

Abstract
Autophagy is a catabolic process in response to starvation or other stress conditions to sustain cellular homeostasis. At present, histone deacetylase inhibitors (HDACIs) are known to induce autophagy in cells through inhibition of mechanistic target of rapamycin (MTOR) pathway. FOXO1, an important transcription factor regulated by AKT, is also known to play a role in autophagy induction. At present, the role of FOXO1 in the HDACIs-induced autophagy has not been reported. In this study, we first observed that HDACIs increased the expression of FOXO1 at the mRNA and protein level. Second, we found that FOXO1 transcriptional activity was enhanced by HDACIs, as evidenced by increased FOXO1 nuclear accumulation and transcriptional activity. Third, suppression of FOXO1 function by siRNA knockdown or by a chemical inhibitor markedly blocked HDACIs-induced autophagy. Moreover, we found that FOXO1-mediated autophagy is achieved via its transcriptional activation, leading to a dual effect on autophagy induction: (i) enhanced expression of autophagy-related (ATG) genes, and (ii) suppression of MTOR via transcription of the SESN3 (sestrin 3) gene. Finally, we found that inhibition of autophagy markedly enhanced HDACIs-mediated cell death, indicating that autophagy serves as an important cell survival mechanism. Taken together, our studies reveal a novel function of FOXO1 in HDACIs-mediated autophagy in human cancer cells and thus support the development of a novel therapeutic strategy by combining HDACIs and autophagy inhibitors in cancer therapy.
Keywords: autophagy, cancer, cell death, FOXO1, histone deacetylase inhibitors, MTOR
Abbreviations
- ATG
autophagy-related
- BAF
bafilomycin A1
- CQ
chloroquine
- FOXO
forkhead box O
- GFP
green fluorescent protein
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- MTOR
mechanistic target of rapamycin
- PI3K
phosphoinositide 3-kinase
- siRNA
short interfering RNA
- SAHA
suberoylanilide hydroxamic acid
- TSA
trichostatin A
- TSC
tuberous sclerosis
Introduction
Autophagy is an evolutionarily conserved self-digestive process via which cells adapt to nutrient starvation and other stress conditions.1,2 Autophagy plays important roles in many physiological processes such as development, innate immune defense, protein quality control, cell survival, and cell death, as well in the pathogenesis of important diseases including cancer, neurodegenerative diseases, and metabolic disorders.3,4 During autophagy, portions of cytoplasmic materials, including macromolecules and organelles, are engulfed into specialized double-membrane structures, autophagosomes, which then fuse with lysosomes to form autolysosomes for degradation of their cargos and regeneration of nutrients.5 Under normal growth conditions when nutrients are abundant, autophagy is kept at a basal level mainly for housekeeping purposes such as degradation of long-lived proteins and turnover of damaged cellular organelles. Under stress conditions like nutrient starvation, autophagy is induced to provide cells with additional internal nutrient supplies. This induction is largely mediated via suppression of mechanistic target of rapamycin (MTOR), the key negative regulator of autophagy through suppression of the ULK1 complex consisting of ULK1, RB1CC1/FIP200, and ATG13.6,7 At present, several important upstream pathways controlling MTOR activation have been identified,8,9 including the class I phosphoinositide 3-kinase (PI3K)-AKT pathway activated by growth factors, the amino acids-Rag GTPase pathway activated by the intracellular amino acids, and the adenosine monophosphate activated protein kinase (AMPK) pathway activated by disruption of the energy homeostasis. The first 2 pathways are positive regulators of MTOR, while the AMPK pathway is a negative regulator of MTOR.10,11
Histone deacetylase inhibitors (HDACIs) are a class of compounds that interfere with the function of histone deacetylase. HDACIs exert their antitumor activities through the induction of cell cycle arrest, differentiation, and apoptosis.12,13 Several important HDACIs have been well studied, including suberoylanilide hydroxamic acid (SAHA), trichostatin A (TSA), and depsipeptide.14 Some of them have already been in clinical trials or approved as cancer therapeutic agents.15 The main antitumor activity of HDACIs is believed to be via induction of apoptotic cell death in a variety of cancer cells.16 HDACIs are known to induce apoptosis via multiple mechanisms, such as increasing the expression of BIM and other related genes.17,18 Moreover, recent studies have demonstrated that HDACIs, such as SAHA and TSA, are able to induce autophagy in human cancer cells, an effect related to their anticancer property.19,20 At present, the molecular mechanisms underlying HDACIs-mediated autophagy are still not clear. Furthermore, the contribution of autophagy to cell death remains controversial and, most likely, is context-dependent. Some groups report that autophagy serves as a cell death mechanism in HDACIs-caused cancer cell death,19-22 whereas other groups have found that autophagy acts as a cell survival mechanism in HDACIs-mediated cancer cell death.20-24
The forkhead box proteins (FOXOs) are a family of transcription factors that play important roles in genes regulation involved in cell growth, proliferation, differentiation, and longevity.25 There are 4 FOXO family members in humans, FOXO1, FOXO3, FOXO4, and FOXO6. Among them, FOXO1 is the most widely studied member. Post-translational modification of FOXO1 is an important mechanism that regulates its ability to activate distinct gene sets, involved in cell cycle arrest, apoptosis, defense against oxidative stress, and DNA repair.26-28 AKT phosphorylates FOXO1 at multiple sites and drives FOXO1 into the cytoplasm, where it is then ubiquitinated and degraded.29,30 In addition, FOXO1 acetylation has been reported to play an important role in regulating its biological functions such as apoptosis and autophagy by dissociation from SIRT2, a member of the family of class III NAD+-dependent deacetylases.14,31 FOXO1 acetylation is also found in autophagy mediated by benzyl isothiocyanate and curcumin.32,33 Whether FOXO1 acetylation is also involved in HDACIs-mediated autophagy is not clear.
In this study, we aimed to study the regulatory circuits underlying interplay between FOXO1, MTOR, and autophagy induced by HDACIs. Here, data from our study provide strong evidence that HDACIs induced autophagy through FOXO1-dependent pathway and such autophagy served as a prosurvival mechanism in HDACIs-mediated cell death in human cancer cells. Our findings thus provide novel insights into the molecular mechanisms underlying HDACIs-induced autophagy involving FOXO1.
Results
HDACIs induce autophagy
TSA is known to effectively inhibit HDAC enzyme activity at nanomolar concentrations, suppress cell growth, and induce cell death.34,35 Here, we treated cancer cells with this inhibitor and investigated the effect of TSA on autophagy. After treatment with TSA, there was an accumulation of LC3-II in HCT116 cells (Fig. 1A) and an increase of GFP-LC3 puncta representing autophagic vacuoles in MEFs with stable expression of GFP-LC3 (Fig. 1B and C). Meanwhile, autophagy flux was determined by bafilomycin A1/BAF (a vacuolar-type H+-ATPase inhibitor that blocks autophagosome and lysosome fusion). TSA led to further increase of LC3-II level (Fig. 1A) and GFP-LC3 puncta in the presence of BAF (Fig. 1B and C), suggesting that TSA increases autophagy flux level. The autophagy flux was further confirmed by the decrease of SQSTM1 protein level, a well-established autophagy substrate (Fig. 1A). In addition, we also tested the effect of SAHA, another HDACI that has been approved by FDA for treatment of T cell lymphoma,15 on HCT116 and HepG2 cells and found similar results (Fig. S1).
Figure 1.

HDACIs induce autophagy. (A) HCT116 cells were treated with trichostatin A (TSA) (0.5 μM) alone or in combination with 15 nM BAF for 12 h. Cell lysates were lysed, collected, and immunoblotted using western blotting for LC3 and SQSTM1 levels. (B) MEFs with stable expression of GFP-LC3 were treated with 1 μM TSA in the presence or absence of 15 nM BAF for 12 h. The cells were examined and representative cells were photographed using a confocal microscope (Scale bar: 10 μm). (C) The number of GFP-LC3 puncta/cell was counted and presented (*P < 0.05, **P < 0.01).
