

Abstract
The DNA-dependent protein kinase (DNA-PK) is a serine/threonine protein kinase composed of a large catalytic subunit (DNA-PKcs) and the Ku70/80 heterodimer. Over the past two decades, significant progress has been made in elucidating the role of DNA-PK in non-homologous end joining (NHEJ), the major pathway for repair of ionizing radiation-induced DNA double strand breaks in human cells and recently, additional roles for DNA-PK have been reported. In this review, we will describe the biochemistry, structure and function of DNA-PK, its roles in DNA double strand break repair and its newly described roles in mitosis and other cellular processes.
Keywords: DNA-dependent protein kinase, non-homologous end joining, mitosis
1. Introduction
Almost thirty years have passed since Anderson and colleagues first reported that human cells contain a DNA-activated protein kinase (Walker et al., 1985). This protein kinase, now known as the DNA-dependent protein kinase (DNA-PK), is composed of a large catalytic subunit (DNA-PKcs, gene name PRKDC) and the Ku70/80 heterodimer. In this review, we will describe the discovery, biochemistry, structure, and function of DNA-PK, emphasizing work from our laboratory on the roles of DNA-PK in DNA double strand break (DSB) repair and its newly described roles in mitosis. This mini-review is based on a presentation by SPLM at the Zing Conference on Dynamic Structures in DNA Damage Responses and Cancer, held in Cancun, Mexico, February, 12–15, 2014.
2. Discovery of DNA-PK
The first clues that cells contain a DNA-activated protein kinase came from the work of Carl Anderson and colleagues who showed that addition of sonicated calf thymus DNA to extracts from human cells enhanced the phosphorylation of a number of endogenous proteins, including the 90 kDa heat shock protein, hsp90 (Walker et al., 1985). Using hsp90 as a substrate, we isolated a protein fraction from HeLa cells that was highly enriched for DNA-activated protein kinase activity and showed that it contained the Ku70 and Ku80 polypeptides as well as a large (approximately 350 kDa) ATP-binding polypeptide that we termed p350 (Lees-Miller et al., 1990). Ku70 and Ku80 had previously been identified as a heterodimer that bound double-stranded DNA and was an auto immune antigen (Mimori and Hardin, 1986) but its function, as well as the identity of p350, was unknown. Independently, Carter and colleagues identified and purified the same large polypeptide (p350) from HeLa cells and showed that it phosphorylated casein in a DNA-dependent manner in vitro (Carter et al., 1990; Carter et al., 1988). This newly discovered DNA-activated protein kinase activity phosphorylated hsp90 at threonine followed by glutamine, i.e. a TQ motif, a previously unknown protein kinase consensus site (Lees-Miller and Anderson, 1989). It also phosphorylated transcription factor Sp1 (Jackson et al., 1990) and the tumour suppressor, p53 (Lees-Miller et al., 1990; Lees-Miller et al., 1992), and p53 was phosphorylated on serines followed by glutamines (i.e. SQ motifs) (Lees-Miller et al., 1992) (Supplemental Table 1). The preference for SQ/TQ sequences was subsequently confirmed in combinatorial peptide assays using purified proteins (O’Neill et al., 2000).
Independently, William Dynan and colleagues identified p350 and the Ku heterodimer as components of a DNA-activated serine/threonine protein kinase that phosphorylated the C-terminal domain of RNA polymerase (Dvir et al., 1992; Dvir et al., 1993). These studies, in combination with the seminal work of Stephen Jackson and colleagues, demonstrated that the Ku70/80 heterodimer is the DNA binding subunit that targets p350 (now referred to as the DNA-dependent protein kinase catalytic subunit, DNA-PKcs) to ends of dsDNA (Gottlieb and Jackson, 1993). DNA-PKcs, assembled with Ku on dsDNA is commonly referred to as DNA-PK, for the DNA-dependent protein kinase (Suwa et al., 1994).
In another twist along the path to the discovery and initial characterization of DNA-PK, in a herculean effort, Jackson and colleagues cloned the DNA-PKcs cDNA and showed that the catalytic domain had amino acid similarity to the p110 subunit of the phosphatidyl inositol 3 kinase, PI3K (Hartley et al., 1995). Moreover, DNA-PKcs and the p110 subunit of PI3K shared amino acid similarity in their catalytic domains to the newly discovered product of the ataxia-telangiectasia mutated (ATM) gene (Savitsky et al., 1995). The protein kinase activities of both DNA-PK and ATM were inhibited by the PI3K inhibitor, wortmannin (Banin et al., 1998; Canman et al., 1998; Hartley et al., 1995) and a new class of atypical serine/threonine protein kinases (Manning et al., 2002), the phosphatidyl inositol 3 kinase-like protein kinases or PIKKs was born (Hunter, 1995; Lavin et al., 1995).
3. Ku and its interaction with DNA-PKcs
The Ku70/80 heterodimer was discovered as an autoimmune antigen with the unusual property of binding with high affinity to ends of dsDNA in a DNA sequence independent manner (Blier et al., 1993; Mimori and Hardin, 1986; Mimori et al., 1990). The elucidation of the crystal structure of the Ku70/80 DNA binding core revealed that Ku70 and Ku80 subunits interact to form a basket shaped structure, the arms of which create a preformed ring that surrounds dsDNA, and interact with the DNA through electrostatic interactions (Walker et al., 2001). This elegant structure explained decades of biochemical studies revealing how the Ku heterodimer binds to dsDNA in a sequence independent manner and also how Ku can slide along dsDNA in an ATP-independent manner. The structure also raised the critical question of how Ku is removed from the DNA ends, and potential mechanisms involving backing off and proteolytic cleavage were proposed (Walker et al., 2001). This remains a critical question in the field and one that is only beginning to be understood. Recently, ubiquitination of Ku has been proposed as a mechanism to remove Ku from DNA ends in both Xenopus extracts (Postow, 2011; Postow et al., 2008) and in human cells (Feng and Chen, 2012), discussed in (Wang and Lees-Miller, 2013).
It is now established that the Ku70/80 heterodimer binds ends of dsDNA and recruits DNA-PKcs to DNA double strand breaks (DSBs) in vitro (Chan et al., 1996; Gottlieb and Jackson, 1993; Suwa et al., 1994) and in vivo (Uematsu et al., 2007) (Figure 1). Alone highly purified DNA-PKcs has weak protein kinase activity that, in the presence of dsDNA, is stimulated 5–10 fold in the presence of Ku. Maximum kinase activity is achieved with approximately 1 mole of Ku heterodimer and 1 mole of DNA-PKcs, suggesting that the stoichiometry of the interaction is 1:1 (Chan et al., 1996).
Figure 1. The role of DNA-PKcs in the non-homologous end joining (NHEJ) pathway for the repair of ionizing radiation induced DNA double strand breaks (DSBs).
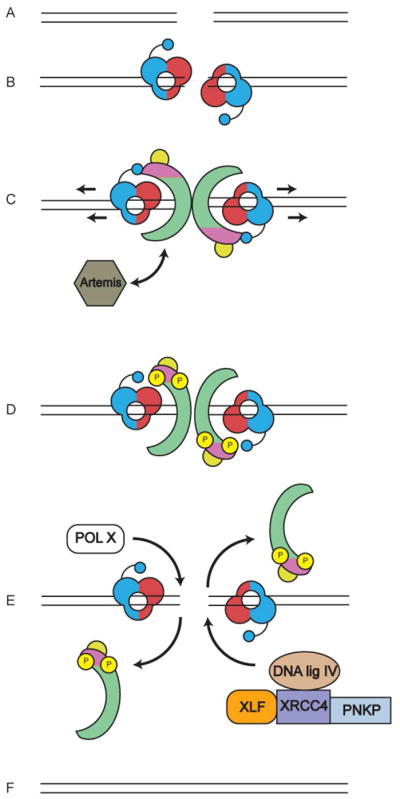
A. DSBs can be formed either by DNA damaging agents such as ionizing radiation (IR) or during the process of V(D)J recombination in the vertebrate immune system.
B. DSBs are detected by the Ku70/80 heterodimer, which binds to the ends of the DSB. Ku70 (shown in red) is positioned proximal to the DSB end and Ku80 (shown in blue) is distal to the end (Walker et al., 2001). The C-terminal domain of Ku80 is composed of a flexible linker with a globular domain and a C-terminal region that interacts directly with DNA-PKcs (Gell and Jackson, 1999), (shown as small blue circle). Significantly, SAXS revealed that this flexible domain extends towards Ku70, positioning the DNA-PKcs interacting region close to the end of the DSB (Hammel et al., 2010b). Ku plays a key role in NHEJ, being responsible for recruitment of multiple downstream proteins, including DNA-PKcs and XLF, to the DSB (reviewed in (Radhakrishnan et al., 2014; Wang and Lees-Miller, 2013)).
C. Upon activation, DNA-PKcs is recruited to the DSB end, causing Ku to move inwards (Frit et al., 2000) (indicated by arrows) such that DNA-PKcs is now positioned at the extreme end of the DSB (Yoo and Dynan, 1999). dsDNA interacts with the central region of DNA-PKcs (Boskovic et al., 2003; Williams et al., 2008). It has been suggested that DNA-PKcs may open or fray the ends of the dsDNA which may allow single stranded DNA to bind in a channel in the head domain of DNA-PKcs (DeFazio et al., 2002) (see also Figure 2B). DNA-PKcs also interacts with Artemis (shown in gray), an endonuclease that, in the presence of DNA-PKcs, facilitates DNA hairpin opening at coding joints in V(D)J recombination (Goodarzi et al., 2006). The role of Artemis in repair of IR-induced DSBs is not known. DNA-PKcs is coloured as in Figure 2A, where the N-terminal HEAT repeats are in green, the FAT and FATC domains in magenta and the kinase domain in yellow, as described by (Sibanda et al., 2010).
D. DNA-PKcs undergoes extensive autophosphorylation (yellow circles with P), which in vitro has been shown to induce large conformational changes and promote release of phosphorylated DNA-PKcs from the Ku-DNA complex (Chan and Lees-Miller, 1996; Dobbs et al., 2010; Hammel et al., 2010b). A recent study suggests that recruitment of the XRCC4-DNA ligase IV complex to the DSB is required for phosphorylation of DNA-PKcs (Cottarel et al., 2013). Artemis has also been reported to interact with DNA ligase IV (Malu et al., 2012; Ochi et al., 2013) (not shown in figure).
