

Background: LPS increases NALP3 levels, but the mechanisms remain unknown.
Results: LPS prolongs the lifespan of NALP3 protein by reducing E3 ligase (SCFFBXL2)-mediated ubiquitination.
Conclusion: Proinflammatory cytokine release is reduced by a small molecule that restores cellular SCFFBXL2 levels.
Significance: We identified a novel pathway of inflammasome priming that may serve as a springboard for future translational studies.
Keywords: cytokine induction, inflammasome, inflammation, innate immunity, IL-1, NALP3
Abstract
The inflammasome is a multiprotein complex that augments the proinflammatory response by increasing the generation and cellular release of key cytokines. Specifically, the NALP3 inflammasome requires two-step signaling, priming and activation, to be functional to release the proinflammatory cytokines IL-1β and IL-18. The priming process, through unknown mechanisms, increases the protein levels of NALP3 and pro-IL-1β in cells. Here we show that LPS increases the NALP3 protein lifespan without significantly altering steady-state mRNA in human cells. LPS exposure reduces the ubiquitin-mediated proteasomal processing of NALP3 by inducing levels of an E3 ligase component, FBXO3, which targets FBXL2. The latter is an endogenous mediator of NALP3 degradation. FBXL2 recognizes Trp-73 within NALP3 for interaction and targets Lys-689 within NALP3 for ubiquitin ligation and degradation. A unique small molecule inhibitor of FBXO3 restores FBXL2 levels, resulting in decreased NALP3 protein levels in cells and, thereby, reducing the release of IL-1β and IL-18 in human inflammatory cells after NALP3 activation. Our findings uncover NALP3 as a molecular target for FBXL2 and suggest that therapeutic targeting of the inflammasome may serve as a platform for preclinical intervention.
Introduction
The inflammasome is a large multiprotein cytosolic complex that consists of three components: a sensor (NACHT, LRR, and PYD domain-containing protein (NALP)),3 an adaptor (apoptosis-associated speck-like protein containing a carboxy-terminal caspase-recruitment domain (ASC)), and an effector (pro-caspase 1). Inflammasomes have emerged as key complexes that, when assembled properly, result in the release of IL-1β and IL-18, which partake in illnesses characterized by severe inflammation. Some inflammasomes are activated by pore-forming toxins, ATP, or hypoxic cellular injury when these conditions are primed with microbial ligands or endogenous cytokines (1). When assembled, inflammasomes cleave and activate the potent proinflammatory cytokines IL-1β and IL-18, which are associated with a higher risk and worse outcomes of disorders such as acute respiratory distress syndrome (2, 3). NALP3, the most studied inflammasome so far, requires two-step signaling to engage in cytokine release: priming and activation. The Gram-negative bacterial endotoxin LPS is known to prime the NALP3 inflammasome by increasing NALP3 protein and pro-IL-1β levels in a dose- and time-dependent fashion in mice (4). However, the molecular mechanisms whereby LPS priming increases NALP3 protein abundance in cells remain elusive. This question is critical because the identification of molecular targets that transcriptionally or posttranslationally mediate increased NALP3 concentrations in cells could be important in pharmacotherapeutic designs to control innate immune responses.
Protein ubiquitination is a highly conserved, irreversible, posttranslational modification among vertebrates that governs cellular protein concentrations. The ubiquitin proteasome system selectively targets proteins in humans by a series of enzymatic steps requiring a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin E3 ligase. E3 ligases are multisubunit complexes that confer a relatively high specificity to ubiquitination by bringing in close proximity a substrate to an E2 enzyme that facilitates the transfer of an activated ubiquitin moiety from the E2 to the substrate (5). Hundreds of human genes encode RING domain-containing E3 ligases, and, among these, the Skp-Cullin-F box (SCF) family is among the best studied (6). The SCF superfamily regulates diverse biological events, including cell growth, proliferation, DNA repair, and inflammation (7).