HDACIs increase FOXO1 expression
It has been previously reported that acetylated FOXO1 is required for starvation-induced autophagy.31 However, it is unknown if acetylation of FOXO1 is also involved in HDACIs-induced autophagy. Therefore, we first investigated the expression of FOXO1 in HDACIs-treated cells. As shown in Figure 2A, FOXO1 protein level and its target gene CDKN1A/p21 were significantly increased in TSA-treated HCT116 and HepG2 cells in a dose- and time-dependent manner. Similar effects were also found with SAHA in these 2 cell lines (Fig. S2). Acetylation changes after TSA treatment were investigated using anti-acetylated-FOXO1 antibody and a time- and dose-dependent increase of acetylated FOXO1 was also observed in TSA-treated HCT116 and HepG2 cells (Fig. 2A).
Figure 2.
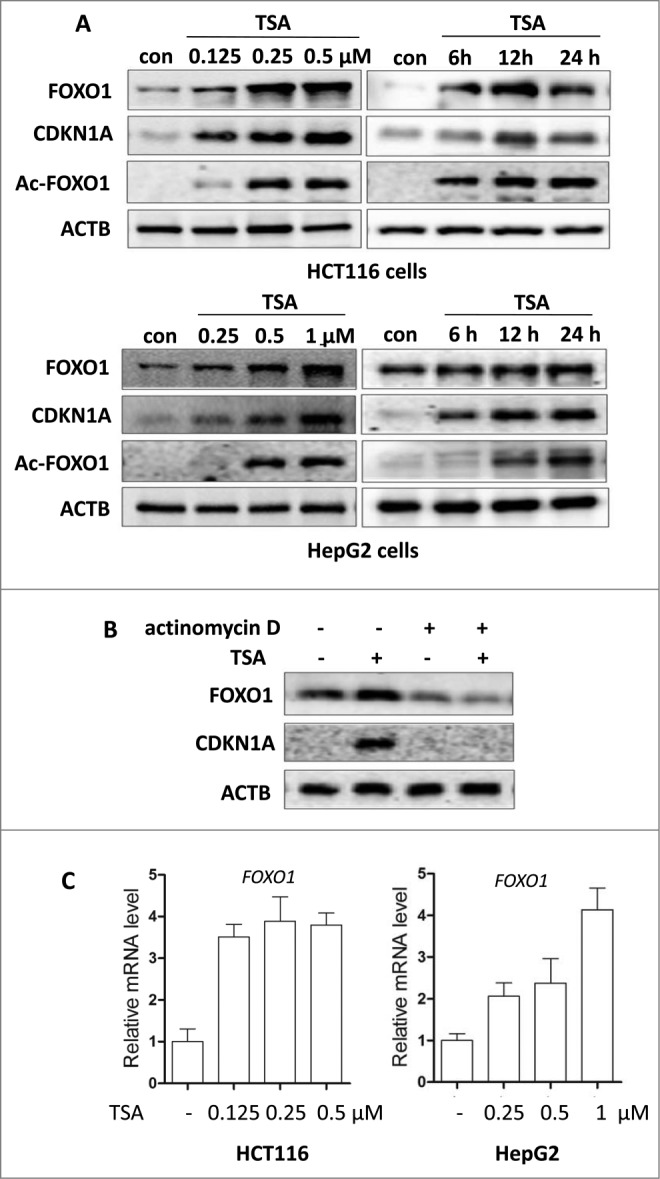
HDACIs increase FOXO1 expression at the mRNA and protein levels. (A) HCT116 cells were treated with TSA (0.125, 0.25 or 0.5 μM for 24 h; or 0.5 μM for 6, 12, or 24 h); HepG2 cells were treated with TSA (0.25, 0.5 or 1 μM for 24 h; or 1 μM for 6, 12, or 24 h). The cells were then harvested and subjected to western blotting analysis to evaluate FOXO1, CDKN1A and acetylated FOXO1 (Ac-FOXO1) expression. (B) HCT116 cells were treated with TSA (0.25 μM) in the absence or presence of actinomycin D (1 μg/ml) for 16 h. Cell lysates were analyzed for protein levels of FOXO1 and CDKN1A. (C) HCT116 cells were treated with TSA (0.125, 0.25 or 0.5 μM) for 24 h; HepG2 cells were treated with TSA (0.25, 0.5 or 1 μM) for 24 h. Total mRNA was extracted and real-time PCR was performed to evaluate changes in FOXO1 mRNA level. GAPDH was used as a loading control for real-time PCR.
Next, we investigated whether the increased FOXO1 protein level results from the transcriptional upregulation in HCT116 and HepG2 cells after TSA treatment. As shown in Figure 2B, actinomycin D abrogated the upregulation of FOXO1 protein level by TSA, demonstrating that this is a direct transcriptional response. We further performed real-time PCR to measure the mRNA level of FOXO1 and found that TSA treatment induced a 3- to 4-fold increase of FOXO1 mRNA level in HCT116 cells and HepG2 cells (Fig. 2C). In addition, we also determined the FOXO3 and FOXO4 expression and TSA also induced an increase in FOXO3 and FOXO4 expression at the mRNA and protein levels (Fig. S3).
TP53, another tumor suppressor, also plays an important role in cell cycle arrest, apoptosis, and genomic stability through the induction of target genes.36 Therefore, we wanted to know whether the increase of CDKN1A by TSA is caused by TP53. A pair of isogenic TP53+/+ and tp53−/− HCT116 cells were treated with TSA for 24 h. However, the TP53 status had no obvious impact on the expression of FOXO1 or CDKN1A caused by TSA, based on the fact that similar changes were found in both TP53+/+ and tp53−/− HCT116 cells (Fig. S4).
HDACIs increase the transcriptional activity of FOXO1
The PI3K-AKT pathway has been well established to regulate the phosphorylation of FOXO proteins.37 Once phosphorylated by AKT, FOXOs translocate into cytosolism from the nucleus, leading to transcriptional downregulation of their target genes.38 Here, we determined the translocation of FOXO1 after TSA treatment. TUBA4A (tubulin, α 4a) and LMNA (lamin A/C) proteins were detected as markers of cytosolic and nuclear fractions, respectively. Our results showed that most FOXO1 protein was in the nuclei and there was a time-dependent increase of nuclear FOXO1 in TSA-treated both HCT116 and HepG2 cells (Fig. 3A), indicating that the transcriptional activity of FOXO1 could be increased.
Figure 3.
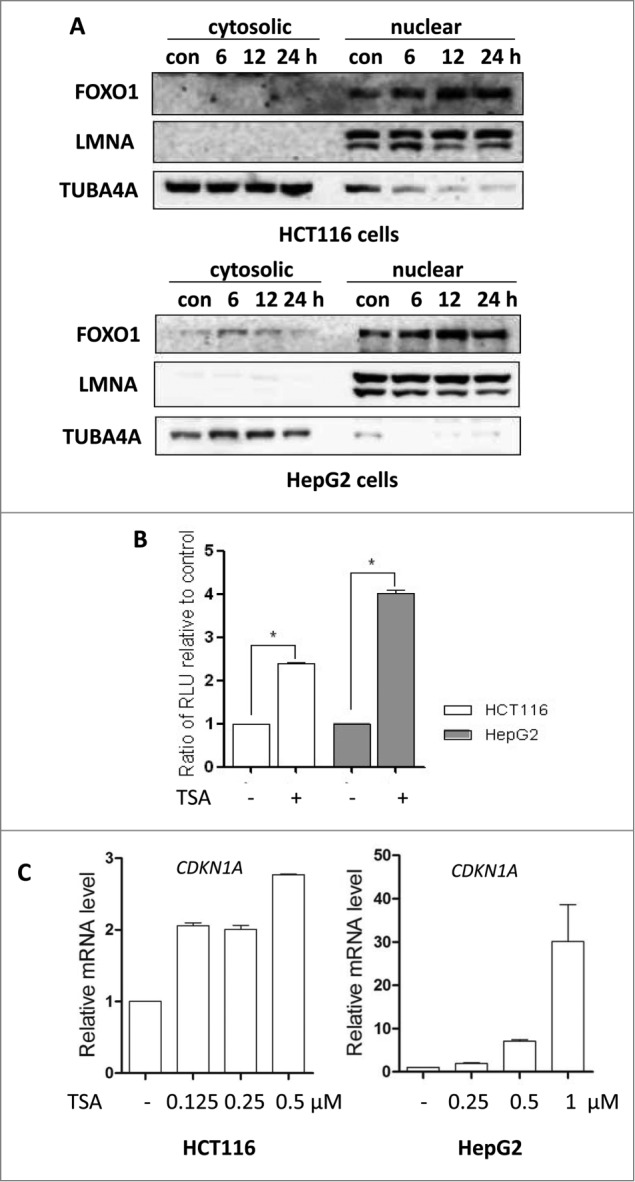
FOXO1 transcriptional activity is increased by HDACIs. (A) Effect of TSA on nuclear retention of FOXO1. HCT116 cells were treated with TSA (0.5 μM for 6, 12, or 24 h); HepG2 cells were treated with TSA (1 μM for 6, 12, or 24 h). To track the subcellular localization of FOXO1, nuclear and cytosolic proteins from control and TSA-treated cells were probed for FOXO1. The same membrane was then stripped and reprobed for TUBA4A or LMNA to ensure equal protein loading. (B) The luciferase reporter plasmid under the control of the FOXO1 promoter was transfected into HCT116 and HepG2 cells. After 24 h, the cells were treated with TSA (0.5 or 1 μM) for another 12 h and the relative luciferase activity was measured. RLU refers to relative luciferase units. Error bars represent the standard deviation. (C) HCT116 cells were treated with TSA (0.125, 0.25 or 0.5 μM) for 24 h; HepG2 cells were treated with TSA (0.25, 0.5 or 1 μM) for 24 h. Total mRNA was extracted and real-time PCR was performed to determine changes in CDKN1A mRNA. GAPDH was used as a loading control.
