

Summary
Proteins begin to fold as they emerge from translating ribosomes. The kinetics of ribosome transit along a given mRNA can influence nascent chain folding, but the extent to which individual codon translation rates impact proteome integrity remains unknown. Here, we show that slower decoding of discrete codons elicits widespread protein aggregation in vivo. Using ribosome profiling, we find that loss of anticodon wobble uridine (U34) modifications in a subset of tRNAs leads to ribosome pausing at their cognate codons in S. cerevisiae and C. elegans. Cells lacking U34 modifications exhibit gene expression hallmarks of proteotoxic stress, accumulate aggregates of endogenous proteins, and are severely compromised in clearing stress-induced protein aggregates. Overexpression of hypomodified tRNAs alleviates ribosome pausing, concomitantly restoring protein homeostasis. Our findings demonstrate that modified U34 is an evolutionarily conserved accelerator of decoding and reveal an unanticipated role for tRNA modifications in maintaining proteome integrity.
Graphical Abstract
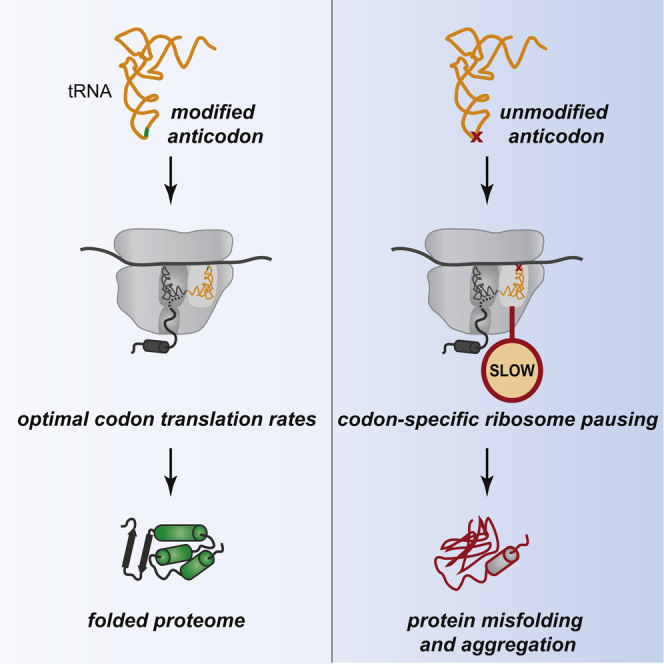
Highlights
-
•
tRNA anticodon modification loss slows translation at cognate codons in vivo
-
•
Codon-specific translational pausing triggers protein misfolding in yeast and worms
-
•
Codon translation rates and protein homeostasis are restored by tRNA overexpression
Optimal codon translation rates—ensured by the presence of nucleoside modifications in the tRNA anticodon—are critical for maintaining proteome integrity.
Introduction
The rate of peptide synthesis by ribosomes is not uniform along mRNAs and is often punctuated by transient pauses. This can impact protein folding, as proteins begin to attain their active conformations in concert with their synthesis (Pechmann et al., 2013). In particular, the differential use of synonymous codons may define local elongation speed and, in turn, aid cotranslational protein folding (Komar, 2009; Pechmann and Frydman, 2013; Thanaraj and Argos, 1996). Though synonymous substitutions in individual genes can indeed have detrimental effects on protein function (Kimchi-Sarfaty et al., 2007; Xu et al., 2013; Zhou et al., 2013), the factors that control translation rates at individual codons in vivo are poorly understood (Tarrant and von der Haar, 2014), and evidence linking global changes in these rates to protein misfolding in living cells is so far lacking.
A key factor modulating codon translation rates is tRNA selection. During each round of peptide elongation, the ribosome decodes the mRNA codon in its acceptor (A) site by selecting a matching (cognate) aminoacyl-tRNA from a large pool of tRNA molecules. The abundance of the cognate aminoacyl-tRNA and competing near-cognate ones, the cellular demand for a tRNA species, and the nature of codon-anticodon base pairing all impact the speed of decoding (Fluitt et al., 2007; Pechmann and Frydman, 2013; Stadler and Fire, 2011; Varenne et al., 1984). Within the ribosomal A site, cognate codon-anticodon interactions are identified based on Watson-Crick base pairing. The third codon position can form non-standard (wobble) pairs, allowing some tRNAs to be used in decoding multiple codons. In all kingdoms of life, the tRNA wobble nucleoside (position 34) frequently carries chemical modifications that enhance codon binding in vitro (Agris et al., 2007). The most striking example of this is the wobble uridine (U34), which is almost always modified (Grosjean et al., 2010), and the genes required for this in eukaryotes are conserved from yeast to mammals (Glatt and Müller, 2013; Shigi, 2014).
Loss of U34 modifications decreases fitness through poorly defined mechanisms. Although deletion of genes dedicated to modifying U34 does not reduce tRNA stability or aminoacylation (Johansson et al., 2008), it is embryonic lethal in flies and mice (Chen et al., 2009b; Walker et al., 2011), decreases resistance to challenging environments in yeast (Dewez et al., 2008; Huang et al., 2005; Leidel et al., 2009), and causes developmental and neuronal dysfunction in nematodes (Chen et al., 2009a). In humans, genetic variants and loss-of-function mutations within the orthologous genes are associated with neurodegeneration and intellectual disabilities (reviewed in Torres et al., 2014).
A number of observations suggest that cellular dysfunction upon loss of U34 modifications may be tied to inefficient translation via a subset of tRNAs with modified U34. In eukaryotes, the U34 base of 11 cytoplasmic tRNAs carries a 5-methoxycarbonylmethyl (mcm5) or 5-carbamoylmethyl (ncm5). The addition of these moieties requires the six-subunit Elongator (Elp) complex (Huang et al., 2005; Johansson et al., 2008) (Figure 1A). Following mcm5U34 addition in three species—tEUUC, tKUUU, and tQUUG—the base is further decorated with a 2-thio group (s2) via a sulfur-relay pathway that requires Uba4, Urm1, Ncs2, and Ncs6 (Leidel et al., 2009; Nakai et al., 2008; Schlieker et al., 2008) (Figure 1A). Thiolation of U34 in tEUUC, tKUUU, and tQUUG is universally conserved, forming a part of the core translational apparatus in extant cells (Grosjean et al., 2014). Several studies have hinted at a disproportionate functional importance of U34 modifications in these three isoacceptors. The stress-induced growth defects of yeast lacking Elp subunits are nearly identical to those observed upon loss of U34 2-thiolation (Nakai et al., 2008) and can be rescued by increasing cellular levels only of species that normally carry mcm5s2U34: tEUUC, tKUUU, and tQUUG (Bauer et al., 2012; Björk et al., 2007; Dewez et al., 2008; Esberg et al., 2006; Leidel et al., 2009). Long repeats of the codons pairing with these three tRNAs (GAA, AAA, and CAA) decrease protein output from highly expressed reporter genes in urm1Δ yeast (Rezgui et al., 2013), and ribosomes accumulate at these codons in ncs6Δ and uba4Δ yeast grown under nutrient-replete conditions (Zinshteyn and Gilbert, 2013). The physiological relevance of this codon-specific ribosome pausing is unclear, given that the mutants show little growth impairment under such conditions (Huang et al., 2005; Leidel et al., 2009). In contrast, their growth is severely impaired by environmental stress, such as exposure to the sulfhydryl-oxidizing agent diamide, or TOR pathway inhibition by rapamycin (Björk et al., 2007; Leidel et al., 2009). A prominent hypothesis of why U34 modifications become more critical during stress suggests that codons read via U34-modified tRNAs may be more frequent than average in stress response transcripts, decreasing their translation upon modification loss (Begley et al., 2007). However, this hypothesis has not been directly tested, and whether the absence of U34 modifications has any distinct consequences for translation during stress remains unknown.
Figure 1.

Loss of U34 Modifications Leads to Codon-Specific Ribosome Pausing in Yeast and Nematodes
(A) Pathways for wobble uridine (U34) modification in the eukaryotic cytoplasm.
(B) Codon-specific changes in A-site ribosome occupancy in S. cerevisiae strains with aberrant U34 modification in comparison to WT (mean ± SD; n = 3). Codons cognate for tRNAs with mcm5s2U34 (yellow), mcm5U34 (blue), and ncm5U34 (cyan) are boxed. Symbol size reflects the relative frequency of each codon within the A site in WT (small, <0.005; medium, 0.005–0.015; large, >0.015).
