

Abstract
Background
The clinical consequences of antibodies to red blood cells (RBC) have been studied for a century. Most clinically relevant antibodies can be detected by sensitive in vitro assays. Several mechanisms of antibody-mediated hemolysis are well understood. Such hemolysis following transfusion is reliably avoided in a donor/recipient pair, if one individual is negative for the cognate antigen to which the other has the antibody.
Study design and results
Mechanisms of antibody-mediated hemolysis were reviewed based on a presentation at the Strategies to Address Hemolytic Complications of Immune Globulin Infusions Workshop addressing intravenous immunoglobulin (IVIG) and ABO antibodies. The presented topics included the rates of intravascular and extravascular hemolysis; IgM and IgG isoagglutinins; auto- and alloantibodies; antibody specificity; A, B, A,B and A1 antigens; A1 versus A2 phenotypes; monocytes/macrophages, other immune cells and complement; monocyte monolayer assay (MMA); antibody-dependent cell-mediated cytotoxicity (ADCC); and transfusion reactions due to ABO and other antibodies.
Conclusion
Several clinically relevant questions remained unresolved, and diagnostic tools were lacking to routinely and reliably predict the clinical consequences of RBC antibodies. Most hemolytic transfusion reactions associated with IVIG were due to ABO antibodies. Reducing the titers of such antibodies in IVIG may lower the frequency of this kind of adverse event. The only way to stop these events is to have no anti-A or anti-B antibodies in the IVIG products.
… except for the immunohematologist’s work on the erythrocyte, little has appeared in the literature on membrane-bound complement components of other cells.1
George Garratty, 1980.
Introduction
The immune mediated destruction of circulating red blood cells (RBC) is described by two distinct mechanisms: one is the intravascular destruction of RBC by complement lysis, which is initiated by antibodies that are often, but not exclusively, of IgM class. The second mechanism is extravascular destruction by immune cells, which recognize IgG and complement bound to RBC. If an IVIG product is free of IgM, intravascular hemolysis should be rare. There is, however, evidence that IgG, purposefully in high abundance in IVIG products, can induce intravascular hemolysis.
If IgM is bound to a RBC in the circulation, the complement cascade may become activated and punctures the membrane, causing intravascular hemolysis. The required complement factors are contributed by the patient and not found in IVIG products. Unless large amounts of C3b are generated to combine with C5, complement activation does not proceed to lysis. IgG bound to a RBC can mark the RBC for extravascular destruction, which occurs in the reticuloendothelial system (RES, also known as mononuclear phagocyte system) of primarily the spleen and liver and is effected by macrophages (Fig. 1).2
Figure 1.
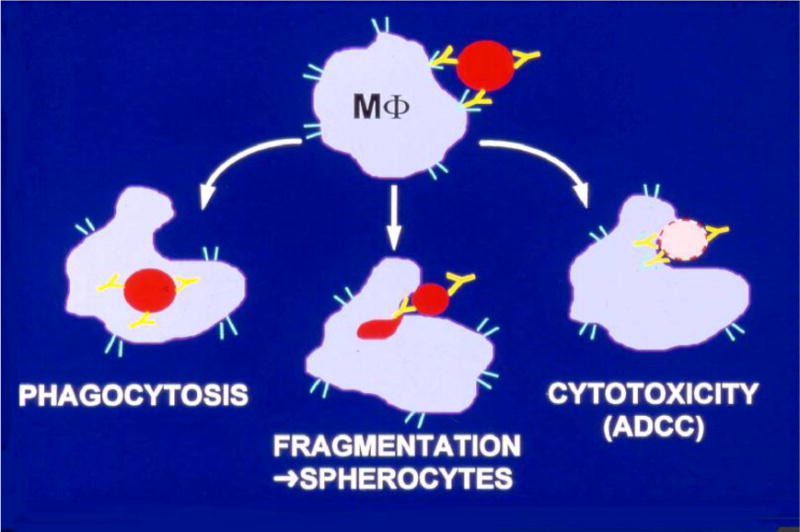
Antibody-mediated destruction or alteration of a red blood cell (RBC). An effector cell recognizes an RBC by antibodies that are bound to the RBC’s cell membrane. Three mechanisms can lead to the RBC’s destruction or alteration: (i) An RBC is engulfed by a macrophage (MΦ) and lysed intracellularly (phagocytosis). (ii) An RBC is partially phagocytized (fragmentation), but the altered RBC (spherocytes) escapes the immune attack by the macrophage (MΦ) and remains circulating. (iii) An RBC is attacked by a macrophage (MΦ) and lysed extracellularly (ADCC, antibody-dependent cell-mediated cytotoxicity). Modified from Garratty.2
In theory, IgM antibodies are expected to activate complement more readily than IgG antibodies.3 However, two IgG molecules close together on the RBC surface can activate complement. Hence, in practice the nature and distribution of antigen sites seems to be more important than immunoglobulin class, because the ability of RBC alloantibodies to bind complement is closely related to blood group specificity.4
Rate of hemolysis
The clinically important difference between the 2 mechanisms of hemolysis is the maximum rate of RBC destruction. Extravascular hemolysis is limited to 0.25 ml packed RBC/kg/hour by the capacity of the RES. For example in a 70 kg patient, 18 ml of packed RBC can be destroyed in 1 hour and more than 400 ml in 24 hours. Such an amount of cell destruction may impair the ability of the organ systems to cope with the patient’s underlying condition. While this hemolysis burdens the system, it is a rather slow process and typically not life threatening. In contrast, intravascular hemolysis can destroy 200 ml of RBC or more in 1 hour. A drop of hemoglobin by 5 g/dl may occur within hours, which can be fatal if not rapidly adjusted by compatible RBC transfusions.
Not many antibodies destroy RBC intravascularly by a complement-mediated mechanism.5 Only anti-A and anti-B destroy RBC commonly this way; other antibodies, such as anti-Jka, -Jkb, Vel, -PP1Pk (formerly -Tja), and -Lea are capable of causing intravascular lysis on rare occasions, but few others are capable of activating complement efficiently enough to form the membrane attack complex.5
IgM- or IgG-mediated intravascular hemolysis can cause anemia at 10 g/dL without any need for transfusion, but transfusion should be considered at 7 g/dl or less. Even if a patient’s hemoglobin concentration drops precipitously from 10 g/dl to 5 g/dl, there may not necessarily be an urgent need to transfuse depending on the patient’s vital signs. The situation differs drastically if the hemoglobin concentration is 7 g/dl before hemolysis and a drop to 2 g/dl is imminent; in this scenario transfusion will be needed urgently. Such clinical occurrences are not rare, but because they do not have to be routinely reported, they are not systematically recorded in aggregate form.6
In practice, the risk of reaching critically low hemoglobin concentrations will increase as patient blood management (PBM) prompts us to lower the transfusion trigger to transfuse less and patients with 7 g/dl are seen more frequently. Consequently, the clinical response time to an incipient intravascular hemolysis will be shortened in patients with significantly lower hemoglobin concentrations. These considerations exemplify that the topic of this Workshop was very timely7–12 and may become even more relevant over time. Also, the incidence of immune hemolysis induced by IVIG has increased13,14 and is of particular concern in smaller patients, especially children.
Monocytes and molecules involved in hemolysis
Cell-mediated hemolysis requires recognition of RBC and binding to monocytes or macrophages, which control extravascular hemolysis. Several proteins are involved in marking RBCs for recognition by macrophages (Table 1).15 The Fc-receptor is critical in binding RBC coated with IgG. Accumulation of activated complement proteins on the RBC surface, so effective in causing antibody-mediated intravascular hemolysis, also contributes to cell-mediated hemolysis as the 2 complement proteins C3b and iC3b are recognized by macrophages and facilitate RBC binding.
Table 1.
