

Abstract
In this study, we investigated the impact of the cell membrane composition of E. faecalis on its recognition by the host immune system. To this end, we employed an E. faecalis deletion mutant (ΔbgsA) that does not synthesize the major cell membrane glycolipid diglycosyl-diacylglycerol (DGlcDAG). Proteomic analysis revealed that 13 of a total of 21 upregulated surface-associated proteins of E. faecalis ΔbgsA were lipoproteins. This led to a total lipoprotein content in the cell membrane of 35.8% in ΔbgsA compared to only 9.4% in wild-type bacteria. Increased lipoprotein content strongly affected the recognition of ΔbgsA by mouse macrophages in vitro with an increased stimulation of TNF-α production by heat-fixed bacteria and secreted antigens. Inactivation of the prolipoprotein diacylglycerol transferase (lgt) in ΔbgsA abrogated TNF-α induction by a ΔbgsA_lgt double mutant indicating that lipoproteins mediate increased activation of mouse macrophages by ΔbgsA. Heat-fixed ΔbgsA bacteria, culture supernatant, or cell membrane lipid extract activated transfected HEK cells in a TLR2-dependent fashion; the same was not true of wild-type bacteria. In mice infected intraperitoneally with a sublethal dose of E. faecalis we observed a 70% greater mortality in mice infected with ΔbgsA compared with wild-type-infected mice. Increased mortality due to ΔbgsA infection was associated with elevated plasma levels of the inflammatory cytokines TNF-α, IL-6 and MIP-2. In summary, our results provide evidence that an E. faecalis mutant lacking its major bilayer forming glycolipid DGlcDAG upregulates lipoprotein expression leading to increased activation of the host innate immune system and virulence in vivo.
Introduction
In invasive bacterial infections, host inflammation may vary from low-grade to a strong systemic response associated with multi-organ failure and severe sepsis. The differences in the host response are thought to result mainly from activation of the innate immune system by pathogen- and danger-associated molecular patterns. In Gram-positive sepsis, a variety of microbial compounds such as peptidoglycan and its derivatives, bacterial DNA, lipoteichoic acid, and lipoproteins are believed to activate the host immune system [1]. Numerous studies in mice have underlined the role of Toll-like receptor 2 (TLR2) as a major sensor of Gram-positive bacteria, yet its role in vivo is strongly dependent on the specific infectious microorganism [2–6]. In contrast, no clear association has been established between TLR2 variants and susceptibility to Gram-positive infection in humans [7,8].
Several TLR2 ligands have been identified in Gram-positive bacteria, including peptidoglycan, LTA, and lipoproteins/lipopeptides [9]. Studies with mutants of the lipoprotein-acyl transferase (lgt) gene and considerations regarding the structure-function relationship suggest that lipopeptides/lipoproteins are the predominant agonists of the TLR2/TLR6 dimer [10,11]. Lipoproteins/lipopeptides are important amphiphiles of the cell membrane in Gram-positive bacteria. They are found both in the cell envelope and culture supernatant [12]. In Mycobacterium tuberculosis, membrane-derived vesicles have been described as a vehicle to release lipoproteins into the environment and similar vesicles have also been described for S. aureus [13,14]. Together with phospholipids, glycolipids, and lipoteichoic acid they constitute the lipid bilayer of the cell membrane.
We have previously studied the impact of cell membrane composition on the virulence of E. faecalis using mutants deficient in glycolipid biosynthesis. For this purpose we constructed two deletion mutants in E. faecalis strain 12030 (ΔbgsA and ΔbgsB) that are defective in the glycosylation of glycolipids [15,16]. Inactivation of bgsA leads to a complete loss of DGlcDAG from the cell membrane and accumulation of high concentrations of its precursor molecule monoglycosyl-diacylglycerol (MGlcDAG) [15]. Inactivation of ΔbgsB, on the other hand, results in a cell membrane devoid of glycolipids [16]. Both ΔbgsA and ΔbgsB elaborate a longer poly-glycerophosphate polymer of LTA than wild-type bacteria and show impaired biofilm formation and attachment to colonic epithelial cells. In a mouse bacteremia model, both mutants were cleared more rapidly from the bloodstream [15,16]. Interestingly, defects in glycolipid biosynthesis in ΔbgsA and ΔbgsB were not associated with changes in the bacterial cell shape or ultrastructure, in the growth rate, or in sensitivity to osmotic stress. This finding was surprising, since the ratio of the bilayer-forming DGlcDAG and the nonbilayer-prone MGlcDAG was shown to be critical for cell membrane architecture and curvature stress in studies using Acholeplasma laidlawii [17,18].
Here we examined the consequences of the altered glycolipid composition in ΔbgsA on the cell-surface proteome of the bacteria and studied the impact of these changes on the interaction between bacteria and the host immune system. For the investigation of the virulence of glycolipid-deficient E. faecalis strains we used a mouse peritonitis model that has been validated in several previous studies [19–22]. Our results show that in the absence of DGlcDAG, lipoprotein expression is upregulated in E. faecalis, which substantially increases the activation of TLR2 and virulence in vivo.
Materials and Methods
Bacterial strains, growth conditions, and medium
The bacterial strains and plasmids used in this study are listed in Table 1. Enterococci were cultured in tryptic soy broth (TSB, Merck), M17 broth (Difco Laboratories), Caso Bouillon (Carl Roth), or TSB plus 1% glucose (TSBG) as indicated. In addition, tryptic soy agar or M17 agar plates were used. Escherichia coli DH5α and TOP10 (Invitrogen) were cultivated aerobically in LB-broth. For cell culture stimulation studies, bacteria were grown in chemically defined medium (CDM) prepared from endotoxin-free water [23].
Table 1. Bacterial strains used and plasmids used in this study.
| strain or plasmid | characterization | reference |
|---|---|---|
| strains | ||
| E. faecalis 12030 | Clinical isolate, strong biofilm producer | [54] |
| E. faecalis 12030ΔbgsA | (EF2891) bsgA deletion mutant | [15] |
| E. faecalis 12030ΔbgsB | (EF2890) bsgB deletion mutant | [25] |
| E. faecalis 12030Δlgt | (EF 1748) lgt deletion mutant | this study |
| E. faecalis 12030ΔbgsA_lgt | double bgsA-lgt deletion mutant | this study |
| E. faecalis 12030ΔbgsB_lgt | double bgsB-lgt deletion mutant | this study |
| Escherichia coli DH5α | Gram-negative cloning host | Invitrogen |
| Escherichia coli TOP10 | Gram-negative cloning host | Invitrogen |
| plasmids | ||
| pCASPER | Gram-positive, temperature-sensitive mutagenesis vector | [55] |
| pCRII-TOPO | Gram-negative cloning vector | Invitrogen |
| pCASPER/Δlgt | pCASPER carrying a lgt deletion | this study |
Construction of deletion mutant Δlgt
A non-polar deletion of a portion of gene lgt (EF1748 in E. faecalis V583, GenBank ID accession number NP_815451) was created using the method described by Cieslewicz et al., [24] with the following modifications: primers 1 and 2 (Table 2) were used to amplify a 503-bp fragment from the region upstream of gene lgt, and also the end part of EF1747. Primers 3 and 4 were used to amplify a 546-bp fragment downstream of the lgt gene and the beginning of EF1749. Primers 2 and 3 contain a 21-bp complementary sequence (underlined in Table 2). Overlap extension PCR was used to create a PCR product lacking a portion of gene EF1748. The resulting fragment was cloned into Gram-negative cloning vector pCRII-TOPO (Invitrogen) and cut with the restriction enzyme EcoRI (Invitrogen); the resulting fragment was then inserted into shuttle vector pCASPER containing a temperature-sensitive origin of replication. The resulting plasmid, pCASPER/Δlgt, was transformed into E. faecalis 12030 by electroporation, and integrants were selected at a non-permissive temperature (42°C) on TSA plates with kanamycin (1 mg/ml). A single colony was picked, and insertion of plasmid into the chromosome was confirmed by PCR. The integrant was passaged 10 times in liquid culture without antibiotic at the permissive temperature (30°C), and colonies were replica-plated to screen for loss of kanamycin resistance. The excision of the plasmid either creates a reconstituted wild-type strain or leads to an allelic replacement with the deleted sequence in the chromosome. The deletion mutant created was designated E. faecalis 12030Δlgt, containing a 507-bp (169 amino acids) in-frame deletion. The genotype was confirmed by PCR and automated sequencing.