E. DNA-end processing enzymes such as polynucleotide kinase/phosphatase (PNKP), which interacts with XRCC4 (Koch et al., 2004) and DNA polymerases mu and/or lambda of the pol X family are also recruited to the DSB, as is the XRCC4-DNA ligase IV-XLF complex, which seals the DSB ends. XRCC4 and XLF form long helical filaments (not shown in figure) that may facilitate bridging of ends prior to ligation (Hammel et al., 2011; Hammel et al., 2010a; Mahaney et al., 2013).
F. NHEJ is completed when the processed ends of the DSB are covalently joined. How and when Ku is removed from DSB ends is not known precisely but may involve ubiquitination (Feng and Chen, 2012; Postow, 2011; Postow et al., 2008).
A critical and as yet unresolved question is how does the interaction of DNA-PKcs with the Ku-DNA complex results in activation of DNA-PKcs’ protein kinase activity. In the absence of Ku, the interaction of DNA-PKcs with dsDNA is transient, characterized by an extremely fast off-rate. Interaction with Ku decreased the off rate, stabilizing the formation of the DNA-PKcs-Ku-DNA complex (West et al., 1998). Accordingly, in electrophoretic mobility shift assays (EMSA), addition of crosslinking agents stabilizes formation of the DNA-PKcs-Ku-DNA complex (Ting et al., 1999; Ting et al., 1998). Ku binds to dsDNA such that Ku70 is proximal to the dsDNA termini while Ku80 is located distal to the end (Walker et al., 2001; Yoo and Dynan, 1999; Yoo et al., 1999). Binding of DNA-PKcs promotes inward movement of Ku, replacing Ku at the extreme DNA termini with DNA-PKcs (Frit et al., 2000; Yoo and Dynan, 1999; Yoo et al., 1999) (Figure 1). In vitro, in the absence of DNA-PKcs, Ku has the ability to translocate inwards for several hundred base pairs from the dsDNA end in an ATP-independent manner (Paillard and Strauss, 1991), however, recent studies using high resolution microscopy suggest that only one Ku heterodimer resides either side of the DSB in cells (Britton et al., 2013), casting doubt on whether extensive translocation of Ku along naked dsDNA is physiologically relevant.
The final 13 amino acids of Ku80 (residues 720–732) have been shown to interact directly with DNA-PKcs in vitro (Falck et al., 2005; Gell and Jackson, 1999). Moreover, the C-terminal 189 amino acids of Ku80 form a flexible extension suitable for interacting with other proteins, such as DNA-PKcs (Hammel et al., 2010b) (Figure 1). However, although the Ku80 C-terminal deletion mutants were radiosensitive, DNA-PK activity was reduced, but not abolished, suggesting that additional regions of Ku are important for DNA-PKcs recruitment and activation (Weterings et al., 2009). The region of DNA-PKcs required for interaction with Ku has been mapped to the C-terminal ~1000 amino acids (Jin et al., 1997) (Figure 2A) but precisely how interaction of DNA-PKcs with Ku and DNA leads to stimulation of DNA-PKcs’ activity remains poorly understood.
Figure 2. Structure of DNA-PKcs.

A. Cartoon of architecture of DNA-PKcs showing the N terminal HEAT repeat region (green), and the FAT and FAT-C domains (magenta) that flank the kinase domain (yellow). The major in vivo phosphorylation sites discussed in this review are indicated above the figure and the domain boundaries by the numbers below. See also (Dobbs et al., 2010). Mitotic phosphorylation sites (indicated in red) are from (Douglas et al., 2014) and (Lee et al., 2011). The region of DNA-PKcs implicated in interaction with Ku and DNA are from (Jin et al., 1997) and (Gupta and Meek, 2005), respectively. Also shown are the caspase cleavage sites (green) reported in (Song et al., 1996).
B. Representation of the cryo-electron microscopy structure of DNA-PKcs from (Williams et al., 2008) showing the potential path of double stranded DNA through the large central cavity, and a model for the single stranded ends of frayed DNA through the putative upper central channel of the DNA-PKcs molecule. On the left, DNA-PKcs is shown with the crown (now known to contain the FAT-kinase and FATC domains) on the top, and the HEAT repeats on the sides, forming the arms that surround the large central cavity. In the representation shown, dsDNA is shown to the left in the central cavity. The structure on the right is rotated by 90°, illustrating the potential path of single stranded DNA into the head domain as suggested in (Williams et al., 2008). This figure is reproduced from (Williams et al., 2008) with permission.
C. Upper two panels: Representations of the crystal structure of DNA-PKcs from (Sibanda et al., 2010) coloured as in panel A (HEAT repeats in green, FAT-kinase and FATC in magenta-yellow-magenta), superimposed on the SAXS envelopes of non-phosphorylated (cream) and autophosphorylated (brown) DNA-PKcs from (Hammel et al., 2010b) and (Dobbs et al., 2010). The lower two panels represent the structures rotated by 90°.
D. Hypothetical models for DNA-PKcs in its non-phosphorylated and autophosphorylated forms. In the lower panel, the hypothetical structures are rotated by 90°. We suggest that autophosphorylation of DNA-PKcs may induce opening of the putative gap at the base of DNA-PKcs, as reported in (Sibanda et al., 2010) (top panels) or a conformational change, perhaps with opening or elongation of the concave surface created by the HEAT domain (lower panels). Colours are as in panel A. See text and (Dobbs et al., 2010; Hammel et al., 2010b) for details.
4. Structure of DNA-PKcs and the DNA-PK complex
DNA-PKcs is composed of a large N-terminal region, predicted to consist largely of HEAT (Huntingtin, Elongation factor 3, regulatory subunit A of PP2A, TOR1) repeats and other α-helical regions and a C-terminal region containing the FAT (FRAP, ATM, TRRAP), kinase and FAT-C domains (Figure 2A). This overall architecture is conserved in other members of the PIKK family, such as ATM and ATM- and Rad3-related (ATR) (Baretic and Williams, 2014; Lempiainen and Halazonetis, 2009), suggesting that these family members may have conserved mechanisms of activation in response to different DNA damage stimuli. The junction between the α-helical HEAT repeat region and the C-terminal region FAT domain is likely flexible and/or solvent exposed, as it contains caspase cleavage sites at residues Asp2713 and Asp2983 (Song et al., 1996) (Figure 2A) and is readily cleaved by trypsin and other proteases in vitro (Yu and Lees-Miller, unpublished).
Cryo-electron microscopy (cryo-EM) studies suggested that DNA-PKcs has dimensions of approximately 120 Å by 150 Å and is composed of a head domain and a base separated by arms that surround a central open cavity (Rivera-Calzada et al., 2005; Williams et al., 2008) (Figure 2B). The low-resolution of the cryo-EM structures prevented definitive assignment of the kinase domain or other regions (Dore et al., 2004), however, based on modelling with biotinylated/gold labelled double stranded (ds)-DNA, the central cavity was proposed to accommodate dsDNA (Boskovic et al., 2003; Williams et al., 2008) (Figure 2B), although, recent cryo-EM studies suggest that dsDNA may interact closer to the base of DNA-PKcs (Villarreal and Stewart, 2014). A smaller cavity in the head domain was suggested to bind single-stranded DNA (Williams et al., 2008), in keeping with biochemical studies that suggested that DNA-PKcs separates the ends of dsDNA (DeFazio et al., 2002) (Figure 2B).
The elegant X-ray crystallographic studies by Blundell and colleagues of DNA-PKcs in complex with the C-terminal region of Ku80 revealed that the HEAT repeats of DNA-PKcs form an α-solenoid, the arms of which surround the central cavity, while the FAT, kinase and FAT-C domains are located on the top or crown of the molecule (Figure 2C/D). The structure also suggested the presence of a gap at the base of the molecule, close to the putative N-terminus (Sibanda et al., 2010) (Figure 2C). However, assignment of individual residues, amino acid side chains and atomic details of the DNA-PKcs structure awaits a higher resolution structure. The recent high-resolution crystal structure of the PIKK family member mTOR revealed that the PIKK kinase domain is very similar to that of the canonical serine/threonine protein kinase family, typified by cAMP-dependent protein kinase, and revealed that the FAT domain plays an integral role in stabilizing both mTOR and DNA-PKcs catalytic domains (Baretic and Williams, 2014; Yang et al., 2013).
The majority of the DNA-PKcs structure (74%) is composed of the α-helical HEAT repeat domain (Figure 1), which surrounds the central cavity (Figure 2A and C). When the structure is rotated by 90°, the HEAT repeats are shown to adopt a concave structure (Baretic and Williams, 2014; Lara-Gonzalez et al., 2012; Sibanda et al., 2010) (Figure 2C and D). It seems likely that the unique architecture of the HEAT repeat region of DNA-PKcs, and presumably the other PIKK family members, is critical to their function. A possible clue comes from protein phosphatase 2 A (PP2A) A-subunit, a scaffolding protein composed entirely of 15 tandem HEAT repeats (Groves et al., 1999). Intriguingly, the HEAT repeats of the PP2A-A subunit respond to pressure, acting, in effect, as compressible molecular springs (Grinthal et al., 2010). It is tempting to speculate that the HEAT repeat region of DNA-PKcs, and possibly other PIKKs, may serve a similar function, perhaps acting as a molecular spring responding to or maintaining tension between ends of DNA. Interestingly, a recent publication from the Foiani laboratory has shown that the related protein ATR (ATM- and Rad3-related), which like DNA-PKcs and ATM is composed of a large N-terminal domain consisting of multiple HEAT domains (Perry and Kleckner, 2003) and a C-terminal kinase domain, is activated in the absence of DNA damage by mechanical forces that arise due to chromosome dynamics and torsional stress on nuclear membranes (Kumar et al., 2014). Clearly, understanding the role of the HEAT repeats in DNA-PKcs will be critical to better understanding its function.