(C) Codon-specific changes in A-site ribosome occupancy upon U34 thiolation loss in C. elegans compared to WT (n = 2). Codons cognate for tRNAs with mcm5s2U34 are boxed in yellow; symbol size as in (B).
See also Figures S1 and S2.
To define how U34 modifications maximize cellular fitness, we explored the consequences of their absence for codon translation dynamics in Saccharomyces cerevisiae and Caenorhabditis elegans. Using ribosome profiling (Ingolia et al., 2009), we found that tRNAs with mcm5s2U34 rely on modified U34 for efficient decoding of their cognate codons in vivo. Surprisingly, the codon-specific ribosome pausing we detected upon loss of U34 modifications in yeast was not augmented by environmental stresses. Instead, we discovered that slower decoding via hypomodified tRNAs elicits the aggregation of many essential proteins and profoundly impairs the ability of cells to balance protein homeostasis during stress. Our findings rationalize how loss of U34 modifications triggers cellular dysfunction and reveal a hitherto unappreciated role of codon translation rates for proteome integrity.
Results
U34 Modifications Increase Translation Rates at a Subset of Cognate Codons in Yeast
To establish the impact of U34 modifications on codon translation rates in vivo, we used ribosome profiling, which comprises deep sequencing of ∼30 nt mRNA fragments protected from nuclease digestion by bound ribosomes (Ingolia et al., 2009). This method provides a quantitative snapshot of ribosome occupancy along endogenous transcripts and allows the identification of codons in the ribosomal exit (E), peptidyl (P), and A sites (Ingolia et al., 2009; Stadler and Fire, 2011) (Figures S1A and S1B). As ribosome pausing typically increases the likelihood of capturing a footprint by sequencing, variations in codon-specific ribosome occupancy can be used to infer changes in translation rates (Ingolia et al., 2011; Li et al., 2012; Stadler and Fire, 2011). To stabilize translating ribosomes in a stage of the elongation cycle prior to peptide bond formation (Lareau et al., 2014) and to prevent runoff elongation during cell harvesting and ribosomal footprint isolation, cells were briefly treated with cycloheximide.
Figure S1.

Effects of U34 Modification Loss on Ribosome Occupancy at Codons Flanking the A Site In Vivo, Related to Figure 1
(A) Cumulative coverage of 5′ nucleotides from ribosome footprint reads mapping near start codons across all transcripts in S. cerevisiae (left) and C. elegans (right). Reads of length between 29 and 31 nucleotides (nt) mapped without mismatches are shown. The peak located 12-13 nt upstream of start sites is inferred to represent ribosomes poised for translation initiation that contain the AUG codon in their P-site.
(B) Approach for determining codon representation in tRNA-binding sites within ribosome footprints inferred from (A).
(C) Codon occupancy within P, E, and +1 sites in yeast with U34 modification defects compared to WT (mean ± SD; n = 3).
(D) Codon-specific changes in A-site ribosome occupancy in ncs2Δ cells compared to WT when cycloheximide (CHX) was omitted from all steps of the ribosome profiling protocol (mean ± SD; n = 3). Data from CHX-treated ncs2Δ cells is from Figure 1B.
(E) Cumulative distribution of A-site ribosome occupancy at individual AAA, CAA, GAA, and GCU codons in WT and ncs2Δelp6Δ yeast. To calculate single-codon occupancy, data from three biological replicates were pooled, and the number of A-site reads at a particular codon was normalized to the average per-codon A-site read density in the ORF containing it (p values are from a one-sample Kolmogorov–Smirnov test).
(F) Codon-specific changes in A-site ribosome occupancy in S. cerevisiae strains lacking U34 modifications and ribosome rescue factors (mean ± SD; n = 3). Note that the elevated occupancy at codons such as CCG and CGC in dom34Δncs2Δelp6Δ and hbs1Δncs2Δelp6Δ cells is likely caused by additive effects, as occupancy at these codons is increased in hbs1Δ yeast.
(G) Exponentially growing cultures from the indicated strains were serially diluted and spotted on medium without additives (YPD) or containing 1.2 mM diamide or 1.9 nM rapamycin. Plates were imaged after 2 days of incubation at 30°C.
(H) Cultures from the indicated strains carrying an empty vector or overexpressing isoacceptors for E, K, and Q with U34 (tEUUC, tKUUU, and tQUUG) or C34 (tECUC, tKCUU, and tQCUG) were grown to exponential phase, serially diluted, and spotted on the indicated plates. Images were taken after 2 days of incubation at 30°C.
We examined the contributions of mcm5/ncm5 and s2 moieties at U34 to translation by comparing codon-level ribosome occupancy in wild-type (WT) yeast to that of mutants with various U34 modification defects. Complete loss of U34 modifications is lethal in the W303 strain (Björk et al., 2007), but we found that simultaneous knockout of Urm1 pathway components and Elp complex genes was compatible with cell viability in the S288c background. Thus, we performed ribosome profiling on ncs2Δelp6Δ yeast (lacking all U34 modifications), as well as ncs2Δ (lacking 2-thiolation), and elp6Δ (lacking mcm5/ncm5 modifications and with lower 2-thiolation levels).
Our analysis revealed strikingly distinct effects of U34 modification loss on codon occupancy. CAA and AAA triplets, read by the mcm5s2U34-containing tKUUU and tQUUG, were reproducibly enriched (20%–36%) within the ribosomal A site in all mutants (Figures 1B and S1C), suggesting they are translated more slowly when U34 is partially or fully unmodified. The increase in ribosome occupancy at these codons was larger and markedly more robust than previously reported for U34 thiolation-deficient strains (Zinshteyn and Gilbert, 2013). Occupancy at CAA and AAA was also consistently elevated when ribosome profiling was carried out in the absence of cycloheximide (Figure S1D), although the magnitude of the effect was smaller, likely due to translational runoff during sample preparation. There was little effect on ribosome occupancy at most other codons cognate for hypomodified tRNAs, including GAA, which is also decoded by a tRNA with mcm5s2U34 (Figure 1B). Curiously, a small but reproducible decrease in A-site ribosome occupancy was evident at AGA and GGA in elp6Δ and ncs2Δelp6Δ cells (Figure 1B), indicating that these codons are translated faster when their cognate tRNAs (tRUCU and tGUCC) lack mcm5 at U34. Isoacceptors with U34 are unlikely to contribute to decoding via U:G wobble in yeast, evidenced by the minor effects of loss of U34 modifications on ribosome occupancy at G-ending codons (Figure 1B).
Next, we asked whether U34 modifications mediate decoding specificity, reasoning that if aberrant U34 modification relaxes the specificity of codon binding, hypomodified tRNAs could compete for binding at near-cognate codons with mismatches at the third position, eliciting translational pausing. Such events could decrease decoding accuracy in split codon boxes, where two different amino acids are specified by a purine or a pyrimidine at the third position. Ribosome occupancy of AAC/U, CAC/U, or GAC/U codons was not altered in ncs2Δ yeast (Figure 1B), suggesting that U34 2-thiolation does not restrict the decoding specificity of tEUUC, tKUUU, and tQUUG. In contrast, the mcm5 group in U34 of tQUUG may be a prerequisite for accurate decoding in the Glu/His split codon box, as the A-site occupancy of CAC and CAU codons was reproducibly increased in elp6Δ and ncs2Δelp6Δ cells (Figure 1B). Ribosome occupancy of AAC/U, GAC/U, and AGC/U codons was largely unaffected in elp6Δ and ncs2Δelp6Δ cells, suggesting that a decrease in decoding accuracy is not a universal consequence of ablating mcm5U34.
To assess if the global increase in ribosome occupancy at CAA and AAA in U34 modification-deficient yeast stems from slowed elongation at many instances of these codons, we compared the A-site ribosome occupancy at individual codons in wild-type and ncs2Δelp6Δ yeast. We found that a greater proportion of CAA and AAA codons had high A-site occupancy in ncs2Δelp6Δ cells (Figure S1E), indicative of widespread translational slowdown. By contrast, single-codon A-site occupancy at GAA and GCU was largely comparable in both strains, consistent with our global codon occupancy measurements (Figure 1B). We also asked whether codon occupancy in cells with unmodified U34 is modulated by the absence of known ribosome rescue factors. The Dom34-Hbs1 complex facilitates the dissociation of stalled ribosomes (Shoemaker and Green, 2012), and GTPBP2, a binding partner of the Dom34 homolog Pelota, was recently shown to resolve codon-specific translational stalling caused by depletion of a specific tRNA in the mouse brain (Ishimura et al., 2014). However, deletion of DOM34 or HBS1 in ncs2Δelp6Δ yeast did not appreciably augment ribosome occupancy at CAA and AAA (Figure S1F) and had minimal phenotypic consequences (Figure S1G). Taken together, these findings argue against stalled ribosomes contributing significantly to the phenotypes of U34 modification mutants and suggest that increased codon occupancy in the mutant strains results from ribosome pausing, rather than from stalling.