Examples for molecules involved in the bonding between RBC and monocytes/macrophages
| Receptor on monocytes/macrophages* | Proteins that can be bound to the RBC surface |
|---|---|
| Fc | IgG1, IgG3, IgA, possibly IgG2 |
| CR1 | C3b, iC3b† |
| CR3 | iC3b |
| CR4 | iC3b |
Monocytes, floating in the blood stream (intravascular), and macrophages, residing in tissues (extravascular), are the same cell type.
iC3b is an inactive derivative of C3b and degrades to C3d,g, which is the last step in vivo and can remain attached to the RBC surface for the rest of the RBC’s life span. In vitro, C3d,g can be further cleaved, using trypsin, to C3d.4
Fc – Fc-receptors. They exist in variations.15
CR1 – complement receptor 1, CD35, blood group system Knops
CR3 – complement receptor 3, CD11b + CD18, integrin alpha-M
CR4 – complement receptor 4, DC11c + CD18, intergrin alpha-X
Once a macrophage has bound a RBC, it has 3 ways to effect hemolysis of the RBC (Fig. 1): (i) the RBC is engulfed and destroyed inside the macrophage (MΦ); (ii) the RBC is fragmented as the macrophage nibbles fragments off the RBC membrane, leaving the rest of the RBC to eventually detach and float in the blood, where it can be visualized on a smear as a spherocyte, which may have shortened survival; and (iii) the RBC stays outside of the macrophage and is lysed by antibody-dependent cell-mediated cytotoxicity (ADCC),16,17 in which the macrophage secretes toxic substances lytic for the attached RBC.
All 3 pathways begin with the adherence of RBC to macrophages. The Monocyte Monolayer Assay (MMA) can be applied in clinical diagnostics8,18–22 and measures adherence or phagocytosis or both, but not ADCC. A few other cell types may mediate hemolysis with or without involvement of antibodies (Table 2),1,16,17,23,24–26,27–30,31–34,35–37 whose effects are not as well studied as those of monocytes. The ADCC assay, which can be either lymphocyte- or monocyte-driven,28–30 has been used to measure the 3rd pathway, but is not often applied in clinical diagnostics.38
Table 2.
Immune cells known to cause hemolysis
| Cell type | Hemolysis
|
Cell type carries Fc-receptors | |
|---|---|---|---|
| Phagocytosis & Fragmentation | ADCC | ||
| Monocytes/macrophages* | Yes1,16,17,23 | Yes23 | Yes |
| Granulocytes† | Yes24 | Yes25 | Yes |
| Dendritic cells | Yes26 | No26‡ | Yes |
| Natural Killer (NK) cells | No27§ | Yes28–30 | Yes |
| Cytotoxic T cells‖ | No31 | Yes32–34 | Yes |
Macrophage names vary depending on the tissue: Kupffer cell (liver), Langerhans cell (dermis), microglia (brain), and osteoclast (bone). They also occur as subcapsular sinusoidal or medullary macrophages (lymph node) or splenic, bone marrow, intestinal, alveolar and intraocular macrophages.35 Monocytes can differentiate into dendritic cells, which is a different cell type.36
Neutrophils, eosinophils and basophils are called mast cells when found in tissues.
There is no ADCC by dendritic cells (APC – antigen presenting cells), forcing them to engulf cells, such as RBC, necessary for their antigen processing function.
NK cells do not phagocytize RBC, but can function as phagocytes for other cell types.37
also known as ‘Killer’ cytotoxic T cells
If complement proteins attach to the RBC surface without inducing hemolysis directly or by cell recruitment, the complement degrades and the C3d,g component remains attached to the RBC for the remainder of its life span without much impairing of its survival. Up to 2 – 3 months after the event, complement-coated RBC continue to be detected by anti-C3d in the direct antiglobulin test (DAT),39 a very sensitive, if unspecific, assay for a previous hemolysis that involved the activation of complement on the RBC surface.
Pathogenicity of antibodies
The clinical effect that an antibody can exert depends on many factors, including antibody class, antibody subclass, antibody specificity, thermal range, complement activating efficiency, and affinity. Only a few of these characteristics are routinely tested.8 Even when such factors are quantified, correlation with clinical outcomes is often weak because of the complexity of the factors and their mutual interaction. Polymorphism of proteins, for instance, even when rare in a population for any single protein, may become relevant when a large number of proteins interact. Personalized medicine may allow further insight in the future.
The relationship between thermal range and complement activating efficiency is on example of this type of interaction. The thermal range of antibodies, such as cold agglutinins (often IgM, reacting at 4 °C up to 25 °C) or warm antibody (often IgG, best reactive at 37 °C but may react at 22 °C or below),40 correlates with the efficiency of complement activation, known to require antibody binding at 25 °C or above, and can be tested when indicated. Most autoimmune hemolytic anemias (AIHA) are caused by warm-reactive autoantibodies of IgG type (warm AIHA)41 and approximately 15% by cold-reactive antibodies of IgM type (CAD – cold agglutinin disease).42 Cold-induced IgG intravascular hemolytic anemia (PCH – paroxysmal cold hemoglobinuria) is now rare and based on a special type of cold-reactive (Donath-Landsteiner) antibody, binding best to RBC at less than 20 °C but activating complement efficiently above 25 °C.15 Approximately 0.1% of donors have warm-reactive autoantibodies, which are not clinically relevant when diluted in plasma pools, and approximately 1 % of donors have cold-reactive autoantibodies, most of which are clinically irrelevant. Specific tests for antibody class (IgG, IgM etc.), subclass (IgG1 etc.) and affinity are not widely applied in transfusion medicine laboratories. More reliable and clinically relevant, however, is the antibody specificity, which is widely tested in the routine clinical setting.
Antibody specificity
The antibody specificity is most informative because it defines the (cognate) antigen that the antibody binds and, as much is known about the distribution of antigens in tissues or body fluids and their quantity on the RBC membrane and other cells, specificity can therefore predict pathogenicity. A few of the 35 blood group systems recognized today are restricted to RBC only, although many are expressed on other tissues as well (Fig. 2).43–45 ABO antigens are widely expressed and often to a greater degree on tissue cells than on RBC. These antigens are also soluble and present in plasma and other bodily fluids in persons who “secrete” their ABO antigens, e.g. 80% of Europeans.
Figure 2.
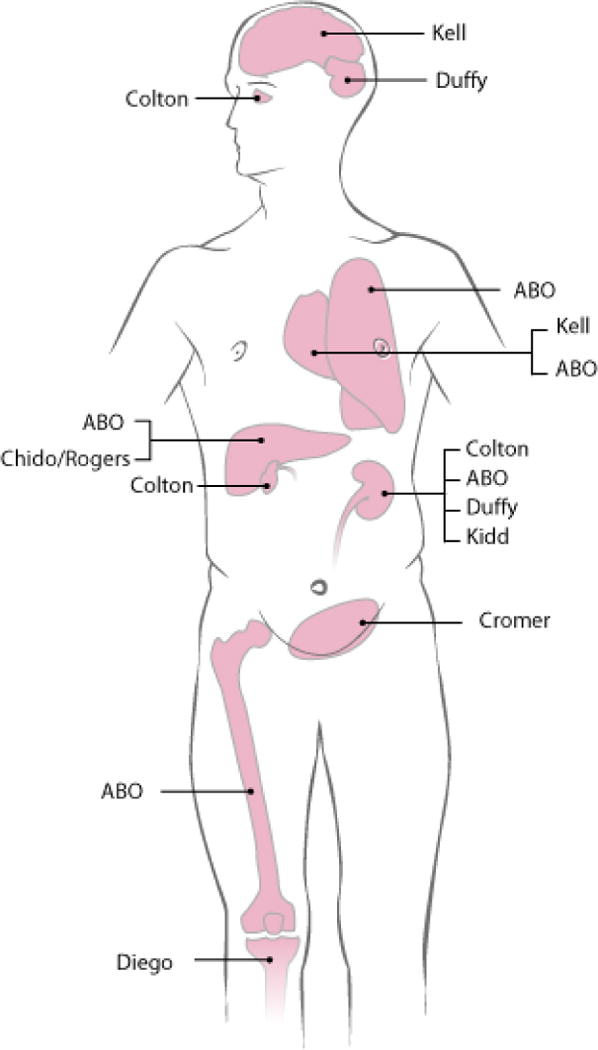
Organ distribution of blood group antigens. A schematic representation for the antigen distribution of some clinically relevant blood group systems is shown. ABO antigens are formed by carbohydrates expressed on the surfaces of many cells in the human body. When an individual carries active ABO genes, the ABO antigens are expressed on red blood cells (RBC) and even more strongly on some tissues other than RBC, such as kidney, lung, liver and intestine. The ABO antigens are also soluble in the plasma of persons and then attach passively to cells that do not express the ABO antigens by themselves.43 Modified from Nydegger et al.44 and reprinted with permission.