As discussed above, how interaction of DNA-PKcs with the Ku-dsDNA complex results in stimulation of DNA-PKcs’ protein kinase activity remains poorly understood. This is due in large part to the large size of the DNA-PK complex, composed of the 469 kDa DNA-PKcs plus the ~150 kDa Ku heterodimer in complex with dsDNA. Some progress has been made in this regard using small angle X-ray scattering (SAXS) and cryo-electron microscopy. Indeed, stable complexes of DNA-PKcs assembled with Ku on dsDNA have been detected and partially characterized using both techniques (Hammel et al., 2010b; Rivera-Calzada et al., 2007; Spagnolo et al., 2006), however, the relatively low resolution of both approaches has limited our ability to draw firm conclusions. Improvements in the resolution of these technologies as well as application of other technologies such as mass spectrometry (Pi and Sael, 2013) may hold promise in the elucidation of the molecular structure of large, dynamic nucleo-protein complexes such as DNA-PK.
5. DNA-PKcs function in cells
Given the interaction of DNA-PKcs and Ku with ends of dsDNA, in early studies it was suggested that DNA-PKcs might be involved in transcription, DNA repair and/or detection of viral DNA (Anderson, 1993; Anderson and Lees-Miller, 1992). Indeed, there is now evidence for involvement of DNA-PKcs in each of these, as well as other, cellular processes (discussed below). In the following section, we will focus on the role of DNA-PKcs in the detection and repair of DSBs, such as those resulting from cellular exposure to ionizing radiation (IR) as well as its newly reported roles in mitosis. Finally, we will briefly mention recent studies that reveal additional functions for DNA-PKcs in eukaryotic cells.
5.1 DNA double strand break repair
The first clues that DNA-PKcs and Ku were involved in DSB repair came from findings that cells that lacked either DNA-PKcs or Ku were highly sensitive to IR and other DSB-inducing agents (Getts and Stamato, 1994; Jeggo et al., 1996; Lees-Miller et al., 1995; Taccioli et al., 1994). Animals lacking either DNA-PKcs or Ku were also immune deficient (Jeggo et al., 1996; Kirchgessner et al., 1995; Taccioli et al., 1994), revealing roles in V(D)J recombination (a process required for immunoglobulin and T cell receptor gene processing and consequently mature T and B cells (Helmink and Sleckman, 2012; Lieber, 2010)). We now know that Ku and DNA-PKcs are required for repair of DSBs via non-homologous end joining (NHEJ), the major pathway for the repair of IR-induced DSBs in human cells and for repair of programmed DSBs during V(D)J recombination (Lieber, 2008; Mahaney et al., 2009; Radhakrishnan et al., 2014; Wang and Lees-Miller, 2013) (Figure 1).
The function of DNA-PKcs in NHEJ is tightly linked to its protein kinase activity. Small molecule inhibitors of DNA-PKcs, such as NU7441 (Zhao et al., 2006) as well as mutations in the kinase domain of DNA-PKcs, radiosensitize cells and inhibit DSB repair (Kienker et al., 2000; Kurimasa et al., 1999). These observations prompted our lab, as well as others, to search for physiological substrates of DNA-PKcs. We showed that DNA-PKcs phosphorylates many components of the NHEJ pathway in vitro including Ku70 and Ku80 (Chan et al., 1999; Douglas et al., 2005), XRCC4 (Yu et al., 2003), XLF (Yu et al., 2008), Artemis (Goodarzi et al., 2006), PNKP (Zolner et al., 2011), as well as DNA-PKcs itself (Douglas et al., 2007; Douglas et al., 2002; Meek et al., 2007) (Supplementary Table 1). In these experiments, potential substrates were either purified from human cells [Ku70/80 (Chan et al., 1999; Douglas et al., 2005), and DNA-PKcs (Douglas et al., 2002)] or recombinant human proteins were expressed in and purified from bacteria [Artemis (Goodarzi et al., 2006), PNKP (Zolner et al., 2011), XRCC4 and XLF (Yu et al., 2003) and (Yu et al., 2008)]. Substrate proteins were phosphorylated in vitro by purified DNA-PK and phosphorylation sites were identified by a variety of techniques, including radiochemical sequencing, mass spectrometry and mutagenesis and were confirmed using phosphospecific antibodies. Surprisingly, many of the in vitro phosphorylation sites identified by this approach did not correspond to the expected SQ/TQ consensus, rather, in many cases, phosphorylated serines and threonines were followed by other amino acids, such as leucine, tyrosine or methionine (Supplementary Table 1). Similar results were observed by others for Artemis and DNA-PKcs (Ma et al., 2005), DNA Ligase IV (Wang et al., 2004) and non-NHEJ protein SAF-A/hnRNP-U (Berglund and Clarke, 2009; Britton et al., 2009) revealing that, in vitro at least, DNA-PK recognises substrate determinants other than SQ and TQ sequences (Supplementary Table 1).
Many of the identified DNA-PK phosphorylation sites in NHEJ proteins are phosphorylated in vivo in response to DNA damage, however, in several cases (Artemis, PNKP, XRCC4 and XLF), phosphorylation requires the related protein kinase ATM rather than, or in addition to, DNA-PK (Supplementary Table 1). One clear target of DNA-PKcs however is DNA-PKcs itself (Figure 2A). DNA-PKcs undergoes autophosphorylation at multiple SQ/TQ (T2609, S2612, T2638 and T2647) as well as non-SQ/TQ sites (S2624, S3205) in vitro (Douglas et al., 2002) and many of these residues are phosphorylated in a DNA damage-dependent manner in vivo [Supplementary Table 1 and (Dobbs et al., 2010)]. Moreover, phosphorylation of many of these sites (S2056, T2609, S2612, T2620, S2624, T2638, T2647 as well as T3950) is DNA-PK-dependent in vivo (Douglas et al., 2007; Meek et al., 2007), suggesting that DNA-PKcs undergoes autophosphorylation in vivo in response to DNA damage (Supplementary Table 1). T2609 of DNA-PKcs can also be targeted by ATM and ATR in vivo (Chen et al., 2007; Yajima et al., 2009) and we have shown that S3205 is phosphorylated in an ATM-dependent manner in IR-treated cells (Neal et al., 2011), thus DNA-PKcs is also a substrate of other PIKKs in vivo (Supplementary Table 1). Other in vivo phosphorylation sites on DNA-PKcs revealed from phosphoproteomics studies are shown in (Dobbs et al., 2010). Interested readers may wish to check the Phosphosite database for updates (http://www.phosphosite.org/proteinAction.do?id=2072&showAllSites=true). Proteomics studies have also revealed that DNA-PKcs is modified by extensive acetylation (Choudhary et al., 2009) and ubiquitination in vivo (Kim et al., 2011) but how these post-translational modifications affect its activity and function are unknown.
In sum, our understanding of the full complement of physiological targets of DNA-PKcs and the function of DNA-PKcs-mediated phosphorylation events in the cell remains incomplete. The availability of highly selective inhibitors of DNA-PK such as NU7441 (Zhao et al., 2006) as well as cell lines lacking DNA-PKcs, make identification of the DNA-PK phosphoproteome by mass spectrometry an exciting and realistic possibility that will further clarify the role of DNA-PKcs in the cell.
5.1.1 Role of DNA-PKcs autophosphorylation in DSB repair
Early studies showed that DNA-PKcs undergoes autophosphorylation in vitro (Lees-Miller et al., 1990) and this results in dissociation of autophosphorylated DNA-PKcs from DNA-bound Ku (Block et al., 2004; Chan and Lees-Miller, 1996). Using SAXS, we showed that autophosphorylation of DNA-PKcs causes a dramatic conformational change that we speculate may promote autophosphorylation-induced release of DNA-PKcs from DNA-bound Ku (Dobbs et al., 2010; Hammel et al., 2010b) (Figure 1 and Figures 2C and D).
One of the most extensively studied phosphorylation events in DNA-PKcs is autophosphorylation of the ABCDE cluster (T2609, S2612, T2620, S2624, T2638 and T2647) (Figure 2A). Rodent cells expressing DNA-PKcs with ablation of the ABCDE cluster are highly radiation sensitive and are severely defective in V(D)J recombination coding joint formation (Ding et al., 2003). The rare coding joints formed in these cells indicate minimal nucleotide loss, suggesting that inability of DNA-PKcs to autophosphorylate at the ABCDE cluster impairs subsequent processing by downstream enzymes, possibly by interfering with autophosphorylation-induced dissociation of DNA-PKcs from DNA ends (Ding et al., 2003). In vitro studies also suggest that phosphorylation of the ABCDE cluster may contribute to autophosphorylation-induced dissociation as purified DNA-PKcs expressing alanine at the six sites in the ABCDE cluster is less efficient at dissociating from Ku-DNA complexes than wild type DNA-PKcs (Figure 3).
Figure 3. Ablation of ABCDE phosphorylation sites in DNA-PKcs impairs its ability to dissociate from DNA-Ku complexes.

A. Human wild type-DNA-PKcs and DNA-PKcs in which serines and threonines at the ABCDE phosphorylation sites (T2609, S2612, T2620, S2624, T2638, T2647) were mutated to alanine, were stably expressed in DNA-PKcs-deficient V3 rodent cells, purified and incubated with biotin-labelled dsDNA either in the absence of ATP, in the presence of ATP, or the presence of the non-hydrolysable ATP analogue, AMP-PNP as described in (Hammel et al., 2010b). After washing with buffer, beads were resuspended in SDS sample buffer and analysed by SDS PAGE and immunoblot using antibodies to DNA-PKcs or Ku80 as shown. The results show that phosphorylation results in loss of wild-type DNA-PKcs from Ku-DNA complexes whereas more DNA-PKcs is retained with Ku-DNA complexes in the presence of ATP in the autophosphorylation defective mutant (ABCDE sites mutated to alanine). See text and (Hammel et al., 2010b) for details. The upper panel (interaction of wt-DNA-PKcs with Ku and DNA) is taken from Supplementary Figure 10 in (Hammel et al., 2010b), permission applied for. The experiment in the lower panel (purified DNA-PKcs with ABCDE phosphorylation sites mutated to alanine, ABCDE>A) was carried out at the same time as the experiment in the upper panel and equivalent exposures are shown.