Overall, our data reveal a surprising heterogeneity in the outcomes of U34 hypomodifications for codon translation speed in yeast: while mcm5s2U34 in tKUUU and tQUUG is required for accurate and efficient translation elongation, U34 modifications play only minor functional roles in most other cytoplasmic tRNAs. Our observations are consistent with the phenotypic rescue of U34 modification defects by elevated levels of tKUUU and tQUUG, with a minor contribution of tEUUC (Björk et al., 2007; Esberg et al., 2006; Leidel et al., 2009) (Figure S1H).
Codon Translation Rates Are Augmented by U34 Modifications in C. elegans
To determine whether U34 modifications modulate codon translation rates in a multicellular organism, we compared the codon-specific ribosome occupancies of wild-type C. elegans (N2) and tut-1(tm1297), a U34 2-thiolation-deficient strain (Chen et al., 2009a; Dewez et al., 2008; Leidel et al., 2009). The mutant displayed a robust 1.5- to 2.5-fold increase in A-site ribosome occupancy at AAA, CAA, and GAA (Figures 1C and S2). No enrichment of near-cognate codons was detectable in the A site within ribosome footprints from tut-1(tm1297) worms, similarly to ncs2Δ yeast (Figure 1B). Therefore, the function of U34 2-thiolation in promoting efficient translation of cognate codons is conserved from yeast to metazoans.
Figure S2.

Codon Occupancy within P, E, and +1 Sites in U34 Thiolation-Deficient tut-1(tm1297) Nematodes Compared to WT, n = 2, Related to Figure 1
Symbol size as in Figure 1C.
U34 Modifications Are Dispensable for Stress Responses in Yeast
To test whether codon-specific ribosome pausing increases during stress, we exposed wild-type and ncs2Δ yeast to diamide or rapamycin and subjected them to ribosome profiling, while monitoring changes in mRNA abundance (Figure 2A). Treatment with either drug induced a dramatic remodeling of gene expression in comparison to normal growth. Hundreds of genes displayed significant changes in transcript levels and ribosome occupancy that were remarkably concordant (Figures 2B–2E; Table S1), emphasizing the key role of mRNA abundance for regulating gene expression upon stress (Preiss et al., 2003). Gene expression signatures associated with the environmental stress response in yeast (Gasch et al., 2000) were prominent in both ncs2Δ and wild-type yeast. The induced genes comprised mediators of oxidative stress responses, protein folding and degradation, and respiration, whereas genes associated with energetically costly processes, such as ribosome biogenesis and translation, were profoundly repressed (Table S1; data not shown). Reduced translation in response to both compounds was also evident in a decreased polysome-to-monosome ratio (Figure S3A). Surprisingly, ncs2Δ cells were not overtly compromised in mounting a response to either treatment, since the extent and magnitude of changes in mRNA abundance and ribosome occupancy were similar in wild-type and the mutant (Figures 2B–2F). These data indicate that loss of U34 modifications does not impact the reprogramming of gene expression initiated in response to environmental stress.
Figure 2.

Codon-Specific Pausing in U34 Modification-Deficient Yeast Is Not Enhanced during Stress
(A) Experimental design to assess the effect of U34 modification loss during adaptation to stress in vivo.
(B–E) Log2 fold changes in mRNA abundance and gene-level ribosome occupancy in WT and ncs2Δ cells exposed to diamide or rapamycin in comparison to WT grown in YPD (n = 3). Genes with statistically significant changes (Benjamini-corrected p < 0.01) for mRNA levels (orange), ribosome occupancy (gray), or both (red) are indicated. Pearson correlation coefficient (r) between ribosome occupancy and mRNA abundance changes is shown.
(F) Hierarchically clustered heat map of ribosome occupancy data in (B)–(E).
(G) Codon-specific changes in A-site ribosome occupancy in ncs2Δ yeast after diamide or rapamycin exposure (mean ± SD; n = 3). Data for ncs2Δ cells grown in YPD are from Figure 1B.
(H) Codon-specific changes in A-site ribosome occupancy in elp6Δ yeast after rapamycin exposure (mean ± SD; n = 3). Data for elp6Δ cells grown in YPD are from Figure 1B.
Figure S3.

Effects of Diamide and Rapamycin on Global Translation and Codon Occupancy, Related to Figure 2
(A) Polysome profiles of WT and ncs2Δ cells grown in YPD or after treatment with diamide or rapamycin. Extracts from samples subjected to ribosome profiling in Figures 1 and 2 were analyzed. The polysome/monosome ratio (P/M) was calculated by quantification of integrated peak areas.
(B) Codon occupancy within P, E, and +1 sites in ncs2Δ cells exposed to diamide or rapamycin (mean ± SD; n = 3). Values for ncs2Δ grown in YPD are derived from Figure S1C.
As the composition of the transcript pool bound by ribosomes was dramatically different between rapidly growing and stressed cells but highly similar in wild-type and ncs2Δ yeast grown under the same conditions (Figure 2F), we could directly assess whether codon-specific translational pausing is magnified during stress. The codon-specific differences in ribosome occupancy in ncs2Δ exposed to diamide or rapamycin were comparable to those observed during normal growth (Figures 2G and S3B), showing that global demand for U34-modified tRNAs does not change upon environmental stress. We obtained analogous results when comparing A-site occupancy in elp6Δ yeast during normal growth or after rapamycin exposure (Figure 2H). The only discernible quantitative difference was a moderate increase of A-site occupancy at GAA during stress (Figures 2G and 2H); given the minor importance of tEUUC for phenotypic suppression (Esberg et al., 2006; Leidel et al., 2009) (Figure S1H), we consider this increase unlikely to be functionally relevant. Taken together, these data demonstrate that neither the extent of codon-specific ribosome pausing nor the ability to mount stress responses are altered in U34 modification-deficient cells.
U34 Modification Defects Trigger Proteotoxic Stress in Yeast
To explore alternative explanations for the diminished stress tolerance of yeast with aberrantly modified U34, we examined whether they display adaptive responses during normal growth. Indeed, when compared to wild-type, numerous genes in the mutants showed statistically significant changes both in mRNA abundance and gene-level ribosome occupancy (Figures 3A–3C). These changes were highly concordant (Pearson r = 0.74–0.85), indicating that reprogramming of gene expression in the mutants occurs predominantly via altered transcript levels. We noted a considerable overlap among the differentially expressed genes in ncs2Δ, elp6Δ, and ncs2Δelp6Δ cells (Figures 3E and 3F), implying a common trigger for the cellular response to aberrant U34 modification. To characterize this response, we focused on genes in ncs2Δelp6Δ yeast with significantly altered ribosome occupancy.
Figure 3.

Cells with U34 Modification Defects Exhibit Gene Expression Hallmarks of Proteotoxic Stress
(A–C) Log2 fold changes (mutant/WT) in mRNA abundance and gene-level ribosome occupancy in U34 modification-deficient strains grown in YPD (n = 3). Color annotation is as in Figure 2B.
(D) Hierarchically clustered heat map of ribosome occupancy data from (A)–(C).
(E) Venn diagram of the overlap between significantly downregulated (Benjamini-corrected p < 0.01) genes in U34 modification-deficient strains.
(F) GO term enrichment in genes significantly downregulated in ncs2Δ, elp6Δ, and ncs2Δelp6Δ strains was summarized and visualized in semantic similarity-based scatter plots using REViGO. x axis: likeliness of meaning between GO terms; y axis: Benjamini-corrected p values of statistical significance for GO category enrichment; symbol size: frequency of GO term in database.
(G) Venn diagram of the overlap between significantly upregulated genes.
(H) GO term enrichment in genes upregulated in ncs2Δelp6Δ calculated as in (F).
(I) Growth defects of strains lacking Rpn4, which controls expression of the UPS via the proteasome-associated control element (PACE).
The functional enrichment in downregulated genes was remarkably reminiscent of the core response to environmental stress in yeast: repression of genes related to protein synthesis (Figure 3F; Table S2) aimed at conserving energy (Gasch et al., 2000). Decreased abundance of proteins from these functional groups has previously been argued to occur without concomitant changes in transcript levels and to result from inefficient translation of codons for K, Q, and E in U34 modification-deficient yeast (Laxman et al., 2013; Rezgui et al., 2013). However, we found that decreased ribosome occupancy in ncs2Δelp6Δ cells correlated strongly with a reduction in mRNA abundance (r = 0.85; Figure 3C; Table S2) and is thus unlikely due to defects in translation.