To further confirm the transcriptional activity of FOXO1, a FOXO1 promoter-driven luciferase reporter construct was transfected into both HepG2 and HCT116 cells, followed by TSA treatment for 12 h. As shown in Figure 3B, the relative luciferase activity of the FOXO1 was significantly increased by TSA. Nuclear retention of FOXO1 is expected to upregulate the transcription of FOXO1-responsive gene such as CDKN1A.39 Our results also clearly showed that TSA treatment substantially enhanced CDKN1A mRNA level in HepG2 and HCT116 cells (Fig. 3C), indicating that TSA is capable of activating the transcriptional activity of FOXO1.
Transcriptional activity of FOXO1 is implicated in HDACIs-induced autophagy
FOXO1 has recently been identified as a critical regulator of autophagy induced by H2O2 and serum starvation.31 It is also reported that the proautophagy function of acetylated FOXO1 is associated with its increased interaction with ATG7.31,33 We thus proceeded to test the possible involvements of FOXO1 in autophagy induced by TSA. As shown in Figure 4A, Foxo1 knockdown with siRNA led to a significant decrease of GFP-LC3 puncta in MEF cells treated with TSA. To further confirm the role of FOXO1 in HDACIs-mediated autophagy, we tested the effects of AS1842856, a known FOXO1 inhibitor that preferentially inhibits the transcription activity of FOXO1 through direct binding to the active FOXO1.40 As expected, inhibition of FOXO1 by AS1842856 markedly reduced autophagy level induced by TSA, demonstrated by reduced GFP-LC3 puncta (Fig. 4B).
Figure 4.

For figure legend, see page 634.Figure 4 (See previous page). Foxo1/FOXO1 knockdown or inhibition impairs HDACIs-induced autophagy. (A) Effect of Foxo1 knockdown on autophagy caused by TSA. MEFs with stable expression of GFP-LC3 were transiently transfected with the Foxo1-specific siRNA and then cells were subsequently treated with 1 μM TSA for 12 h. The cells were examined and representative cells were photographed using a confocal microscope (Scale bar: 10 μm). The number of GFP-LC3 puncta/cell was counted and presented (** P < 0.01). (B) MEFs with stable expression of GFP-LC3 were treated with 1 μM TSA in the presence or absence of FOXO1 inhibitor AS1842856 (100 nM) for 24 h. The cells were examined and representative cells were photographed using a confocal microscope (Scale bar: 10 μm). The number of GFP-LC3 puncta/cell was counted and presented (**P < 0.01). (C) HCT116 cells were transiently transfected with a nonspecific siRNA or the FOXO1-specific siRNA according to the manufacturer's protocol. HCT116 cells were subsequently treated with 0.5 μM TSA with or without BAF (15 nM) for 12 h. Cell lysates were lysed, collected, and immunoblotted for FOXO1, LC3, and SQSTM1 levels. (D) HCT116 cells were treated with 0.25 μM TSA in the presence or absence of FOXO1 inhibitor AS1842856 (100 nM) for 24 h. Protein from HCT116 cell lysates was then immunoblotted with FOXO1, LC3, and SQSTM1 antibodies. (E) and (F) Similarly as described earlier in (C) and (D), total RNA was isolated from treated HCT116 cells. The mRNA levels of ATGs were also determined by real-time PCR, including ATG4B, ATG12, PIK3C3, BECN1, and MAP1LC3B. Fold change in mRNA levels was calculated by normalizing to GAPDH. Meanwhile, the mRNA level of FOXO1 was measured by real-time PCR.
Similarly, knockdown of FOXO1 markedly reduced the autophagic flux level shown by changes of LC3-II and SQSTM1 protein level in both HCT116 and HepG2 cells treated with TSA (Fig. 4C and S5). In HCT116 cells, FOXO1 inhibition by AS1842856 decreased LC3-II and increased SQSTM1 protein level (Fig. 4D). All these results clearly suggest that FOXO1 plays a critical role in TSA-mediated autophagy. In addition, knockdown FOXO3 or FOXO4 also caused a decrease in LC3-II and an increase in SQSTM1 (Fig. S6), indicating a reduced autophagy level.
Based on an earlier report that many autophagy-related genes, such as Atg4b, Atg12, Pik3c3, Becn1, and Map1lc3b, are transcriptionally regulated by FOXO proteins and involved in autophagic protein degradation in atrophying muscle cells.41 In this part of our study, using real-time PCR, we examined whether these genes were also transcriptionally induced by activated FOXO1 in HCT116 cells after TSA treatment. Indeed, after knockdown or inhibition of FOXO1 by AS1842856 (Fig. 4E and F), these genes were downregulated in TSA-treated HCT116 cells, indicating that the activation of autophagy and induction of many ATGs are in fact due to FOXO1-dependent transcriptional activation.
In order to further confirm the transcriptional regulation of ATGs by FOXO1, we performed the CHIP assay to assess the binding of FOXO1 protein to the promoter of various ATGs. As shown in Figure S7, TSA treatment significantly increased FOXO1 binding to the promoter of these ATGs in HCT116 cells, indicating that FOXO1 transcriptionally upregulates expression of ATGs.
FOXO1 is required for MTOR suppression by HDACIs
It has been reported that HDACIs can downregulate AKT-MTOR signaling and induce autophagy.19 Here, we determined whether FOXO1 is involved in HDACIs-mediated MTOR suppression. We examined the effects of FOXO1 knockdown in HCT116 in response to TSA. As shown in Figure 5A, TSA markedly reduced phospho-RPS6KB, phospho-RPS6 and phospho-EIF4EBP1 in HCT116 cells. Similar results were also found in HepG2 cells (Fig. S8), indicating suppression of MTOR activity. Interestingly, FOXO1 knockdown partly reversed such a trend caused by TSA. Similar results were also found in cells treated with FOXO1 inhibitor AS1842856 (Fig. 5B). These data thus suggest that HDACIs-induced MTOR inhibition is partly FOXO1-dependent.
Figure 5.

For figure legend, see page 636. Figure 5 (See previous page). Role of FOXO1 in HDACIs-induced MTOR suppression. (A) FOXO1 knockdown increased MTOR activity. HCT116 were transiently transfected with a nonspecific siRNA or the FOXO1-specific siRNA followed by TSA treatment (0.5 μM) for 12 h. (B) HCT116 cells were treated with 0.25 μM TSA in the absence or presence of FOXO1 inhibitor AS1842856 (100 nM) for 24 h. Total protein was extracted and subjected to immunoblotting for FOXO1, phospho-RPS6KB (T389), RPS6KB, phospho-RPS6 (Ser235/236), RPS6, phospho-EIF4EBP1 (T37/46), and EIF4EBP1 antibodies. (C) Tsc2+/+ and tsc2−/− MEFs were treated with 1 μM TSA for 2 different time points (6 or 12 h). Cell lysates were lysed, collected, and immunoblotted for TSC2, FOXO1, Ac-FOXO1, phospho-RPS6 ribosomal protein (Ser235/236), RPS6, phospho-EIF4EBP1 (T37/46), EIF4EBP1, LC3, and SQSTM1 levels. (D) Tsc2+/+ and tsc2−/− MEFs were treated with 1 μM TSA for 12 h and total RNA was isolated from cells. The mRNA levels of Atgs were also determined by real-time PCR, including Atg4b, Atg12, Pik3c3, Becn1, and Map1lc3b. Fold change in mRNA levels was calculated by normalizing to Gapdh.