B. Bands corresponding to DNA-PKcs in panel A were quantitated using Quantity One software (Biorad), normalized to Ku80 and expressed as a percentage of the sample in lane 1 (incubated with no ATP).
Further support for the role of the ABCDE cluster in dissociation of DNA-PKcs from DSBs comes from elegant UV laser microirradiation studies of Chen and colleagues. These studies showed that wild type DNA-PKcs, kinase dead DNA-PKcs and DNA-PKcs with ablation of the ABCDE phosphorylation cluster were recruited to DSBs with similar kinetics, but whereas over 80% of wild type DNA-PKcs dissociated from damage sites within 2 hours, kinase dead DNA-PKcs and DNA-PKcs unable to phosphorylate at ABCDE sites were retained at DSBs substantially longer (Uematsu et al., 2007). Together these studies support a model in which autophosphorylation of DNA-PKcs at multiple sites, including the ABCDE cluster, is required for release of DNA-PKcs from DNA damage sites in vivo (Dobbs et al., 2010). Inability to undergo efficient autophosphorylation at the ABCDE sites may also interfere with the alternative DSB repair pathway, homologous recombination repair (HRR), as Rad51 foci (an indication of resection and initiation of HRR) were delayed in cells expressing ABCDE/alanine mutants (Shibata et al., 2011). Further evidence for the importance of the ABCDE sites comes from mice expressing DNA-PKcs with alanine at threonines 2605, 2634, and 2643 (corresponding to T2609, T2638, and T2647 in human DNA-PKcs). These mice were radiation sensitive and died from massive bone marrow failure shortly after birth (Zhang et al., 2011).
The physiological significance of other autophosphorylation sites has been extensively studied, primarily by our long-time collaborator Dr Kathy Meek (Michigan State University). S2056 was identified as an in vitro DNA-PK phosphorylation site (Cui et al., 2005) and an in vivo autophosphorylation site (Chen et al., 2005; Meek et al., 2007). Indeed, phosphorylation of S2056 in response to DNA damage is widely accepted as a reliable indicator of DNA-PK activation in cells. Ablation of serine 2056 and other residues in the PQR cluster (Figure 2A) increased processing of V(D)J coding ends indicating that phosphorylation of this cluster of sites has a DNA end protective effect, and suggests that phosphorylation at the different clusters (ABCDE and PQR) may have opposite effects on DNA-PKcs function and accessibility of DNA ends for processing (Cui et al., 2005).
Replacement of T3950 in the putative activation loop in the kinase domain of DNA-PKcs (Fig 2A) with the phosphomimic aspartic acid resulted in loss of DNA-PK activity, radiation sensitivity and severe defects in both coding and signal joint formation, suggesting that dephosphorylation of T3950 is required for DNA-PK activity and that a negative charge at T3950, possibly mimicking constitutive phosphorylation of DNA-PKcs, has severe, inhibitory effects on NHEJ and V(D)J recombination (Douglas et al., 2007).
By mass spectrometry, we have tentatively identified 40 in vitro DNA-PKcs autophosphorylation sites (Yu and Lees-Miller, unpublished), several of which have been characterized in vivo (Neal et al., 2011). Cells expressing DNA-PKcs in which S56 and S72 in the N-terminal domain were converted to aspartic acid (phosphomimic) were highly radiosensitive and displayed severe defects in signal and coding joint formation, however, the rare signal and coding joints that were formed did not show end processing defects (Neal et al., 2011). The presence of the phosphomimic at serines 56 and 72 abrogated DNA-PK kinase activity, but these mutations did not promote dissociation of DNA-PKcs, which along with other phosphorylation data suggests that phosphorylation at either the extreme N terminus (S72 and/or 56) or C terminal kinase domain (T3950) inactivates DNA-PK kinase activity without disrupting stability of the DNA-PKcs-Ku-DNA complex (Neal et al., 2011). For further in depth discussion of the role of autophosphorylation in DNA-PK function the reader is referred to several excellent and detailed reviews on this topic (Meek et al., 2008; Neal and Meek, 2011).
Clearly, the kinase activity of DNA-PKcs is critical for many of its important functions in DSB repair, but it is worth noting that the kinase domain accounts for less than 10% of the total residues in DNA-PKcs, 27% if one considers the adjacent FAT and FAT-C domains, which likely play important roles in stabilizing the kinase domain (Yang et al., 2013). As discussed above, it seems probable that other domains within DNA-PKcs, such as the N-terminal HEAT repeat region, are also critical for its function.
5.2 Interaction of DNA-PKcs with protein phosphatase 6 (PP6)
In order to better understand the function of DNA-PKcs in DSB repair and beyond, we immunoprecipitated DNA-PKcs from unirradiated and irradiated human cells and, in collaboration with Dr. Nick Morrice (then at the University of Dundee), identified interacting proteins, one of which was protein phosphatase 6 (PP6) (Douglas et al., 2010) (Figure 4A). PP6 is composed of catalytic (PP6c) and regulatory subunits, including PP6R1, PP6R2 and PP6R3, (also called SAPS1, SAPS2 and SAPS3, respectively) (Stefansson et al., 2008). DNA-PKcs was shown to co-immunoprecipitate with each of these PP6 subunits and to interact with each independently with each subunit in GST-pull down assays (Douglas et al., 2010). Independently, Larner and Brautigan reported that DNA-PKcs interacts with PP6c (Mi et al., 2009) and that this interaction is primarily through PP6R1 (Hosing et al., 2012). Moreover, PP6 was shown to be required for DNA-PKcs activation after DNA damage (Mi et al., 2009). An obvious question was whether PP6 dephosphorylated any of the known DNA-PKcs autophosphorylation sites, however, siRNA depletion of PP6c did not affect phosphorylation of DNA-PKcs S2056 or T2609 (Douglas et al., 2010; Mi et al., 2009). Interestingly, PP6 was shown to contribute to dephosphorylation of γ-H2AX after IR (Figure 4A), confirming a role in the DNA damage response (Douglas et al., 2010).
Figure 4. Possible roles of DNA-PKcs in mitosis.

A. DNA-PKcs is autophosphorylated (green arrows) on multiple sites in mitosis (Douglas et al., 2014; Lee et al., 2011). DNA-PKcs also interacts with protein phosphatase 6 (PP6) (double headed black arrow) in asynchronously growing and nocodazole-treated mitotic cells (Douglas et al., 2010; Mi et al., 2009). PP6 dephosphorylates T288 of mitotic protein kinase Aurora A in mitosis (Zeng et al., 2010) and serine 139 of H2AX after IR (Douglas et al., 2010) (dashed lines), but whether these two events are related to its ability to interact with DNA-PKcs is unknown. DNA-PKcs interacts with polo-like kinase (PLK1) (double headed black arrow) and PLK1 phosphorylates DNA-PKcs on serine 3205 in mitosis (red arrow). S3205 is dephosphorylated by PP6 (dashed lines) in mitosis and after IR (Douglas et al., 2014). Also shown is DNA-PKcs-dependent phosphorylation of Chk2 on threonine 68 and histone H2AX on serine 139 in mitosis (red arrows) (see Figure 5). In vivo DNA-PKcs may phosphorylate histone H2AX either directly or indirectly through phosphorylation and activation of Chk2 (Tu et al., 2013). DNA-PK-dependent activation of Chk2 also leads to phosphorylation of BRCA1 on serine 988 in mitosis, which is required for accurate mitosis and genomic stability (Shang et al., 2014).
B. Depletion or inhibition of DNA-PKcs results in misaligned mitotic chromosomes and abnormal nuclear morphologies suggesting that DNA-PKcs is required for correct alignment of mitotic chromosomes at the metaphase plate (left) and to prevent abnormal segregation of nuclear contents in cytokinesis.
5.3 New roles for DNA-PKcs in mitosis
In 2010, Barr and Gruneberg reported that PP6 dephosphorylates T288 of Aurora A kinase (Figure 4A), regulating its activity, and that siRNA depletion of PP6 causes profound mitotic defects (Zeng et al., 2010). This finding prompted us to ask whether DNA-PKcs also has a role in mitosis. We and others showed that siRNA-mediated depletion of DNA-PKcs, or inhibition of DNA-PKcs kinase activity with NU7441, led to a number of mitotic defects including increased number of misaligned mitotic chromosomes, abnormal nuclear morphologies, and lagging chromosomes (Figure 4B), and that DNA-PKcs is phosphorylated in a DNA-PK-dependent manner on S2056, S2609, T2647 and T3950 in mitosis (Douglas et al., 2014; Lee et al., 2011) (Figure 4A). Moreover, DNA-PKcs phosphorylated at T2609, S2056 or T2647 localized to centrosomes and DNA-PKcs T2609 phosphorylation was observed at kinetochores and at the midbody during cytokinesis (Douglas et al., 2014; Lee et al., 2011; Shang et al., 2010). Similarly, DNA-PKcs phosphorylated at T3950 localized to centrosomes during prophase and metaphase and at the midbody in cytokinesis (Douglas et al., 2014).
As discussed above, DNA-PKcs autophosphorylates on S3205 in vitro (Douglas et al., 2002), and S3205 is phosphorylated in an ATM-dependent manner after IR (Neal et al., 2011). In mitosis however, DNA-PKcs is phosphorylated on S3205 by polo-like kinase 1 (PLK1) (Douglas et al., 2014) (Figure 4A). Consistent with phosphoproteomics studies (Olsen et al., 2010), S3205 phosphorylation was enhanced in mitosis but was very low in interphase suggesting regulated phosphorylation in mitosis (Douglas et al., 2014). We showed that S3205 is dephosphorylated by PP6 in mitosis (Douglas et al., 2014) (Figure 4A). Like T2609-phosphorylated DNA-PKcs, S3205-phosphorylated DNA-PKcs was located at the midbody in cytokinesis (Douglas et al., 2014). The effects, if any, of DNA-PKcs S3205 phosphorylation on mitosis remain to be determined.