Table 2. Primers used in this study.
| name | sequence (5´-3´) a | |
|---|---|---|
| 1 | pEF1748delF | CCTTGTTCGAGCCCTTTACTT |
| 2 | pEF1748OEF | ACTAGCGCGGCCGCTTGCTCCGTTCGTGGCAGCAATTGTTAT |
| 3 | pEF1748delR | ACGTCATGAACCTGTTTGGAG |
| 4 | pEF1748OER | GGAGCAAGCGGCCGCGCTAGTTAATCTTGCCATTGAAAAGCG |
aLinkers are underlined.
Construction of a ΔbgsA_lgt and ΔbgsB_lgt double mutant
For construction of the double mutants bgsA_lgt and bgsB_lgt the plasmid pCASPER/Δlgt was transformed into prepared electroporation-competent cells of E. faecalis 12030ΔbgsA or ΔbgsB following the procedure described above for the construction of the single Δlgt deletion mutant.
Preparation of E. faecalis antigens for stimulation experiments
E. faecalis strains were grown for 16 h in CDM to stationary phase, collected by centrifugation and washed twice in phosphate buffered saline (PBS). The multiplicity of infection (MOI) for the cell culture experiments was calculated by quantification of colony-forming units (CFU) of serially diluted live bacteria of the respective strain on agar plates, with subsequent adjustment of the suspension of heat-fixed cells to the desired MOI. Cell culture supernatant was filter-sterilized with a 0.2-μm membrane, dialyzed for 24 h against endotoxin-free water, and lyophilized. Bacteria were fixed at 65°C for 60 min. Extraction according to the method of Bligh and Dyer was used to obtain total cell membrane lipids as described previously [15,25]. The concentration of the supernatant and total lipid extracts in the stimulation experiments is expressed as weight per volume.
Lipoproteins were enriched from cell membrane fractions by phase-partitioning with Triton X-114 (TX114) [11,26,27]. To this end, bacterial cells were grown in CDM medium as described above and quantified by serial dilutions and viable counts after culture on agar plates. Bacterial cells were disrupted by vibration with glass beads as described previously [15]. Cell debris and glass beads were pelleted by centrifugation and the supernatant was diluted in TN-buffer (20 mM Tris, 100 mM NaCl, pH 8.0) and passed through a 0.2 μm membrane. For separation of the cell membrane fraction, the filtered supernatant was ultracentrifuged at 50,000 rpm at 4°C for 1 h. The supernatant was discarded and the cell membrane fraction redissolved in TN-buffer plus 2% TX114 (v/v) and incubated at 4°C for 2 h, followed by a second incubation step at 37°C for 30 min. The solution was centrifuged (6,000 rpm, 10 min, 37°C) for phase separation. The lower detergent-soluble phase was carefully collected. The detergent-soluble phase was further purified by adjusting the TX114 concentration to 2% and repeating the phase partitioning, as described above. For removal of TX114, the detergent phase was mixed with 90% ethanol, incubated at -20°C for 18 h and precipitated proteins were collected by centrifugation. After resuspension in TN-buffer the protein concentration of the lipoprotein extracts was determined photometrically and normalized to bacterial cfu.
Metabolic labeling, isolation of surface-associated proteins
Surface-associated proteins were isolated by biotin labeling and affinity chromatography. Proteins were quantified with the spike-in of a stable isotope-labeled standard. For metabolic labeling the method of Becher et al. was used with modifications [28]. E. faecalis 12030 wild-type and ΔbgsA were each grown separately in both labeled and unlabeled rich growth media for E. coli, (E.coli-OD5, Silantes) supplemented with 2% glucose, vitamins (p-amino benzoic acid, biotin, folic acid, niacinamide, pantothenate, riboflavin, thiamine), and nucleotides. Cultures were grown at 37°C for 18 h while shaking at 140 rpm. As an internal standard, equal volumes of wild-type and ΔbgsA grown to the same OD600nm in 15N-labeled medium were mixed. Subsequently, equivalent OD units of wild-type and ΔbgsA cell culture grown in unlabeled medium were each mixed with the internal standard and centrifuged.
Cell pellets of wild-type and ΔbgsA were each washed once with PBS (pH 8) plus PMSF (1 mM). Cells (0.15 g) were resuspended in 1 ml PBS/PMSF, and Sulfo-N-hydroxysulfosuccinimide-disulfide-Biotin (Sulfo-NHS-SS-Biotin; Thermo Scientific) was added to produce a final concentration of 1 mM. The cell suspension was shaken carefully on ice for 1 h. After centrifugation, biotinylation was stopped by washing cells three times in 1 ml PBS plus 500 mM glycine. Next, cells were resuspended in 1 ml PBS and disrupted with glass beads (0.1 mm diameter) in two cycles at 6,800 rpm for 30 s using a ribolyser (Roche). To obtain cell membrane proteins, cell debris was washed six times with PBS and centrifuged at 45,000 rpm for 1 h at 4°C. Cell debris was resuspended in 0.4 ml PBS/I2-Iodoacetamide (5%) and homogenized with glass beads (0.1 mm diameter) using a ribolyser (6,800 rpm; 2x 30 s). To each sample 100 μl PBS plus 20% CHAPS and 20% Amidosulfobetaine-14 was added, homogenized in a ribolyser (6,800 rpm; 2 x 30 s) and shaken gently for 1 h at 20°C. Cell debris was removed again by centrifugation (14,000 rpm; 15 min; 4°C). Biotinylated proteins were recovered by incubation of the protein lysate with NeutrAvidin-agarose-beads equilibrated in PBS/Nonidet P40 (1%) for 1 h on ice while shaking. Unbound proteins were removed by washing beads four times with PBS/NP-40 plus 6% CHAPS and two times with 1 ml PBS/NP-40 plus 2% SDS. For elution of bound proteins, the disulfide bond of Sulfo-NHS-SS-Biotin was cleaved by incubation with 5% β-mercaptoethanol in deionized water for 1 h at 20°C. NeutrAvidin-Agarose-Beads were removed by centrifugation and the supernatant was transferred to 8 ml acetone (-20°C). Elution buffer was once again added to each sample, centrifuged and the resulting supernatant was also added to the acetone-elution-buffer-mix. Proteins were precipitated with acetone overnight at -20°C. Precipitated proteins were centrifuged (8,500 rpm; 30 min; 4°C) and washed with ethanol (98%; 4°C). Finally, the pellet was dried in 6 M urea/2 M thiourea under vacuum (SpeedVac; Bachofer).
Protein identification and quantification by ESI-LC-MS/MS
Subsequent to preparation of the mixed and labeled protein extracts, samples were subjected to SDS-gel electrophoresis. Resulting gel lanes were cut into equidistant pieces, followed by tryptic in-gel digestion as described elsewhere [29]. The resulting proteolytic digests were subjected to LC-MS/MS analyses as described elsewhere [30]. In brief, peptides were applied to reversed-phase C18 chromatography on an EASYnLC system (Thermo Fisher) online coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher). For LC-MS analyses a full survey scan in the Orbitrap (m/z 300–2000) with a resolution of 30,000 was followed by MS/MS experiments of the five most abundant precursor ions acquired in the linear trap quadrupole (LTQ) via collision-induced dissociation (CID).
Database searching for light and heavy extracts and subsequent quantitation were done as described in Otto et al. [30]. For unambiguous identification data, database searching by Sorcerer- Sequest (version 3.5) relied on an E. faecalis target-decoy protein sequence database (E. faecalis V583 (NC_004668.1) including a set of common contaminants). The following parameters were set for database searching: enzyme type, trypsin maximum of two missed trypsin cleavage sites; peptide tolerance, 10 ppm; tolerance for fragment ions, 1amu.; b- and y-ion series; variable modification, oxidation of methionine (15.99 Da). For database searches for 15N-labeled peptides, the mass shift of all amino acids completely labeled with15N-nitrogen was taken into account. Relative quantification was carried out as described previously [31]. Protein identifications were considered significant for the biological system if the protein was identified in at least two out of three samples in wild type or the mutant. The crude search results served as the base for the further analysis using Census software to obtain quantitative data of 14N peaks (sample) and 15N peaks (standard) [32].