Immunoglobulin binding to any of the blood group antigens foreign to the carrier is called an alloantibody. Anti-A, anti-B and anti-A,B are more commonly called isoagglutinins.46,47 They were formerly labeled as hemagglutinins48,49 or isohemagglutinins46 for their agglutination effect in vitro and isolysins50 or hemolysins,48–50 if they showed hemolytic activity in vitro as well.
Currently, 35 blood group systems and 342 antigens are recognized. The antigens of 7 blood group systems are constituted by carbohydrates; 4 other blood group systems involve the complement cascade, the CR1 complement receptor and 2 proteins (CD55 and CD59) that attenuate the complement attack on the RBC surface (Table 3).51 The complement component C4 (Chido/Rodgers) is actually not synthesized by erythroid cells; rather the soluble protein is adsorbed onto the RBC surface from plasma of all persons expressing the protein.
Table 3.
Known blood group systems constituted by carbohydrates or complement-related proteins
| Blood group
|
Carbohydrate or involved protein | Defined antigens
|
||
|---|---|---|---|---|
| ISBT number | System name | Number | Examples | |
| 1 | ABO | Sugar | 4 | A, B, A,B and A1 |
| 3 | P1PK | Sugar | 4 | P1PK1, P1PK3, NOR |
| 7 | LE | Sugar | 6 | Lea, Leb |
| 17 | Chido/Rodgers | C4a/C4b | 9 | Ch1, Rg1 |
| 18 | H | Sugar | 1 | H |
| 21 | Cromer | CD55* | 18 | Cra |
| 22 | Knops | CD35† | 9 | Kna, McCa, Sl1, Yka |
| 27 | I | Sugar | 1 | I |
| 28 | GLOB | Sugar | 1 | P |
| 31 | FORS | Sugar | 1 | FORS1 |
| 35 | CD59 | CD59‡ | 1 | CD59.1 |
DAF – Decay Accelerating Factor
CR1 – complement receptor 1
previously described as HRF20 – homologous restriction factor, MIRL – membrane inhibitor of reactive hemolysis, and MACIF – membrane attack complex inhibitory factor
RBC lacking Cromer (CD55)52 or CD5953,54 cannot attenuate a complement attack and are more prone to hemolysis.55 Patients with inherited deficiencies are rare, but those harboring a paroxysmal nocturnal hemoglobinuria (PNH) clone with a subclinical or smoldering susceptibility to hemolysis are more common, lack the 2 aforementioned complement regulators and may manifest themselves when their pathologic RBC are exposed to an antibody challenge,15 such as by IVIG infusion.
Antibody quantity
While not the sole determinant of an antibody’s pathogenicity, the clinical effect of any particular antibody will invariably be greater when it presents in larger quantity, typically gauged as a “high titer”.47 A “titer” is defined by the agglutination strength (reciprocal of serial dilution) of an antibody preparation.14 Much data have been gathered recently for the titers of ABO antibodies in blood products7,13,14,56–58 mainly in an effort to limit the adverse effects of platelet apheresis products.10,59 Few donors have high titers (non-Europeans or previously pregnant), which may vary over time,60 but those with excessive titers contribute inordinately to adverse effects12,61 and eventually to the ultimate amount of ABO antibodies in IVIG products.
Only 1% of healthy donors may be expected to have antibodies against common blood group antigens other than ABO. These antibodies can be routinely tested for and products eliminated if titers are high or clinically relevant.7,58,59,62,63 Other healthy donors may have antibodies to blood group antigens of low prevalence (1% or less in the population).64 The titers can be high and clinically relevant.65,66 Because antibodies to a low prevalence antigen are not systematically screened,64 they are bound to occur in large donor pools. Such an IVIG batch administered to a large patient cohort can eventually result in an inadvertent match between the antibody and a patient with the low prevalence antigen. Antibody-mediated hemolysis by non-ABO antibodies can easily be missed without a proper evaluation of adverse effects.67,68 Hitherto unknown side effects continue to be recognized.69
While antibodies cannot be eliminated by pooling, they do become diluted in large pools of donor plasma and titers are reduced in predictable ways. The limit of dilution is defined by the frequency of donors with antibodies,65 particularly donors with high titers.12,47 Enlarging the donor pool can only reduce the titers to some extent. Eventually one donor’s plasma carrying a high titer is added to the pool and thwarts the effect of dilution. The process to reduce antibodies becomes more efficient if donors are tested for antibodies;70 then, all donors with high titer or against low prevalence antigens can be excluded.
ABO antigens and A1 and A2 phenotypes
The ABO antigens A and B exist in several variations on the RBC surface.46,71 The A or B type 1 antigens can be acquired from the plasma onto RBC in “secretor” persons. A and B type 2 antigens are the dominant ABO antigens on RBC and the primary target of clinically significant isoagglutinins. The A type 3 exists on all A cells (there is no B antigen equivalent) and A type 4 only on those of the A1 phenotype; antibodies directed against these low quantity RBC antigens are clinically insignificant.
The chemical basis of the A1 and A2 phenotypes has been resolved recently.72 While the A1 phenotype is characterized by increased amounts of A antigen, there also exists a minor structural difference compared to the A2 phenotype: A1 individuals carry small quantities of A type 4 antigen and A2 individuals appear to lack this antigen (Fig. 3).71,72
Figure 3.
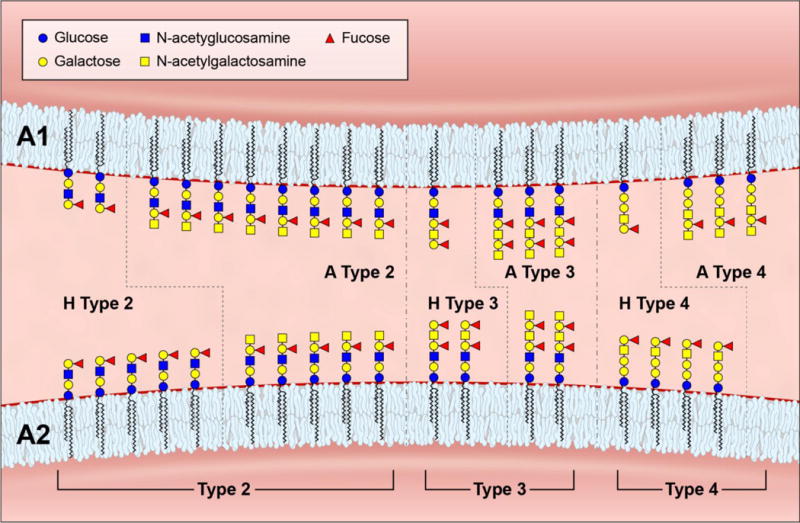
Schematic simplified model of the glycosylation patterns in the A1 and A2 blood group phenotypes. The elongated, branched and repetitive carbohydrate structures on the RBC surface end in various types of H and A antigens: In an A1 phenotype (upper panel), most of the H antigens (types 2 to 4) are transformed to their A antigen analogues (types 2 to 4). The A type 4 antigen represents the A1 antigen.72 In an A2 phenotype (lower panel), many H type 2 antigens remain unchanged and are the targets of anti-H lectins; also, most H type 4 antigens remain unchanged. Hence, the number of A type 2 and 3 antigens is reduced and the A type 4 antigen is lacking in an A2 phenotype. The Secretor/Lewis antigens (H and A type 1 antigens, not shown) are adsorbed from plasma onto the RBC surface in Le(a-b-) secretors and occur in ratios like the H and A type 4 antigens. The antigens are shown as glycolipids in their simplest form; the same antigens also exist on glycoproteins and commonly as large extended glycans with greater than 60 residues (not shown). The ratios among the antigens are not absolute but for demonstration purposes only. Modified from Flegel and Wagner71 according to Svensson et al.72
The antibody commonly known as anti-A1 is directed against the A type 4 antigen and could be used to differentiate the A1 and A2 phenotypes. However, the routinely used laboratory reagents are usually diluted lectins that discriminate the A1 and A2 phenotypes purely on a quantitative basis, with A1 individuals having more A antigens and less H antigens than A2 individuals. A2 is a blood group phenotype, not an antigen. Reagents designed to detect the A2 phenotype are in fact anti-H reagents and a label “anti-A2 reagent” is formally incorrect. The amounts of the H and A types 2 and 3 and the presence of A type 4 determine the difference between the A1 (Fig. 3, upper panel) and A2 phenotypes (Fig. 3, lower panel).