We have demonstrated previously that an SCF subunit, F box protein, F-box O3 (FBXO3), is a proinflammatory molecule that is activated in leukocytes by endotoxin (8). FBXO3 ubiquitinates and mediates degradation of another F box protein, F-box L2 (FBXL2). FBXL2 is an anti-inflammatory molecule that mediates the degradation of TNF receptor-associated factors (TRAFs), resulting in a reduction of inflammatory cytokine release. The aim of our study is to investigate how LPS priming increases NALP3 inflammasome protein levels. Here we show that NALP3 is also a target for FBXL2. Moreover, we demonstrate a unique mechanism whereby LPS priming decreases FBXL2-mediated NALP3 ubiquitination and degradation by activating the FBXL2 inhibitor FBXO3 in cells. A small-molecule FBXO3 antagonist, BC-1215, potently reduces the severity of NALP3-mediated inflammation by increasing FBXL2 protein levels, therefore antagonizing actions of endotoxin on the inflammasomes.
Experimental Procedures
Antibodies and Reagents
Antibodies against FBXL8, anti-mouse IgG, and anti-rabbit IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against FBXL2 were from Aviva Systems Biology (San Diego, CA). Antibodies against F-box L7 (FBXL7) and NALP6 were obtained from Abcam (Cambridge, MA). Ubiquitin and IL-1β antibodies were purchased from Cell Signaling Technology (Danvers, MA). NALP3 antibodies were obtained from Adipogen (San Diego, CA). GAPDH and β-actin antibodies were obtained from Sigma. IL-18 human antibodies were purchased from MBL (Woburn, MA). QuikChange II site-directed mutagenesis kits were purchased from Agilent Technologies (Santa Clara, CA). Pierce protein A/G-agarose beads were purchased from Thermo Scientific (Rockford, IL). Cycloheximide (CHX), MG132, and leupeptin were from Calbiochem (Darmstadt, Germany). Mouse V5 antibodies, pcDNA3.1D cloning kits, and the pENTR directional TOPO cloning kits were purchased from Invitrogen. ATP was obtained from Sigma. Miniprep spin kits were obtained from Qiagen (Valencia, CA) and Nucleobond Xtra Maxi prep kits were obtained from Macherey-Nagel (Düren, Germany). In vitro transcription and translation (TnT) kits were from Promega. BC-1215 was synthesized to >98% purity.
Cell Culture
Human monocyte U937 and THP1 cells were purchased from Sigma-Aldrich (St. Louis, MO). Human erythroleukemia K562 cells, human bronchial epithelial A549 cells, Beas2B cells, mouse lung epithelial (MLE-12) cells, and human cervical epithelial HeLa cells were obtained from the ATCC. Human primary macrophages and their culture medium were purchased from Celprogen (San Pedro, CA). A bronchoscopy was performed to collect bronchoalveolar lavage fluid after obtaining informed consent. Human primary alveolar macrophages from the fluid were cultured in culture medium (Celprogen) as described previously (9). The study was approved by the University of Pittsburgh Institutional Review Board. DMEM and RPMI 1640 medium were obtained from Gibco (Life Technologies) and supplemented with fetal bovine serum (FBS) from Gemini (Sacramento, CA). MLE cells were cultured in HITES (hydrocortisone, insulin, transferrin, estrogen, and selenium) medium supplemented with 10% FBS. Prior to CHX treatment, cells were starved for 1 h with 0% FBS HITES medium. U937, THP1, and K562 cells were cultured in RPMI 1640 medium supplemented with 10% FBS. A549 and Beas2B cells were cultured in DMEM with 10% FBS. HeLa cells were cultured in Eagle's minimum essential medium (Gibco) supplemented with 10% FBS. CHX treatment was carried out at a concentration of 40 μg/ml at varying time points in 0% FBS medium with or without MG132 (20 μg/ml) or leupeptin (20 μg/ml).
Immunoprecipitation and Immunoblotting
Cells were lysed with lysis buffer (0.05% Triton X-100 in PBS and 1:1000 protease inhibitor mixture) and sonicated. After centrifugation at 13,200 rpm for 10 min, cell lysates (containing 2 mg of protein) were incubated and rotated with 5 μg of anti-ubiquitin, anti-NALP3, or FBXL2 antibody at 4 °C for 3 h and then incubated with 30 μl of protein A/G-agarose beads overnight. The next day, the beads were spun down and washed with lysis buffer three times. The washed beads were mixed with SDS-PAGE loading dye prior to SDS-PAGE and immunoblot analysis. Immunoblotting was performed as described previously (10). We precipitated extracellular media with trichloroacetic acid and subsequently measured cytokine levels by immunoblotting.