Relative quantification of surface-associated proteins by spectral counting
For estimation of relative proportions of proteins within the surface proteome of E. faecalis, the normalized spectral abundance factor (NSAF) for the light mass traces (14N) were calculated as described elsewhere [33]. For calculation of NSAF, spectra from raw data were extracted and searched as described above taking into account only light masses. Scaffold (version Scaffold_4.3.2, Proteome Software Inc.) was then used to validate MS/MS based peptide and protein identifications. Peptide identifications were only accepted if they exceeded specific database search engine thresholds. Sequest identifications required at least deltaCn scores of greater than 0.10 and XCorr scores of greater than 2.2, 3.3, and 3.8 for doubly, triply, and quadruply charged peptides, respectively. Protein identifications were accepted if they contained at least two identified peptides. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Calculation of NSAF was carried out as described elsewhere [34].
RAW 264.7 mouse macrophage stimulation
RAW 264.7 macrophages were seeded at a density of 1 x 105 cells/ml in 24-well dishes in endotoxin-free DMEM containing 10% fetal calf serum. Cultures were stimulated with either heat-killed bacteria, lyophilized supernatant or lipoprotein TX114-extracts from bacterial culture for 16 h at 37°C in a 5% humidified CO2 environment. After the incubation period, cell culture supernatant was collected and TNF-α production measured by commercial ELISA (R&D Systems) according to the manufacturer’s protocols. A commercial LPS preparation from E. coli 0111:B4 (Sigma Chemicals) was used as positive control.
Reporter gene analysis
HEK293 cells (Sigma Aldrich) and the stable cell line HEK-TLR2YFP were used in reporter gene studies as described [2]. In brief, cells were seeded into 96-well tissue-culture plates at a density of 5 x 105 cells/well. Cells were transiently transfected 16 hours later with an ELAM.luc reporter construct with TransIT-LT1 transfection reagent (Mirus Bio). Plasmid pcDNA was used to assure equal amounts of transfected DNA. The following day cells were incubated for 6 h with bacterial cells or cell wall extracts as indicated. After incubation, cultured cells were lysed in passive lysis buffer (Promega) and reporter gene activity was measured using a plate reader luminometer (MicroLumat Plus; Berthold).
Mouse peritonitis model
The virulence of E. faecalis strains was evaluated in a mouse peritonitis model. The mice were housed in groups of 5 per cage. All procedures were carried out between 8 a.m. and 18 p.m. in the animals home cage. Fourteen female BALB/C mice (Charles River Laboratories) 6–8 weeks old per group were assigned randomly to infection by intra-peritoneal injection of E. faecalis strains as indicated. The inoculum for the infection model was grown in TSB for 16 h to stationary phase, washed in PBS, adjusted to the desired concentration and injected intraperitoneally (i.p.) in 200 μl of PBS. The bacterial inoculum was confirmed by plating serial dilutions on agar plates. Mice were monitored twice daily for mortality or signs of illness. If mice had reached an unconscious or moribund state they were euthanized by carbon dioxide inhalation and counted as dead. A moribund condition was defined as impaired mobility, the inability to reach food and water or to keep an upright position, labored breathing or cyanosis, or a hunched position for more than 48 h. Moribund mice were placed in a chamber and CO2 was introduced at a displacement rate approximately 20% of the chamber volume per min. The CO2 flow was maintained for at least 1 minute after respiratory arrest of the animal. For quantification of bacterial counts and measurement of cytokines, mice (six per group) were sacrificed as described above. Peritoneal lavage fluid (PLF) was obtained by injecting 2 ml of sterile PBS with a 18-gauge needle. Blood was drawn under sterile precautions by cardiac puncture and transferred to heparin tubes. Next, the abdomen was opened, and the right kidney was harvested. All samples were directly placed on ice and processed immediately. The number of E. faecalis CFU in the PLF, blood, and kidney homogenate was determined. Kidneys were weighed and homogenized at 4°C in 2 ml of TSB with a tissue homogenizer. Serial 10-fold dilutions were made of each sample in TSB, 10 μl of each dilution was plated onto TSA plates and CFUs were enumerated after 18 h of incubation. The leukocytes in the PLF were counted as described elsewhere [22]. PLF supernatant and plasma were stored at -20°C until measurement of the cytokines.
Ethics statement
All animal experiments were performed in compliance with the German animal protection law (TierSchG). The mice were housed and handled in accordance with good animal practice as defined by FELASA and the national animal welfare body GV-SOLAS. The animal welfare committees of the University of Freiburg (Regierungspraesidium Freiburg Az 35/9185.81/G-07/15) approved all animal experiments. The institutional review board of the University of Freiburg approved the study protocol.
Statistical analysis
Statistical significance for two-way comparisons was determined by an unpaired t-test as indicated. Analysis of variance (ANOVA) for multigroup comparisons was used on log-transformed data, and the Tukey’s multiple-comparison test was used for posthoc analysis for pairwise comparisons. Survival data were compared using the log-rank (Mantel-Cox) test. To identify the up- and downregulated proteins of the mutant compared to the wild-type, the log2 ratio of each protein quantification was calculated, subtracting the median log2 ratio of the replicates of the wild-type from that of the mutant. The relative quantification of proteins by spectral counting was deemed reliable when its ratio was ascertained in two replicates with not less than two peptides in at least one of the replicates [35]. Statistical results were calculated using the Prism 3 software package. Statistical significance for the NSAF values (mutant versus wild-type) was determined by a t-test in the software Scaffold (version Scaffold_4.3.2, Proteome Software Inc., Portland, OR) with a significance threshold alpha = 0.05.
Results
Analysis of enterococcal surface-associated proteins by the proteomics approach
To gain insight into the protein composition of the enterococcal cell-surface, wild-type bacteria and ΔbgsA surface-exposed proteins were biotinylated and purified by affinity chromatography prior to SDS-PAGE followed by in-gel digestion (IGD) and analysis by LC-MS/MS (GeLC-MS). Before surface proteins were selectively coupled to Sulfo-NHS-SS-Biotin, 14N15N metabolic protein labeling was performed during cultivation in CDM. Biotin-labeled proteins were purified by affinity-chromatography and eluted by cleavage of the disulfide bond of the Sulfo-NHS-SS-Biotin by reduction.
This approach led to the unambiguous identification of a total of 210 proteins from wild-type bacteria in 2/3 replicates (S1 Table). Determined by the theoretical prediction rules of subcellular localization (reference 53, 36 and the LocateP DataBase was used (http://www.cmbi.ru.nl/locatep-db/cgi-bin/locatepdb.py)) the surface proteome fraction included 23 lipoproteins, 16 membrane proteins, and seven extracellular proteins containing a signal peptide. One hundred and sixty-four of the biotinylated proteins obtained were annotated with a cytoplasmic subcellular localization.
In ΔbgsA the production of lipoproteins is upregulated
The altered composition of cell membrane glycolipids in ΔbgsA led to a profound change in the pattern of surface-associated proteins as compared to isogenic wild-type E. faecalis. A total of 88 proteins were significantly up- or downregulated proteins in ΔbgsA according to the statistical testing. Forty showed a significantly increased amount in ΔbgsA compared with wild-type bacteria, while 48 proteins were decreased (Table 3). Twenty-one predicted surface-associated proteins were upregulated in ΔbgsA. Of those, 13 were predicted to be lipoproteins, six cell membrane proteins and two extracellular proteins (Fig 1). Only six of the downregulated proteins were surface-associated proteins. Strikingly, lipoproteins represented 35.8% of the surface-associated proteins of ΔbgsA compared to only 9.4% in wild-type bacteria as quantified by spectral abundance factors (detailed information see S2 Table). Of the five most overexpressed surface-associated proteins, all were lipoproteins and they were upregulated 1.95–12.22-fold in ΔbgsA compared to wild type bacteria (Table 3). Altogether, our data suggest that the inactivation of bgsA disturbs the equilibrium in the cell membrane that in consequence leads to an increased lipoprotein content.
Table 3. Surface-associated proteins present in significantly different amounts in ΔbgsA compared to the wild-type.