Blood donors of blood group phenotypes O or B always have anti-A directed against all variations of the A antigen. Donors of A2 or A2B phenotype can have anti-A1 (anti-A type 4), sometimes of IgG type.73 Most anti-A1 are not clinically relevant. Anti-A,B binds to the common regions shared between the A and B antigens. A plasma pool from a large number of donors would naturally contain a mixture of antibodies to all variations of the A and B antigens (Table 3). Hence, ABO antigen minutiae74 may be considered if antibodies are to be removed completely from an IVIG product by absorption to ABO antigens. Fortunately, most of these antibodies show significant cross-reactivity and a single adsorbent may remove them, be they directed against A types 1, 2, 3 and 4 or B types 1 and 2.
Complement and immune cells
Apart from antibodies, the 2 remaining critical players in hemolysis are complement and immune cells (Table 2). Activation of complement proteins and their binding to the RBC surface is the major cause of intravascular hemolysis,39,75 and can also lead to permanent extravascular sequestration by the reticuloendothial system (RES) with subsequent phagocytosis by macrophages. A temporary sequestration in the RES may shorten RBC survival. If the complement activation is aborted and only C3d,g residues remain on the RBC surface (Table 1), the RBC survival is considered normal, although C3d has been reported to facilitate phagocytosis by monocytes.76,77 Complement fragments induce a multitude of other effects including cell activation, which may led to life-threatening systemic inflammatory response syndrome (SIRS).78
Cell-mediated hemolysis is modulated by the activity of the RES and its macrophages. The macrophages can be hyperactive in clinical situations such as infection, inflammation,79 autoimmune hemolysis, sickle cell disease, thalassemia, or cytokine release.8,78,79 They can become hypoactive, when their Fc-receptors are blocked by immune complexes (systemic lupus erythematosus), maternal anti-HLA or by certain drugs (corticosteroids).
Monocyte Monolayer Assay (MMA)
Originally described in the early 1980s, the MMA was applied to determine the pathogenicity of alloantibodies18,19 and autoantibodies.20,21 Optimization and validation allowed the assay to distinguish between clinically significant and insignificant alloantibodies of IgG type.80 IgM cannot itself provide a second signal for phagocytosis (Table 1), and IgM-mediated RBC-bound complement is an inefficient stimulus for phagocytosis.4
Dr. Garratty summarized his 20 years’ experience with the MMA in 2004:81 An MMA was considered negative when ≤ 5% reactivity was observed, that is monocytes with adhering RBC or phagocytized RBC or both. A negative MMA indicated that incompatible blood could be given without risk of an overt hemolytic transfusion reaction, although it does not guarantee a normal long-term survival of the transfused RBC. Only specialized laboratories offer MMA, and even health care systems in large Western countries may have no routine access to the assay. Assays for hemolysis induced by other cells (Table 2) are not readily available for clinical use and, even if antibody-mediated, hemolysis effected by cells other than monocytes and macrophages remains difficult to gauge. Adverse reactions to IVIG that cannot be explained solely due to ABO incompatibility may yield insight into the other cellular mechanisms, if systematically evaluated by specialized laboratories.7,8,79 Such research might improve our understanding of hemolysis and contribute to blood product quality generally, even beyond IVIG products.
Transfusion reactions caused by non-ABO antibodies
The appearance of a non-ABO antibody within one month of a transfusion may represent either its passive infusion from the donor plasma, or an anamnestic immune response in the recipient. The latter phenomenon is also known as a delayed hemolytic transfusion reaction (DHTR) or, if there is no apparent hemolysis, a delayed serologic transfusion reaction (DSTR). The proportion of anamnestic immune responses to transfusions that cause hemolysis is not always easy to determine, but has been estimated in several studies to be approximately one third.82,83 However, the presence of hemolysis can be obscured by other disorders with laboratory signs mimicking hemolysis, for example liver disease, or by blood loss.39 When hemolysis does occur, it is often considered to be of minimal clinical consequence, but clinicians may fail to recognize the subtle effects such hemolysis may have on the patient’s underlying condition. Only if the antibody-mediated hemolysis is the single or clearly attributable factor for an adverse effect or death, does the transfusion reaction become reportable to the Food and Drug Administration (FDA). This may explain a possible under-reporting of fatalities secondary to non-ABO antibodies, as is widely perceived by practitioners of transfusion medicine.9,84
Despite a possible under-reporting, non-ABO antibodies still represent the second most commonly reported cause of fatal transfusion reactions in the hemovigilance systems of many large Western countries,9,85–87 including the US.88 If under-reporting was considered,9 non-ABO antibodies might represent the most common cause of all fatal transfusion reactions today, exceeding by far the fatalities due to transfusion transmitted infections.
Parenthetically, in the US the most common cause of fatal transfusion reactions is transfusion-related acute lung injury (TRALI), a clinical syndrome reportable without any evidence of antibodies in the blood product.89 Because no causative agent needs to be discerned in the product, there may be a fatality properly reported as TRALI and attributed to blood products which did not actually cause any transfusion reaction. Such false attributions cannot occur with antibody-mediated hemolysis in the current reporting practice, and it may therefore be conjectured that TRALI is in fact not a more common cause of transfusion-related mortality than immune-mediated hemolysis.
Hemolysis caused by ABO antibodies
Transfusion of ABO incompatible RBC
The third most common cause of fatal transfusion reactions are hemolytic transfusion reactions caused by ABO antibodies following ABO incompatible RBC transfusion.88 These could theoretically be eliminated completely, as all ABO incompatible RBC transfusions occur by error.90 Such transfusions are lethal for the patient in approximately 10% of cases.91 Fortunately, approximately half of the involved patients have no adverse effects91 and remain asymptomatic.87,92 The clinical outcome depends very much on the volume of ABO incompatible RBC transfused. When recognized early by a severe reaction, and stopped after a few milliliters, survival is excellent. RBC volumes of greater than 50 mL are associated with a lethality of 20%.91 If transfusion continues because reactions are masked, such as by anesthesia, severe and lethal transfusion reactions become very likely. Even the many patients who survive may well suffer permanent organ damage, which is important to consider. Regarding any ABO incompatible exposure, the objective is not only that the patient survives or has no obvious reportable side effects; we aim to avoid all permanent side effects from incompatible ABO antigens and antibodies.
Transfusion of ABO incompatible plasma or IVIG
Exposure to the antibodies of mixed Ig types (IgM, IgG and IgA)93 in ABO incompatible plasma is rarely fatal.12,61,62 The risk from IVIG products may differ as the predominant antibodies are IgG type. When incompatible antibodies are transfused by plasma, platelet products or IVIG, the soluble A and B antigens compete with the cellular A and B antigens in “secretor” persons, less so in “non-secretor” persons,8,11,94,95 and may inhibit the hemolytic effect of IgM antibodies, less so of IgG antibodies,96 in some patients.79 Incompatible ABO antibodies are also captured by various tissue cells (Fig. 2), which further dampens a hemolytic effect. However, many clinical effects remain unknown;8,46 whether they may reduce glycosylated plasma protein concentrations, generate immune complexes,97 or damage endothelia is yet to be demonstrated.98
The theoretical risk of hemolysis following the infusion of ABO incompatible antibodies may be stratified based on the recipient’s ABO phenotype and the attenuation by soluble A and B antigens in secretors. This predicted risk does not differ much among ethnicities, despite the wide variability of ABO and Secretor phenotypes (Table 4).43,45,95,99,100,101 Up to 40% of recipients may have a high risk to develop hemolysis according to this model, while more than 40% have no risk at all. The risk can be evaluated individually by ABO, FUT2 (SE)99,100 and FUT3 (LE) genotyping99 using molecular immunohematology methods, which are particularly useful after recent transfusions.102
Table 4.
Risk model for hemolysis caused by anti-A and anti-B in blood products, such as IVIG, based on ABO and Secretor phenotypes in the recipient
| Risk | Recipient ABO and Secretor phenotype | Population*
|
|||||
|---|---|---|---|---|---|---|---|
| East Asian
|
European
|
Sub-Saharan African
|
|||||
| Frequency | Total | Frequency | Total | Frequency | Total | ||
| High | A1 or A1B, non-secretor† | 3% | 35% | 7% | 40% | 7% | 31% |
| A2, B or A2B, non-secretor | 3% | 4% | 9% | ||||
| A1 or A1B, secretor | 29% | 29% | 15% | ||||
| Low | A2, B or A2B, secretor | 22% | 22% | 16% | 16% | 20% | 20% |
| None | O, secretor or non-secretor | 43% | 43% | 44% | 44% | 49% | 49% |
| Total | 100% | 100% | 100% | ||||
Calculations based on published population data for ABO and Secretor genes: Secretor phenotype in sub-Saharan Africans was set at 90% (Europeans 80%, East Asians 90%99).95,100
The highest risk in the general population may occur in non-secretor (genotype FUT2*se homozygous) recipients of A1 Le(a-b-) phenotype (genotype ABO*A1 homo- or heterozygous and FUT3*le homoyzgous).43,45,101 An even higher risk is found in patients carrying ABO antigens on their RBC but not on their tissue cells nor soluble in their plasma,11 which is a rare acquired condition such as after hematopoietic progenitor cell transplantation.8
Do we understand immune RBC destruction?