Consistent with a role in mitosis, DNA-PKcs interacts with PLK1 (Douglas et al., 2014; Huang et al., 2014) and phosphorylates checkpoint kinase 2 (Chk2) on T68 in mitosis (Douglas et al., 2014; Tu et al., 2013) (Figure 4A). This is contrast to phosphorylation of T68 of Chk2 in response to DNA damage, which is ATM-dependent (Ahn et al., 2000; Matsuoka et al., 1998). Moreover, DNA-PK-dependent phosphorylation of Chk2 on T68 and consequent activation of Chk2 in mitosis is required for phosphorylation of BRCA1 on S988 (Figure 4A), which in turn regulates mitotic spindle formation and maintains genome stability (Shang et al., 2014). Interestingly, phosphorylation of histone H2AX on serine 139 in nocodazole-treated cells requires DNA-PK, with ATM playing a lesser role (Figure 5). DNA-PK is also required for phosphorylation and activation of Chk2 in mitosis, and it has been suggested that DNA-PKcs may phosphorylate H2AX indirectly through phosphorylation and activation of Chk2 in mitosis (Tu et al., 2013) (question marks in Figure 4A).
Figure 5. Serine 139 phosphorylation of histone H2AX in nocodazole-treated cells requires the protein kinase activity of DNA-PK.
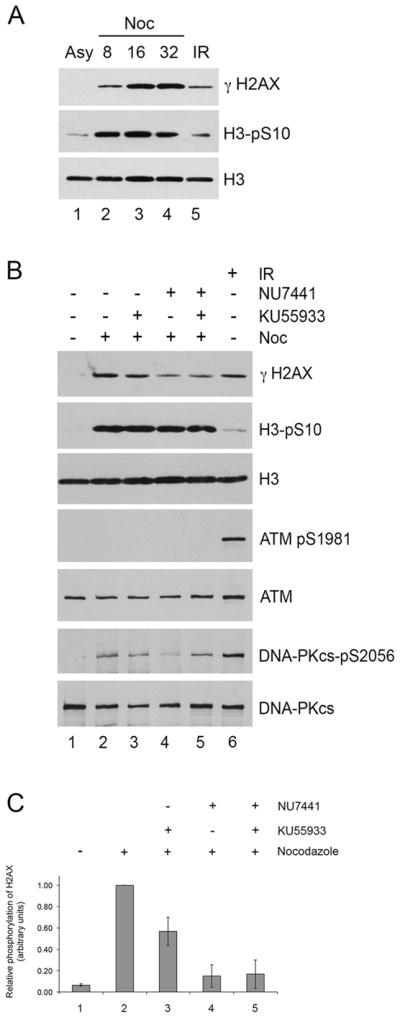
A. HeLa cells were either grown under asynchronous conditions (Asy, lane 1), or incubated with nocodazole (Noc, 40ng/ml) for 8, 16 or 32 hours as indicated (lanes 2–5). For lanes 2–5, mitotic cells were collected by mitotic shake off and allowed to recover in the absence of nocodazole for 35 minutes. Cells were lysed in NETN buffer and H2AX was extracted from the chromatin pellets with 1% SDS as described in (Douglas et al., 2014; Douglas et al., 2010). 50 μg of extract was run on SDS PAGE, transferred to membrane and probed with antibodies to serine 139-phosphorylated histone H2AX (γ-H2AX), serine 10 phosphorylated histone H3 (H3pS10) (a marker of mitosis) and total H3 as indicated. Lane 5 contains 50 μg of extract from asynchronously growing HeLa cells harvested 1 hour after exposure to 10 Gy IR as a control.
B. Hela cells were either grown under asynchronous conditions (lane 1) or treated with nocodazole (40 ng/ml) (lanes 2–5). After 15 hours, either DMSO control (lane 2), the ATM inhibitor KU55933 at 5 μM (lane 3), the DNA-PK inhibitor NU7441 at 8 μM (lane 4), or both KU55933 and NU7441 (lane 5) was added to the media. After 1 hour, mitotic cells were harvested by mitotic shake off as described in (Douglas et al., 2014) and mitotic cells were incubated for a further 1 hour with DMSO, KU55933 and/or NU7441 as above but in the absence of nocodazole. Cells were lysed in NETN buffer, H2AX was extracted from the chromatin pellet of NETN lysates as described above and 50 μg of protein was analysed on SDS PAGE gels followed by immunoblot as described previously (Douglas et al., 2010). Lane 6 contained 50 μg of protein from NETN extracts generated from asynchronously growing cells that had been irradiated 10 Gy, and allowed to recover for 1 hour as a control. See (Douglas et al., 2014) for details.
C. Serine 139-H2AX phosphorylation from panel B was quantitated as in Figure 3, and normalized to total histone H3 staining. Relative phosphorylation of histone γ-H2AX in lanes 1–5 was normalized to phosphorylation with nocodazole treatment alone (lane 2). Results show the average of three independent experiments with standard deviation. Statistical analysis was carried out using the Student’s T test. In the presence of the ATM inhibitor KU55933, H2AX phosphorylation decreased by 43.3 +/− 13.0% (p = 0.029), in the presence of the DNA-PK inhibitor it decreased by 85.1+/− 10.6% (p = 0.005), and in the presence of both inhibitors combined it decreased by 83.3 +/− 13.5% (p = 0.008). Absence of ATM serine 1981 phosphorylation and presence of nocodazole-induced phosphorylation of DNA-PKcs on serine 2056 confirm results reported in (Douglas et al., 2014).
Interestingly, DNA-PK dependent phosphorylation of H2AX occurred in the apparent absence of DNA damage (Tu et al., 2013). Moreover, DNA-PKcs-dependent phosphorylation of S2056 (autophosphorylation) in mitosis and DNA-PK-dependent phosphorylation of Chk2 on T68 in mitosis were found to be largely independent of Ku, since they were unaffected by siRNA depletion of Ku70/80 and were observed in nocodazole-treated xrs6 cells, which lack Ku70/80 expression (Douglas et al., 2014). These observations suggest that the mechanism of activation of DNA-PKcs in mitosis may be fundamentally different from its well-established Ku-dependent activation after DNA damage.
It should also be noted that other components of the DSB damage response have also been shown to function and/or are regulated in mitosis. The NHEJ scaffolding protein XRCC4 is phosphorylated by cyclin-dependent protein kinase (CDK1) and PLK1 in mitosis and this phosphorylation suppresses DSB repair, preventing anaphase bridge formation and genomic instability (Terasawa et al., 2014). Moreover, ATM, which is activated after DNA damage by the MRN complex (Williams et al., 2010), is activated by Aurora B in mitosis in the absence of either DNA damage or the MRN complex (Yang et al., 2011). Aurora B phosphorylates ATM on serine 1403, which leads to phosphorylation of Bub1 on serine 314, which in turn is required for activation of the spindle assembly checkpoint (SAC) (Yang et al., 2011). ATM also phosphorylates Mad1 (Yang et al., 2014), and, with MDC1, is required for localization of mitotic checkpoint proteins Mad2 and Cdc20 (Eliezer et al., 2014), which are required for activation of the SAC. Thus, like DNA-PKcs, ATM also plays an important role in regulation of mitosis, likely facilitating its role in promoting genome stability. However, since cells lacking either ATM or DNA-PKcs are viable, ATM and DNA-PKcs likley have overlapping or redundant roles in mitosis.
These observations raise an important question; why are DNA DSB repair proteins involved in mitosis? The answer may lie in observations made over 60 years ago, that showed that repair of DSBs is attenuated in mitosis (Zirkle and Bloom, 1953). As discussed above, one mechanism for this may be through mitotic phosphorylation and inactivation of XRCC4 by mitotic protein kinases CDK1 and PLK1 (Terasawa et al., 2014). Another mechanism is through suppression of DSB repair by PLK1 and CDK1-dependent phosphorylation of the E3 ubiquitin ligase RNF8 and the NHEJ promoting protein, 53BP1 (Giunta et al., 2010; Orthwein et al., 2014). Moreover, inability to inactivate RNF8 and 53BP1-dependent pathways in mitosis leads to telomeric fusions and genomic instability (Orthwein et al., 2014). Whether phosphorylation of DNA-PKcs in mitosis contributes to inactivation of DSB repair in mitosis or whether DNA-PKcs has additional, DSB repair independent roles in mitosis remains to be determined.
5.4 Other cellular functions of DNA-PKcs
In addition to its well-established role in DSB repair, and its newly emerging role in mitosis, other recent studies have revealed exciting and provocative new roles of DNA-PK in the cell. These include roles in transcription, viral infection, maintenance of telomeres and other processes described briefly below as well as in a recent review on non-DNA repair functions of DNA-PKcs (Goodwin and Knudsen, 2014).
5.4.1 Transcription
DNA-PKcs and Ku were found, along with poly ADP ribose polymerase 1 (PARP-1) and topoisomerase II beta, to localize to the promoters of estrogen-inducible and lipogenic genes (Ju et al., 2006; Wong et al., 2009; Wong and Sul, 2009; Wong and Sul, 2010). Topo II beta induces transient DSBs that are required for active transcription of estrogen responsive and lipogenic genes, possibly initiating chromatin alterations required for transcription initiation.
5.4.2 Viral Infection
Early studies showed that DNA-PKcs was targeted for proteasome-mediated destruction in Herpes Simplex Virus infected cells, suggesting that it acts as a barrier to viral replication or function (Lees-Miller et al., 1996; Parkinson et al., 1999). More recently DNA-PKcs has been linked to HIV-1 infection (Cooper et al., 2013a; Cooper et al., 2013b; Skalka, 2013). HIV infection leads to death of activated CD4+ T cells, the root cause of AIDS. Cooper et al showed that HIV infection activates DNA-PKcs, triggering p53 dependent apoptosis, while inhibition of DNA-PKcs kinase activity with NU7026 blocked HIV-induced cell death in CD4+ T cells, thus DNA-PK inhibitors may have potential in preserving T cell function in AIDS (Cooper et al., 2013b), reviewed in (Cooper et al., 2013a; Skalka, 2013).
5.4.3 Telomeres
Like many other DSB damage response proteins (O’Sullivan and Karlseder, 2010), DNA-PKcs plays a major role in protection of telomeres, the DNA sequences at the ends of chromosomes, and is required for telomere capping (Bailey et al., 2004; Bailey et al., 1999; Gilley et al., 2001; Le et al., 2013; Williams et al., 2009).