The log2ratio depicts the change of ΔbgsA compared to the wild-type.
| Protein accession number | log2 (ΔbgsA/wt) | Annotation | Localization a |
|---|---|---|---|
| EF3041 | 12.222 | Pheromone binding protein | lipoprotein |
| EF1641 | 3.114 | Iron compound ABC transporter, iron compound binding protein | lipoprotein |
| EF1534 | 2.870 | Peptidyl prolyl cis/trans isomerase | lipoprotein |
| EF3198 | 2.258 | Lipoprotein. YaeC family | lipoprotein |
| EF1354 | 2.230 | Pyruvate dehydrogenase complex. E1 component. beta subunit | cytoplasmic |
| EF1353 | 2.207 | Pyruvate dehydrogenase complex. E1 component. alpha subunit | cytoplasmic |
| EF2191 | 1.983 | dTDP-4-dehydrorhamnose reductase | cytoplasmic |
| EF3082 | 1.955 | Iron compound ABC transporter. substrate binding protein | lipoprotein |
| EF1111 | 1.842 | Signal peptidase I | cell membrane |
| EF1212 | 1.828 | Transcriptional regulator LytR | cell membrane |
| EF2156 | 1.813 | Uncharacterized protein | extracellular |
| EF3062 | 1.768 | Cell shape determining protein MreC | extracellular |
| EF3120 | 1.740 | Serine threonine protein kinase | cytoplasmic |
| EF3037 | 1.602 | Glutamyl aminopeptidase | cytoplasmic |
| EF1677 | 1.601 | Lipoprotein. putative | lipoprotein |
| EF1340 | 1.562 | Pheromone cAM373 lipoprotein | lipoprotein |
| EF1191 | 1.526 | DegV family protein | cytoplasmic |
| EF1759 | 1.491 | Phosphate ABC transporter. phosphate binding protein | lipoprotein |
| EF3027 | 1.420 | Serine protease DO | cell membrane |
| EF1523 | 1.324 | Conserved domainl protein | cytoplasmic |
| EF2656 | 1.315 | Flavoprotein family protein | cytoplasmic |
| EF0176 | 1.300 | Basic membrane protein family | lipoprotein |
| EF1416 | 1.201 | Glucose-6-phosphate isomerase | cytoplasmic |
| EF2697 | 1.159 | Conserved domain protein | cell membrane |
| EF1753 | 1.141 | Uncharacterized protein | cytoplasmic |
| EF1045 | 1.111 | 6-phosphofructokinase | cytoplasmic |
| EF0761 | 1.097 | Amino acid ABC transporter. amino acid binding permease protein | cell membrane |
| EF2496 | 1.085 | Lipoprotein | lipoprotein |
| EF0685 | 1.082 | Foldase protein PrsA | cell membrane |
| EF0784 | 1.079 | S-adenosylmethionine synthase | cytoplasmic |
| EF0949 | 1.040 | Phosphotransacetylase | cytoplasmic |
| EF2608 | 0.904 | ATP synthase subunit beta | cytoplasmic |
| EF2610 | 0.903 | ATP synthase subunit alpha | cytoplasmic |
| EF0863 | 0.887 | Glycine betaine carnitine choline ABC transporter. glycine betaine carnitine choline binding protein | lipoprotein |
| EF2609 | 0.794 | ATP synthase gamma chain | cytoplasmic |
| EF1355 | 0.793 | Pyruvate dehydrogenase complex E2 component. dihydrolipoamide acetyltransferase | cytoplasmic |
| EF3255 | 0.637 | Thiamin biosynthesis lipoprotein ApbE. putative | lipoprotein |
| EF1961 | 0.478 | Enolase | cytoplasmic |
| EF2549 | 0.473 | Uracil phosphoribosyltransferase | cytoplasmic |
| EF2903 | 0.190 | ABC transporter. substrate binding protein | lipoprotein |
| EF3256 | -0.569 | Pheromone cAD1 lipoprotein | lipoprotein |
| EF1548 | -0.577 | Ribosomal protein S1 | cytoplasmic |
| EF1402 | -0.580 | Conserved domain protein | cytoplasmic |
| EF1193 | -0.592 | DNA binding response regulator VicR | cytoplasmic |
| EF2932 | -0.600 | AhpC TSA family protein | cytoplasmic |
| EF1584 | -0.607 | Cysteine synthase | cytoplasmic |
| EF0997 | -0.696 | Cell division protein FtsZ | cytoplasmic |
| EF1138 | -0.871 | Oxidoreductase. aldo-keto reductase family | cytoplasmic |
| EF2355 | -0.934 | Chaperone protein ClpB | cytoplasmic |
| EF1744 | -0.956 | General stress protein. putative | extracellular |
| EF1963 | -0.992 | Phosphoglycerate kinase | cytoplasmic |
| EF1560 | -1.012 | Uncharacterized protein | cytoplasmic |
| EF3054 | -1.051 | Lipoprotein. putative | lipoprotein |
| EF0944 | -1.059 | protein. putative | extracellular |
| EF0715 | -1.107 | Trigger factor | cytoplasmic |
| EF1522 | -1.118 | RNA polymerase sigma factor RpoD | cytoplasmic |
| EF1764 | -1.171 | Ribosomal subunit interface protein | cytoplasmic |
| EF3230 | -1.174 | 30S ribosomal protein S9 | cytoplasmic |
| EF2607 | -1.236 | ATP synthase epsilon chain | cytoplasmic |
| EF2397 | -1.314 | Elongation factor Ts | cytoplasmic |
| EF0228 | -1.380 | Adenylate kinase | cytoplasmic |
| EF2612 | -1.382 | ATP synthase subunit b | cytoplasmic |
| EF2866 | -1.384 | Probable transcriptional regulatory protein | cytoplasmic |
| EF0671 | -1.412 | Xaa-his dipeptidase | cytoplasmic |
| EF0287 | -1.442 | Elongation factor P | cytoplasmic |
| EF2718 | -1.468 | 50S ribosomal protein L1 | cytoplasmic |
| EF0453 | -1.605 | mC-Ohr family protein | cell membrane |
| EF0105 | -1.613 | Ornithine carbamoyltransferase. catabolic | cytoplasmic |
| EF0770 | -1.628 | Uncharacterized protein | cytoplasmic |
| EF0020 | -1.639 | PTS system. mannose specific IIAB components | cytoplasmic |
| EF2715 | -1.642 | 50S ribosomal protein L7 L12 | cytoplasmic |
| EF2729 | -1.689 | Transcription termination antitermination protein nusG | cytoplasmic |
| EF0709 | -1.708 | Phosphocarrier protein HPr | cytoplasmic |
| EF2415 | -1.738 | Uncharacterized protein | cytoplasmic |
| EF0220 | -1.846 | 30S ribosomal protein S8 | cytoplasmic |
| EF2719 | -1.847 | 50S ribosomal protein L11 | cytoplasmic |
| EF0079 | -1.896 | Gls24 protein | cytoplasmic |
| EF0820 | -1.993 | 50S ribosomal protein L25 | cytoplasmic |
| EF2552 | -2.036 | Sua5 YciO YrdC YwlC family protein | cytoplasmic |
| EF2395 | -2.093 | Ribosome recycling factor | cytoplasmic |
| EF0394 | -2.117 | Secreted antigen. putative | extracellular |
| EF1307 | -2.132 | Protein GrpE | cytoplasmic |
| EF0012 | -2.132 | 50S ribosomal protein L9 | cytoplasmic |
| EF0080 | -2.160 | Gls24 protein | cytoplasmic |
| EF2925 | -2.233 | Cold shock domain family protein | cytoplasmic |
| EF2633 | -2.306 | 60 kDa chaperonin | cytoplasmic |
| EF1308 | -2.341 | Chaperone protein DnaK | cytoplasmic |
| EF0466 | -2.345 | Glucosamine-6-phosphate deaminase | cytoplasmic |
a Transmembrane domains were predicted with the TMHMM 2.0 algorithm [53]. Lipoproteins were classified according to Reffuveille et al. [36]. For prediction of cell wall, cytoplasmatic and extracellular proteins Locate P was used (http://www.cmbi.ru.nl/locatep-db/cgi-bin/locatepdb.py). For detailed data see S1 Table.
Fig 1. Altered proteins in ΔbgsA classified according to subproteomes by bioinformatic identification.
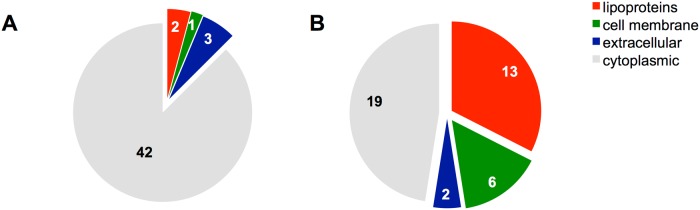
Proteins significantly downregulated in ΔbgsA compared to E. faecalis 12030 wild type (A). Upregulated proteins in ΔbgsA compared to the wild-type (B). Transmembrane domains were predicted with the TMHMM 2.0 algorithm [53]. Lipoproteins were classified according to Reffuveille et al. [36]. For prediction of cell wall, cytoplasmic and extracellular proteins Locate P was used (http://www.cmbi.ru.nl/locatep-db/cgi-bin/locatepdb.py).