Dr. Garratty summarized his experience once by stating, “Nevertheless, after more than 30 years researching this area, I am sometimes embarrassed to realize how much I cannot explain” 2 and listed questions that are pending answers by ongoing research.
Why do some auto- and alloantibodies in blood products with low titers cause severe hemolysis, whereas some antibodies of the same specificity in blood products with high titers do not? Do differences in the clinical severity and treatment response relate to the relative efficiency of macrophage-induced phagocytosis versus cytotoxicity? Despite many proposed mechanisms, why is there no conclusive test predicting a hyperhemolytic transfusion reaction in patients with sickle cell disease?
In addition, how do we define “clinical significance”? Is it safe to always consider delayed serologic transfusion reactions as clinically in significant? What defines the threshold to consider a hemolytic transfusion reaction as clinical significant? After all, both types of transfusion reactions damage tissue to some degree and it depends on the patients and their clinical conditions as to how well the inflicted damage is tolerated acutely and long term.
Concluding remarks
Most reported hemolytic transfusion reactions associated with IVIG are due to ABO alloantibodies, and although reducing the titer of the anti-A and anti-B in IVIG will help lower the number of cases with hemolytic anemia, there will remain a few cases associated with low titer in the blood product. The only way to stop ABO mediated hemolytic transfusion reactions is to have no antibodies against any of the 4 ABO antigens A, B, A,B and A1 in the blood products. Perhaps cases of IVIG-associated hemolysis will continue to occur even if the products contain no anti-A and anti-B? It may turn out that there are non-ABO antibodies or other factors in the products causing hemolysis, and this possibility should eventually be explored.
Supplementary Material
Acknowledgments
The author dedicates this publication to the memory of Professor George Garratty PhD FRCPath, who died on March 17, 2014 at the age of 78. Being an iconic figure in the areas of serology and hemolysis, George had a lifetime of contributions to these fields until his last year of life. He shared much of his insight as editor, thus contributing to the improvement of very many publications. George will be remembered most of all by his sincerity.
The author expresses his appreciation to the FDA for the honor of having been chosen to deliver Dr Garratty’s presentation, who had to decline participation on the weekend before the Workshop, and thanks Stephen M Henry and Franz F Wagner for advice in preparing Figure 3; and also Donald R Branch, Gregory A Denomme, Harvey G Klein, Urs E Nydegger, Kshitij Srivastava and Ingeborg von Zabern for review and helpful comments; Allan B Hoofring and David A Stiles for help with Figure 3; and Elizabeth J Furlong for English edits.
This work was supported by the Intramural Research Program of the NIH Clinical Center.
Footnotes
Conflict of interest disclosure: The author does not have a conflict of interest relevant to this article.
Publisher's Disclaimer: Statement of Disclaimer: The views expressed do not necessarily represent the view of the National Institutes of Health, the Department of Health and Human Services, or the U.S. Federal Government.
Authorship contribution: This manuscript represents the author’s thoughts on the subject and his interpretation of a set of slides prepared by Dr George Garratty (Supplementary Material) as presented in the transcript of the Strategies to Address Hemolytic Complications of Immune Globulin Infusions Workshop organized by FDA/PPTA/NHLBI in Bethesda on January 28, 2014.
Additional Supporting Information may be found in the online version of this article:
Slide set. George Garratty: Pathogenesis and mechanisms of immune hemolytic anemia. January 2014
References
- 1.Garratty G. Erythrocyte-bound complement components in health and disease. Prog Clin Biol Res. 1980;43:133–55. [PubMed] [Google Scholar]
- 2.Garratty G. The James Blundell Award Lecture 2007: do we really understand immune red cell destruction? Transfus Med. 2008;18:321–34. doi: 10.1111/j.1365-3148.2008.00891.x. [DOI] [PubMed] [Google Scholar]
- 3.Garratty G. The significance of IgG on the red cell surface. Transfus Med Rev. 1987;1:47–57. doi: 10.1016/s0887-7963(87)70005-4. [DOI] [PubMed] [Google Scholar]
- 4.Freedman J. The significance of complement on the red cell surface. Transfus Med Rev. 1987;1:58–70. doi: 10.1016/s0887-7963(87)70006-6. [DOI] [PubMed] [Google Scholar]
- 5.Petz LD, Garratty G. Immune Hemolytic Anemias. Churchill Livingstone/Elsevier Science; 2004. pp. 133–4. [Google Scholar]
- 6.Flegel WA, Natanson C, Klein HG. Does prolonged storage of red blood cells cause harm? Br J Haematol. 2014;165:3–16. doi: 10.1111/bjh.12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinti I, Pulvirenti F, Milito C, Granata G, Giovannetti G, La Marra F, Pesce AM, Farrugia A, Coluzzi S, Girelli G. Hemolysis in patients with antibody deficiencies on immunoglobulin replacement treatment. Transfusion. 2014 doi: 10.1111/trf.12939. [DOI] [PubMed] [Google Scholar]
- 8.Michelis FV, Branch DR, Scovell I, Bloch E, Pendergrast J, Lipton JH, Cserti-Gazdewich CM. Acute hemolysis after intravenous immunoglobulin amid host factors of ABO-mismatched bone marrow transplantation, inflammation, and activated mononuclear phagocytes. Transfusion. 2014;54:681–90. doi: 10.1111/trf.12329. [DOI] [PubMed] [Google Scholar]
- 9.Desborough MJ, Miller J, Thorpe SJ, Murphy MF, Misbah SA. Intravenous immunoglobulin-induced haemolysis: a case report and review of the literature. Transfus Med. 2014;24:219–26. doi: 10.1111/tme.12083. [DOI] [PubMed] [Google Scholar]
- 10.Fontaine MJ, Mills AM, Weiss S, Hong WJ, Viele M, Goodnough LT. How we treat: risk mitigation for ABO-incompatible plasma in plateletpheresis products. Transfusion. 2012;52:2081–5. doi: 10.1111/j.1537-2995.2012.03596.x. [DOI] [PubMed] [Google Scholar]
- 11.Pendergrast JM, Pavenski K, Hannach B. Does deficiency of plasma A/B substances increase the risk of IVIG-mediated hemolysis? Transfusion. 2007;47:197A. [Google Scholar]
- 12.Daniel-Johnson J, Leitman S, Klein H, Alter H, Lee-Stroka A, Scheinberg P, Pantin J, Quillen K. Probiotic-associated high-titer anti-B in a group A platelet donor as a cause of severe hemolytic transfusion reactions. Transfusion. 2009;49:1845–9. doi: 10.1111/j.1537-2995.2009.02208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kahwaji J, Barker E, Pepkowitz S, Klapper E, Villicana R, Peng A, Chang R, Jordan SC, Vo AA. Acute hemolysis after high-dose intravenous immunoglobulin therapy in highly HLA sensitized patients. Clin J Am Soc Nephrol. 2009;4:1993–7. doi: 10.2215/CJN.04540709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakem B, Matozan K, Nydegger UE, Weigel G, Griesmacher A, Risch L. Anti-red blood cell antibodies, free light chains, and antiphospholipid antibodies in intravenous immunoglobulin preparations. Isr Med Assoc J. 2013;15:617–21. [PubMed] [Google Scholar]