RT-PCR
RNA was isolated from cells using RNeasy mini kits (Qiagen) according to the protocol provided. Isolated RNAs were immediately converted to cDNA using high-capacity RNA-to-cDNA kits (Life Technologies) after their concentrations were measured. Real-time PCR assays were performed using SYBR® Select Master Mix for CFX (2×, Life Technologies) per the protocol provided in C1000 Thermal Cycler (Bio-Rad).
Cloning and Mutagenesis
Human NALP3 cDNA was cloned into a pcDNA3.1D/V5-His vector. Site-directed mutagenesis of lysine to arginine residues was carried out via a PCR-based approach using the QuikChange II XL kit from Agilent Technologies and appropriate primers. Primers used for lysine to arginine mutagenesis were as follows: K689R forward (5′-CATAACATGCCCAGGGAGGAAGAGGAG-3′) andreverse (5′-CTCCTCTTCCTCCCTGGGCATGTTATG-3′); K696R forward (5′-GAAGAGGAGGAGGAAAGGGAAGGCCGACACCTTG-3′) and reverse (5′-CAAGGTGTCGGCCTTCCCTTTCCTCCTCCTCTTC-3′); K86R forward (5′-GAGAGACCTTTATGAGAGAGCAAAAAGAGATGAG-3′) and reverse (5′-CTCATCTCTTTTTGCTCTCTCATAAAGGTCTCTC-3′); and K742R forward (5′-GGTCCTCAGCAGCAACCAGAGGCTGGTGGAGCTGGACCTGAG-3′) and reverse(5′-CTCATCTCTTTTTGCTCTCTCATAAAGGTCTCTC-3′).Generated mutants were sequence-confirmed (10).
Transfection
2.5 × 106 MLE cells were suspended in 100 μl of 20 mm HEPES and mixed with 4 μg of DNA in a cuvette. Cells were nucleofected on the T-013 protocol using an Amaxa Nucleofector II device (Basel, Switzerland). Immediately following nucleofection, 0.5 ml of 10% HITES medium was added to the cuvette, and the cell solution was plated in 1.5 ml of 10% HITES culture medium in a 6-well plate. Cells were allowed to grow until 50% confluency prior to half-life experiments (48–72 h). For HeLa cells, the PolyJetTM DNA in vitro transfection reagent was used following the protocols of SignaGen (Rockville, MD). Cells were then treated with 40 μg/ml of CHX at different time points for half-life studies and were harvested in lysis buffer using a rubber policeman for subsequent immunoblot analysis. siRNA to FBXL2, purchased from Integrated DNA Technologies (Coralville, IA), was introduced into primary human macrophages using GenMuteTM siRNA transfection reagent based on protocols from SignaGen's.
In Vitro Ubiquitination Conjugation Assay
The details of this assay have been described previously (11). Briefly, the ubiquitination of NALP3 was performed in a volume of 25 μl containing 50 mm Tris (pH 7.6); 5 mm MgCl2; 0.6 mm DTT; 2 mm ATP; 1.5 ng/μl E1; 10 ng/μl Ubc5; 10 ng/μl Ubc7; 1 μg/μl ubiquitin (Calbiochem); 1 μm ubiquitin aldehyde; 4–16 μl of purified Cullin1; Skp1, Rbx1, and in vitro-synthesized FBXL2. Reaction products were processed for NALP3 immunoblotting.
In Vitro Protein Binding Assays
FBXL2 protein was immunoprecipitated from 1 mg of cell lysate using FBXL2 antibody and coupled to protein A/G-agarose resin. FBXL2 beads were then incubated with in vitro-synthesized products (50 μl) expressing NALP3-V5 mutants. After washing, the proteins were eluted and processed for NALP3-V5 immunoblotting.