Intriguingly, the genes induced in the ncs2Δelp6Δ strain were strongly enriched for components of the protein degradation and refolding machinery (Figures 3G and 3H), suggesting that protein homeostasis may be perturbed. Genes encoding catalytic and regulatory subunits of the proteasome (PRE2, PRE3, PRE10, PUP1, PUP3, RPT1–RPT5, and others), heat shock proteins from the cytosolic disaggregation and refolding machinery (HSP104, HSP42, HSP82, SSA3, and SSA4), and membrane chaperones (HSP12 and HSP30) all had significantly higher mRNA levels and ribosome occupancy in ncs2Δelp6Δ cells (Table S2). The induction of ubiquitin-proteasome system (UPS) genes in the mutant falls in the range observed upon acute proteotoxic insults, such as diamide exposure (∼2-fold; Table S1) (Gasch et al., 2000). Given the high intracellular abundance of UPS components, this increased expression is likely to incur substantial metabolic costs, prompting us to examine its significance for cellular fitness. Decreasing UPS levels via genetic ablation of the transcription factor Rpn4 (Xie and Varshavsky, 2001) profoundly delayed the growth of ncs2Δelp6Δ (Figure 3I) and exacerbated the stress sensitivity of ncs2Δ, ncs6Δ, elp2Δ, and elp6Δ strains (Figures S4A and S4B), whereas it had little effect on growth in a wild-type background. We conclude that the ability of U34 modification-deficient strains to induce and maintain high UPS levels is critical for cellular fitness.
Figure S4.

Phenotypes of U34 Modification-Deficient Yeast Strains Lacking RPN4 or GCN4, Related to Figure 3
(A) RPN4 deletion significantly increases the doubling time of ncs2Δ and elp6Δ in YPD (mean ± SD; n = 3, ∗∗:p ≤ 0.01 using an unpaired two-tailed Student’s t test).
(B and C) Cultures from the indicated strains were grown to exponential phase, serially diluted, and spotted on the indicated plates. Imaging was performed after 3 days of incubation at 30°C.
Several other gene ontology (GO) terms were enriched among the transcripts induced in U34 modification-deficient cells, albeit to a much lower extent (Figure 3G). Among these were amino-acid biosynthesis genes and their transcription activator GCN4, which we found to be translationally upregulated in cells with hypomodified U34 (Table S2), consistent with previous observations (Zinshteyn and Gilbert, 2013). The absence of GCN4, however, did not overtly compromise the growth or stress sensitivity of yeast strains with hypomodified U34 (Figure S4C), demonstrating that activation of the Gcn4 pathway does not enhance cellular fitness in the absence of U34 modification.
Endogenous Proteins Aggregate upon Loss of U34 Modifications in Yeast
The prominent induction of the UPS, coupled with the upregulation of cytosolic chaperones, raises the intriguing possibility that loss of U34 modifications may perturb the conformational integrity of proteins. To test this, we performed a quantitative isolation of endogenous protein aggregates from exponentially growing wild-type and U34 modification-deficient cells (Koplin et al., 2010). Strikingly, we found that aberrant U34 modification elicits the aggregation of endogenous proteins, while aggregated species were virtually undetectable in the wild-type (Figure 4A; Table S3). The pattern of insoluble proteins was comparable in all mutants, but the extent of aggregation was most pronounced in the ncs2Δelp6Δstrain.
Figure 4.

Loss of U34 Modifications Elicits Protein Aggregation in Yeast and Nematodes
(A) Detergent-insoluble protein aggregates isolated from WT and mutant strains were visualized by SDS-PAGE and Coomassie staining (left: total extracts; right: aggregates).
(B) Protein aggregation patterns in the absence of ribosome-associated chaperones Ssb1/Ssb2 and upon loss of U34 modifications.
(C) Quantitative mass spectrometry (MS) identification of proteins aggregating in ncs2Δelp6Δ and ssb1Δssb2Δ yeast.
(D) Fraction of essential proteins in aggregates from ncs2Δelp6Δ and ssb1Δssb2Δ.
(E and F) GO term enrichment in aggregates from ncs2Δelp6Δ (D) and ssb1Δssb2Δ (E) cells calculated as in Figure 3F.
(G) Q35-YFP aggregation in body-wall muscle cells is enhanced by loss of U34 modifications in C. elegans. Representative images of the head region from 1-day-old adult control and tut-1(tm1297), elpc-1(tm2149) animals are shown; Q35-YFP aggregates are indicated with arrows. Box plots depict the median aggregate number per animal (solid line), the 25% and 75% quartiles (box), and the 1.5× interquartile range (dashed lines) in data pooled from five independent experiments (n ≥ 20 animals per experiment). ∗∗∗∗p ≤ 10−15, Wilcoxon test.
See also Figures S5 and S6 and Table S3.
Since U34 modification defects lead to codon-specific translational pausing (Figure 1B), we considered that pausing might perturb cotranslational protein folding. Consistent with this possibility, we found that the absence of the ribosome-associated chaperones Ssb1 and Ssb2 aggravates the phenotypes of strains with hypomodified U34 (Figure S5A). Deletion of SSB1 and SSB2 is known to also elicit protein aggregation (Koplin et al., 2010; Willmund et al., 2013). Remarkably, we found that the extent of aggregation and the pattern of insoluble species in ncs2Δelp6Δ and ssb1Δssb2Δ cells were very similar (Figure 4B). These data indicate that loss of U34 modification is detrimental for the solubility of many proteins that are destabilized also in the absence of cotranslationally acting chaperones. Using quantitative mass spectrometry, we identified 610 proteins as significantly enriched in insoluble fractions from ncs2Δelp6Δ, and 70% of these also aggregated in ssb1Δssb2Δ cells (Figure 4C). Approximately 40% of the aggregated proteins from both ncs2Δelp6Δ and ssb1Δssb2Δ mutants are essential for yeast viability (Figure 4D). Importantly, increased aggregation propensity did not result from higher abundance of the insoluble proteins in the mutants when compared to wild-type (Figures S5B and S5C).
Figure S5.

Loss of U34 Modifications Elicits Protein Aggregation in Yeast and Nematodes, Related to Figure 4
(A) Exponentially growing cultures from the indicated strains were serially diluted and spotted on the indicated plates. Imaging was performed after 3 days of incubation at 30°C.
(B and C) Comparison of log2 fold enrichment of protein species in aggregates versus log2 fold changes in abundance in total protein for ncs2Δelp6Δ (B) and ssb1Δssb2Δ yeast (C). Pearson correlation coefficient (r) is indicated.
(D and E) Analysis of the fraction of glutamines encoded by CAA (D) and lysines encoded by AAA (E) in proteins aggregating only in ncs2Δelp6Δ, ssb1Δssb2Δ, or in both mutants (overlap) compared to all yeast verified and uncharacterized ORFs. Boxplots depict the median (solid line), the 25% and 75% quartiles (box) and 1.5 x the interquartile range (dashed lines). Statistical significance was defined as p ≤ 0.01 from a Wilcoxon test. ∗∗p ≤ 10−6, ∗∗∗p ≤ 10−12
(F) The number of Q35-YFP aggregates in muscle cells of one-day-old nematodes is increased by knockdown of tut-1 by RNAi. Animals were fed bacteria carrying an empty L4440 vector (control) or dsRNA targeting tut-1 throughout development. Data from three independent experiments (n = 50 animals) were pooled. Solid lines indicate the median number of aggregates, boxes delimit 25% and 75% quartiles, and dashed lines show 1.5 x the interquartile range. ∗∗p ≤ 10−6, Wilcoxon test.
(G) Fold changes in abundance of transcripts encoding cytosolic heat shock response genes (hsp-16.41, hsp-70) and chaperones resident in mitochondria (hsp-6, hsp-60) and the ER (hsp-3, hsp-4) in adult tut-1(tm1297), elpc-1(tm2149) nematodes compared to age-matched wild-type N2 animals. Data from two biological replicates was analyzed by DESeq and the cut-off for statistical significance was defined as a Benjamini-corrected p value of less than 0.05 (ns = not significant).