- 15.Rosse WF, Hillmen P, Schreiber AD. Immune-mediated hemolytic anemia. Hematology Am Soc Hematol Educ Program. 2004:48–62. doi: 10.1182/asheducation-2004.1.48. [DOI] [PubMed] [Google Scholar]
- 16.Perlmann P, Holm G. Cytotoxic effects of lymphoid cells in vitro. Adv Immunol. 1969;11:117–93. doi: 10.1016/s0065-2776(08)60479-4. [DOI] [PubMed] [Google Scholar]
- 17.Holm G. Lysis of antibody-treated human erythrocytes by human leukocytes and macrophages in tissue culture. Int Arch Allergy Appl Immunol. 1972;43:671–82. doi: 10.1159/000230883. [DOI] [PubMed] [Google Scholar]
- 18.Hunt JS, Beck ML, Hardman JT, Tegtmeier GE, Bayer WL. Characterization of human erythrocyte alloantibodies by IgG subclass and monocyte interaction. Am J Clin Pathol. 1980;74:259–64. doi: 10.1093/ajcp/74.3.259. [DOI] [PubMed] [Google Scholar]
- 19.Schanfield MS, Stevens JO, Bauman D. The detection of clinically significant erythrocyte alloantibodies using a human mononuclear phagocyte assay. Transfusion. 1981;21:571–6. doi: 10.1046/j.1537-2995.1981.21582040822.x. [DOI] [PubMed] [Google Scholar]
- 20.Conley CL, Lippman SM, Ness PM, Petz LD, Branch DR, Gallagher MT. Autoimmune hemolytic anemia with reticulocytopenia and erythroid marrow. N Engl J Med. 1982;306:281–6. doi: 10.1056/NEJM198202043060507. [DOI] [PubMed] [Google Scholar]
- 21.Gallagher MT, Branch DR, Mison A, Petz LD. Evaluation of reticuloendothelial function in autoimmune hemolytic anemia using an in vitro assay of monocyte-macrophage interaction with erythrocytes. Exp Hematol. 1983;11:82–9. [PubMed] [Google Scholar]
- 22.Garratty G. Predicting the clinical significance of red cell antibodies with in vitro cellular assays. Transfus Med Rev. 1990;4:297–312. doi: 10.1016/s0887-7963(90)70272-6. [DOI] [PubMed] [Google Scholar]
- 23.van der Meulen FW, van der Hart M, Fleer A, von dem Borne AE, Engelfriet CP, van Loghem JJ. The role of adherence to human mononuclear phagocytes in the destruction of red cells sensitized with non-complement binding IgG antibodies. Br J Haematol. 1978;38:541–9. doi: 10.1111/j.1365-2141.1978.tb01079.x. [DOI] [PubMed] [Google Scholar]
- 24.Hernandez JA, Steane SM. Erythrophagocytosis by segmented neutrophils in paroxysmal cold hemoglobinuria. Am J Clin Pathol. 1984;81:787–9. doi: 10.1093/ajcp/81.6.787. [DOI] [PubMed] [Google Scholar]
- 25.Wagner C, Iking-Konert C, Denefleh B, Stegmaier S, Hug F, Hansch GM. Granzyme B and perforin: constitutive expression in human polymorphonuclear neutrophils. Blood. 2004;103:1099–104. doi: 10.1182/blood-2003-04-1069. [DOI] [PubMed] [Google Scholar]
- 26.Fanger NA, Voigtlaender D, Liu C, Swink S, Wardwell K, Fisher J, Graziano RF, Pfefferkorn LC, Guyre PM. Characterization of expression, cytokine regulation, and effector function of the high affinity IgG receptor Fc gamma RI (CD64) expressed on human blood dendritic cells. J Immunol. 1997;158:3090–8. [PubMed] [Google Scholar]
- 27.Roos D, Spits H, Hack CE. Innate immunity – phagocytes, natural killer cells and the complement system. In: Nijkamp FP, Parnham MJ, editors. Principles of Immunopharmacology. Basel: Birkhäuser Verlag; 2005. pp. 63–80. [Google Scholar]
- 28.Urbaniak SJ. Lymphoid cell dependent (K-cell) lysis of human erythrocytes sensitized with Rhesus alloantibodies. Br J Haematol. 1976;33:409–13. doi: 10.1111/j.1365-2141.1976.tb03558.x. [DOI] [PubMed] [Google Scholar]
- 29.Northoff H, Grätz W, Schultze D. Anti-D vermittelte zelluläre Zytotoxizität. Ärztl Lab. 1977;23:481–6. [Google Scholar]
- 30.Northoff H, Kluge A, Resch K. Antibody dependent cellular cytotoxicity (ADCC) against human erythrocytes, mediated by human blood group alloantibodies: a model for the role of antigen density in target cell lysis. Z Immunitätsforsch. 1978;154:15–33. [PubMed] [Google Scholar]
- 31.Groscurth P, Filgueira L. Killing mechanisms of cytotoxic T lymphocytes. News Physiol Sci. 1998;13:17–21. doi: 10.1152/physiologyonline.1998.13.1.17. [DOI] [PubMed] [Google Scholar]
- 32.Handwerger BS, Kay NE, Douglas SD. Lymphocyte-mediated antibody-dependent cytolysis: role in immune hemolysis. Vox Sang. 1978;34:276–80. doi: 10.1111/j.1423-0410.1978.tb02482.x. [DOI] [PubMed] [Google Scholar]
- 33.Olovnikova NI, Belkina EV, Nikolaeva TL, Miterev GY, Chertkov IL. Lymphocyte antibody-dependent cytotoxicity test for evaluation of clinical role of monoclonal anti-D-antibodies for prevention of rhesus sensitization. Bull Exp Biol Med. 2006;141:57–61. doi: 10.1007/s10517-006-0093-4. [DOI] [PubMed] [Google Scholar]
- 34.Kurlander RJ, Rosse WF, Ferreira E. Quantitative evaluation of antibody-dependent lymphocyte-mediated lysis of human red cells. Am J Hematol. 1979;6:295–311. doi: 10.1002/ajh.2830060402. [DOI] [PubMed] [Google Scholar]
- 35.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohgimoto K, Ohgimoto S, Ihara T, Mizuta H, Ishido S, Ayata M, Ogura H, Hotta H. Difference in production of infectious wild-type measles and vaccine viruses in monocyte-derived dendritic cells. Virus Res. 2007;123:1–8. doi: 10.1016/j.virusres.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Voigt J, Hunniger K, Bouzani M, Jacobsen ID, Barz D, Hube B, Loffler J, Kurzai O. Human natural killer cells acting as phagocytes against Candida albicans and mounting an inflammatory response that modulates neutrophil antifungal activity. J Infect Dis. 2014;209:616–26. doi: 10.1093/infdis/jit574. [DOI] [PubMed] [Google Scholar]
- 38.Zupanska B. Assays to predict the clinical significance of blood group antibodies. Curr Opin Hematol. 1998;5:412–6. doi: 10.1097/00062752-199811000-00010. [DOI] [PubMed] [Google Scholar]
- 39.Salama A, Mueller-Eckhardt C. Delayed hemolytic transfusion reactions; evidence for complement activation involving allogeneic and autologous red cells. Transfusion. 1984;24:188–93. doi: 10.1046/j.1537-2995.1984.24384225018.x. [DOI] [PubMed] [Google Scholar]
- 40.Arndt P, Garratty G. Evaluation of the optimal incubation temperature for detecting certain IgG antibodies with potential clinical significance. Transfusion. 1988;28:210–3. doi: 10.1046/j.1537-2995.1988.28388219144.x. [DOI] [PubMed] [Google Scholar]
- 41.Pruss A, Salama A, Ahrens N, Hansen A, Kiesewetter H, Koscielny J, Dorner T. Immune hemolysis-serological and clinical aspects. Clin Exp Med. 2003;3:55–64. doi: 10.1007/s10238-003-0009-4. [DOI] [PubMed] [Google Scholar]
- 42.Swiecicki PL, Hegerova LT, Gertz MA. Cold agglutinin disease. Blood. 2013;122:1114–21. doi: 10.1182/blood-2013-02-474437. [DOI] [PubMed] [Google Scholar]
- 43.Kelton JG, Hamid C, Aker S, Blajchman MA. The amount of blood group A substance on platelets is proportional to the amount in the plasma. Blood. 1982;59:980–5. [PubMed] [Google Scholar]
- 44.Nydegger UE, Tevaearai H, Berdat P, Rieben R, Carrel T, Mohacsi P, Flegel WA. Histo-blood group antigens as allo- and autoantigens. Ann N Y Acad Sci. 2005;1050:40–51. doi: 10.1196/annals.1313.006. [DOI] [PubMed] [Google Scholar]