ELISA
Quantikine ELISA kits were purchased from R&D Systems (Minneapolis, MN) to measure human IL-1β and human IL-18 levels. The quantitative assays were performed following protocols provided by the manufacturer using cell culture supernatants after LPS/ATP exposure with or without BC-1215.
Statistical Analysis
Descriptive statistics were reported as mean ± S.D. or S.E. as indicated. As our sample sizes of each experimental group were all less than 10, which limits the ability to study a normal sample distribution, we employed appropriate non-parametric methods such as a Mann-Whitney U test and a Kruskal-Wallis equality of populations rank test to compare multiple groups within and between experiments. Also, a Wilcoxon-type test for trend was used where appropriate to check trends in data significance. Using these methods provided a conservative analysis to determine statistical significance. All analyses were performed using Stata Statistical Software, Release 10.1 (StataCorp 2007, College Station, TX).
Results
LPS Priming Up-regulates Steady-state NALP3 Protein Levels by Prolonging Its Half-life
Human monocyte U937 cells express NALP3, as evidenced by immunoblotting with an NALP3 antibody. NALP3 protein levels increased in a dose- and time-dependent fashion with LPS exposure, whereas NALP6, another NOD (nucleotide-binding oligomerization domain)-like receptor, did not (Fig. 1, A and B). There were modest, if any, increases in mRNA expression of NALP3 with LPS exposure (Fig. 2, A and B), and the changes were not statistically significant. These data suggest that LPS up-regulates NALP3 protein mainly through posttranslational mechanisms. Therefore, we measured the half-life of NALP3 protein both at baseline and after LPS exposure by blocking new protein synthesis with CHX. The half-life of NALP3 is ∼4 h in an unchallenged condition and prolonged to >6 h after LPS exposure (Fig. 2, C and D). NALP3 degradation was reduced with inclusion of MG132 in the medium but not with a lysosomal inhibitor, leupeptin, indicating that the degradation of NALP3 occurs via the ubiquitin proteasome system rather than the lysosome (Fig. 2, E and F).
FIGURE 1.

LPS priming increases NALP3 protein in human monocyte U937 cells. A and B, immunoblots for NALP3, NALP6 (negative control), and β-actin in lysates from human monocytes, U937 cells (total 3 × 106 cells), treated with LPS for 16 h as indicated (A) or treated with LPS (200 ng/ml) for the indicated periods of time (B). The relative densitometries of NALP3 protein for each immunoblot are shown in the bottom panel. Data are mean ± S.E. of three to five independent experiments. The p values were determined by a nonparametric test for trend.
FIGURE 2.

LPS priming increases NALP3 protein levels by prolonging its half-life. Box plots of messenger RNA expression –fold increase of NALP3 in LPS primed human monocytes, U937 cells. A and B, cells were primed with LPS for 16 h as indicated (A) or with LPS (200 ng/ml) for the indicated periods of time (B). Data are from four independent experiments. The p values were determined by a Kruskal-Wallis test. C, U937 cells were incubated with CHX (40 μg/ml) for various times with or without LPS priming (200 ng/ml for 16 h). D, densitometric plot of NALP3 protein decay versus time of CHX exposure with a best fit line. The half-life was ∼4 h in an unchallenged condition and >6 h after LPS priming. E, U937 cells were incubated with CHX (40 μg/ml) and MG132 (20 μg/ml) or leupeptin (20 μg/ml), and immunoblotting for NALP3 was performed. F, decay curves over time in the presence of MG132 or leupeptin were best fitted. Data are mean ± S.E. of five to eight independent experiments. The p values were determined by a Mann-Whitney test comparing two groups at each time point. *, p < 0.05; **, p < 0.01.
FBXL2 Mediates NALP3 Ubiquitination and Degradation, an Effect Inhibited by LPS
To confirm that NALP3 is degraded through the ubiquitin proteasome system, we transfected MLE cells with either control plasmids or HA-ubiquitin plasmids. High-level ubiquitin cellular expression correlated with lower levels of NALP3 protein in cells (Fig. 3A). NALP3-ubiquitin binding, measured by coimmunoprecipitation, was decreased by 40% after LPS exposure when densitometrically controlled for input loading (Fig. 3, B and C). Therefore, NALP3 is ubiquitinated, an effect reduced by LPS.
