1016/s0092-8674(03)00557-9. [DOI] [PubMed] [Google Scholar]
- Heller B, Adu-Gyamfi E, Smith-Kinnaman W, Babbey C, Vora M, Xue Y, Bittman R, Stahelin RV, Wells CD. Amot recognizes a juxtanuclear endocytic recycling compartment via a novel lipid binding domain. J Biol Chem. 2010;285:12308–12320. doi: 10.1074/jbc.M109.096230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JM, Nagatomo I, Suzuki E, Mizuno T, Kumagai T, Berezov A, Zhang H, Karlan B, Greene MI, Wang Q. YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene. 2013;32:2220–2229. doi: 10.1038/onc.2012.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Nusrat A, Parkos CA. Endocytosis of the apical junctional complex: mechanisms and possible roles in regulation of epithelial barriers. BioEssays. 2005;27:356–365. doi: 10.1002/bies.20203. [DOI] [PubMed] [Google Scholar]
- Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9:534–546. doi: 10.1101/gad.9.5.534. [DOI] [PubMed] [Google Scholar]
- Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci USA. 2011;108:11930–11935. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitt KN, Hernandez-Deviez D, Ballantyne SD, Spiliotis ET, Casanova JE, Wilson JM. Rab14 regulates apical targeting in polarized epithelial cells. Traffic. 2008;9:1218–1231. doi: 10.1111/j.1600-0854.2008.00752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai ZC, Wei X, Shimizu T, Ramos E, Rohrbaugh M, Nikolaidis N, Ho LL, Li Y. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 2005;120:675–685. doi: 10.1016/j.cell.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Li D, Mrsny RJ. Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J Cell Biol. 2000;148:791–800. doi: 10.1083/jcb.148.4.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling C, Zheng Y, Yin F, Yu J, Huang J, Hong Y, Wu S, Pan D. The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc Natl Acad Sci USA. 2010;107:10532–10537. doi: 10.1073/pnas.1004279107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Bilder D. Endocytic control of epithelial polarity and proliferation in Drosophila. Nat Cell Biol. 2005;7:1232–1239. doi: 10.1038/ncb1324. [DOI] [PubMed] [Google Scholar]
- Mana-Capelli S, Paramasivam M, Dutta S, McCollum D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol Biol Cell. 2014;25:1676–1685. doi: 10.1091/mbc.E13-11-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH, Turner JR. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter SD, Johnson DL, Kitt KN, Donohue C, Adams A, Wilson JM. Regulation of tight junction assembly and epithelial polarity by a resident protein of apical endosomes. Traffic. 2010;11:856–866. doi: 10.1111/j.1600-0854.2010.01052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI, Yap AS. Contact inhibition (of proliferation) redux. Curr Opin Cell Biol. 2012;24:685–694. doi: 10.1016/j.ceb.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Nishimura N, Terai T, Manabe S, Yamamoto Y, Shinahara W, Miyake H, Tashiro S, Shimada M, Sasaki T. Rab13 mediates the continuous endocytic recycling of occludin to the cell surface. J Biol Chem. 2005;280:2220–2228. doi: 10.1074/jbc.M406906200. [DOI] [PubMed] [Google Scholar]
- Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA. 2006;103:12405–12410. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais M, Chen X, Perez-Moreno M, Gumbiner BM. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol Biol Cell. 2007;18:2013–2025. doi: 10.1091/mbc.E06-04-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- Robinson BS, Huang J, Hong Y, Moberg KH. Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr Biol. 2010;20:582–590. doi: 10.1016/j.cub.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Speelman BA, Allen K, Grounds TL, Neutra MR, Kirchhausen T, Wilson JM. Molecular characterization of an apical early endosomal glycoprotein from developing rat intestinal epithelial cells. J Biol Chem. 1995;270:1583–1588. doi: 10.1074/jbc.270.4.1583. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai T, Nishimura N, Kanda I, Yasui N, Sasaki T. JRAB/MICAL-L2 is a junctional Rab13-binding protein mediating the endocytic recycling of occludin. Mol Biol Cell. 2006;17:2465–2475. doi: 10.1091/mbc.E05-09-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Vergnes L, Lee JM, Chin RG, Auwerx J, Reue K. Diet1 functions in the FGF15/19 enterohepatic signaling axis to modulate bile acid and lipid levels. Cell Metab. 2013;17:916–928. doi: 10.1016/j.cmet.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Huang J, Chen J. Angiomotin-like proteins associate with and negatively regulate YAP1. J Biol Chem. 2011;286:4364–4370. doi: 10.1074/jbc.C110.205401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, Kulkarni S, Gish G, Virag C, Lim C, et al. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535–548. doi: 10.1016/j.cell.2006.02.045. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Colton TL. Targeting of an intestinal apical endosomal protein to endosomes in nonpolarized cells. J Cell Biol. 1997;136:319–330. doi: 10.1083/jcb.136.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, de Hoop M, Zorzi N, Toh BH, Dotti CG, Parton RG. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol Biol Cell. 2000;11:2657–2671. doi: 10.1091/mbc.11.8.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Whitney JA, Neutra MR. Identification of an endosomal antigen specific to absorptive cells of suckling rat ileum. J Cell Biol. 1987;105:691–703. doi: 10.1083/jcb.105.2.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Grawe F, Knust E. CRUMBS is involved in the control of apical protein targeting during Drosophila epithelial development. Mech Dev. 1993;44:175–187. doi: 10.1016/0925-4773(93)90066-7. [DOI] [PubMed] [Google Scholar]
- Wodarz A, Hinz U, Engelbert M, Knust E. Expression of crumbs confers apical character on plasma membrane domains of ectodermal epithelia of Drosophila. Cell. 1995;82:67–76. doi: 10.1016/0092-8674(95)90053-5. [DOI] [PubMed] [Google Scholar]
- Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14:388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Wyatt L, Wadham C, Crocker LA, Lardelli M, Khew-Goodall Y. The protein tyrosine phosphatase Pez regulates TGFb, epithelial–mesenchymal transition, and organ development. J Cell Biol. 2007;178:1223–1235. doi: 10.1083/jcb.200705035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Chen J, Feng H, Peng S, Adams U, Bai Y, Huang L, Li J, Huang J, Meng S, Yuan Z. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013;73:3615–3624. doi: 10.1158/0008-5472.CAN-12-3793. [DOI] [PubMed] [Google Scholar]
- Yamamura R, Nishimura N, Nakatsuji H, Arase S, Sasaki T. The interaction of JRAB/MICAL-L2 with Rab8 and Rab13 coordinates the assembly of tight junctions and adherens junctions. Mol Biol Cell. 2008;19:971–983. doi: 10.1091/mbc.E07-06-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC, Liu Q, Shimono A, Sudol M, Holmgren L, et al. The p130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci Signal. 2013;6:ra77. doi: 10.1126/scisignal.2004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Smolen GA, Haber DA. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008;68:2789–2794. doi: 10.1158/0008-5472.CAN-07-6205. [DOI] [PubMed] [Google Scholar]
- Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011;25:51–63. doi: 10.1101/gad.2000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP) Genes Dev. 2010;24:72–85. doi: 10.1101/gad.1843810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi X, Zhao D, Zhou Z, Liu R, Chen C. YAP promotes breast cell proliferation and survival partially through stabilizing the KLF5 transcription factor. Am J Pathol. 2012;180:2452–2461. doi: 10.1016/j.ajpath.2012.02.025. [DOI] [PubMed] [Google Scholar]
- Zondag GC, Koningstein GM, Jiang YP, Sap J, Moolenaar WH, Gebbink MF. Homophilic interactions mediated by receptor tyrosine phosphatases mu and kappa. A critical role for the novel extracellular MAM domain. J Biol Chem. 1995;270:14247–14250. doi: 10.1074/jbc.270.24.14247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.