5.4.4 Cytoplasmic consequences of DNA damage
An intriguing new study reports that DNA damage causes DNA-PK-dependent phosphorylation of Golgi protein GOLPH3 on threonines 143 and 148 leading to fragmentation of Golgi and its dispersal in the cytoplasm. This pathway is required for cell survival following DNA damage (Farber-Katz et al., 2014). Moreover, GOLPH3 is over expressed in many human cancers and its overexpression confers resistance to DNA damaging agents, suggesting that this novel DNA-PK-dependent pathway could have therapeutic relevance (Farber-Katz et al., 2014), reviewed in (Baumann, 2014; Foiani and Bartek, 2014).
5. Conclusions
In summary, while the role of DNA-PKcs in DSB repair and V(D)J recombination has been well-studied, several important questions remain. Chief for structural biology is the structure of the DNA-PKcs-Ku-DNA complex and the understanding of how interaction of DNA-PKcs with Ku and dsDNA results in activation of DNA-PKcs’ protein kinase activity. Also, how phosphorylation regulates DNA-PKcs activity and function remains to be determined, as does a complete understanding of how the Ku heterodimer is released from ends of DSB prior to or after DNA end ligation. Meanwhile, the functions of DNA-PKcs in mitosis, transcription and the stability of the Golgi body are only beginning to emerge and much more remains to be learned about the role of this intriguing, multifunctional protein kinase in the cell.
Supplementary Material
The table lists several DNA-PKcs substrates that are NHEJ proteins, focusing primarily on work from the authors laboratory but is not an exhaustive list of DNA-PK substrates. The phosphorylated and adjacent amino acid in each amino acid sequences are indicated in bold face.
Acknowledgments
SPLM wishes to thank to all lab members, past and present, in particular Dr. Pauline Douglas for her work on DNA-PK in NHEJ and mitosis, Sarvan Radhakrishnan and Dr. Edward Bartlett for helpful discussions, Dr. B. Mahaney for assistance with Supplementary Table 1, and R. Ye for the experiments shown in Figure 3 and 5. We also thank our collaborators and colleagues, in particular, Drs. Michal Hammel and John Tainer (Lawrence Berkeley National Laboratory) and Katheryn Meek (Michigan State University). Work in the authors laboratory was funded by the Canadian Institutes of Health Research (grant # MOP13639), the Alberta Heritage Foundation for Medical Research, the Engineered Air Chair in Cancer Research, NIH program project grant PO1 CA92584 and the Cancer Research Society.
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ATM-and Rad3-related
- ds
double-stranded
- cryo-EM
cryo-electron microscopy
- DSB
DNA double strand break
- DNA-PK
DNA-activated/dependent protein kinase
- DNA-PKcs
DNA-PK catalytic subunit
- FAT
FRAP, ATM and TRRAP
- HEAT
Huntingtin, Elongation factor, PP2A-A subunit, TOR)
- IR
ionizing radiation
- PI3K
phosphatidyl inositol 3 kinase
- PIKK
phosphatidyl inositol-3 kinase-like protein kinase
- PP6
protein phosphatase 6
- SAXS
small angle X ray scattering
Footnotes
The authors declare they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–6. [PubMed] [Google Scholar]
- Anderson CW. DNA damage and the DNA-activated protein kinase. Trends Biochem Sci. 1993;18:433–7. doi: 10.1016/0968-0004(93)90144-c. [DOI] [PubMed] [Google Scholar]
- Anderson CW, Lees-Miller SP. The nuclear serine/threonine protein kinase DNA-PK. Crit Rev Eukaryot Gene Expr. 1992;2:283–314. [PubMed] [Google Scholar]
- Bailey SM, Cornforth MN, Ullrich RL, Goodwin EH. Dysfunctional mammalian telomeres join with DNA double-strand breaks. DNA Repair (Amst) 2004;3:349–57. doi: 10.1016/j.dnarep.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc Natl Acad Sci U S A. 1999;96:14899–904. doi: 10.1073/pnas.96.26.14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Baretic D, Williams RL. PIKKs - the solenoid nest where partners and kinases meet. Curr Opin Struct Biol. 2014;29C:134–142. doi: 10.1016/j.sbi.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Baumann K. DNA damage: Dispersing Golgi. Nat Rev Mol Cell Biol. 2014;15:153. doi: 10.1038/nrm3762. [DOI] [PubMed] [Google Scholar]
- Berglund FM, Clarke PR. hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem Biophys Res Commun. 2009;381:59–64. doi: 10.1016/j.bbrc.2009.02.019. [DOI] [PubMed] [Google Scholar]
- Blier PR, Griffith AJ, Craft J, Hardin JA. Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J Biol Chem. 1993;268:7594–601. [PubMed] [Google Scholar]
- Block WD, Yu Y, Merkle D, Gifford JL, Ding Q, Meek K, Lees-Miller SP. Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Res. 2004;32:4351–7. doi: 10.1093/nar/gkh761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskovic J, Rivera-Calzada A, Maman JD, Chacon P, Willison KR, Pearl LH, Llorca O. Visualization of DNA-induced conformational changes in the DNA repair kinase DNA-PKcs. EMBO J. 2003;22:5875–82. doi: 10.1093/emboj/cdg555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Coates J, Jackson SP. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol. 2013;202:579–95. doi: 10.1083/jcb.201303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Froment C, Frit P, Monsarrat B, Salles B, Calsou P. Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle. 2009;8:3717–22. doi: 10.4161/cc.8.22.10025. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Carter T, Vancurova I, Sun I, Lou W, DeLeon S. A DNA-activated protein kinase from HeLa cell nuclei. Mol Cell Biol. 1990;10:6460–71. doi: 10.1128/mcb.10.12.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter TH, Kopman CR, James CB. DNA-stimulated protein phosphorylation in HeLa whole cell and nuclear extracts. Biochem Biophys Res Commun. 1988;157:535–40. doi: 10.1016/s0006-291x(88)80282-1. [DOI] [PubMed] [Google Scholar]
- Chan DW, Lees-Miller SP. The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J Biol Chem. 1996;271:8936–41. doi: 10.1074/jbc.271.15.8936. [DOI] [PubMed] [Google Scholar]
- Chan DW, Mody CH, Ting NS, Lees-Miller SP. Purification and characterization of the double-stranded DNA-activated protein kinase, DNA-PK, from human placenta. Biochem Cell Biol. 1996;74:67–73. doi: 10.1139/o96-007. [DOI] [PubMed] [Google Scholar]
- Chan DW, Ye R, Veillette CJ, Lees-Miller SP. DNA-dependent protein kinase phosphorylation sites in Ku 70/80 heterodimer. Biochemistry. 1999;38:1819–28. doi: 10.1021/bi982584b. [DOI] [PubMed] [Google Scholar]
- Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280:14709–15. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Lobrich M, Shiloh Y, Chen DJ. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–7. doi: 10.1074/jbc.M611605200. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV integration and T cell death: additional commentary. Retrovirology. 2013a;10:150. doi: 10.1186/1742-4690-10-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature. 2013b;498:376–9. doi: 10.1038/nature12274. [DOI] [PubMed] [Google Scholar]
- Cottarel J, Frit P, Bombarde O, Salles B, Negrel A, Bernard S, Jeggo PA, Lieber MR, Modesti M, Calsou P. A noncatalytic function of the ligation complex during nonhomologous end joining. J Cell Biol. 2013;200:173–86. doi: 10.1083/jcb.201203128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNAdependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol. 2005;25:10842–52. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002;21:3192–200. doi: 10.1093/emboj/cdf299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Reddy YV, Wang W, Woods T, Douglas P, Ramsden DA, Lees-Miller SP, Meek K. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol Cell Biol. 2003;23:5836–48. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs TA, Tainer JA, Lees-Miller SP. A structural model for regulation of NHEJ by DNAPKcs autophosphorylation. DNA Repair (Amst) 2010;9:1307–14. doi: 10.1016/j.dnarep.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore AS, Drake AC, Brewerton SC, Blundell TL. Identification of DNA-PK in the arthropods. Evidence for the ancient ancestry of vertebrate non-homologous end-joining. DNA Repair (Amst) 2004;3:33–41. doi: 10.1016/j.dnarep.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Douglas P, Cui X, Block WD, Yu Y, Gupta S, Ding Q, Ye R, Morrice N, Lees-Miller SP, Meek K. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol Cell Biol. 2007;27:1581–91. doi: 10.1128/MCB.01962-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Gupta S, Morrice N, Meek K, Lees-Miller SP. DNA-PK-dependent phosphorylation of Ku70/80 is not required for non-homologous end joining. DNA Repair (Amst) 2005;4:1006–18. doi: 10.1016/j.dnarep.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, Meek K, Alessi DR, Lees-Miller SP. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J. 2002;368:243–51. doi: 10.1042/BJ20020973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Ye R, Trinkle-Mulcahy L, Neal JA, De Wever V, Morrice NA, Meek K, Lees-Miller SP. Polo-like kinase 1 (PLK1) and protein phosphatase 6 (PP6) regulate DNA-dependent protein kinase catalytic subunit (DNA-PKcs) phosphorylation in mitosis. Biosci Rep. 2014;34 doi: 10.1042/BSR20140051. pii: e00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Zhong J, Ye R, Moorhead GB, Xu X, Lees-Miller SP. Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates gamma-H2AX. Mol Cell Biol. 2010;30:1368–81. doi: 10.1128/MCB.00741-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir A, Peterson SR, Knuth MW, Lu H, Dynan WS. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc Natl Acad Sci U S A. 1992;89:11920–4. doi: 10.1073/pnas.89.24.11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir A, Stein LY, Calore BL, Dynan WS. Purification and characterization of a templateassociated protein kinase that phosphorylates RNA polymerase II. J Biol Chem. 1993;268:10440–7. [PubMed] [Google Scholar]
- Eliezer Y, Argaman L, Kornowski M, Roniger M, Goldberg M. Interplay between the DNA damage proteins MDC1 and ATM in the regulation of the spindle assembly checkpoint. J Biol Chem. 2014;289:8182–93. doi: 10.1074/jbc.M113.532739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing M, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM, Fuchs GJ, Zhou H, Field SJ. DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell. 2014;156:413–27. doi: 10.1016/j.cell.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Chen J. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat Struct Mol Biol. 2012;19:201–6. doi: 10.1038/nsmb.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiani M, Bartek J. Golgi feels DNA’s pain. Cell. 2014;156:392–3. doi: 10.1016/j.cell.2014.01.030. [DOI] [PubMed] [Google Scholar]