- 45.Rachkewich RA, Crookston MC, Tilley CA, Wherrett JR. Evidence that blood group A antigen on lymphocytes is derived from the plasma. J Immunogenet. 1978;5:25–9. doi: 10.1111/j.1744-313x.1978.tb00627.x. [DOI] [PubMed] [Google Scholar]
- 46.Branch DR. Anti-A and anti-B: what are they and where do they come from? Transfusion. 2015;55 doi: 10.1111/trf.13087. this issue. [DOI] [PubMed] [Google Scholar]
- 47.Siani B, Willimann K, Wymann S, Antunes AM, Widmer E. Donor screening reduces the isoagglutinin titer in IgG products. Transfusion. 2015;55 doi: 10.1111/trf.13095. this issue. [DOI] [PubMed] [Google Scholar]
- 48.Landsteiner K. Zur Kenntnis der antifermentativen, lytischen und agglutinierenden Wirkungen des Blutserums und der Lymphe. Zbl Bakt Hyg I Abt Orig A. 1900;27:357–62. [Google Scholar]
- 49.Ford WW, Halsey JT. Contributions to the study of hemagglutinins and hemolysins. J Med Res. 1904;11:403–25. [PMC free article] [PubMed] [Google Scholar]
- 50.Daufi L, Rondell P. Universality of anti-A and anti-B human isolysins. Vox Sang. 1971;21:81–5. doi: 10.1111/j.1423-0410.1971.tb00561.x. [DOI] [PubMed] [Google Scholar]
- 51.Anliker M, von Zabern I, Hochsmann B, Kyrieleis H, Dohna-Schwake C, Flegel WA, Schrezenmeier H, Weinstock C. A new blood group antigen is defined by anti-CD59, detected in a CD59-deficient patient. Transfusion. 2014;54:1817–22. doi: 10.1111/trf.12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Telen MJ, Hall SE, Green AM, Moulds JJ, Rosse WF. Identification of human erythrocyte blood group antigens on decay-accelerating factor (DAF) and an erythrocyte phenotype negative for DAF. J Exp Med. 1988;167:1993–8. doi: 10.1084/jem.167.6.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Motoyama N, Okada N, Yamashina M, Okada H. Paroxysmal nocturnal hemoglobinuria due to hereditary nucleotide deletion in the HRF20 (CD59) gene. Eur J Immunol. 1992;22:2669–73. doi: 10.1002/eji.1830221029. [DOI] [PubMed] [Google Scholar]
- 54.Hochsmann B, Dohna-Schwake C, Kyrieleis HA, Pannicke U, Schrezenmeier H. Targeted therapy with eculizumab for inherited CD59 deficiency. N Engl J Med. 2014;370:90–2. doi: 10.1056/NEJMc1308104. [DOI] [PubMed] [Google Scholar]
- 55.Telen MJ. Glycosyl phosphatidylinositol-linked blood group antigens and paroxysmal nocturnal hemoglobinuria. Transfus Clin Biol. 1995;2:277–90. doi: 10.1016/s1246-7820(05)80094-1. [DOI] [PubMed] [Google Scholar]
- 56.Cooling L. ABO and platelet transfusion therapy. Immunohematology. 2007;23:20–33. [PubMed] [Google Scholar]
- 57.Perry H, Henry S. Training students in serologic reaction grading increased perception of self-efficacy and ability to recognize serologic reactions but decreased grading accuracy. Transfusion. 2015;55 doi: 10.1111/trf.12985. in press. [DOI] [PubMed] [Google Scholar]
- 58.Hill EA, Bryant BJ. Comparison of antibody titers in donor specimens and associated AS-1 leukoreduced donor units. Transfusion. 2014;54:1580–4. doi: 10.1111/trf.12486. [DOI] [PubMed] [Google Scholar]
- 59.Quillen K, Sheldon SL, Daniel-Johnson JA, Lee-Stroka AH, Flegel WA. A practical strategy to reduce the risk of passive hemolysis by screening plateletpheresis donors for high-titer ABO antibodies. Transfusion. 2011;51:92–6. doi: 10.1111/j.1537-2995.2010.02759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Henry S, Clark P, Woodfield G. High titre IgG ABO antibodies in group O Polynesian and European blood donors: incidence, variability, racial and gender differences. NZ J Med Lab Science. 1995;49:164–5. [Google Scholar]
- 61.Pierce RN, Reich LM, Mayer K. Hemolysis following platelet transfusions from ABO-incompatible donors. Transfusion. 1985;25:60–2. doi: 10.1046/j.1537-2995.1985.25185116506.x. [DOI] [PubMed] [Google Scholar]
- 62.Stainsby D. ABO incompatible transfusions–experience from the UK Serious Hazards of Transfusion (SHOT) scheme Transfusions ABO incompatible. Transfus Clin Biol. 2005;12:385–8. doi: 10.1016/j.tracli.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 63.Thorpe SJ, Fox BJ, Dolman CD, Lawrence J, Thorpe R. Batches of intravenous immunoglobulin associated with adverse reactions in recipients contain atypically high anti-Rh D activity. Vox Sang. 2003;85:80–4. doi: 10.1046/j.1423-0410.2003.00336.x. [DOI] [PubMed] [Google Scholar]
- 64.Garratty G. How concerned should we be about missing antibodies to low incidence antigens? Transfusion. 2003;43:844–7. doi: 10.1046/j.1537-2995.2003.00492.x. [DOI] [PubMed] [Google Scholar]
- 65.Cherian G, Search S, Thomas E, Poole J, Davies SV, Massey E. An acute haemolytic transfusion reaction caused by anti-Wr. Transfus Med. 2007;17:312–4. doi: 10.1111/j.1365-3148.2007.00739.x. [DOI] [PubMed] [Google Scholar]
- 66.Boctor FN. Overt immediate hemolytic transfusion reaction attributable to anti-Wr(a) Immunohematology. 2008;24:113–5. [PubMed] [Google Scholar]
- 67.Oberman HA, Beck ML. Red blood cell sensitization due to unexpected Rh antibodies in immune serum globulin. Transfusion. 1971;11:382–4. doi: 10.1111/j.1537-2995.1971.tb04433.x. [DOI] [PubMed] [Google Scholar]
- 68.Rushin J, Rumsey DH, Ewing CA, Sandler SG. Detection of multiple passively acquired alloantibodies following infusions of IV Rh immune globulin. Transfusion. 2000;40:551–4. doi: 10.1046/j.1537-2995.2000.40050551.x. [DOI] [PubMed] [Google Scholar]
- 69.Narra S, Al-Kawas F, Carroll JE, Sandler SG. Biliary sludge and obstruction: another sign of a delayed hemolytic transfusion reaction. Transfusion. 2011;51:686–7. doi: 10.1111/j.1537-2995.2010.03054.x. [DOI] [PubMed] [Google Scholar]
- 70.Novaretti MC, Gallucci AO, Dorlhiac-Llacer PE, Chamone DA. Rapid detection of high titer anti-A/anti-B in blood donors using an Olympus PK 7200 equipment. Transfusion. 2009;49:116A. [Google Scholar]
- 71.Flegel WA, Wagner FF. Blutgruppen: Alloantigene auf Erythrozyten. In: Kiefel V, editor. Transfusionsmedizin und Immunhamatologie. Berlin: Springer; 2010. pp. 133–68. [Google Scholar]
- 72.Svensson L, Rydberg L, de Mattos LC, Henry SM. Blood group A and A revisited: an immunochemical analysis. Vox Sang. 2009;96:56–61. doi: 10.1111/j.1423-0410.2008.01112.x. [DOI] [PubMed] [Google Scholar]
- 73.Northoff H, Wölpl A, Sugg U, Junginger W, Meinke J, Welter J, Schneider W. An unusual sample of irregular anti-A1, probably causing an early delayed transfusion reaction. Blut. 1986;52:317–21. doi: 10.1007/BF00320795. [DOI] [PubMed] [Google Scholar]
- 74.Obukhova P, Korchagina E, Henry S, Bovin N. Natural anti-A and anti-B of the ABO system: allo- and autoantibodies have different epitope specificity. Transfusion. 2012;52:860–9. doi: 10.1111/j.1537-2995.2011.03381.x. [DOI] [PubMed] [Google Scholar]
- 75.Salama A, Hugo F, Heinrich D, Höge R, Müller R, Kiefel V, Mueller-Eckhardt C, Bhakdi S. Deposition of terminal C5b-9 complement complexes on erythrocytes and blood leukocytes during cardiopulmonary bypass. N Engl J Med. 1988;318:408–14. doi: 10.1056/NEJM198802183180704. [DOI] [PubMed] [Google Scholar]