ΔbgsA promotes vigorous activation of RAW 264.7 mouse macrophages
To evaluate the effect of the altered cell-surface composition of ΔbgsA on inflammatory responses in vitro, we stimulated RAW 264.7 macrophages in the presence of live and heat-fixed E. faecalis and measured TNF-α in the cell culture supernatant after 3 h (live bacteria) and 16 h (heat-fixed bacteria and supernatant). In addition to ΔbgsA and wild-type bacteria, we used ΔbgsB as a second mutant defective in glycolipid biosynthesis in these experiments. At multiplicities of infection between 1:1 and 10:1, ΔbgsA induced significantly higher TNF-α concentrations compared to wild-type bacteria and ΔbgsB, respectively (Fig 2A and 2B).
Fig 2. Induction of TNF-α in RAW 264.7 mouse macrophages by E. faecalis strains.

Macrophages were stimulated with live and heat-fixed bacteria at multiplicities of infection (MOI) as indicated (A and B) or with cell-free, dialyzed E. faecalis supernatants (C). RAW 264.7 macrophages were seeded at a density of 1 x 105 cells/ml in 24-well dishes in endotoxin-free DMEM containing 10% fetal calf serum. Cultures were stimulated at 37°C in a 5% humidified CO2 environment for 3 h (A) and 16 h (B), respectively, and supernatant from macrophage culture was analyzed for TNF-α by ELISA. The strains used for stimulation are indicated in the legend. LPS was used as positive control. Data represent mean ± SEM of triplicates. ND = not detected. * p < 0.001 12030 ΔbgsA versus 12030 WT, ** p < 0.001 12030 ΔbgsA versus 12030 ΔbgsB, + p < 0.001 12030 ΔbgsB versus 12030 WT. § p < 0.001 12030 wild type versus 12030 Δlgt, $ p < 0.001 12030 ΔbgsA versus 12030 ΔbgsA_lgt, & p < 0.001 12030 ΔbgsB versus 12030 ΔbgsB_lgt. Results were compared by 2-way ANOVA with Bonferroni post-test for pairwise comparisons.
Staphylococcus aureus lipoproteins are known to be released during growth into the culture medium [12]. We therefore also stimulated RAW 264.7 macrophages with dialyzed culture supernatant of E. faecalis strains. Supernatant from ΔbgsA was a potent inducer of TNF-α production of RAW 264.7 cells in vitro (Fig 2C). While culture supernatant of wild-type bacteria was inactive even at concentrations as high as 10,000 ng/ml (dry weight per volume), ΔbgsA stimulated TNF-α production at concentrations a 100-fold lower (Fig 2). Low amounts of TNF-α were also induced by cell culture supernatant from ΔbgsB (Fig 2). Together with the proteome analysis, these results demonstrate that lack of DGlcDAG in the cell membrane of ΔbgsA not only changes the composition of cell-surface proteome, but also enhances the activation of innate immunity.
Lipoprotein-enriched cell membrane fractions of ΔbgsA but not wild-type cells stimulate TNF-α production of RAW 264.7 macrophages
Due to their amphiphilic properties, lipoproteins can be purified from cell membrane fractions by the detergent Triton X-114 [11,26]. MS-shotgun analysis of Triton-extracted total membrane protein fractions from ΔbgsA and wild-type bacteria confirmed a high concentration of lipoproteins of 85% and 63%, respectively, in the extracts (data not shown). The Triton-X114 extracts from ΔbgsA induced an increased TNF-α production compared to the wild-type extracts (Fig 3). Taken together, cell membrane fractions of ΔbgsA highly enriched in lipoprotein are more potent inducers of TNF-α than those from the wild-type strain.
Fig 3. Stimulation of TNF-α production in RAW 264.7 mouse macrophages by lipoprotein-enriched cell membrane fractions of E. faecalis wild type and ΔbgsA.

RAW 264.7 cells were incubated with lipoprotein-enriched Triton X-114 extracts from total membrane protein fractions derived from the indicated E. faecalis strains. The concentration of lipoprotein extracts was measured photometrically and normalized to a bacterial cfu:RAW 264.7 cell ratio of 10,000:1. At 16 h, supernatants were collected and TNF-α concentrations were quantified by ELISA. LPS at a concentration of 100 ng/ml was used as positive control. Data represent mean ± SEM of triplicates. * p < 0.001 12030 ΔbgsA versus 12030 WT.
Inactivation of lipoprotein acylation abrogates induction of TNF-α by wild-type E. faecalis and ΔbgsA
In total, 90 genes that harbor the type II signal sequence typical for lipoproteins have been identified in E. faecalis, representing about 2.7% of the genome [36]. Similar to other Gram-positive bacteria, inactivation of the prolipoprotein diacylglycerol transferase (lgt) in E. faecalis causes the arrest of lipoprotein maturation at the stage of acylation of the protein, yielding non-acylated lipoproteins [37,38]. In E. faecalis, the deletion of lgt had no effect on bacterial morphology, growth rate, and sensitivity to sodium chloride, different pH condition, or exposure to antibiotics [38]. To corroborate our findings, we constructed a deletion mutant of the lgt gene in E. faecalis 12030 and created the double-deletion mutants EF 12030ΔbgsA_lgt and EF 12030ΔbgsB_lgt. Activation of TNF-α production in macrophages stimulated with lgt-mutants or wild-type E. faecalis was analyzed (Fig 2). Similar to S. aureus and group B streptococci [2,12,39], impaired protein-lipidation in E. faecalis 12030 led to a reduction of macrophage activation (Fig 2). Likewise, deletion of lgt in ΔbgsA and ΔbgsB decreased the production of TNF-α by RAW 264.7 macrophages compared to the respective single mutant (Fig 2). Inactivation of the lgt-gene in ΔbgsA had even stronger effects on TNF-α production in the RAW 264.7 macrophage activation assay if culture supernatant was used as stimulant (Fig 2).
ΔbgsA induces a strong activation of TLR2
Next, we wanted to identify the cognate receptor for lipoproteins in ΔbgsA. To this end, wild-type bacteria, ΔbgsA and ΔbgsB were analyzed for NF-κB activation in a NF-κB-dependent luciferase reporter assay in epithelial cells (HEK 293) that stably express the human TLR2 receptor [40]. Whole E. faecalis wild-type bacteria did not activate NF-κB even at high concentrations (Fig 4). In contrast, ΔbgsA induced NF-κB activation at concentrations as low as 1 μg/ml (dry weight). ΔbgsB also induced NF-κB, but was a less potent stimulus than ΔbgsA (Fig 4). Dialyzed, cell-free bacterial cell culture supernatant of wild-type bacteria also did not induce NF-κB activation, while supernatant from ΔbgsA was a strong inducer. Cells were also stimulated with bacterial cell envelope compounds extracted by the Bligh-Dyer method. This method extracts lipids and lipid-containing biomolecules from bacterial or eukaryotic cells [41]. Again, extracts of ΔbgsA but not of the wild-type stimulated TLR2 activation (Fig 4). Compared to whole bacterial cells, cell culture supernatant and lipophilic antigens extracts were about 10-fold less potent agonists of the TLR2 receptor (Fig 4). These data establish that E. faecalis ΔbgsA cells and its culture supernatant contain higher levels of TLR2 agonists compared to wild-type bacteria.
Fig 4. Stimulation of HEK-TLR2 cells transfected with an NF-κB-dependent ELAM-luciferase reporter gene with E. faecalis antigens.

HEK cells were stimulated for 6 h with escalating antigen concentrations as indicated, lysed and luciferase activity was determined by luminometry. The concentration of bacterial cells is expressed as dry weight per ml. Fold-induction denotes stimulated versus non-stimulated luciferase activity. Stimulation with whole bacterial cells (A). Stimulation with dialyzed cell-free culture supernatant (B). Stimulation with E. faecalis cell membrane total lipid extracts (C). Data are expressed as mean ± SEM of triplicates. ND = not detected. * p < 0.001 12030 ΔbgsA versus 12030 WT, ** p < 0.001 12030 ΔbgsA versus 12030 ΔbgsB, +++ p < 0.001 12030 ΔbgsB versus 12030 wild type by 2-way ANOVA with Bonferroni post-test for pairwise comparisons.