- Frit P, Li RY, Arzel D, Salles B, Calsou P. Ku entry into DNA inhibits inward DNA transactions in vitro. J Biol Chem. 2000;275:35684–91. doi: 10.1074/jbc.M004315200. [DOI] [PubMed] [Google Scholar]
- Gell D, Jackson SP. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 1999;27:3494–502. doi: 10.1093/nar/27.17.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts RC, Stamato TD. Absence of a Ku-like DNA end binding activity in the xrs doublestrand DNA repair-deficient mutant. J Biol Chem. 1994;269:15981–4. [PubMed] [Google Scholar]
- Gilley D, Tanaka H, Hande MP, Kurimasa A, Li GC, Oshimura M, Chen DJ. DNA-PKcs is critical for telomere capping. Proc Natl Acad Sci U S A. 2001;98:15084–8. doi: 10.1073/pnas.261574698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to doublestrand breaks during mitosis. J Cell Biol. 2010;190:197–207. doi: 10.1083/jcb.200911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Harer C, Marchetti C, Morrice N, Jeggo PA, Lees-Miller SP. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J. 2006;25:3880–9. doi: 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JF, Knudsen KE. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014;4:1126–39. doi: 10.1158/2159-8290.CD-14-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell. 1993;72:131–42. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- Grinthal A, Adamovic I, Weiner B, Karplus M, Kleckner N. PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proc Natl Acad Sci U S A. 2010;107:2467–72. doi: 10.1073/pnas.0914073107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96:99–110. doi: 10.1016/s0092-8674(00)80963-0. [DOI] [PubMed] [Google Scholar]
- Gupta S, Meek K. The leucine rich region of DNA-PKcs contributes to its innate DNA affinity. Nucleic Acids Res. 2005;33:6972–81. doi: 10.1093/nar/gki990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Rey M, Yu Y, Mani RS, Classen S, Liu M, Pique ME, Fang S, Mahaney BL, Weinfeld M, Schriemer DC, Lees-Miller SP, Tainer JA. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J Biol Chem. 2011;286:32638–50. doi: 10.1074/jbc.M111.272641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure. 2010a;18:1431–42. doi: 10.1016/j.str.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammel M, Yu Y, Mahaney BL, Cai B, Ye R, Phipps BM, Rambo RP, Hura GL, Pelikan M, So S, Abolfath RM, Chen DJ, Lees-Miller SP, Tainer JA. Ku and DNA-dependent protein kinase dynamic conformations and assembly regulate DNA binding and the initial nonhomologous end joining complex. J Biol Chem. 2010b;285:1414–23. doi: 10.1074/jbc.M109.065615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley KO, Gell D, Smith GC, Zhang H, Divecha N, Connelly MA, Admon A, Lees-Miller SP, Anderson CW, Jackson SP. DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell. 1995;82:849–56. doi: 10.1016/0092-8674(95)90482-4. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosing AS, Valerie NC, Dziegielewski J, Brautigan DL, Larner JM. PP6 regulatory subunit R1 is bidentate anchor for targeting protein phosphatase-6 to DNA-dependent protein kinase. J Biol Chem. 2012;287:9230–9. doi: 10.1074/jbc.M111.333708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Shang ZF, Li B, Wang Y, Liu XD, Zhang SM, Guan H, Rang WQ, Hu JA, Zhou PK. DNA-PKcs associates with PLK1 and is involved in proper chromosome segregation and cytokinesis. J Cell Biochem. 2014;115:1077–88. doi: 10.1002/jcb.24703. [DOI] [PubMed] [Google Scholar]
- Hunter T. When is a lipid kinase not a lipid kinase? When it is a protein kinase. Cell. 1995;83:1–4. doi: 10.1016/0092-8674(95)90225-2. [DOI] [PubMed] [Google Scholar]
- Jackson SP, MacDonald JJ, Lees-Miller S, Tjian R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell. 1990;63:155–65. doi: 10.1016/0092-8674(90)90296-q. [DOI] [PubMed] [Google Scholar]
- Jeggo PA, Jackson SP, Taccioli GE. Identification of the catalytic subunit of DNA dependent protein kinase as the product of the mouse scid gene. Curr Top Microbiol Immunol. 1996;217:79–89. doi: 10.1007/978-3-642-50140-1_6. [DOI] [PubMed] [Google Scholar]
- Jin S, Kharbanda S, Mayer B, Kufe D, Weaver DT. Binding of Ku and c-Abl at the kinase homology region of DNA-dependent protein kinase catalytic subunit. J Biol Chem. 1997;272:24763–6. doi: 10.1074/jbc.272.40.24763. [DOI] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- Kienker LJ, Shin EK, Meek K. Both V(D)J recombination and radioresistance require DNAPK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Res. 2000;28:2752–61. doi: 10.1093/nar/28.14.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. Systematic and quantitative assessment of the ubiquitinmodified proteome. Mol Cell. 2011;44:325–40. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, Oettinger MA, Brown JM. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science. 1995;267:1178–83. doi: 10.1126/science.7855601. [DOI] [PubMed] [Google Scholar]
- Koch CA, Agyei R, Galicia S, Metalnikov P, O’Donnell P, Starostine A, Weinfeld M, Durocher D. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. Embo J. 2004;23:3874–85. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Mazzanti M, Mistrik M, Kosar M, Beznoussenko GV, Mironov AA, Garre M, Parazzoli D, Shivashankar GV, Scita G, Bartek J, Foiani M. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell. 2014;158:633–46. doi: 10.1016/j.cell.2014.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung CS, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol. 1999;19:3877–84. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22:R966–80. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Khanna KK, Beamish H, Spring K, Watters D, Shiloh Y. Relationship of the ataxia-telangiectasia protein ATM to phosphoinositide 3-kinase. Trends Biochem Sci. 1995;20:382–3. doi: 10.1016/s0968-0004(00)89083-0. [DOI] [PubMed] [Google Scholar]
- Le PN, Maranon DG, Altina NH, Battaglia CL, Bailey SM. TERRA, hnRNP A1, and DNAPKcs Interactions at Human Telomeres. Front Oncol. 2013;3:91. doi: 10.3389/fonc.2013.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Lin YF, Chou HY, Yajima H, Fattah KR, Lee SC, Chen BP. Involvement of DNA-dependent protein kinase in normal cell cycle progression through mitosis. J Biol Chem. 2011;286:12796–802. doi: 10.1074/jbc.M110.212969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Anderson CW. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J Biol Chem. 1989;264:17275–80. [PubMed] [Google Scholar]
- Lees-Miller SP, Chen YR, Anderson CW. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol. 1990;10:6472–81. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Godbout R, Chan DW, Weinfeld M, Day RS, 3rd, Barron GM, Allalunis-Turner J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science. 1995;267:1183–5. doi: 10.1126/science.7855602. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol. 1996;70:7471–7. doi: 10.1128/jvi.70.11.7471-7477.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Sakaguchi K, Ullrich SJ, Appella E, Anderson CW. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol Cell Biol. 1992;12:5041–9. doi: 10.1128/mcb.12.11.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28:3067–73. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA endjoining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Lu H, Niewolik D, Schwarz K, Lieber MR. The DNA-dependent protein kinase catalytic subunit phosphorylation sites in human Artemis. J Biol Chem. 2005;280:33839–46. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- Mahaney BL, Hammel M, Meek K, Tainer JA, Lees-Miller SP. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem Cell Biol. 2013;91:31–41. doi: 10.1139/bcb-2012-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA doublestrand breaks by non-homologous end-joining. Biochem J. 2009;417:639–50. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malu S, De Ioannes P, Kozlov M, Greene M, Francis D, Hanna M, Pena J, Escalante CR, Kurosawa A, Erdjument-Bromage H, Tempst P, Adachi N, Vezzoni P, Villa A, Aggarwal AK, Cortes P. Artemis C-terminal region facilitates V(D)J recombination through its interactions with DNA Ligase IV and DNA-PKcs. J Exp Med. 2012;209:955–63. doi: 10.1084/jem.20111437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–7. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- Meek K, Douglas P, Cui X, Ding Q, Lees-Miller SP. trans Autophosphorylation at DNAdependent protein kinase’s two major autophosphorylation site clusters facilitates end processing but not end joining. Mol Cell Biol. 2007;27:3881–90. doi: 10.1128/MCB.02366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi J, Dziegielewski J, Bolesta E, Brautigan DL, Larner JM. Activation of DNA-PK by ionizing radiation is mediated by protein phosphatase 6. PLoS One. 2009;4:e4395. doi: 10.1371/journal.pone.0004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimori T, Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986;261:10375–9. [PubMed] [Google Scholar]
- Mimori T, Ohosone Y, Hama N, Suwa A, Akizuki M, Homma M, Griffith AJ, Hardin JA. Isolation and characterization of cDNA encoding the 80-kDa subunit protein of the human autoantigen Ku (p70/p80) recognized by autoantibodies from patients with sclerodermapolymyositis overlap syndrome. Proc Natl Acad Sci U S A. 1990;87:1777–81. doi: 10.1073/pnas.87.5.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, Meek K. Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol. 2011;31:1719–33. doi: 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal JA, Meek K. Choosing the right path: does DNA-PK help make the decision? Mutat Res. 2011;711:73–86. doi: 10.1016/j.mrfmmm.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, Abraham RH, Lai JH, Hill D, Shiloh Y, Cantley LC, Rathbun GA. Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem. 2000;275:22719–27. doi: 10.1074/jbc.M001002200. [DOI] [PubMed] [Google Scholar]
- O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11:171–81. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Gu X, Blundell TL. Structure of the catalytic region of DNA ligase IV in complex with an Artemis fragment sheds light on double-strand break repair. Structure. 2013;21:672–9. doi: 10.1016/j.str.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science. 2014;344:189–93. doi: 10.1126/science.1248024. [DOI] [PubMed] [Google Scholar]