(H) Kaplan-Meier survival curves of wild-type N2, tut-1(tm1297), and tut-1(tm1297), elpc-1(tm2149) animals (N2 median lifespan = 16 days versus tut-1(tm1297) median lifespan = 13 days, p < 0.0001, and versus tut-1(tm1297), elpc-1(tm2149) median lifespan = 9 days, p < 0.0001, Mantel-Cox log-rank test).
As the major effect of aberrant U34 modification on translation speed is pausing at CAA and AAA (Figure 1B), we asked whether the usage of these codons was disproportionally high in messages encoding aggregated proteins. Although we found that the fraction of glutamines encoded by CAA was increased in the ncs2Δelp6Δ/ssb1Δssb2Δ overlap dataset (Figure S5D), the fact that these proteins aggregated in both strains suggests that the correlation with CAA codon content is spurious. Moreover, lysines in aggregated proteins were marginally less likely to be encoded by AAA (Figure S5E). Given these results, and the large overlap between proteins aggregating upon such diverse conditions as U34 modification defects and chaperone loss (Figure 4C), we propose that many of the aggregated species abundant enough to be detected by mass spectrometry are metastable and thus highly vulnerable to perturbations in the cellular folding environment. Consistent with this idea, aggregates from both mutant strains contained many constituents of multisubunit assemblies (Figures 4E and 4F; Table S3), and the species we detected only in aggregates from ncs2Δelp6Δ yeast were mostly additional subunits of complexes aggregating in both mutants (Table S3). Proteins that participate in large complexes have many interaction surfaces that can also cause aberrant associations (Pechmann et al., 2009), and their relative abundance exceeds their solubility in cells (Ciryam et al., 2013). Both properties have been suggested to predispose them to aggregation upon disruption of protein homeostasis. Indeed, during aging or upon proteotoxic challenge, constituents of the translational machinery and proteasomes aggregate in yeast, nematodes, and human cells (Ciryam et al., 2013; David et al., 2010; Kirstein-Miles et al., 2013; Koplin et al., 2010; Willmund et al., 2013). Thus, the widespread aggregation of endogenous metastable proteins upon loss of U34 modifications indicates an imbalance in protein homeostasis.
Loss of U34 Modifications Impairs Protein Homeostasis in C. elegans
To examine whether loss of U34 modifications disrupts protein homeostasis in C. elegans, we employed an aggregation-prone chimeric protein consisting of a stretch of 35 glutamine residues (Q35) fused to YFP. When expressed in body-wall muscle cells, Q35-YFP is soluble and diffusely localized throughout development, but begins to aggregate with the onset of adulthood (Morley et al., 2002). Aggregation of Q35-YFP is a sensitive sensor for protein folding defects in vivo, since it is enhanced in the presence of misfolded proteins (Gidalevitz et al., 2006). Quantification of Q35-YFP aggregate numbers in young Q35-YFP transgenic animals with unmodified U34 (tut-1(tm1297), elpc-1(tm2149); Chen et al., 2009a) revealed that aggregate burden was 2.5-fold higher in the mutant than in control animals (Figure 4G). Q35-YFP aggregation was similarly augmented after silencing of tut-1 by RNAi (Figure S5F). The aggregate burden is comparable to the one observed upon silencing of the ribosome-bound chaperone NAC (3- to 4-fold; Kirstein-Miles et al., 2013). Moreover, U34 modification deficiency correlated with increased expression of cytosolic heat shock protein genes (Figure S5G) and shorter life span (Figure S5H) (Taylor and Dillin, 2011). Taken together, our results demonstrate that defects in U34 modification impair protein homeostasis in both nematodes and yeast.
Overexpression of Hypomodified tEUUC, tKUUU, and tQUUG Restores Codon Translation Rates and Protein Homeostasis in U34 Modification-Deficient Cells
To establish whether proteotoxic stress (Figure 3H) and aggregate accumulation (Figure 4A) in cells with aberrantly modified U34 stem from codon-specific translational pausing (Figure 1B), we examined whether translation speed and protein homeostasis can be restored by tRNA overexpression. We used ribosome profiling to test whether ribosomal pausing at their cognate codons is mitigated in U34 modification-deficient yeast overexpressing tEUUC, tKUUU, and tQUUG. Indeed, while tRNA overexpression had no effect on growth or A-site occupancy in wild-type (Figures S6A and S6B), it markedly decreased occupancy at CAA and AAA in ncs2Δ and elp6Δ cells (Figures 5A and S6C). These data suggest that in U34 modification-deficient cells, ribosomes accept the hypomodified tRNAs less efficiently as a match to their cognate codons during decoding, leading to a perceived scarcity of these species when present at endogenous levels. To test this model further, we compared codon occupancy in ncs2Δelp6Δ and wild-type cells after exposure to paromomycin, which increases near-cognate tRNA acceptance (Kramer et al., 2010). Pausing at CAA and AAA codons was mitigated by paromomycin (Figure 5B), providing further evidence that impaired codon-anticodon interactions within the ribosomal A site account for the slower codon translation rates in the absence of U34 modifications.
Figure S6.

Effect of tRNA Overexpression on Codon Occupancy, Related to Figure 4
(A) Doubling time of cultures from the indicated strains carrying an empty vector or overexpressing tEUUC, tKUUU, and tQUUG (+tEKQ) (mean ± SD; n = 3; ∗∗: p ≤ 0.01 from an unpaired two-tailed Student’s t test; n.s. denotes not significant).
(B and C) Effect of overexpression of tEUUC, tKUUU and tQUUG on A-site codon occupancy in WT (B) and elp6Δ (C). Note that the least used codons in yeast (CGA, CGG) exhibit larger fluctuations in the elp6Δ data sets due to lower read depth.
Figure 5.

Elevated Levels of Hypomodified tEUUC, tKUUU, and tQUUG Alleviate Ribosome Pausing and Relieve Proteotoxic Stress in U34 Modification-Deficient Yeast
(A) A-site ribosome occupancy changes in ncs2Δ cells carrying an empty plasmid or overexpressing tEUUC, tKUUU, and tQUUG (tEKQ) in comparison to WT (mean ± SD; n = 3).
(B) Effect of paromomycin on A-site codon occupancy in ncs2Δelp6Δ yeast (mean ± SD; n = 3). Values for the ncs2Δelp6Δ strain grown in YPD are derived from Figure 1B.
(C) Effect of tEUUC, tKUUU, and tQUUG overexpression on protein aggregation in ncs2Δ and elp6Δ cells.
(D) Heat map of log2 fold changes (compared to WT) for genes with statistically significant differences in ribosome occupancy in ncs2Δ (left panel) and elp6Δ (right panel) cells carrying an empty plasmid or overexpressing tEUUC, tKUUU, and tQUUG.
Next, we asked whether restoration of codon translation rates is sufficient to improve protein homeostasis in yeast with hypomodified U34. Remarkably, we found that when intracellular levels of tEUUC, tKUUU, and tQUUG were augmented, the aggregate burden in ncs2Δ and elp6Δ cells decreased substantially (Figure 5C). Concomitantly, gene expression in the mutants reverted close to wild-type patterns upon tRNA overproduction (Figure 5D). The restoration of codon-specific translation speed therefore not only ameliorates protein aggregation but also obviates the need for increased protein quality control and repression of growth-related genes. Collectively, our findings indicate that protein homeostasis imbalance in U34 modification-deficient cells is elicited by slower decoding of CAA and AAA.
Cells with Unmodified U34 Are Deficient in Resolving Stress-Induced Protein Aggregates
Protein aggregation in cells with aberrantly modified U34 (Figure 4A) can reduce cellular fitness via loss of function of essential proteins (Figure 4D) and aggregate toxicity (Geiler-Samerotte et al., 2011). The chronic accumulation of protein aggregates is also known to compromise the capacity of cells to cope with misfolding triggered by acute proteotoxic stress (Hipp et al., 2014). We therefore asked whether impaired clearance of stress-induced protein aggregates might contribute to the stress sensitivity of cells with unmodified U34. For this, we monitored the subcellular localization of Hsp104, an aggregate-remodeling chaperone, in wild-type and ncs2Δelp6Δ cells upon diamide exposure. Hsp104 is diffusely distributed throughout cells during normal growth, but upon proteotoxic insults, it localizes to distinct cytoplasmic foci containing spatially sequestered misfolded proteins (Escusa-Toret et al., 2013). GFP-tagged Hsp104 was uniformly distributed in exponentially growing wild-type and ncs2Δelp6Δ cells. Within 1 hr of diamide exposure however >80% of cells from both strains harbored multiple Hsp104-GFP foci (Figures 6A and 6B), demonstrating that diamide is a potent inducer of protein aggregation in vivo. Whereas aggregates were rapidly eliminated from wild-type cells upon transfer to fresh medium, they persisted significantly longer in ncs2Δelp6Δ cells (Figure 6B). Aggregate elimination in the mutant was impaired to an even greater extent upon continuous diamide exposure: virtually all ncs2Δelp6Δ cells still harbored Hsp104-GFP foci 4 hr after addition of the drug, as opposed to only 40% of wild-type cells (Figure 6C). Thus, the clearance of diamide-induced protein aggregates is severely delayed in cells with aberrantly modified U34, a result that correlates well with their increased sensitivity to this compound (Figures S1H and S3B). Given that diamide does not augment codon-specific ribosome pausing (Figure 2G), our findings suggest that chronic protein aggregation in U34 modification-deficient cells severely compromises their ability to restore proteome integrity upon acute proteotoxic insults.