FIGURE 3.

NALP3 is degraded through the ubiquitin proteasome system. A, MLE cells were transfected with either the control plasmid or HA-ubiquitin (HA-Ubi) plasmid for 24 h. Cells were collected and assayed for HA, NALP3, and β-actin (loading control) by immunoblotting. B, A549 cells were incubated with or without LPS (200 ng/ml for 16 h), and cells were lysed and used to detect NALP3 or ubiquitin (left panels, input). Top right panel, ubiquitin was immunoprecipitated (IP), followed by NALP3 immunoblotting (IB) showing reduced intensity of several bands (arrows). C, the relative densitometric intensities of NALP3 bands are shown reflecting reduced association between NALP3 and ubiquitin by 40% after LPS exposure. The bar graph represents mean ± S.D. The p value was determined by Mann-Whitney test. Data are representative of at least two to three independent experiments.
Because our prior studies have demonstrated that FBXO3 targets FBXL2 for its disposal in cells and that an FBXO3 inhibitor reduces IL-1β levels after LPS treatment (8), we tested the hypothesis that FBXL2 targets NALP3 to mediate its degradation. Indeed, the ubiquitination of NALP3 is increased after expression of FBXL2. As shown in Fig. 4A, ectopic expression with the FBXL2 plasmid in MLE cells decreases levels of NALP3. Furthermore, NALP3 protein levels in both MLE cells and primary human macrophages were increased by using one of the several lentivirally delivered shRNAs or siRNAs to FBXL2 (Fig. 4B). To determine whether FBXL2 interacts with NALP3, we performed coimmunoprecipitation studies showing that FBXL2 binds NALP3 selectively because it does not associate with the NALP6 inflammasome (Fig. 4C). When V5-NALP3 was overexpressed in cells, in coimmunoprecipitation studies it was associated with FBXL2 but not other F-box proteins, FBXL7 or FBXL8. In vitro ubiquitin assays demonstrated that FBXL2 robustly catalyzes NALP3 ubiquitination (Fig. 4D). The binding between NALP3 and FBXL2 was also decreased after LPS exposure, as was NALP3 ubiquitination (Fig. 4, E and F). These observations suggest that NALP3 is a molecular target for FBXL2-mediated degradation, a process impaired by endotoxin.
FIGURE 4.

FBXL2 triggers NALP3 degradation. A, MLE cells were transfected with increasing amounts of FBXL2 plasmid for 24–48 h. Cells were collected and assayed for V5 (FBXL2), NALP3, and β-actin by immunoblotting. B, MLE cells were transfected with several FBXL2 lentiviral shRNAs (4 μg). Human primary macrophages were also transfected with several siRNAs to FBXL2 (500 nm) for gene silencing. Shown are protein levels of FBXL2 after transfection of shRNAs or siRNAs and corresponding levels of NALP3 protein. Compared with a control RNA, despite a modest reduction in FBXL2 levels, increased NALP3 levels in the shRNA or siRNA groups were observed. C, U937 cells were lysed, and FBXL2 proteins were coimmunoprecipitated (IP) followed by NALP3, FBXL2, or NALP6 immunoblotting (IB, top panel). In addition, Beas2B cells were transfected with NALP3-V5 prior to IP for V5 and then, NALP3 and various F box proteins were immunoblotted (bottom blot). IgG was used as a control in all IP experiments, and inputs are the IBs of 10% of the cell lysates before performing IP. Note that an upper IgG chain band was detected in the FBXL7 immunoblot. D, in vitro ubiquitination assays showing that NALP3 is a substrate for FBXL2-mediated ubiquitination. E, A549 cells were incubated with or without LPS (200 ng/ml) for 16 h, and cells were lysed. FBXL2 protein was coimmunoprecipitated; first IP for FBXL2 followed by NALP3, FBXL2, or β-actin immunoblotting. F, the relative association between FBXL2 and NALP3 after LPS was quantified using densitometric analysis of the bands in E. The binding capacity between NALP3 and FBXL2 is reduced by ∼45–50% after LPS exposure. Data are representative of at least two to three independent experiments.