- Paillard S, Strauss F. Analysis of the mechanism of interaction of simian Ku protein with DNA. Nucleic Acids Res. 1991;19:5619–24. doi: 10.1093/nar/19.20.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol. 1999;73:650–7. doi: 10.1128/jvi.73.1.650-657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry J, Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell. 2003;112:151–5. doi: 10.1016/s0092-8674(03)00033-3. [DOI] [PubMed] [Google Scholar]
- Pi J, Sael L. Mass spectrometry coupled experiments and protein structure modeling methods. Int J Mol Sci. 2013;14:20635–57. doi: 10.3390/ijms141020635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L. Destroying the ring: Freeing DNA from Ku with ubiquitin. FEBS Lett. 2011;585:2876–82. doi: 10.1016/j.febslet.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L, Ghenoiu C, Woo EM, Krutchinsky AN, Chait BT, Funabiki H. Ku80 removal from DNA through double strand break-induced ubiquitylation. J Cell Biol. 2008;182:467–79. doi: 10.1083/jcb.200802146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan SK, Jette N, Lees-Miller SP. Non-homologous end joining: emerging themes and unanswered questions. DNA Repair (Amst) 2014;17:2–8. doi: 10.1016/j.dnarep.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Calzada A, Maman JD, Spagnolo L, Pearl LH, Llorca O. Three-dimensional structure and regulation of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) Structure. 2005;13:243–55. doi: 10.1016/j.str.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Rivera-Calzada A, Spagnolo L, Pearl LH, Llorca O. Structural model of full-length human Ku70-Ku80 heterodimer and its recognition of DNA and DNA-PKcs. EMBO Rep. 2007;8:56–62. doi: 10.1038/sj.embor.7400847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Shang Z, Yu L, Lin YF, Matsunaga S, Shen CY, Chen BP. DNA-PKcs activates the Chk2- Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis. 2014;3:e85. doi: 10.1038/oncsis.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD, Wang Y, Zhou PK. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 Phosphorylation in response to DNA damage. Cancer Res. 2010;70:3657–66. doi: 10.1158/0008-5472.CAN-09-3362. [DOI] [PubMed] [Google Scholar]
- Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, Jeggo PA. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–92. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118–21. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalka AM. HIV: Integration triggers death. Nature. 2013;498:305–6. doi: 10.1038/nature12254. [DOI] [PubMed] [Google Scholar]
- Song Q, Lees-Miller SP, Kumar S, Zhang Z, Chan DW, Smith GC, Jackson SP, Alnemri ES, Litwack G, Khanna KK, Lavin MF. DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO J. 1996;15:3238–46. [PMC free article] [PubMed] [Google Scholar]
- Spagnolo L, Rivera-Calzada A, Pearl LH, Llorca O. Three-dimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol Cell. 2006;22:511–9. doi: 10.1016/j.molcel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Stefansson B, Ohama T, Daugherty AE, Brautigan DL. Protein phosphatase 6 regulatory subunits composed of ankyrin repeat domains. Biochemistry. 2008;47:1442–51. doi: 10.1021/bi7022877. [DOI] [PubMed] [Google Scholar]
- Suwa A, Hirakata M, Takeda Y, Jesch SA, Mimori T, Hardin JA. DNA-dependent protein kinase (Ku protein-p350 complex) assembles on double-stranded DNA. Proc Natl Acad Sci U S A. 1994;91:6904–8. doi: 10.1073/pnas.91.15.6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taccioli GE, Gottlieb TM, Blunt T, Priestley A, Demengeot J, Mizuta R, Lehmann AR, Alt FW, Jackson SP, Jeggo PA. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 1994;265:1442–5. doi: 10.1126/science.8073286. [DOI] [PubMed] [Google Scholar]
- Terasawa M, Shinohara A, Shinohara M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 2014;10:e1004563. doi: 10.1371/journal.pgen.1004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting NS, Chan DW, Lintott LG, Allalunis-Turner J, Lees-Miller SP. Protein-DNA complexes containing DNA-dependent protein kinase in crude extracts from human and rodent cells. Radiat Res. 1999;151:414–22. [PubMed] [Google Scholar]
- Ting NS, Kao PN, Chan DW, Lintott LG, Lees-Miller SP. DNA-dependent protein kinase interacts with antigen receptor response element binding proteins NF90 and NF45. J Biol Chem. 1998;273:2136–45. doi: 10.1074/jbc.273.4.2136. [DOI] [PubMed] [Google Scholar]
- Tu WZ, Li B, Huang B, Wang Y, Liu XD, Guan H, Zhang SM, Tang Y, Rang WQ, Zhou PK. gammaH2AX foci formation in the absence of DNA damage: mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013;587:3437–43. doi: 10.1016/j.febslet.2013.08.028. [DOI] [PubMed] [Google Scholar]
- Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol. 2007;177:219–29. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal SA, Stewart PL. CryoEM and image sorting for flexible protein/DNA complexes. J Struct Biol. 2014;187:76–83. doi: 10.1016/j.jsb.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Walker AI, Hunt T, Jackson RJ, Anderson CW. Double-stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat-shock protein in animal cell extracts. EMBO J. 1985;4:139–45. doi: 10.1002/j.1460-2075.1985.tb02328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–14. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- Wang C, Lees-Miller SP. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Radiat Oncol Biol Phys. 2013;86:440–9. doi: 10.1016/j.ijrobp.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YG, Nnakwe C, Lane WS, Modesti M, Frank KM. Phosphorylation and regulation of DNA ligase IV stability by DNA-dependent protein kinase. J Biol Chem. 2004;279:37282–90. doi: 10.1074/jbc.M401217200. [DOI] [PubMed] [Google Scholar]
- West RB, Yaneva M, Lieber MR. Productive and nonproductive complexes of Ku and DNAdependent protein kinase at DNA termini. Mol Cell Biol. 1998;18:5908–20. doi: 10.1128/mcb.18.10.5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weterings E, Verkaik NS, Keijzers G, Florea BI, Wang SY, Ortega LG, Uematsu N, Chen DJ, van Gent DC. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol Cell Biol. 2009;29:1134–42. doi: 10.1128/MCB.00971-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Lee KJ, Shi J, Chen DJ, Stewart PL. Cryo-EM structure of the DNAdependent protein kinase catalytic subunit at subnanometer resolution reveals alpha helices and insight into DNA binding. Structure. 2008;16:468–77. doi: 10.1016/j.str.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ES, Klingler R, Ponnaiya B, Hardt T, Schrock E, Lees-Miller SP, Meek K, Ullrich RL, Bailey SM. Telomere dysfunction and DNA-PKcs deficiency: characterization and consequence. Cancer Res. 2009;69:2100–7. doi: 10.1158/0008-5472.CAN-08-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010;9:1299–306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009;136:1056–72. doi: 10.1016/j.cell.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RH, Sul HS. DNA-PK: relaying the insulin signal to USF in lipogenesis. Cell Cycle. 2009;8:1977–8. doi: 10.4161/cc.8.13.8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RH, Sul HS. Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Curr Opin Pharmacol. 2010;10:684–91. doi: 10.1016/j.coph.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima H, Lee KJ, Zhang S, Kobayashi J, Chen BP. DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J Mol Biol. 2009;385:800–10. doi: 10.1016/j.jmb.2008.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Hao J, Kong D, Cui X, Zhang W, Wang H, Guo X, Ma S, Liu X, Pu P, Xu B. ATM-mediated Mad1 Serine 214 phosphorylation regulates Mad1 dimerization and the spindle assembly checkpoint. Carcinogenesis. 2014;35:2007–13. doi: 10.1093/carcin/bgu087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Tang X, Guo X, Niikura Y, Kitagawa K, Cui K, Wong ST, Fu L, Xu B. Aurora-B mediated ATM serine 1403 phosphorylation is required for mitotic ATM activation and the spindle checkpoint. Mol Cell. 2011;44:597–608. doi: 10.1016/j.molcel.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–23. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Dynan WS. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 1999;27:4679–86. doi: 10.1093/nar/27.24.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Kimzey A, Dynan WS. Photocross-linking of an oriented DNA repair complex. Ku bound at a single DNA end. J Biol Chem. 1999;274:20034–9. doi: 10.1074/jbc.274.28.20034. [DOI] [PubMed] [Google Scholar]
- Yu Y, Mahaney BL, Yano K, Ye R, Fang S, Douglas P, Chen DJ, Lees-Miller SP. DNAPK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair (Amst) 2008;7:1680–92. doi: 10.1016/j.dnarep.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang W, Ding Q, Ye R, Chen D, Merkle D, Schriemer D, Meek K, Lees-Miller SP. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair (Amst) 2003;2:1239–52. doi: 10.1016/s1568-7864(03)00143-5. [DOI] [PubMed] [Google Scholar]
- Zeng K, Bastos RN, Barr FA, Gruneberg U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol. 2010;191:1315–32. doi: 10.1083/jcb.201008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Yajima H, Huynh H, Zheng J, Callen E, Chen HT, Wong N, Bunting S, Lin YF, Li M, Lee KJ, Story M, Gapud E, Sleckman BP, Nussenzweig A, Zhang CC, Chen DJ, Chen BP. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J Cell Biol. 2011;193:295–305. doi: 10.1083/jcb.201009074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, Calvert AH, Newell DR, Smith GC, Curtin NJ. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66:5354–62. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- Zirkle RE, Bloom W. Irradiation of parts of individual cells. Science. 1953;117:487–93. doi: 10.1126/science.117.3045.487. [DOI] [PubMed] [Google Scholar]
- Zolner AE, Abdou I, Ye R, Mani RS, Fanta M, Yu Y, Douglas P, Tahbaz N, Fang S, Dobbs T, Wang C, Morrice N, Hendzel MJ, Weinfeld M, Lees-Miller SP. Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxiatelangiectasia mutated regulates its association with sites of DNA damage. Nucleic Acids Res. 2011;39:9224–37. doi: 10.1093/nar/gkr647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The table lists several DNA-PKcs substrates that are NHEJ proteins, focusing primarily on work from the authors laboratory but is not an exhaustive list of DNA-PK substrates. The phosphorylated and adjacent amino acid in each amino acid sequences are indicated in bold face.