- 76.Gaither TA, Vargas I, Inada S, Frank MM. The complement fragment C3d facilitates phagocytosis by monocytes. Immunology. 1987;62:405–11. [PMC free article] [PubMed] [Google Scholar]
- 77.Bajic G, Yatime L, Sim RB, Vorup-Jensen T, Andersen GR. Structural insight on the recognition of surface-bound opsonins by the integrin I domain of complement receptor 3. Proc Natl Acad Sci U S A. 2013;110:16426–31. doi: 10.1073/pnas.1311261110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.von Zabern I, Ehlers M, Grunwald U, Mauermann K, Greinacher A. Release of mediators of systemic inflammatory response syndrome in the course of a severe delayed hemolytic transfusion reaction caused by anti-D. Transfusion. 1998;38:459–68. doi: 10.1046/j.1537-2995.1998.38598297215.x. [DOI] [PubMed] [Google Scholar]
- 79.Pendergrast J, Willie-Ramharack K, Riden SL, Laroche V, Branch DR. The role of inflammation in intravenous immune globulin (IVIG)-mediated hemolysis. Transfusion. 2015;55 doi: 10.1111/trf.13097. this issue. [DOI] [PubMed] [Google Scholar]
- 80.Branch DR, Gallagher MT, Mison AP, Sy Siok Hian AL, Petz LD. In vitro determination of red cell alloantibody significance using an assay of monocyte-macrophage interaction with sensitized erythrocytes. Br J Haematol. 1984;56:19–29. doi: 10.1111/j.1365-2141.1984.tb01268.x. [DOI] [PubMed] [Google Scholar]
- 81.Arndt PA, Garratty G. A retrospective analysis of the value of monocyte monolayer assay results for predicting the clinical significance of blood group alloantibodies. Transfusion. 2004;44:1273–81. doi: 10.1111/j.1537-2995.2004.03427.x. [DOI] [PubMed] [Google Scholar]
- 82.Ness PM, Shirey RS, Thoman SK, Buck SA. The differentiation of delayed serologic and delayed hemolytic transfusion reactions: incidence, long-term serologic findings, and clinical significance. Transfusion. 1990;30:688–93. doi: 10.1046/j.1537-2995.1990.30891020325.x. [DOI] [PubMed] [Google Scholar]
- 83.Pineda AA, Vamvakas EC, Gorden LD, Winters JL, Moore SB. Trends in the incidence of delayed hemolytic and delayed serologic transfusion reactions. Transfusion. 1999;39:1097–103. doi: 10.1046/j.1537-2995.1999.39101097.x. [DOI] [PubMed] [Google Scholar]
- 84.Flegel WA, Johnson ST, Keller MA, Klapper EB, Khuu HM, Moulds JM, Seltsam AW, Stack G, St-Louis M, Tormey CA, Wagner FF, Weinstock C, Yazer MH, Denomme GA. Molecular immunohematology round table discussions at the AABB Annual Meeting, Boston 2012. Blood Transf. 2014;12:280–6. doi: 10.2450/2013.0022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keller-Stanislawski B, Lohmann A, Gunay S, Heiden M, Funk MB. The German Haemovigilance System–reports of serious adverse transfusion reactions between 1997 and 2007. Transfus Med. 2009;19:340–9. doi: 10.1111/j.1365-3148.2009.00947.x. [DOI] [PubMed] [Google Scholar]
- 86.Stainsby D, Jones H, Asher D, Atterbury C, Boncinelli A, Brant L, Chapman CE, Davison K, Gerrard R, Gray A, Knowles S, Love EM, Milkins C, McClelland DB, Norfolk DR, Soldan K, Taylor C, Revill J, Williamson LM, Cohen H, Group SS. Serious hazards of transfusion: a decade of hemovigilance in the UK. Transfus Med Rev. 2006;20:273–82. doi: 10.1016/j.tmrv.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 87.Robillard P, Nawej KI, Jochem K. The Quebec hemovigilance system: description and results from the first two years. Transfus Apher Sci. 2004;31:111–22. doi: 10.1016/j.transci.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 88.Vamvakas EC, Blajchman MA. Blood still kills: six strategies to further reduce allogeneic blood transfusion-related mortality. Transfus Med Rev. 2010;24:77–124. doi: 10.1016/j.tmrv.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reddy DR, Guru PK, Blessing MM, Stubbs JR, Rabinstein AA, Wijdicks EF. Transfusion-Related Acute Lung Injury After IVIG for Myasthenic Crisis. Neurocrit Care. 2015 doi: 10.1007/s12028-015-0115-z. [DOI] [PubMed] [Google Scholar]
- 90.Linden JV, Wagner K, Voytovich AE, Sheehan J. Transfusion errors in New York State: an analysis of 10 years’ experience. Transfusion. 2000;40:1207–13. doi: 10.1046/j.1537-2995.2000.40101207.x. [DOI] [PubMed] [Google Scholar]
- 91.Janatpour KA, Kalmin ND, Jensen HM, Holland PV. Clinical outcomes of ABO-incompatible RBC transfusions. Am J Clin Pathol. 2008;129:276–81. doi: 10.1309/VXY1ULAFUY6E6JT3. [DOI] [PubMed] [Google Scholar]
- 92.Robillard P, Nawej KI, Garneau N. Trends in red cell-associated ABO mistransfusion, acute and delayed hemolytic and delayed serologic transfusion reactions in the Quebec Hemovigilance System. Transfus Apher Sci. 2004;31:111–22. doi: 10.1016/j.transci.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 93.Rieben R, Buchs JP, Fluckiger E, Nydegger UE. Antibodies to histo-blood group substances A and B: agglutination titers, Ig class, and IgG subclasses in healthy persons of different age categories. Transfusion. 1991;31:607–15. doi: 10.1046/j.1537-2995.1991.31791368336.x. [DOI] [PubMed] [Google Scholar]
- 94.Holburn AM, Masters CA. The radioimmunoassay of serum and salivary blood group A and Lea glycoproteins. Br J Haematol. 1974;28:157–67. doi: 10.1111/j.1365-2141.1974.tb06650.x. [DOI] [PubMed] [Google Scholar]
- 95.Gaensslen RE, Bell SC, Lee HC. Distributions of genetic markers in United States populations: I. Blood group and secretor systems. J Forensic Sci. 1987;32:1016–58. [PubMed] [Google Scholar]
- 96.Holburn AM, Masters CA. The reactions of IgG and IgM anti-A and anti-B blood group antibodies with 125I-labelled blood group glycoproteins. Vox Sang. 1974;27:115–23. doi: 10.1111/j.1423-0410.1974.tb02399.x. [DOI] [PubMed] [Google Scholar]
- 97.Zimring JC. Possible product risks related to presence of immune complexes, complement/IG association, and other factors. Transfusion. 2015;55 this issue. [Google Scholar]
- 98.O’Donghaile D, Kelley W, Klein HG, Flegel WA. Recommendations for transfusion in ABO-incompatible hematopoietic stem cell transplantation. Transfusion. 2012;52:456–8. doi: 10.1111/j.1537-2995.2011.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Henry S. The Sew FUT2 mutation A385T does not result in a nonsecretor allele. Transfusion. 2014;54:3255. doi: 10.1111/trf.12909. [DOI] [PubMed] [Google Scholar]
- 100.Ferrer-Admetlla A, Sikora M, Laayouni H, Esteve A, Roubinet F, Blancher A, Calafell F, Bertranpetit J, Casals F. A natural history of FUT2 polymorphism in humans. Mol Biol Evol. 2009;26:1993–2003. doi: 10.1093/molbev/msp108. [DOI] [PubMed] [Google Scholar]
- 101.Tilley CA, Crookston MC, Brown BL, Wherrett JR. A and B and A1Leb substances in glycosphingolipid fractions of human serum. Vox Sang. 1975;28:25–33. doi: 10.1111/j.1423-0410.1975.tb02737.x. [DOI] [PubMed] [Google Scholar]
- 102.Castilho L, Rios M, Bianco C, Pellegrino J, Jr, Alberto FL, Saad ST, Costa FF. DNA-based typing of blood groups for the management of multiply-transfused sickle cell disease patients. Transfusion. 2002;42:232–8. doi: 10.1046/j.1537-2995.2002.00029.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.