Inactivation of bgsA enhances virulence in a mouse bacteremia model
TLR-mediated activation of cellular innate immunity during infection is tightly regulated, since over- and underactivation can have detrimental consequences to the host. Given the strong engagement of TLR2 by ΔbgsA, we were interested in the effects of the deletion of bgsA on enterococcal virulence in a mouse peritonitis model. The LD50 in this model is 5.0 x 109 bacteria, reflecting the relatively low virulence of E. faecalis in vivo [22]. For the mouse peritonitis model we employed a bacterial dose slightly below the LD50. One of 14 mice died after infection with 1.3 x 109 wild-type bacteria (Fig 5). In contrast, the same inoculum of ΔbgsA killed 10 of 14 mice within 24 h (p = 0.0096). There were no subsequent deaths (total observation time five days). No significant difference in mortality between mice infected with ΔbgsB and those infected with wild-type E. faecalis was observed (Fig 5A). We repeated the experiment at a lower inoculum of 3 x 108 bacteria per mouse. With this lower dose, 2 of 8 mice infected with ΔbgsA died within the observation period (5 days), while all mice infected with the wild-type survived (p = 0.14).
Fig 5. Survival and bacterial load of BALB/c mice after intraperitoneal infection with E. faecalis strains.

Survival after infection with 1.3 x 109 E. faecalis. P = 0.0096 EF 12030 wild-type vs. ΔbgsA determined by the log-rank test (A). Bacterial counts 3 h and 12 h after i.p. challenge (inoculum 2.0 x 109 cfu per mouse, 14 mice per group). The bacterial load is expressed as the log10 (cfu) per ml ± SEM (B). P > 0.05 EF 12030 wild-type vs. ΔbgsA vs. ΔbgsB at 3 h and 12 h by 2-way ANOVA.
Peritonitis caused by EF12030ΔbgsA is associated with increased pro-inflammatory cytokines
To better understand excess mortality in peritonitis induced by ΔbgsA, we infected mice with an inoculum similar to that used in the survival model (2.0 x 109) and quantified the bacterial concentration in the peritoneal cavity, blood, and kidneys at 3 and 12 hours after infection (Fig 5B). No significant difference in bacterial load was noted at corresponding time points. Thus, ΔbgsA does not appear to impede bacterial clearance during peritonitis, and differences in bacterial load cannot explain the differences in mortality.
We also measured leukocytes and inflammatory cytokines during E. faecalis peritonitis. Three hours after infection, more leukocytes were recruited to the peritoneal cavity in animals infected with ΔbgsA than in those infected with wild-type bacteria or ΔbgsB (Fig 6). To determine whether a dysregulated inflammatory response was involved in the increased mortality caused by ΔbgsA, we measured cytokine concentrations in the peritoneal fluid and blood. One hour after infection with ΔbgsA, plasma concentrations of TNF-α, IL-6, and MIP-2 were significantly increased in mice infected with ΔbgsA as compared to mice infected with wild-type bacteria or ΔbgsB (Fig 7). Infection with ΔbgsA resulted also in significantly increased TNF-α concentrations in the peritoneal lavage fluid 3 h after infection compared to mice with peritonitis caused by wild-type bacteria or ΔbgsB (Fig 6B). At 12 h after infection, the kinetics of TNF-α production in the peritoneum had reversed: while low amounts of TNF-α were found in mice infected with ΔbgsA, mice infected with the wild-type strain or ΔbgsB displayed elevated levels of this cytokine (Fig 6B). These results show a correlation between mortality caused by ΔbgsA and increased production of inflammatory cytokines at early time points.
Fig 6. Leukocyte count and TNF-α expression in the PLF 1, 3, and 12 h after intraperitoneal infection with E. faecalis strains.

Number of leukocytes in the PLF (A). TNF-α in the PLF (B). * p < 0.05, ** p < 0.001, by 2-way ANOVA. Data represent means ± SEM of individual mice (8 mice per group).
Fig 7. Inflammatory cytokines/chemokines in the plasma 1 h after intraperitoneal infection with E. faecalis strains.

* p < 0.05, ** p < 0.001, by 2-way ANOVA. Data represent means ± SEM of individual cytokine levels of 8 mice.
Discussion
Our current work demonstrates the consequences of a disturbed cell membrane glycolipid homeostasis on the expression of lipoproteins in the cell envelope and the activation of the innate immune system by E. faecalis. Inactivation of the bgsA gene in E. faecalis led to a more than 3-fold higher lipoprotein-content in ΔbgsA compared to wild-type bacteria. ΔbgsA antigens were more potent activators of mouse macrophages and much stronger agonists of the TLR2 receptor. Genetic inactivation of the biosynthesis of lipoprotein strongly reduced the potency of ΔbgsA to stimulate macrophages. Conversely, lipoprotein-enriched Triton X114 extracts from total membrane fractions of ΔbgsA were stronger activators of mouse macrophages than wild type extracts. The alteration of the cell-surface proteome by inactivation of bgsA also enhanced the virulence of E. faecalis in a mouse peritonitis model.
Immune recognition of enterococci during mouse peritonitis has been described in detail by Leendertse and coworkers [20,22]. Peritoneal infection by Enterococcus faecium is sensed predominantly by macrophages via TLR2 which then secrete IL6, TNF-α, MIP-2, and KC during early infection [20,22]. A consecutive influx of neutrophils into the peritoneal cavity then leads to the clearance of the bacteria [20,22]. Working with an infection model similar to the one used by Leendertse, we were able to examine the consequences of overstimulation of the TLR2 system on the natural course of peritoneal infection. Peritonitis due to ΔbgsA led to an excessive induction of TNF-α, IL6, and MIP-2 accompanied by a higher influx of leukocytes in the peritoneum and to increased mortality of infected mice. Our results confirm previous studies which have shown that the overproduction of chemokines such as MIP-2 contributes to mortality in animal models of polymicrobial sepsis [42–44].
It is interesting that the effects of the inactivation of bgsA and a de-repressed lipoprotein production on the virulence of E. faecalis are highly dependent on the type of infection. We reported previously that inactivation of bgsA reduces biofilm formation and adherence to colonic cells, while the mutation improves the ability of E. faecalis to colonize the urinary tract [15,45]. In a mouse bacteremia model, ΔbgsA was cleared more rapidly from the bloodstream than the wild-type strain [15]. Yet during peritoneal infection as reported here, inactivation of bgsA induced stronger inflammation and led to higher mortality. Because of the pleiotropic phenotype of ΔbgsA, these results have to be interpreted with caution [15]. Nevertheless, repression of lipoprotein production by bgsA may be advantageous in some host compartments to escape detection by the host immune system while in others, like the peritoneum, virulence is suppressed. Since colonization of the gastrointestinal tract is the default ecological niche of E. faecalis, repression of lipoprotein production by bgsA probably improves overall fitness by evasion of the mucosal immune system. A similar strategy to subvert recognition by the innate immune system has been described by the intracellular pathogen Francisella novicida [46]. The F. novicida protein FTN_0757 specifically represses the production of its lipoprotein FTN_1103 and deletion of FTN_0757 leads to stronger stimulation of TLR2 and induction of higher levels of inflammatory cytokines compared to wild-type bacteria [46]. Hence, FTN_0757 was suggested to function as an immune escape mechanism in F. novicida. Furthermore, in S. aureus, capsule expression and formation of small colony variants has been described as a means to downregulate TLR2 activation [47]
Our study does not reveal a clear mechanism how the glycolipid mix of the cell membrane leads to an upregulation of lipoprotein concentration. Studies on the role of glycolipids in membrane physiology, however, point to secondary adjustments of the lipid composition in ΔbgsA to restore cell membrane homeostasis [48]. The biophysical properties of membrane lipids are determined by the size of their polar head groups in relation to the hydrophobic acyl-glycerol backbone. MGlcDAG with its smaller polar head group forms inverted nonlamellar structures as opposed to the bilayer conformation of DGlcDAG [49]. This results in a larger negative curvature and increased bilayer curvature stress for membranes composed of MGlcDAG as compared to DGlcDAG [49]. Cell membranes of ΔbgsA only contain MGlcDAG and the presence of additional, bilayer forming amphiphiles are most likely needed to dilute the concentration of the non-bilayer forming glycolipid. In Streptococcus pneumoniae, for example, deletion of glycosyltransferase cpoA leads to an arrest of the synthesis of galactosyl-glucosyl-diacylglycerol and to a secondary increase in the proportion of phosphatidylglycerol to cardiolipin [50]. Lipoproteins also contain large polar head groups and could potentially act as bilayer-prone amphiphiles in the cell membrane of ΔbgsA. Studies in Acholeplasma laidlawii suggest that the activities of the interfacial glycosyltransferases that synthesize MGlcDAG and DGlcDAG are regulated by the physical properties of the membrane containing their substrates [48,51]. Hence, biochemical regulation possibly also governs the compensatory increase of synthesis of lipoproteins in ΔbgsA. This model could also explain why inactivation of ΔbgsB does not cause enhanced activation of the innate immunity or promotes virulence in peritonitis. Mutation of bgsB in E. faecalis causes a complete arrest of glycolipid synthesis [25] and the mutant therefore does not overproduce toxic non-bilayer forming glycolipids. Hence, fewer adaptive changes to the cell envelope maybe needed to maintain membrane homeostasis.