To further establish the role of FOXO1 in MTOR inhibition by TSA, we utilized the tsc2−/− MEFs in which the MTORC1 level is constitutively active.42 As shown in Figure 5C, higher basal FOXO1 protein level was found in tsc2−/− MEFs in comparison to the Tsc2+/+ MEFs, and TSA treatment further increased FOXO1 and Ac-FOXO1 expression. Notably, TSA failed to suppress the MTOR activity in the tsc2−/− MEFs (no further reduction of phospho-RPS6 and phospho-EIF4EBP1). Consequently, TSA failed to induce autophagy in the tsc2−/− MEFs, which was indicated by the lower LC3-II level and higher SQSTM1 level. Furthermore, TSA also failed to induce Atgs expression, such as Atg4b, Atg12, Pik3c3, Becn1, and Map1lc3b in tsc2−/− MEFs, while the increase of Foxo1 mRNA was not affected (Fig. 5D). Although TSA is able to increase Foxo1 mRNA, protein and acetylation level in tsc2−/− MEFs, the transcriptional activity of FOXO1 is not enhanced. These results thus suggest that modulation of FOXO1 transcriptional activity by TSA is dependent on MTOR suppression.
FOXO1 inhibits MTOR activity by elevating SESN3/Sesn3 expression
The Sestrin gene family is comprised of 3 members, SESN1, SESN2, and SESN3 (Sesn1, (Sesn2, Sesn3 in mouse nomenclature). FOXO1 has been reported to exclusively elevate the expression of SESN3/Sesn3 leading to inhibition of MTOR activity.43,44 Here, we determined the expression of SESN3/Sesn3 in different cells treated with TSA. As shown in Figure 6A, TSA markedly increased SESN3 mRNA level, which was significantly blocked by FOXO1 knockdown or inhibition. In MEFs, data from real-time PCR showed that activation of Foxo1 by TSA failed to upregulate Sesn3 in tsc2−/− MEFs (Fig. 6B), indicating the underlying reason for the unresponsiveness of tsc2−/− MEFs to TSA-induced MTOR inhibition. More importantly, SESN3 knockdown can partly reverse the reduction of phospho-RPS6 and phospho-EIF4EBP1 by TSA in HCT116 cells (Fig. 6D), indicating suppression of MTOR activity. The effectiveness of SESN3 knockdown was also confirmed by the reduced SESN3 mRNA level in HCT116 cells (Fig. 6C). Such observations therefore suggest the possibility that SESN3/Sesn3 is implicated in FOXO1-mediated MTOR suppression and induction of autophagy in TSA-treated cells of human and murine origin.
Figure 6.

FOXO1 inhibits MTOR activity by elevating SESN3/Sesn3 expression. (A) HCT116 cells were transiently transfected with the FOXO1-specific siRNA followed by TSA treatment (0.5 μM) for 12 h (left panel). HCT116 cells were treated with 0.25 μM TSA in the absence or presence of FOXO1 inhibitor AS1842856 (100 nM) for 24 h (right panel). (B) Tsc2+/+ and tsc2−/− MEFs were treated with 1 μM TSA for 12 h and total RNA was isolated from cells. Total RNA was isolated from HCT116 cells, Tsc2+/+ and tsc2−/− MEFs, and the SESN3/Sesn3 mRNA level was quantified using real-time PCR. The Foxo1 mRNA level was also measured in Tsc2+/+ and tsc2−/− MEFs. Fold change in mRNA levels was calculated by normalizing to respective Gapdh groups. (C) and (D) HCT116 cells were transiently transfected with a nonspecific siRNA or the SESN3-specific siRNA followed by TSA treatment (0.5 μM) for 12 h. Total protein was extracted and subjected to immunoblotting for FOXO1, phospho-RPS6 (Ser235/236), RPS6, phospho-EIF4EBP1 (T37/46), and EIF4EBP1 antibodies. Total RNA was also isolated from HCT116 cells and the mRNA level of SESN3 was quantified using real-time PCR. Fold change in mRNA levels was calculated by normalizing to GAPDH.
Suppression of autophagy sensitizes HDACI-induced cell death
In this part of our study, we sought to determine the functional role of autophagy in TSA-induced cell death. First, we used 2 pharmacological inhibitors of autophagy, chloroquine (CQ), an inhibitor of the lysosomal pH gradient, and BAF, an inhibitor of vacuolar type H+-ATPase. As shown in Figure 7A, the morphological changes showed that more cell death was observed when TSA was combined with CQ or BAF. Quantification of cell death using the propidium iodide (PI) exclusion test coupled with flow cytometry in both HCT116 and HepG2 cells showed a similar trend (Fig. 7B). Similar results were also found in HCT116 cells treated with SAHA in the presence of CQ or BAF (Fig. S9). Moreover, ATG7 knockdown also significantly enhanced HCT116 cell death by TSA (Fig. S10A and S10B). Such observations thus indicate that TSA-induced autophagy serves as a cell survival mechanism.
Figure 7.

For figure legend, see page 639. Figure 7 (See previous page). Effect of autophagy inhibition on HDACIs-induced cell death. HCT116 cells were treated with TSA (0.5 μM), with or without chloroquine (25 μM) or BAF (25 nM) for 24 h. The concentration of TSA used for HepG2 cells was 1 μM. (A) Morphological changes of HCT116 and HepG2 cells with respective treatments were examined and photographed with an inverted microscope (Scale bar: 200 μm). (B) Cell pellets were subsequently collected and cell death was quantified using propidium iodide (PI) live exclusion staining. Statistical significance (*P < 0.05) is indicated in the bar chart. (C) Cells were then harvested for protein analysis. Cell lysates were resolved in SDS-PAGE and probed with specific antibodies against CASP3 and PARP1.
Next, we examined the format of the cell death with detection of 2 apoptotic markers, namely, the cleavage of both CASP3 and PARP1 (poly [ADP-ribose] polymerase 1). As shown in Figure 7C and Figure S10C, there was evident cleavage of CASP3 and PARP1 after TSA treatment. In addition, in the presence of CQ or BAF, or after ATG7 knockdown, TSA treatment further enhanced CASP3 and PARP1 cleavage, respectively, strongly indicating that autophagy inhibition is able to sensitize TSA-mediated apoptotic cell death. Such observations are indeed consistent with some earlier reports on apoptosis caused by HDACIs.19
Finally, we knocked down FOXO1 in HCT116 cells and then treated the cells with TSA. As shown in Figure S11, after FOXO1 knockdown, the morphological changes showed less cell death by TSA and the cleavage of both CASP3 and PARP1 by TSA was also reduced, suggesting that inhibition of FOXO1 protects from HDACI-caused cell death, which is consistent with an earlier report.15 Therefore, we think that FOXO1 activated by HDACIs may induce proapoptotic genes, such as BCL2L11/BIM and PMAIP1/NOXA,17,18 and finally cause cell death.
Discussion
In this study, we reveal a novel molecular mechanism underlying HDACIs-induced autophagy: HDACIs activate FOXO1 and the transcriptional activation of FOXO1 then promotes autophagy by SESN3-mediated MTOR suppression of and upregulation of ATGs' expression (as illustrated in Fig. 8). Moreover, inhibition of autophagy markedly enhances HDACIs-mediated cell death, indicating that autophagy serves as a cell survival mechanism in HDACIs-treated cancer cells.
Figure 8.