Figure 6.

Clearance of Stress-Induced Protein Aggregates Is Severely Compromised in Yeast with Unmodified U34
(A) Intracellular distribution of Hsp104-GFP in WT and ncs2Δelp6Δ yeast grown in YPD (left) or treated with 1.5 mM diamide for 60 min (right).
(B) Percentage of WT (black squares) and ncs2Δelp6Δ cells (red diamonds) with Hsp104-GFP-containing aggregates upon transient exposure to diamide. Exponentially growing cells were treated with 1.5 mM diamide for 60 min and returned to fresh YPD medium. At least 100 cells from three different fields were counted per time point (mean ± SD; n = 3).
(C) Percentage of WT and ncs2Δelp6Δ cells with Hsp104-GFP-containing aggregates in the continuous presence of 1.5 mM diamide. Quantification was performed as in (B).
(D) Model for the molecular consequences of aberrant U34 modification.
Discussion
The non-uniform rate of peptide synthesis by ribosomes has been suggested to impact the folding of nascent polypeptides (Pechmann et al., 2013; Thanaraj and Argos, 1996). Experimental support for this model has been difficult to obtain due to the poorly defined determinants of codon translation rates in living cells. By probing the consequences of tRNA anticodon modification loss for translation in vivo, we show that codon-anticodon interactions are an evolutionarily conserved modulator of decoding speed in eukaryotes (Figures 1B and 1C). We further demonstrate that translational pausing at a small subset of codons severely disrupts protein homeostasis and triggers the widespread aggregation of endogenous proteins (Figures 3H and 4A). Thus, the stability of codon-anticodon interactions mediated by anticodon modifications emerges as a major determinant of codon translation rates and the integrity of the eukaryotic proteome.
Modifications at U34 Accelerate Decoding In Vivo
The magnitude of ribosome pausing triggered by U34 modification loss differs in yeast and nematodes (Figures 1B and 1C), since it is likely modulated by additional factors, such as the abundance of near-cognate tRNAs that can compete for A-site codon binding, mRNA topology, and the nature of the nascent chain. Pausing most likely results from inefficient decoding, since it was relieved by elevated levels of hypomodified tRNAs (Figures 5A and S6C) and paromomycin (Figure 5B). Unexpectedly, loss of mcm5U34 or ncm5U34 had only minor effects on decoding speed in yeast, both during normal growth and during stress (Figures 2G and 2H). Since U34 modifications are dispensable for tRNA stability and charging (Johansson et al., 2008), our data suggest that 5-carbon moieties in many tRNAs have little function (Phizicky and Alfonzo, 2010). The importance of mcm5s2U34 for decoding efficiency may have been the driving force behind the evolution of enzymes modifying the 5-carbon of U34, and the minor functional consequences of these modifications for most other tRNAs with U34 might have decreased the evolutionary pressure for higher substrate specificity. Alternatively, 5-carbon moieties at U34 could play a more prominent role in decoding in other eukaryotes; however, the full repertoire of tRNA species with mcm5U34 or ncm5U34 is only known in budding yeast so far (Johansson et al., 2008).
Decoding Speed: A Safeguard against Protein Aggregation
To our surprise, a reduction in global translation rates of only two codons—CAA and AAA (Figure 1B)—led to widespread protein aggregation and imbalanced protein homeostasis (Figures 3H, 4A, and 4C), defects alleviated solely by restoring decoding speed (Figures 5A, 5C, and 5D; S6C). Slower decoding may increase the probability of rare translation events, such as misreading or frameshifting (Drummond and Wilke, 2008; Fluitt et al., 2007; Komar, 2009), or trigger cotranslational misfolding if a nascent chain is trapped in an off-target folding intermediate (O’Brien et al., 2014). Thus, we expect that ubiquitous translational pausing at CAA and AAA (Figures 1B and S1E) will impose different functional costs on different proteins, depending on codon and amino acid context. As one example, lysines flanking aggregation-prone amino acid stretches are thought to oppose aggregation (Rousseau et al., 2006), and slower translation of AAA codons that specify such “gatekeeper” residues might be especially detrimental for protein folding. Future measurements of amino-acid misincorporation rates in endogenous proteins will clarify the frequency and type of translational errors in cells with unmodified U34. However, as such errors are likely to occur in only a fraction of molecules from a protein species, it will be challenging to define how the presence of a subset of erroneously synthesized polypeptides will impact the folding or function of a given protein in the cell.
Regardless of its effect on individual proteins, ribosome pausing at CAA and AAA codons (Figure 1B) clearly destabilizes the conformation of many proteins with essential cellular functions (Figures 4C and 4D). Given that the presence of a single aggregation-prone polypeptide is sufficient to promote aberrant interactions between metastable endogenous proteins (Olzscha et al., 2011), it is conceivable that misfolding of only very few species can significantly reduce the capacity of cells with unmodified U34 to maintain proteome integrity. Misfolding would not necessarily be correlated with a high frequency of CAA or AAA codons, as even a single codon substitution in a structurally important region can substantially alter protein conformation (Kimchi-Sarfaty et al., 2007). Proteins or nascent chains that misfold upon translational pausing may occupy critical regulators of protein homeostasis and sequester them from conformationally dynamic species that depend on these regulators for proper folding. This model (Figure 6D) would account for the widespread aggregation of metastable proteins we observed in yeast with unmodified U34 (Figures 4C and 4D; Figure 6D), as well as the enhanced aggregation of the Q35-YFP reporter in U34 modification-deficient C. elegans (Figures 4G and S5F).
Interestingly, loss of U34 modifications did not activate the endoplasmic reticulum unfolded protein response (UPR) in yeast, as judged by the lack of induction of HAC1, its key regulator, in the ncs2Δelp6Δ strain (Table S2). UPR activation was not generally impaired in cells with hypomodified U34, because HAC1 ribosome occupancy was upregulated ∼10-fold in diamide-treated ncs2Δ cells, and mutant strains were not hypersensitive to inducers of ER stress like tunicamycin or dithiothreitol (data not shown). It is plausible that the integrity of ER proteins is less affected by changes in decoding speed of single codons during their synthesis due to the entirely different folding constraints imposed by co-translational translocation across the ER membrane. Sensing of co-translational misfolding may also be more efficient at the ER to avoid retro-translocating damaged lumenal proteins to the cytoplasm.
Cellular Dysfunction upon Loss of U34 Modifications Is Caused by Disruption of Protein Homeostasis
Previous models attempting to rationalize the stress sensitivity of yeast mutants with aberrantly modified U34 have proposed that frequent usage of codons read via U34-modified tRNAs in genes encoding stress response regulators may decrease their translational output in such mutants (Bauer et al., 2012; Begley et al., 2007). However, our ribosome-profiling data are inconsistent with a general increase in the translational demand for U34-modified tRNAs during stress, as ribosome pausing elicited by hypomodified tRNAs in yeast was similar in rapidly growing and stressed cells (Figures 2G and 2H). Moreover, U34 modification-deficient cells reprogrammed gene expression in response to both drugs in a manner similar to wild-type (Figure 2F). Taken together, these data argue against a specific role for U34-modified tRNAs in the regulation of stress responses. Instead, we propose that chronic proteotoxic stress causes the decreased fitness of cells lacking U34 modifications. The extensive aggregation of endogenous proteins and the induction of protein quality control pathways in these cells are consistent with this hypothesis (Figures 3H and 4C). Both events correlated with ribosomal pausing at CAA and AAA codons (Figures 5A, 5C, and 5D), suggesting they are triggered by translation and folding errors that arise from decreased codon translation rates. Notably, we found that the capacity of cells with unmodified U34 to re-establish protein homeostasis upon acute stress is strongly diminished (Figures 6B and 6C); this is a common consequence of chronic protein misfolding and probably occurs via overload of the chaperone and degradation machineries (Hipp et al., 2014). Failure of protein homeostasis is thus a major contributor to cellular dysfunction upon defects in U34 modification (Figure 6D).