Lys-689 is a NALP3 Ubiquitination Acceptor Site
Protein ubiquitination involves the generation of an isopeptide bond between an acceptor lysine on a target protein (e.g. NALP3) and the C-terminal glycine of the incoming ubiquitin moiety. Human NALP3 protein has 68 Lys residues. Using the UbPred software (12), potential ubiquitination sites in NALP3 protein were predicted among the Lys residues: one with high probability (Lys-689), five with medium probability (Lys-93, Lys-192, Lys-324, Lys-430, and Lys-696), and six residues with low probability (Lys-86, Lys-194, Lys-437, Lys-510, Lys-742, and Lys-958) (Fig. 5A). We cloned the human NALP3 cDNA into a pcDNA3.1D/V5-His vector and generated mutants substituting arginine at each of the predicted Lys residues within the NALP3 protein. MLE cells were transfected with wild-type and mutant plasmids and were exposed to CHX to measure the half-life of ectopically expressed NALP3 (Fig. 5B). The half-life of wild-type ectopically expressed NALP3 is 4–5 h, similar to that of endogenous NALP3. The K689R NALP3 mutant has an extended half-life (>6 h) in cells compared with wild-type NALP3, resembling the effects of LPS exposure on wild-type NALP3 (Figs. 2C and 5, B and C). Other expressed mutants did not exhibit a significantly prolonged lifespan in cells (Fig. 5D). Cotransfection of NALP3 mutant plasmids with or without FBXL2 plasmid decreased ectopically expressed wild-type NALP3 protein and a variant, K696R. However, levels of the K689R NALP3 mutant were not decreased with overexpressed FBXL2 (Fig. 5E). In vitro ubiquitin assays confirmed that the K689R NALP3 mutant is not ubiquitinated in comparison with wild-type or another mutant NALP3 (Fig. 5F). Therefore, Lys-689 is likely a major ubiquitin acceptor residue within human NALP3 protein.
FIGURE 5.

Lys-689 is a NALP3 ubiquitination acceptor site. A, full-length and point mutant constructs encoding human NALP3 protein were cloned into a pcDNA3.1D/V5-His vector. B, MLE cells were transfected with either the pcDNA-NALP3 full-length plasmid (4 μg) or point mutant plasmid for 48 h. Cells were exposed to CHX (40 μg/ml) at different time points for a half-life study. Cells were collected and assayed for NALP3 (V5) and β-actin (loading control) by immunoblotting. Representative images are shown. C and D, densitometric plots of NALP3 protein decay versus time of CHX exposure with best fit lines. The half-life was prolonged with a K689R mutant by >6 h (C), whereas WT full-length protein or other mutants exhibited a t½ of ∼4 h (D). Data represent the mean ± S.E. of three to four independent experiments. The p values were determined by a Mann-Whitney test comparing two groups at each time point. *, p < 0.05. E, MLE cells were cotransfected with FBXL2/empty (cont) plasmid (2 μg) and either the WT NALP3 plasmid or point mutant plasmids (2 μg) for 48 h. Cells were collected and assayed for NALP3 (V5) and β-actin (loading control) by immunoblotting. F, in vitro ubiquitination assays show that a K689R NALP3 mutant is less polyubiquitinated compared with WT NALP3 or a K696R mutant. The data in E and F are representative of two to three independent experiments.