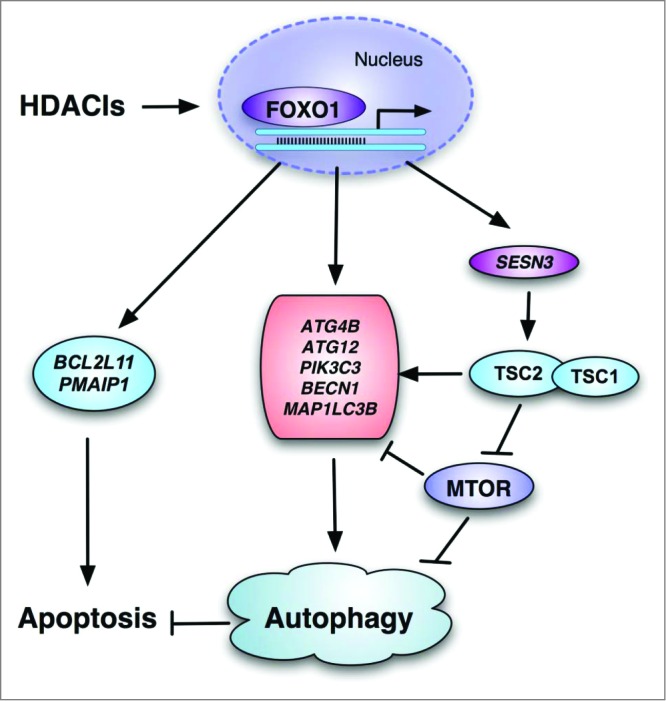
Illustration showing the mechanisms mediating the activation of FOXO1 in HDACI-induced apoptosis and autophagy involving the FOXO1-MTOR signaling and transcriptional regulation of autophagy genes.
Protein acetylation, an important form of post-transcriptional regulation mediated by both acetyltransferase and deacetylases, is involved in regulation of autophagy.45,46 Intriguingly, it is still controversial how protein acetylation modulates autophagy. On the one hand, deacetylation of essential autophagy proteins including ATG5, ATG7, ATG12, and LC3 is implicated in starvation-induced autophagy.45,47 Also, inhibition of acetyltransferases or activation of deacetylases stimulates autophagy.48,49 On the other hand, hyperacetylation of ATGs has been implicated in autophagy induction by starvation, including Atg3 in yeast50 and ULK1 in human cells.51 In this study, we provide clear evidence that suppression of deacetylases by HDACIs (TSA and SAHA) leads to enhanced FOXO1 acetylation and transcriptional activation, which is required for HDACI-induced autophagy. Our data are generally consistent with earlier reports that HDACIs are capable of inducing autophagy in human cancer cells19,20 and thus support the notion that suppression of HDACs or increase of protein acetylation plays a positive role in autophagy.
Our data provide clear evidence establishing the critical role of FOXO1 in HDACIs-induced autophagy. At present, FOXO1 activation has been identified as a mediator of autophagy induced by starvation, or some natural products, such as benzyl isothiocyanate and curcumin.31-33,52,53 A recent study from our own lab also implicates FOXO1 in autophagy following suppression of SCD (stearoyl-CoA desaturase [delta-9-desaturase]).54 It is still controversial whether HDACs activate or inhibit the functions of FOXO family proteins and it is thought that the exact effect might be context-dependent.55,56 For instance, a very recent study shows that HDAC1 activates FOXO proteins in skeletal muscle cells.57 In contrast, suppression of HDACs activates FOXO1 and promotes apoptosis in cancer cells.14 It is known that FOXO family members are subject to regulation by acetylation, in addition to phosphorylation.55,56 FOXOs are able to interact with some histone acetyltransferases, such as CREBBP (CREB binding protein) and EP300 (E1A binding protein p300), as well as histone deacetylases SIRT1 and SIRT2.31,54-59 However, it remains unclear how the acetylation status of FOXO1 determines its involvement in autophagy. An earlier study reported that deacetylation of FOXO1 by HDACs promotes starvation-mediated autophagy in cardiac myocytes.60 In contrast, it has been reported that acetylation of cytosolic FOXO1 by dissociation from SIRT2 plays an important role in autophagy mediated by oxidative stress and starvation, via enhanced interaction with ATG7.31 In our study, following suppression of HDACs by TSA, we observed enhanced FOXO1 acetylation in HCT116 and HepG2 cells (Fig. 2A) and Tsc2+/+ and tsc2−/− MEFs (Fig. 5C). But in tsc2−/− MEFs, FOXO1 transcriptional activity could not be enhanced (no upregulation of Atgs by TSA in Fig. 5D), suggesting that the transcriptional activity of FOXO1 is not associated with its acetylation level.
After confirming the role of FOXO1 in HDACIs-mediated autophagy, we next investigated the mechanisms underlying the regulatory function of FOXO1 in autophagy, and we demonstrated that the transcriptional activation of FOXO1 mediates autophagy via a dual mechanism: (i) transcriptional upregulation of ATGs/Atgs and (ii) suppression of MTOR via expression of SESN3/Sesn3. Our findings are generally in agreement with an earlier report that FOXO3 activation is able to stimulate the expression of many Atgs, leading to muscle wasting upon denervation and fasting.41 Interestingly, the suppressive effect of FOXO1 on MTOR activity has been reported via expression of SESN3/Sesn3.43 In our study, similar mechanisms were also found in HDACIs-treated cancer cells. Activation of FOXO1 by TSA upregulated Sesn3/SESN3 in Tsc2+/+ MEFs and HCT116 cells (Fig. 6A and 6B), whereas TSA treatment failed to induce Sesn3/SESN3 in tsc2−/− MEFs or HCT116 cells after FOXO1 knockdown or inhibition, which may be the reason that tsc2−/− MEFs had no response to TSA-induced MTOR inhibition. Moreover, SESN3 knockdown attenuated MTOR suppression and autophagy induction by TSA in HCT116 cells (Fig. 6D). As it is known, AKT is the principal negative regulator of FOXO1 via direct phosphorylation, followed by nuclear export and cytoplasmic sequestration and proteasomal degradation.30,58 In the present study, TSA treatment caused nuclear retention of FOXO1 and enhanced FOXO1 transcriptional activity (Fig. 3), which is possibly caused by inhibition of activation of AKT by TSA, as reported previously.19 In tsc2−/− MEFs, a higher basal level of FOXO1 was observed (Fig. 5C), which is likely due to the constitutive inactivation of AKT in tsc2−/− cells. It has been reported that the TSC1-TSC2 complex is capable of activating the PI3K-AKT signaling by suppressing MTORC1-RPS6KB activation, thereby preventing RPS6KB-mediated negative feedback on IRS1 and IRS2.61 Therefore, TSC2 deficiency leads to suppression of AKT and activation of FOXO1, as shown in our results.
At present, the role of autophagy in HDACIs-mediated cell death in cancer cells is still controversial. Some studies reported that autophagy serves as a cell death mechanism as either autophagy inhibitors or knockdown of ATGs was able to reduce the cytotoxicity of HDACIs in cancer cells.19,21,22 Our observations are generally consistent with several other studies in which autophagy serves as a prosurvival mechanism.20,23,24 However, FOXO1 knockdown protected against TSA-caused cell death (Fig. S11), which is consistent with the earlier report.14 The pro-apoptotic function of FOXO1 has been well established, through upregulation of its target genes BCL2L11/BIM or PMAIP1/NOXA.17,18 Therefore, we speculate that autophagy may delay the onset of apoptosis during HDACIs treatment through various mechanisms, including clearance of reactive oxygen species that are generated during HDACIs treatment,62 clearance of SQSTM1-containing protein aggregates, which may accumulate during HDAC inhibition, and clearance of damaged mitochondria.63 At present, several HDACIs have been already approved in clinical trials as cancer therapeutic agents.15 Moreover, in different cancer xenograft models, including malignant peripheral nerve sheath tumors,24 breast cancer,64 and colon cancer,23 the combined treatment with HDACIs and autophagy inhibition results in reduced tumor burden and increases the survival of cancer xenografts. These findings strongly support the notion of using a combination of HDACIs and autophagy inhibitors as a new therapeutic strategy to treat cancer patients.
Taken together, data from our study shed new lights into the intricate relationships among FOXO1, MTOR activity, autophagy, and cell death in cancer cells treated with HDACIs. More importantly, our results provide clues for developing more effective cancer therapeutic strategies by the combination of HDACIs and autophagy inhibitors.