Another question raised by our study is, if TLR2 activation by ΔbgsA is mediated by a global upregulation of lipoproteins or by one or more specific lipoproteins that act as dominant inducers of TLR2. Upregulation clearly affected lipoproteins unevenly in ΔbgsA. While a total of 12 lipoproteins were upregulated between 0.19 and 3.11-fold compared to wild-type levels, the expression of EF3041 was increased over 12-fold. Theoretical considerations as well as experimental studies, however, do not support the theory that certain lipoproteins act as dominant inducers of TLR2. Modeling studies of the crystallized structures of the TLR2-TLR6-lipopeptide complex suggest that TLR2 activation by lipopeptides is mediated only by a small and highly conserved structural motif of two ester-bound acyl-chains of at least 12 carbons each and the first two N-terminal amino acids of the polypeptide chain [52]. According to this model, the structure of the polypeptide chain beyond the second amino acid is negligible for the potency of Iipopeptide or lipoprotein to engage TLR2. Furthermore, in vitro studies of the FTN_0757 deletion mutant in F. novicida show that equimolar concentrations of lipoproteins from the wild-type strain and ΔFTN_0757 had a similar potency as TLR2 activators [46].
Taken together, our findings reveal an intimate interplay between the concentration of the bilayer-forming glycolipid DGlcDAG, the expression of lipoproteins, and activation of the host immune system. We show that deletion of bgsA leads to upregulation of bacterial lipoproteins and strongly enhances host inflammatory response and virulence in enterococcal peritonitis.
Supporting Information
The table includes all protein quantification data in “summary”-tab as well as the number of peptides identified in each single analysis in the tabs WT 1/2/3 and bgsA 1/2/3. Protein identifications were considered significant for the biological system if the protein was identified in at least two out of three samples in wild type or the mutant.
(XLSX)
(XLSX)
Acknowledgments
We are grateful to Ioana Toma, Türkan Sankic, and Dominik Anders for expert technical assistance. We would also like to thank Otto Holst for helpful discussions.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the German Federal Ministry of Education and Research (BMBF 01 EO 0803).
References
- 1. Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22: 240–273. 10.1128/CMR.00046-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, Theilacker C, et al. (2008) Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J Immunol 180: 6149–6158. [DOI] [PubMed] [Google Scholar]
- 3. Knapp S, Wieland CW, van 't Veer C, Takeuchi O, Akira S, Florquin S, et al. (2004) Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol 172: 3132–3138. [DOI] [PubMed] [Google Scholar]
- 4. Yimin, Kohanawa M, Zhao S, Ozaki M, Haga S, Nan G, et al. (2013) Contribution of Toll-Like Receptor 2 to the Innate Response against Staphylococcus aureus Infection in Mice. PLoS ONE 8: e74287 10.1371/journal.pone.0074287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Echchannaoui H, Frei K, Schnell C, Leib SL, Zimmerli W, Landmann R (2002) Toll-like receptor 2-deficient mice are highly susceptible to Streptococcus pneumoniae meningitis because of reduced bacterial clearing and enhanced inflammation. J Infect Dis 186: 798–806. 10.1086/342845 [DOI] [PubMed] [Google Scholar]
- 6. Mancuso G, Midiri A, Beninati C, Biondo C, Galbo R, Akira S, et al. (2004) Dual role of TLR2 and myeloid differentiation factor 88 in a mouse model of invasive group B streptococcal disease. J Immunol 172: 6324–6329. [DOI] [PubMed] [Google Scholar]
- 7. Brouwer MC, de Gans J, Heckenberg SGB, Zwinderman AH, Van Der Poll T, van de Beek D (2009) Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. Lancet Infect Dis 9: 31–44. 10.1016/S1473-3099(08)70261-5 [DOI] [PubMed] [Google Scholar]
- 8. Casanova J-L, Abel L, Quintana-Murci L (2011) Human TLRs and IL-1Rs in host defense: natural insights from evolutionary, epidemiological, and clinical genetics. Annu Rev Immunol 29: 447–491. 10.1146/annurev-immunol-030409-101335 [DOI] [PubMed] [Google Scholar]
- 9. Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11: 373–384. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 10. Zähringer U, Lindner B, Inamura S, Heine H, Alexander C (2008) TLR2—promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213: 205–224. 10.1016/j.imbio.2008.02.005 [DOI] [PubMed] [Google Scholar]
- 11. Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, Krikae F, et al. (2006) Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus . J Immunol 177: 3162–3169. [DOI] [PubMed] [Google Scholar]
- 12. Stoll H, Dengjel J, Nerz C, Götz F (2005) Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect Immun 73: 2411–2423. 10.1128/IAI.73.4.2411-2423.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prados-Rosales R, Baena A, Martinez LR, Luque-Garcia J, Kalscheuer R, Veeraraghavan U, et al. (2011) Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J Clin Invest 121: 1471–1483. 10.1172/JCI44261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gurung M, Moon DC, Choi CW, Lee JH, Bae YC, Kim J, et al. (2011) Staphylococcus aureus produces membrane-derived vesicles that induce host cell death. PLoS ONE 6: e27958 10.1371/journal.pone.0027958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Theilacker C, Sanchez-Carballo P, Toma I, Fabretti F, Sava I, Kropec A, et al. (2009) Glycolipids are involved in biofilm accumulation and prolonged bacteraemia in Enterococcus faecalis . Mol Microbiol 71: 1055–1069. 10.1111/j.1365-2958.2009.06587.x [DOI] [PubMed] [Google Scholar]
- 16. Theilacker C, Sava I, Sanchez-Carballo P, Bao Y, Kropec A, Grohmann E, et al. (2011) Deletion of the glycosyltransferase bgsB of Enterococcus faecalis leads to a complete loss of glycolipids from the cell membrane and to impaired biofilm formation. BMC Microbiol 11: 67 10.1186/1471-2180-11-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Edman M, Berg S, Storm P, Wikström M, Vikström S, Ohman A, et al. (2003) Structural features of glycosyltransferases synthesizing major bilayer and nonbilayer-prone membrane lipids in Acholeplasma laidlawii and Streptococcus pneumoniae . J Biol Chem 278: 8420–8428. 10.1074/jbc.M211492200 [DOI] [PubMed] [Google Scholar]
- 18. Wikström M, Xie J, Bogdanov M, Mileykovskaya E, Heacock P, Wislander A, et al. (2004) Monoglucosyldiacylglycerol, a foreign lipid, can substitute for phosphatidylethanolamine in essential membrane-associated functions in Escherichia coli . J Biol Chem 279: 10484–10493. 10.1074/jbc.M310183200 [DOI] [PubMed] [Google Scholar]
- 19. Leendertse M, Willems RJL, Giebelen IAJ, Roelofs JJTH, Bonten MJM, van der Poll T (2009) Neutrophils are essential for rapid clearance of Enterococcus faecium in mice. Infect Immun 77: 485–491. 10.1128/IAI.00863-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leendertse M, Willems RJL, Giebelen IAJ, Roelofs JJTH, Van Rooijen N, Bonten MJM, et al. (2009) Peritoneal macrophages are important for the early containment of Enterococcus faecium peritonitis in mice. Innate Immunity 15: 3–12. 10.1177/1753425908100238 [DOI] [PubMed] [Google Scholar]
- 21. Leendertse M, Willems RJL, Flierman R, de Vos AF, Bonten MJM, van der Poll T (2010) The complement system facilitates clearance of Enterococcus faecium during murine peritonitis. J Infect Dis 201: 544–552. 10.1086/650341 [DOI] [PubMed] [Google Scholar]
- 22. Leendertse M, Willems RJL, Giebelen IAJ, van den Pangaart PS, Wiersinga WJ, de Vos AF, et al. (2008) TLR2-dependent MyD88 signaling contributes to early host defense in murine Enterococcus faecium peritonitis. J Immunol 180: 4865–4874. [DOI] [PubMed] [Google Scholar]