In conclusion, our study establishes a direct link between the kinetics of translation at individual codons and the conformational integrity of the proteome. The widespread protein aggregation elicited by slower decoding of only two codons highlights the extreme sensitivity of cells to local changes in the pace of translation elongation. Such changes can result from loss of U34 modifications via perturbed codon-anticodon interactions, as we described here or from the destabilization of single tRNA species (Ishimura et al., 2014), and both have been associated with neurodegeneration. As neurons are particularly vulnerable to the toxicity of protein aggregates, our findings provide a novel basis for understanding the pathologies that occur when tRNA modification or stability is compromised.
Experimental Procedures
Strains and Growth Conditions
Yeast strains were in the S288c BY4741 background and are listed in Table S4. Cultures were grown to mid-exponential phase (optical density 600 [OD600], ∼0.4–0.5) at 30°C, 200 revolutions per minute in yeast extract peptone dextrose (YPD) (Formedium). For tRNA overexpression, cells were transformed with the high-copy 2μ-based plasmid pRS425 encoding a single copy each of tEUUC, tKUUU, and tQUUG (Leidel et al., 2009) and grown in SD-Leu to maintain plasmid selection. To induce stress, 1.5 mM diamide or 12.5 nM rapamycin was added at OD600 = 0.4 for 30 min.
C. elegans strains were in the N2 (Bristol) wild-type background and were handled using standard methods. The following transgenes and mutants were used: rmIs132[Punc-54::q35::yfp] (Morley et al., 2002), tut-1(tm1297) (Leidel et al., 2009), and elpc-1(tm2149) (Chen et al., 2009a).
Ribosome Profiling and RNA-Seq
Yeast or L4-stage nematodes were pulverized under cryogenic conditions in a freezer mill (Spex) and RNase I-treated extracts were resolved by sucrose gradient centrifugation. Ribosome-protected footprints were purified from monosome fractions by size selection and gel extraction. Sequencing libraries were prepared essentially as described (Ingolia et al., 2012; Stadler and Fire, 2011). Codon occupancy was calculated similarly to Stadler and Fire (2011). Differential expression was tested by DESeq, and GO term enrichment was summarized and visualized with REViGO. Additional details are available in the Supplemental Experimental Procedures.
Analysis of Endogenous Protein Aggregates
Aggregated proteins were isolated as in Koplin et al. (2010), resolved by SDS-PAGE, and visualized with Coomassie staining. Proteins enriched in aggregate fractions were identified by label-free quantitative mass spectrometry. Wild-type and ncs2Δelp6Δ cells expressing Hsp104-GFP from the HSP104 chromosomal locus were grown to exponential phase in YPD, treated as indicated, fixed in 3.7% formaldehyde, and imaged on a Zeiss Axio Imager 2 microscope.
Acknowledgments
We thank K. Buhne for technical support, S. Mitani, S. Tuck, P. Gönczy, and E. Liebau for reagents, H. Drexler and A. Nolte for mass spectrometry, F. Konert and C. Gräf for sequencing, D. Eccles, J.M. Vaquerizas, B. Habermann, R. Luna, and M. Delattre for advice, A. Bertolotti for insightful suggestions, and A.J. te Velthuis, S. Starck, P. Gönczy, G. Rabut, K. Bartscherer, and E.J. Snijder for comments on the manuscript. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by the Max Planck Society and grants from the North Rhine-Westphalian Ministry for Innovation, Science and Research (314-40001009), and the European Research Council (ERC-2012-StG 310489-tRNAmodi) (to S.A.L.); D.D.N. received an EMBO Long-Term Fellowship (ALTF1291-2010).
Published: June 4, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, six figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2015.05.022.
Accession Numbers
Sequencing data are available at the Gene Expression Omnibus (accession number GSE67387).
Supplemental Information
Ribosome occupancy and mRNA abundance changes in WT cells following a 30 min exposure to 1.5 mM diamide or 12.5 nM rapamycin compared to WT cells grown in YPD were computed with DESeq (Anders and Huber, 2010). Systematic gene names were derived from the Saccharomyces Genome Database (SGD). Columns denote fold changes in ribosome occupancy (RO) or mRNA abundance (M) in the treated sample, with the corresponding Benjamini-Hochberg-adjusted p value for statistical significance. Low-coverage genes were filtered out by requiring a value of at least 50 for the mean of normalized reads between the two samples being compared.
Gene-level ribosome occupancy and mRNA abundance changes in ncs2Δelp6Δ cells compared to WT cells grown in YPD were computed with DESeq. Data annotation and filtering are as in Table S1.
Aggregated proteins were isolated from three independent cultures of ncs2Δelp6Δ, ssb1Δssb2Δ, and WT cells. Proteins in the insoluble fractions were identified by mass spectrometry, and species enriched >2-fold (two-sample t test; false discovery rate <5%) in aggregates from ncs2Δelp6 or ssb1Δssb2Δ are listed.
The genotype and source of yeast strains used in this study are listed.
References
- Agris P.F., Vendeix F.A.P., Graham W.D. tRNA’s wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 2007;366:1–13. doi: 10.1016/j.jmb.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Bauer F., Matsuyama A., Candiracci J., Dieu M., Scheliga J., Wolf D.A., Yoshida M., Hermand D. Translational control of cell division by Elongator. Cell Rep. 2012;1:424–433. doi: 10.1016/j.celrep.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley U., Dyavaiah M., Patil A., Rooney J.P., DiRenzo D., Young C.M., Conklin D.S., Zitomer R.S., Begley T.J. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell. 2007;28:860–870. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk G.R., Huang B., Persson O.P., Byström A.S. A conserved modified wobble nucleoside (mcm5s2U) in lysyl-tRNA is required for viability in yeast. RNA. 2007;13:1245–1255. doi: 10.1261/rna.558707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Tuck S., Byström A.S. Defects in tRNA modification associated with neurological and developmental dysfunctions in Caenorhabditis elegans elongator mutants. PLoS Genet. 2009;5:e1000561. doi: 10.1371/journal.pgen.1000561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.T., Hims M.M., Shetty R.S., Mull J., Liu L., Leyne M., Slaugenhaupt S.A. Loss of mouse Ikbkap, a subunit of elongator, leads to transcriptional deficits and embryonic lethality that can be rescued by human IKBKAP. Mol. Cell. Biol. 2009;29:736–744. doi: 10.1128/MCB.01313-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciryam P., Tartaglia G.G., Morimoto R.I., Dobson C.M., Vendruscolo M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013;5:781–790. doi: 10.1016/j.celrep.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D.C., Ollikainen N., Trinidad J.C., Cary M.P., Burlingame A.L., Kenyon C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewez M., Bauer F., Dieu M., Raes M., Vandenhaute J., Hermand D. The conserved Wobble uridine tRNA thiolase Ctu1-Ctu2 is required to maintain genome integrity. Proc. Natl. Acad. Sci. USA. 2008;105:5459–5464. doi: 10.1073/pnas.0709404105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond D.A., Wilke C.O. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esberg A., Huang B., Johansson M.J.O., Byström A.S. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell. 2006;24:139–148. doi: 10.1016/j.molcel.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Escusa-Toret S., Vonk W.I.M., Frydman J. Spatial sequestration of misfolded proteins by a dynamic chaperone pathway enhances cellular fitness during stress. Nat. Cell Biol. 2013;15:1231–1243. doi: 10.1038/ncb2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluitt A., Pienaar E., Viljoen H. Ribosome kinetics and aa-tRNA competition determine rate and fidelity of peptide synthesis. Comput. Biol. Chem. 2007;31:335–346. doi: 10.1016/j.compbiolchem.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch A.P., Spellman P.T., Kao C.M., Carmel-Harel O., Eisen M.B., Storz G., Botstein D., Brown P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiler-Samerotte K.A., Dion M.F., Budnik B.A., Wang S.M., Hartl D.L., Drummond D.A. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc. Natl. Acad. Sci. USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidalevitz T., Ben-Zvi A., Ho K.H., Brignull H.R., Morimoto R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Glatt S., Müller C.W. Structural insights into Elongator function. Curr. Opin. Struct. Biol. 2013;23:235–242. doi: 10.1016/j.sbi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Grosjean H., de Crécy-Lagard V., Marck C. Deciphering synonymous codons in the three domains of life: co-evolution with specific tRNA modification enzymes. FEBS Lett. 2010;584:252–264. doi: 10.1016/j.febslet.2009.11.052. [DOI] [PubMed] [Google Scholar]