Materials and Methods
Reagents and antibodies
The chemicals used in our experiments were: SAHA (Sigma, SML0061), trichostatin A (Sigma, T8552), bafilomycin A1/BAF (Sigma, B1793), chloroquine diphosphate (CQ, Sigma, C6628), FOXO1 inhibitor AS1842856 (Calbiochem, 344355), and actinomycin D (Sigma, A9415).
The antibodies used in our experiments included: FOXO1 (Cell Signaling Technology, 2880), FOXO3 (Cell Signaling Technology, 2497), FOXO4 (Cell Signaling Technology, 2499), CDKN1A/p21 (Cell Signaling Technology, 2947), acetylated-FOXO1 (Santa Cruz, sc-49437), microtubule-associated protein 1 light chain 3/LC3 (Sigma, L7543), SQSTM1 (Sigma, P0067), TSC2 (Cell Signaling Technology, 4308), phospho-RPS6KB (T389; Cell Signaling Technology, 9205), RPS6KB (Cell Signaling Technology, 2708), phospho-RPS6 (S235/236; Cell Signaling Technology, 2211), RPS6 (Cell Signaling Technology, 2217), phospho-EIF4EBP1 (T37/46) (Cell Signaling Technology, 2855), EIF4EBP1 (Cell Signaling Technology, 9452), PARP1 (Cell Signaling Technology, 9542), CASP3 (Cell Signaling Technology, 9662), ACTB (Sigma, A5441), TUBA4A (Sigma, T6199), LMNA/LAMIN A/C (Cell Signaling Technology, 2032).
Cell culture
An isogenic pair of HCT116 colon cancer cell lines (TP53+/+ and tp53−/−) was kindly provided by Dr. Bert Vogelstein (Johns Hopkins University). Tsc2+/+ and tsc2−/− MEFs were obtained from Dr. DJ Kwiatkowski (Harvard University; referred to as Tsc2+/+ and tsc2−/− MEFs hereafter).65 MEFs with stably expressing GFP-LC3 were all kindly provided by Dr. N Mizushima (Tokyo Medical and Dental University).66 HepG2 cells were obtained from American Type Culture Collection (ATCC-HB8065). All cell lines were maintained in DMEM (Sigma, D1152) containing 10% fetal bovine serum (HyClone, SV30160.03) in a 5% CO2 atmosphere at 37°C.
Immunofluorescence staining and confocal microscopy
GFP-LC3-expressing stable MEFs were seeded to a coverglass slide chamber (Lab-Tek, NUNC, 155411), and after the designated treatments, cells were examined and recorded using a confocal microscope (Olympus Fluoview FV1000, Olympus America Inc., PA) and representative cells were selected and photographed.
Luciferase assays
FOXO1 luciferase vector was provided by Prof. Zhu Weiguo (Beijing University, China).31 The transient transfection of FOXO1 luciferase vector was performed in HCT116 and HepG2 cells using Lipofectamine 2000 transfection reagent (Invitrogen, 11668) according to the manufacturer's protocols. After the designated treatments, the luciferase activity was measured 48 h after transfection using a Dual-Luciferase Reporter Assay System (Promega, E1960) based on the protocol provides by the manufacturer.
Cell fraction preparation
HCT116 and HepG2 cells were treated with TSA at different time points. After that, nuclear and cytosolic extracts were then prepared with NE-PER® nuclear and cytoplasmic extraction reagents (Pierce, 78833) according to the manufacturer's protocol.
Small Interfering RNA (siRNA) and transient transfection
The scrambled RNAi oligonucleotides and siRNAs targeting FOXO1 or FOXO3 (Cell Signaling Technology, 6242 or 6302) were transfected into HCT116 and HepG2 cells using the DharmaFECT 4 Transfection Reagent (Dharmacon, T-2001-02) according to the manufacturer's protocol. SMARTpool: ON-TARGETplus human FOXO4 siRNA (Thermo Scientific, L-003016-00-0005) and SMARTpool: ON-TARGETplus human SESN3 siRNA (Thermo Scientific, L-018289-01-0005) were used for FOXO4 and SESN3. After the designated treatments, cell lysates were prepared for western blotting.
Reverse transcription and quantitative real-time PCR
RNA was extracted with the RNeasy kit (Qiagen, 217004). A reverse transcription reaction was performed using 1 μg of total RNA with High Capacity cDNA Reverse Transcription kit (Applied Biosystems, 4368814). The mRNA expression levels were determined by real-time PCR using SsoFast™ EvaGreen® Supermix (Bio-Rad, 172-5201) and CFX96 Touch™ Real-Time PCR Detection System (Bio Rad). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control of RNA integrity. Real-time PCR was performed in triplicate. The primers used were based on previous reports,41,43 including human FOXO1, FOXO3, FOXO4, CDKN1A, ATG4B, ATG12, PIK3C3, BECN1, MAP1LC3B, SESN3, and GAPDH, and mouse Foxo1, Foxo3, Foxo4, Cdkn1a, Atg4b, Atg12, Pik3c3, Becn1, Map1lc3b, Sesn3, and Gapdh.
Western blotting
At the end of the designated treatments, cells were lysed in Laemmli SDS buffer (62.5 mM Tris at pH 6.8, 25% glycerol, 2% SDS, phosphatase inhibitor (Pierce, 78428) and proteinase inhibitor cocktail (Roche Applied Science, 11836153001). An equal amount of protein was resolved by SDS-PAGE and transferred onto PVDF membrane. After blocking with 5% nonfat milk, the membrane was probed with designated primary and secondary antibodies, developed with the enhanced chemiluminescence method, and visualized with the Kodak Image Station 4000R (Kodak, Rochester, NY).
Detection of viable and dead cells
Several methods were used to detect cell death quantitatively and qualitatively, which are (i) morphological changes under phase-contrast microscopy, (ii) propidium iodide (PtdIns) live cell uptake assay coupled with flow cytometry and (iii) western blotting for PARP and CASP3 cleavage. For PI staining, the medium in each well was collected and cells were harvested with trypsin after treatments. Then, cell pellets obtained were resuspended in 1x phosphate-buffered saline (1st Base, Singapore, BUF-2041) containing PtdIns at a final concentration of 5 μg/ml and incubated for 10 min at 37 37°C. Ten thousand cells from each sample were analyzed with FACSCalibur flow cytometry (BD Bioscience, San Jose, CA) using CellQuest software.
Chromatin immunoprecipitation assay
ChIP assay was performed in HCT116 cells using 2 μg of FOXO1 (Abcam, ab39670) antibody using the ChIP assay kit (Upstate-Millipore, 17-295). DNA was amplified with primers specific to human ATGs, including ATG4B, ATG12, PIK3C3, and BECN1. We designed these primers using online Primer3Plus as follows.
Human ATG4B
-
(1)
Forward: AGCTACTTGGGAGGCTGAAG
Reverse: ACCACCCTGTCCCTTATGTG
-
(2)
Forward: TGAGGTCAGGACTTTGAGGC
Reverse: CGCTCCTGGGTTCAAGCTAT
-
(3)
Forward: CATACCATTGCACTCCAGCC
Reverse: ACACAGAGTTTGCCTTAATGAAG
-
(4)
Forward: AGCTACTCTGGAGGCTGAGA
Reverse: GTATCTTCCTGCGCTGGTTG
-
(5)
Forward: GGCTTGAGGGTTCTTTGAGC
Reverse: CCCGCATTCTCCTGTCTCTA
-
(6)
Forward: TGAGAGGTCAGGACTGCATG
Reverse: CAGGCTCCCAAGTAACTGGA
Human ATG12
-
(1)
Forward: ACCACAGCTCCAATCAGTCA
Reverse: CTGCCCAGTGTGACTTTCAG
-
(2)
Forward: CCTCCTGCACTTGTCCATAC
Reverse: TGACTGATTGGAGCTGTGGT
-
(3)
Forward: CTGCACATGGTAGGCATAAATT
Reverse: TTGCTTCCTCCACTAGACCA
-
(4)
Forward: CAAAACTGCCACTTCCCACA
Reverse: GTGGGTTTCCTGGCTAATCC
-
(5)
Forward: GGCTGTGAACGAGAATACTGT
Reverse: TTCCTTTCCATTGCCACGTG
Human BECN1
-
(1)
Forward: TCCCTTGGCCTAGAATGCTC
Reverse: GGAGGCTGGGGTAGGAAAAT
-
(2)
Forward: GCAACAAAGTGAGACCCCAT
Reverse: TCAACCTCCAGTGTAGCAGG
-
(3)
Forward: GCAGATCAGGAGGTCAGGAG
Reverse: AGTCTTGCTCTGTTGCCCA
-
(4)
Forward: ATGATTCCAGTCACCTCCCC
Reverse: TTCCCACCTCCAGAAGTGTC
Human PIK3C3
-
(1)
Forward: AGGCCACTGGAATTTAAACCTG
Reverse: GAGACGCTATCATTTGAGCTTGA
-
(2)
Forward: GGAAGAGCCTACCACACCAA
Reverse: TGGTTGGTAGACTTGAGTATCAG
-
(3)
Forward: ACAGTACCACATGAGCCAGA
Reverse: AAAGCTCCCTAGTCTTGGGC
-
(4)
Forward: ACCAGTTCATGATGGCTCCA
Reverse: TGATTGCTGTCTGACCCACT
Statistical Analysis
All western blotting and image data presented are representatives from at least 3 independent experiments. The numeric data are presented as means ± SD from 3 independent experiments and analyzed using the Student t test.