NALP3 Trp-73 Is a Molecular Recognition Site for FBXL2 Interaction
We next investigated the FBXL2 binding site within NALP3. Several deletion mutants of NALP3 were constructed and cloned in a pcDNA3.1D V5/HIS/TOPO vector (Fig. 6A). Through progressive mapping we determined that FBXL2 binds the N-terminal region of NALP3, spanning residues 70–80 (Fig. 6, B and C). An alanine scan within this region suggested that Trp-73 is a critical residue for FBXL2 binding (Fig. 6D). We tested the half-life of a NALP W73A mutant and a control W68A mutant using CHX treatment. W73A exhibited delayed turnover with an extended half-life from 4 h to >6 h compared with wild-type or a mutant (W68A) NALP3 protein (Fig. 6E). Furthermore, to evaluate the biologic role of these NALP3 proteins in cells, we exposed cells to the FBXO3 inhibitor BC-1215 (8), which, in principle should increase FBXL2 levels, thereby reducing the abundance of its cellular targets, such as NALP3. Indeed, exposure of cells to BC-1215 after cellular transfection of NALP3 wild-type, NALP3 W68A, or NALP3 W73A plasmids revealed that the W73A protein was resistant to BC-1215-driven (FBXL2-mediated) NALP3 degradation (Fig. 6F). Finally, we coexpressed NALP3 wild-type or NALP3 W73A plasmids with FBXL2 and observed that the W73A variant is resistant to degradation (Fig. 6G). These results indicate that Trp-73 is a key molecular dock site for FBXL2 interaction and that its substitution with Ala confers proteolytic resistance within NALP3.
FIGURE 6.

Trp-73 within NALP3 is required for FBXL2 interaction. A, several deletional or point mutants of NALP3 were designed and cloned into a pcDNA3.1D/V5-HIS vector. B–D, FBXL2 protein was immunoprecipitated (IP) from cell lysates using an FBXL2 antibody and coupled to protein A/G-agarose resin. FBXL2 resins were then incubated with in vitro-synthesized products expressing HIS-V5-NALP3 mutants. After washing, proteins were eluted and immunoblotted (IB) for NALP3-V5. E, HeLa cells were transfected with NALP3 WT or mutant plasmids, and the half-life was determined via CHX treatment at 40 μg/ml. F, NALP3 WT or mutants (4 μg) were transfected into HeLa cells and treated with BC-1215 for 16 h. Cells were then collected and processed for V5 immunoblotting. G, HeLa cells were cotransfected with NALP3 WT or W73A mutant (2 μg) with or without the FBXL2 plasmid (2 μg). Cells were then collected and immunoblotted for V5 and β-actin. Cont, control.
An FBXO3 Inhibitor Attenuates LPS Induction of NALP3 Protein
FBXO3 is a proinflammatory E3 ligase subunit that eliminates the anti-inflammatory E3 ligase subunit FBXL2 (8). Therefore, our model suggests that LPS increases FBXO3 protein levels, which, in turn, decreases FBXL2 levels (11). The net effect of LPS in this model is increased NALP3 protein levels. We have previously used in silico modeling to design, synthesize, and test a new genus of small molecules targeting the ApaG motif within the C terminus of FBXO3. In preclinical studies, we observed that BC-1215 binds the F-box protein and significantly decreases the severity of inflammation in multiple murine models (11). Here our results suggest an FBXO3 ⊣ FBXL2 ⊣ NALP3 inflammasome → IL-18 and IL-1β pathway. We validated this pathway by testing BC-1215 in human monocytes. First we verified that BC-1215, in a concentration-dependent manner, increases FBXL2 levels in human monocytic U937 cells (Fig. 7A). In a relatively selective manner, NALP3 protein levels were decreased significantly with BC-1215 treatment with or without LPS exposure in human monocyte U937 and THP1 cells (Fig. 7B). BC-1215 destabilizes NALP3 protein following LPS exposure in a dose- and time-dependent fashion without significant changes in steady-state mRNA expression of NALP3 (Fig. 7, C and D). The prolonged half-life of NALP3 protein after LPS priming is effectively decreased with BC-1215 treatment to baseline levels without LPS priming (Fig. 7E). In the presence of LPS, BC-1215 did not decrease ectopically overexpressed K689R mutant NALP3 protein levels harboring a modified ubiquitin acceptor residue compared with wild-type or a mutant (K696R) NALP3 protein (Fig. 7F). The secretion of IL-1β and IL-18, the downstream products of the NALP3 inflammasome, was also decreased with BC-1215 treatment in human inflammatory cell lines, including THP1 and K562 cells (Fig. 7G). The ratio of active IL-1β versus pro-IL-1β was also decreased with BC-1215 in a time-dependent fashion (Fig. 7H), suggesting that the main mechanism to reduce cytokine release is from decreased conversion from premature to mature forms of cytokines. Lastly, we examined ability of the FBXO3 small molecule inhibitor on primary human alveolar macrophages after LPS priming for the NALP3 inflammasome. BC-1215 effectively decreased LPS induction of NALP3 protein induction by 44% (by densitometry when adjusted for β actin loading) and also reduced the secretion of IL-1β and IL-18 after a second stimulus, ATP (Fig. 7, I and J). Collectively, these observations strongly implicate FBXO3 and FBXL2 as molecular inputs to NALP3 cytokine signaling (Fig. 8).