- Grosjean H., Breton M., Sirand-Pugnet P., Tardy F., Thiaucourt F., Citti C., Barré A., Yoshizawa S., Fourmy D., de Crécy-Lagard V., Blanchard A. Predicting the minimal translation apparatus: lessons from the reductive evolution of mollicutes. PLoS Genet. 2014;10:e1004363. doi: 10.1371/journal.pgen.1004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp M.S., Park S.-H., Hartl F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014;24:506–514. doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Huang B., Johansson M.J.O., Byström A.S. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11:424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R., Nagy G., Dotu I., Zhou H., Yang X.L., Schimmel P., Senju S., Nishimura Y., Chuang J.H., Ackerman S.L. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson M.J.O., Esberg A., Huang B., Björk G.R., Byström A.S. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 2008;28:3301–3312. doi: 10.1128/MCB.01542-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C., Oh J.M., Kim I.-W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- Kirstein-Miles J., Scior A., Deuerling E., Morimoto R.I. The nascent polypeptide-associated complex is a key regulator of proteostasis. EMBO J. 2013;32:1451–1468. doi: 10.1038/emboj.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar A.A. A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 2009;34:16–24. doi: 10.1016/j.tibs.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Koplin A., Preissler S., Ilina Y., Koch M., Scior A., Erhardt M., Deuerling E. A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J. Cell Biol. 2010;189:57–68. doi: 10.1083/jcb.200910074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer E.B., Vallabhaneni H., Mayer L.M., Farabaugh P.J. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 2010;16:1797–1808. doi: 10.1261/rna.2201210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau L.F., Hite D.H., Hogan G.J., Brown P.O. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014;3:e01257. doi: 10.7554/eLife.01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laxman S., Sutter B.M., Wu X., Kumar S., Guo X., Trudgian D.C., Mirzaei H., Tu B.P. Sulfur amino acids regulate translational capacity and metabolic homeostasis through modulation of tRNA thiolation. Cell. 2013;154:416–429. doi: 10.1016/j.cell.2013.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S., Pedrioli P.G.A., Bucher T., Brost R., Costanzo M., Schmidt A., Aebersold R., Boone C., Hofmann K., Peter M. Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA. Nature. 2009;458:228–232. doi: 10.1038/nature07643. [DOI] [PubMed] [Google Scholar]
- Li G.-W., Oh E., Weissman J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley J.F., Brignull H.R., Weyers J.J., Morimoto R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y., Nakai M., Hayashi H. Thio-modification of yeast cytosolic tRNA requires a ubiquitin-related system that resembles bacterial sulfur transfer systems. J. Biol. Chem. 2008;283:27469–27476. doi: 10.1074/jbc.M804043200. [DOI] [PubMed] [Google Scholar]
- O’Brien E.P., Vendruscolo M., Dobson C.M. Kinetic modelling indicates that fast-translating codons can coordinate cotranslational protein folding by avoiding misfolded intermediates. Nat. Commun. 2014;5:2988. doi: 10.1038/ncomms3988. [DOI] [PubMed] [Google Scholar]
- Olzscha H., Schermann S.M., Woerner A.C., Pinkert S., Hecht M.H., Tartaglia G.G., Vendruscolo M., Hayer-Hartl M., Hartl F.U., Vabulas R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- Pechmann S., Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 2013;20:237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S., Levy E.D., Tartaglia G.G., Vendruscolo M. Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc. Natl. Acad. Sci. USA. 2009;106:10159–10164. doi: 10.1073/pnas.0812414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S., Willmund F., Frydman J. The ribosome as a hub for protein quality control. Mol. Cell. 2013;49:411–421. doi: 10.1016/j.molcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky E.M., Alfonzo J.D. Do all modifications benefit all tRNAs? FEBS Lett. 2010;584:265–271. doi: 10.1016/j.febslet.2009.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss T., Baron-Benhamou J., Ansorge W., Hentze M.W. Homodirectional changes in transcriptome composition and mRNA translation induced by rapamycin and heat shock. Nat. Struct. Biol. 2003;10:1039–1047. doi: 10.1038/nsb1015. [DOI] [PubMed] [Google Scholar]
- Rezgui V.A.N., Tyagi K., Ranjan N., Konevega A.L., Mittelstaet J., Rodnina M.V., Peter M., Pedrioli P.G.A. tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc. Natl. Acad. Sci. USA. 2013;110:12289–12294. doi: 10.1073/pnas.1300781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F., Serrano L., Schymkowitz J.W.H. How evolutionary pressure against protein aggregation shaped chaperone specificity. J. Mol. Biol. 2006;355:1037–1047. doi: 10.1016/j.jmb.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Schlieker C.D., Van der Veen A.G., Damon J.R., Spooner E., Ploegh H.L. A functional proteomics approach links the ubiquitin-related modifier Urm1 to a tRNA modification pathway. Proc. Natl. Acad. Sci. USA. 2008;105:18255–18260. doi: 10.1073/pnas.0808756105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigi N. Biosynthesis and functions of sulfur modifications in tRNA. Front. Genet. 2014;5:67. doi: 10.3389/fgene.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker C.J., Green R. Translation drives mRNA quality control. Nat. Struct. Mol. Biol. 2012;19:594–601. doi: 10.1038/nsmb.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M., Fire A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA. 2011;17:2063–2073. doi: 10.1261/rna.02890211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrant D., von der Haar T. Synonymous codons, ribosome speed, and eukaryotic gene expression regulation. Cell. Mol. Life Sci. 2014;71:4195–4206. doi: 10.1007/s00018-014-1684-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R.C., Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a004440. pii: a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanaraj T.A., Argos P. Ribosome-mediated translational pause and protein domain organization. Protein Sci. 1996;5:1594–1612. doi: 10.1002/pro.5560050814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres A.G., Batlle E., Ribas de Pouplana L. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014;20:306–314. doi: 10.1016/j.molmed.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Varenne S., Buc J., Lloubes R., Lazdunski C. Translation is a non-uniform process. Effect of tRNA availability on the rate of elongation of nascent polypeptide chains. J. Mol. Biol. 1984;180:549–576. doi: 10.1016/0022-2836(84)90027-5. [DOI] [PubMed] [Google Scholar]
- Walker J., Kwon S.Y., Badenhorst P., East P., McNeill H., Svejstrup J.Q. Role of elongator subunit Elp3 in Drosophila melanogaster larval development and immunity. Genetics. 2011;187:1067–1075. doi: 10.1534/genetics.110.123893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmund F., del Alamo M., Pechmann S., Chen T., Albanèse V., Dammer E.B., Peng J., Frydman J. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell. 2013;152:196–209. doi: 10.1016/j.cell.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Varshavsky A. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc. Natl. Acad. Sci. USA. 2001;98:3056–3061. doi: 10.1073/pnas.071022298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Ma P., Shah P., Rokas A., Liu Y., Johnson C.H. Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature. 2013;495:116–120. doi: 10.1038/nature11942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Guo J., Cha J., Chae M., Chen S., Barral J.M., Sachs M.S., Liu Y. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013;495:111–115. doi: 10.1038/nature11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B., Gilbert W.V. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet. 2013;9:e1003675. doi: 10.1371/journal.pgen.1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ribosome occupancy and mRNA abundance changes in WT cells following a 30 min exposure to 1.5 mM diamide or 12.5 nM rapamycin compared to WT cells grown in YPD were computed with DESeq (Anders and Huber, 2010). Systematic gene names were derived from the Saccharomyces Genome Database (SGD). Columns denote fold changes in ribosome occupancy (RO) or mRNA abundance (M) in the treated sample, with the corresponding Benjamini-Hochberg-adjusted p value for statistical significance. Low-coverage genes were filtered out by requiring a value of at least 50 for the mean of normalized reads between the two samples being compared.
Gene-level ribosome occupancy and mRNA abundance changes in ncs2Δelp6Δ cells compared to WT cells grown in YPD were computed with DESeq. Data annotation and filtering are as in Table S1.
Aggregated proteins were isolated from three independent cultures of ncs2Δelp6Δ, ssb1Δssb2Δ, and WT cells. Proteins in the insoluble fractions were identified by mass spectrometry, and species enriched >2-fold (two-sample t test; false discovery rate <5%) in aggregates from ncs2Δelp6 or ssb1Δssb2Δ are listed.
The genotype and source of yeast strains used in this study are listed.