Acknowledgments
We would like to thank Dr. Zhu Weiguo (Beijing University, China) for providing the FOXO1-promoter luciferase reporter construct, Dr. Noboru Mizushima (Tokyo Medical and Dental University, Japan) for the MEFs with stable expression of GFP-LC3, Dr. DJ Kwiatkowski (Harvard University, USA) for providing the pair of Tsc2+/+ and tsc2−/− MEFs and Dr. Reshma Taneja (National University of Singapore, Singapore) for kind technical support on CHIP assay.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study was supported in part by research grants from Singapore National Medical Research Council (NMRC/1260/2010 and NMRC/CIRG/1373/2013) to HMS. JZ, SHT, and WJ were supported by research scholarships from NUS.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.He C, Klionsky DJ.. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67–93; PMID:19653858; http://dx.doi.org/ 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y.. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10(7):458–67; PMID:19491929; http://dx.doi.org/ 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- 3.Levine B, Kroemer G.. Autophagy in the pathogenesis of disease. Cell 2008; 132(1):27–42; PMID:18191218; http://dx.doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B.. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12(9):823–30; PMID:20811354; http://dx.doi.org/ 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N. Autophagy: process and function. Genes Dev 2007; 21(22):2861–2873; PMID:18006683; http://dx.doi.org/ 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- 6.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al.. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20(7):1981–91; PMID:19211835; http://dx.doi.org/ 10.1091/mbc.E08-12-1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22(2):132–9; PMID:20056399; http://dx.doi.org/ 10.1016/j.ceb.2009.12.004 [DOI] [PubMed] [Google Scholar]
- 8.Lee DF, Hung MC.. All roads lead to mTOR: integrating inflammation and tumor angiogenesis. Cell Cycle 2007; 6(24):3011–4; PMID:18073529; http://dx.doi.org/ 10.4161/cc.6.24.5085 [DOI] [PubMed] [Google Scholar]
- 9.Zoncu R, Efeyan A, Sabatini DM.. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2010; 12(1):21–35; PMID:21157483; http://dx.doi.org/ 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jewell JL, Russell RC, Guan KL.. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 2013; 14(3):133–9; PMID:23361334; http://dx.doi.org/ 10.1038/nrm3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong PM, Puente C, Ganley IG, Jiang X.. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013; 9(2):124–37; PMID:23295650; http://dx.doi.org/ 10.4161/auto.23323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosato RR, Almenara JA, Dai Y, Grant S.. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol Cancer Ther 2003; 2(12):1273–84; PMID:14707268 [PubMed] [Google Scholar]
- 13.Emanuele S, Lauricella M, Tesoriere G.. Histone deacetylase inhibitors: apoptotic effects and clinical implications (Review). Int J Oncol 2008; 33(4):637–46; PMID:18813776 [PubMed] [Google Scholar]
- 14.Yang Y, Zhao Y, Liao W, Yang J, Wu L, Zheng Z, Yu Y, Zhou W, Li L, Feng J, et al.. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia 2009; 11(4):313–24; PMID:19308286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, et al.. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007; 109(1):31–9; PMID:16960145; http://dx.doi.org/ 10.1182/blood-2006-06-025999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HJ, Bae SC.. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res 2011; 3(2):166–79; PMID:21416059 [PMC free article] [PubMed] [Google Scholar]
- 17.Gillespie S, Borrow J, Zhang XD, Hersey P.. Bim plays a crucial role in synergistic induction of apoptosis by the histone deacetylase inhibitor SBHA and TRAIL in melanoma cells. Apoptosis 2006; 11(12):2251–65; PMID:17051334; http://dx.doi.org/ 10.1007/s10495-006-0283-6 [DOI] [PubMed] [Google Scholar]
- 18.Inoue S, Riley J, Gant TW, Dyer MJ, Cohen GM.. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia 2007; 21(8):1773–82; PMID:17525724; http://dx.doi.org/ 10.1038/sj.leu.2404760 [DOI] [PubMed] [Google Scholar]
- 19.Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC.. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010; 6(8):1057–65; PMID:20962572; http://dx.doi.org/ 10.4161/auto.6.8.13365 [DOI] [PubMed] [Google Scholar]
- 20.Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X.. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A 2012; 109(17):6561–5; PMID:22493;60; http://dx.doi.org/ 10.1073/pnas.1204429109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang PM, Chen CC.. Life or death? Autophagy in anticancer therapies with statins and histone deacetylase inhibitors. Autophagy 2010; 7(1):107–8; PMID:21057221; http://dx.doi.org/ 10.4161/auto.7.1.13988 [DOI] [PubMed] [Google Scholar]
- 22.Hrzenjak A, Kremser ML, Strohmeier B, Moinfar F, Zatloukal K, Denk H.. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J Pathol 2008; 216(4):495–04; PMID:18850582; http://dx.doi.org/ 10.1002/path.2434 [DOI] [PubMed] [Google Scholar]
- 23.Carew JS, Medina EC, Esquivel JA 2nd, Mahalingam D, Swords R, Kelly K, Zhang H, Huang P, Mita AC, Mita MM, et al.. Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J Cell Mol Med 2009; 14(10):2448–59; PMID:19583815; http://dx.doi.org/ 10.1111/j.1582-4934.2009.00832.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez G, Torres K, Liu J, Hernandez B, Young E, Belousov R, Bolshakov S, Lazar AJ, Slopis JM, McCutcheon IE, et al.. Autophagic survival in resistance to histone deacetylase inhibitors: novel strategies to treat malignant peripheral nerve sheath tumors. Cancer Res 2010; 71(1):185–96; PMID:21084276; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eijkelenboom A, Burgering BM.. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol 2013; 14(2):83–97; PMID:23325358; http://dx.doi.org/ 10.1038/nrm3507 [DOI] [PubMed] [Google Scholar]
- 26.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM.. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002; 419(6904):316–21; PMID:12239572; http://dx.doi.org/ 10.1038/nature01036 [DOI] [PubMed] [Google Scholar]
- 27.Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, et al.. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol 2000; 20(24):9138–48; PMID:11094066; http://dx.doi.org/ 10.1128/MCB.20.24.9138-9148.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ.. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 2000; 10(19):1201–4; PMID:11050388; http://dx.doi.org/ 10.1016/S0960-9822(00)00728-4 [DOI] [PubMed] [Google Scholar]
- 29.Huang H, Tindall DJ.. Dynamic FoxO transcription factors. J Cell Sci 2007; 120(Pt 15):2479–87; PMID:17646672; http://dx.doi.org/ 10.1242/jcs.001222 [DOI] [PubMed] [Google Scholar]
- 30.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME.. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96(6):857–68; PMID:10102273; http://dx.doi.org/ 10.1016/S0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG.. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nature cell biology 2010; 12(7):665–75; PMID:20543840; http://dx.doi.org/ 10.1038/ncb2069 [DOI] [PubMed] [Google Scholar]
- 32.Xiao D, Bommareddy A, Kim SH, Sehrawat A, Hahm ER, Singh SV.. Benzyl isothiocyanate causes FoxO1-mediated autophagic death in human breast cancer cells. PLoS One 2012; 7(3): e32597; PMID:22457718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L, Pan Y, Li XJ.. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012; 8(5):812–25; PMID:22622204; http://dx.doi.org/ 10.4161/auto.19471 [DOI] [PubMed] [Google Scholar]
- 34.Michaelis M, Suhan T, Michaelis UR, Beek K, Rothweiler F, Tausch L, Werz O, Eikel D, Zörnig M, Nau H, et al.. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ 2006; 13(3):446–53; PMID:16167071; http://dx.doi.org/ 10.1038/sj.cdd.4401759 [DOI] [PubMed] [Google Scholar]