- 23. Paoletti LC, Ross RA, Johnson KD (1996) Cell growth rate regulates expression of group B Streptococcus type III capsular polysaccharide. Infect Immun 64: 1220–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cieslewicz MJ, Kasper DL, Wang Y, Wessels MR (2001) Functional analysis in type Ia group B Streptococcus of a cluster of genes involved in extracellular polysaccharide production by diverse species of streptococci. J Biol Chem 276: 139–146. 10.1074/jbc.M005702200 [DOI] [PubMed] [Google Scholar]
- 25. Bychowska A, Theilacker C, Czerwicka M, Marszewska K, Huebner J, Holst O, et al. (2011) Chemical structure of wall teichoic acid isolated from Enterococcus faecium strain U0317. Carbohydr Res 346: 2816–2819. 10.1016/j.carres.2011.09.026 [DOI] [PubMed] [Google Scholar]
- 26. Brusca JS, Radolf JD (1994) Isolation of integral membrane proteins by phase partitioning with Triton X-114. Meth Enzymol 228: 182–193. [DOI] [PubMed] [Google Scholar]
- 27. Hashimoto M, Tawaratsumida K, Kariya H, Aoyama K, Tamura T, Suda Y (2006) Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int Immunol 18: 355–362. 10.1093/intimm/dxh374 [DOI] [PubMed] [Google Scholar]
- 28. Becher D, Hempel K, Sievers S, Zühlke D, Pané-Farré J, Otto A, et al. (2009) A proteomic view of an important human pathogen—towards the quantification of the entire Staphylococcus aureus proteome. PLoS ONE 4: e8176 10.1371/journal.pone.0008176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dreisbach A, Otto A, Becher D, Hammer E, Teumer A, Gouw JW, et al. (2008) Monitoring of changes in the membrane proteome during stationary phase adaptation of Bacillus subtilis using in vivo labeling techniques. Proteomics 8: 2062–2076. 10.1002/pmic.200701081 [DOI] [PubMed] [Google Scholar]
- 30. Otto A, Bernhardt J, Meyer H, Schaffer M, Herbst F-A, Siebourg J, et al. (2010) Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis . Nat Commun 1: 137 10.1038/ncomms1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. MacCoss MJ, Wu CC, Liu H, Sadygov R, Yates JR (2003) A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Analytical chemistry 75: 6912–6921. 10.1021/ac034790h [DOI] [PubMed] [Google Scholar]
- 32. Park SK, Venable JD, Xu T, Yates JR (2008) A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods 5: 319–322. 10.1038/nmeth.1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae . J Proteome Res 5: 2339–2347. 10.1021/pr060161n [DOI] [PubMed] [Google Scholar]
- 34. Zhang Y, Wen Z, Washburn MP, Florens L (2010) Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. 82: 2272–2281. Anal Chem 82:2272–2281. 10.1021/ac9023999 [DOI] [PubMed] [Google Scholar]
- 35. Sievers S, Ernst CM, Geiger T, Hecker M, Wolz C, Becher D, et al. (2010) Changing the phospholipid composition of Staphylococcus aureus causes distinct changes in membrane proteome and membrane-sensory regulators. Proteomics 10: 1685–1693. 10.1002/pmic.200900772 [DOI] [PubMed] [Google Scholar]
- 36. Reffuveille F, Leneveu C, Chevalier S, Auffray Y, Rincé A (2011) Lipoproteins of Enterococcus faecalis: bioinformatic identification, expression analysis and relation to virulence. Microbiology 157: 3001–3013. 10.1099/mic.0.053314-0 [DOI] [PubMed] [Google Scholar]
- 37. Hutchings MI, Palmer T, Harrington DJ, Sutcliffe IC (2009) Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold “em, knowing when to fold”em. 17: 13–21. 10.1016/j.tim.2008.10.001 [DOI] [PubMed] [Google Scholar]
- 38. Reffuveille F, Serror P, Chevalier S, Budin-Verneuil A, Ladjouzi R, Bernay B, et al. (2012) The prolipoprotein diacylglyceryl transferase (Lgt) of Enterococcus faecalis contributes to virulence. Microbiology 158: 816–825. 10.1099/mic.0.055319-0 [DOI] [PubMed] [Google Scholar]
- 39. Bubeck Wardenburg J, Williams WA, Missiakas D (2006) Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci USA 103: 13831–13836. 10.1073/pnas.0603072103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Henneke P, Morath S, Uematsu S, Weichert S, Pfitzenmaier M, Takeuchi O, et al. (2005) Role of lipoteichoic acid in the phagocyte response to group B Streptococcus . J Immunol 174: 6449–6455. [DOI] [PubMed] [Google Scholar]
- 41. Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917. [DOI] [PubMed] [Google Scholar]
- 42. Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL (1997) Elevated levels of macrophage inflammatory protein 2 in severe murine peritonitis increase neutrophil recruitment and mortality. Infect Immun 65: 3847–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wengner AM, Pitchford SC, Furze RC, Rankin SM (2008) The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood 111: 42–49. 10.1182/blood-2007-07-099648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ness TL, Hogaboam CM, Strieter RM, Kunkel SL (2003) Immunomodulatory role of CXCR2 during experimental septic peritonitis. J Immunol 171: 3775–3784. [DOI] [PubMed] [Google Scholar]
- 45. Diederich AK, Wobser D, Spiess M Sava IG, Huebner J, Sakinc T (2014) Role of glycolipids in the pathogenesis of Enterococcus faecalis urinary tract infection. Plos One 9: e96295 10.1371/journal.pone.0096295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jones CL, Sampson TR, Nakaya HI, Pulendran B, Weiss DS (2012) Repression of bacterial lipoprotein production by Francisella novicida facilitates evasion of innate immune recognition. Cell Microbiol 14: 1531–1543. 10.1111/j.1462-5822.2012.01816.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hilmi D, Parcina M, Stollewerk D, Ostrop J, Josten M, Meilaender A, et al. (2014) Heterogeneity of host TLR2 stimulation by Staphylocoocus aureus isolates. PLoS ONE 9: e96416 10.1371/journal.pone.0096416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Parsons JB, Rock CO (2013) Bacterial lipids: metabolism and membrane homeostasis. Prog Lipid Res 52: 249–276. 10.1016/j.plipres.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jouhet J (2013) Importance of the hexagonal lipid phase in biological membrane organization. Front Plant Sci 4: 494 10.3389/fpls.2013.00494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Meiers M, Volz C, Eisel J, Maurer P, Henrich B, Hakenbeck R (2014) Altered lipid composition in Streptococcus pneumoniae cpoA mutants. BMC Microbiol 14: 12 10.1186/1471-2180-14-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lind J, Rämö T, Klement MLR, Bárány-Wallje E, Epand RM, Epand RF, et al. (2007) High cationic charge and bilayer interface-binding helices in a regulatory lipid glycosyltransferase. Biochemistry 46: 5664–5677. 10.1021/bi700042x [DOI] [PubMed] [Google Scholar]
- 52. Kang JY, Nan X, Jin MS, Youn S-J, Ryu YH, Mah S, et al. (2009) Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31: 873–884. 10.1016/j.immuni.2009.09.018 [DOI] [PubMed] [Google Scholar]
- 53. Krogh A, Larsson B, von Heijne G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. (2001) J Mol Biol. 305: 567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- 54. Huebner J, Wang Y, Krueger WA, Madoff LC, Martirosian G, Boisot S, et al. (1999) Isolation and chemical characterization of a capsular polysaccharide antigen shared by clinical isolates of Enterococcus faecalis and vancomycin-resistant Enterococcus faecium . Infect Immun 67: 1213–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Callegan MC, Jett BD, Hancock LE, Gilmore MS (1999) Role of hemolysin BL in the pathogenesis of extraintestinal Bacillus cereus infection assessed in an endophthalmitis model. Infect Immun 67: 3357–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table includes all protein quantification data in “summary”-tab as well as the number of peptides identified in each single analysis in the tabs WT 1/2/3 and bgsA 1/2/3. Protein identifications were considered significant for the biological system if the protein was identified in at least two out of three samples in wild type or the mutant.
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.