- 35.Miyashita T, Reed JC.. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995; 80(2):293–9; PMID:7834749; http://dx.doi.org/ 10.1016/0092-8674(95)90412-3 [DOI] [PubMed] [Google Scholar]
- 36.Tokino T, Nakamura Y.. The role of p53-target genes in human cancer. Crit Rev Oncol Hematol 2000; 33(1):1–6; PMID:10714958; http://dx.doi.org/ 10.1016/S1040-8428(99)00051-7 [DOI] [PubMed] [Google Scholar]
- 37.Shankar S, Chen Q, Srivastava RK.. Inhibition of PI3K/AKT and MEK/ERK pathways act synergistically to enhance antiangiogenic effects of EGCG through activation of FOXO transcription factor. J Mol Signal 2008; 3:7; PMID:18355401; http://dx.doi.org/ 10.1186/1750-2187-3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greer EL, Brunet A.. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005; 24(50):7410–25; PMID:16288288; http://dx.doi.org/ 10.1038/sj.onc.1209086 [DOI] [PubMed] [Google Scholar]
- 39.Reagan-Shaw S, Ahmad N.. The role of Forkhead-box Class O (FoxO) transcription factors in cancer: a target for the management of cancer. Toxicol Appl Pharmacol 2007; 224(3):360–8; PMID:17275054; http://dx.doi.org/ 10.1016/j.taap.2006.12.003 [DOI] [PubMed] [Google Scholar]
- 40.Nagashima T, Shigematsu N, Maruki R, Urano Y, Tanaka H, Shimaya A, Shimokawa T, Shibasaki M.. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic db/db mice. Mol Pharmacol 2010; 78(5):961–70; PMID:20736318; http://dx.doi.org/ 10.1124/mol.110.065714 [DOI] [PubMed] [Google Scholar]
- 41.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL.. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6(6):472–83; PMID:18054316; http://dx.doi.org/ 10.1016/j.cmet.2007.11.004 [DOI] [PubMed] [Google Scholar]
- 42.Kwiatkowski DJ. Rhebbing up mTOR: new insights on TSC1 and TSC2, and the pathogenesis of tuberous sclerosis. Cancer Biol Ther 2003; 2(5):471–6; PMID:14614311; http://dx.doi.org/ 10.4161/cbt.2.5.446 [DOI] [PubMed] [Google Scholar]
- 43.Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, Park Y, Hay N.. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell 2010; 18(4):592–604; PMID:20412774; http://dx.doi.org/ 10.1016/j.devcel.2010.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N.. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008; 14(6):458–70; PMID:19061837; http://dx.doi.org/ 10.1016/j.ccr.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banreti A, Sass M, Graba Y.. The emerging role of acetylation in the regulation of autophagy. Autophagy 2013; 9(6):819–29; PMID:23466676; http://dx.doi.org/ 10.4161/auto.23908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, et al.. Regulation of autophagy by cytosolic acetyl-coenzyme A. Molecular cell 2014; 53(5):710–25; PMID:24560926; http://dx.doi.org/ 10.1016/j.molcel.2014.01.016 [DOI] [PubMed] [Google Scholar]
- 47.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T.. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 2008; 105(9):3374–9; PMID:18296641; http://dx.doi.org/ 10.1073/pnas.0712145105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al.. Induction of autophagy by spermidine promotes longevity. Nature cell biology 2009; 11(11):1305–14; PMID:19801973; http://dx.doi.org/ 10.1038/ncb1975 [DOI] [PubMed] [Google Scholar]
- 49.Morselli E, Marino G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Bénit P, Rustin P, Criollo A, et al.. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol 2011; 192(4):615–29; PMID:21339330; http://dx.doi.org/ 10.1083/jcb.201008167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, Zhu L, et al.. Function and molecular mechanism of acetylation in autophagy regulation. Science 2012; 336(6080):474–7; PMID:22539722; http://dx.doi.org/ 10.1126/science.1216990 [DOI] [PubMed] [Google Scholar]
- 51.Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Liu Q, Ruan K, et al.. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 2012; 336(6080):477–81; PMID:22539723; http://dx.doi.org/ 10.1126/science.1217032 [DOI] [PubMed] [Google Scholar]
- 52.Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou J, Fu W, Wei FZ, Wang L, Xie D, et al.. XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell research 2013; 23(4):491–07; PMID:23277279; http://dx.doi.org/ 10.1038/cr.2013.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou J, Liao W, Yang J, Ma K, Li X, Wang Y, Wang D, Wang L, Zhang Y, Yin Y, et al.. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 2012; 8(12):1712–23; PMID:22931788; http://dx.doi.org/ 10.4161/auto.21830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan SH, Shui G, Zhou J, Li JJ, Bay BH, Wenk MR, Shen HM.. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J Biol Chem 2012; 287(18):14364–76; PMID:22408252; http://dx.doi.org/ 10.1074/jbc.M111.294157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al.. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004; 303(5666):2011–15; PMID:14976264; http://dx.doi.org/ 10.1126/science.1094637 [DOI] [PubMed] [Google Scholar]
- 56.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L.. Mammalian SIRT1 represses forkhead transcription factors. Cell 2004; 116(4):551–63; PMID:14980222; http://dx.doi.org/ 10.1016/S0092-8674(04)00126-6 [DOI] [PubMed] [Google Scholar]
- 57.Beharry AW, Sandesara PB, Roberts BM, Ferreira LF, Senf SM, Judge AR.. HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J Cell Sci, 127(Pt 7):1441–53; PMID:24463822; http://dx.doi.org/ 10.1242/jcs.136390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dansen TB, Burgering BM.. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol 2008; 18(9):421–9; PMID:18715783; http://dx.doi.org/ 10.1016/j.tcb.2008.07.004 [DOI] [PubMed] [Google Scholar]
- 59.Nasrin N, Ogg S, Cahill CM, Biggs W, Nui S, Dore J, Calvo D, Shi Y, Ruvkun G, Alexander-Bridges MC.. DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor binding protein 1 promoter in HepG2 cells. Proc Natl Acad Sci U S A 2000; 97(19):10412–7; PMID:10973497; http://dx.doi.org/ 10.1073/pnas.190326997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J.. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res 2010; 107(12):1470–82; PMID:20947830; http://dx.doi.org/ 10.1161/CIRCRESAHA.110.227371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al.. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004; 166(2):213–23; PMID:15249583; http://dx.doi.org/ 10.1083/jcb.200403069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA.. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci U S A 2005; 102(3):673–8; PMID:15637150; http://dx.doi.org/ 10.1073/pnas.0408732102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, Scott CL, Strasser A, Atadja P, Lowe SW, et al.. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009; 114(2):380–93; PMID:19383971; http://dx.doi.org/ 10.1182/blood-2008-10-182758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rao R, Balusu R, Fiskus W, Mudunuru U, Venkannagari S, Chauhan L, Smith JE, Hembruff SL, Ha K, Atadja P, et al.. Combination of pan-histone deacetylase inhibitor and autophagy inhibitor exerts superior efficacy against triple-negative human breast cancer cells. Mol Cancer Ther 2012; 11(4):973–83; PMID:22367781; http://dx.doi.org/ 10.1158/1535-7163.MCT-11-0979 [DOI] [PubMed] [Google Scholar]
- 65.Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Carpenter CL, Kwiatkowski DJ.. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest 2003; 112(8):1223–33; PMID:14561707; http://dx.doi.org/ 10.1172/JCI200317222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N.. The role of autophagy during the early neonatal starvation period. Nature 2004; 432(7020):1032–6; PMID:15525940; http://dx.doi.org/ 10.1038/nature03029 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.