FIGURE 7.

An FBXO3 inhibitor reduces NALP3 abundance, thereby decreasing cytokine release. A, U937 cells were treated with BC-1215 at different concentrations for 16 h. Cells were collected for FBXL2 and β-actin immunoblotting. B, U937 and THP1 cells (3 × 106 cells each) were incubated with LPS (200 ng/ml) or BC-1215 (8 μg/ml) for 16 h. Cells were collected and lysed for NALP3, NALP6 (negative control), and GAPDH/β-actin immunoblotting. C and D, U937 cells (3 × 106 cells) were primed with LPS (200 ng/ml) in combination with different concentrations of BC-1215 for 16 h as indicated (C) or primed with LPS (200 ng/ml for 16 h) and then exposed to BC-1215 (4 μg/ml) in fresh culture medium for the indicated periods of time (D). Cells were collected to measure NALP3 and β-actin protein (top panels) and mRNA levels (bottom panels). Box plots of the -fold increase of steady-state NALP3 mRNA are shown. Data represent four independent experiments. The p values were determined by a Kruskal-Wallis test. E, U937 cells were incubated in LPS (200 ng/ml) with or without BC-1215 (4 μg/ml for 16 h). Cells were then exposed to CHX (40 μg/ml) at different time points for a half-life study. Immunoblotting for NALP3 and GAPDH (loading control) was performed. Densitometric plots of adjusted NALP3 protein decay over time under different conditions were best fitted. The half-life of LPS-primed NALP3 protein was reduced with BC-1215 treatment comparable with native conditions. Data are mean ± S.E. of two independent experiments. F, MLE cells were transfected either with WT or point mutant NALP3 plasmids for 48 h. LPS (40 μg/ml) was then added to the medium with or without BC-1215 (20 μg/ml) for 18 h. Cells were collected and lysed for NALP3 (V5-tagged) and β-actin (loading control) immunoblotting. Levels of K689R, the point mutant for a putative ubiquitin acceptor site within the NALP3 protein, did not decrease with BC-1215 compared with WT or mutant NALP3. Data are representative of two independent experiments. G, THP1 and K562 cells (3 × 106 cells each) were incubated with LPS (200 ng/ml) with or without BC-1215 (8 μg/ml) for 20 h and then pulsed with ATP (5 mm for 20 min). Culture medium was collected for immunoblotting of pro-IL-1β, IL-1β, pro-IL-18, and IL-18. H, THP1 cells (3 × 106 cells each) were incubated with LPS (200 ng/ml) for 20 h and different time periods of BC-1215 (8 μg/ml), as indicated, in the same volume of culture medium. Cells were then exposed to ATP (5 mm for 20 min). Culture medium was collected for immunoblotting of pro-IL-1β and IL-1β. The ratio of IL-1β versus pro-IL-1β by densitometry is shown (bottom panel). Data represent mean ± S.D. of two independent experiments. The p value was determined by nonparametric test for trend. I, primary human alveolar macrophages were obtained from a healthy volunteer. Human alveolar macrophages (3 × 105 cells) were incubated with BC-1215 (4 μg/ml) for 2 h and then exposed to LPS (100 ng/ml) for 16 h. Cells were lysed and collected for immunoblotting for NALP3 and β actin. J, primary human alveolar macrophages (3 × 105 cells) from the same subject as in I were incubated with BC-1215 (4 μg/ml) for 2 h and then exposed to LPS (100 ng/ml) for 16 h. ATP (5 mm) was added for 15 min before collecting cell culture supernatants for ELISA. Data represent the mean ± S.D. of duplicate measurements.
FIGURE 8.
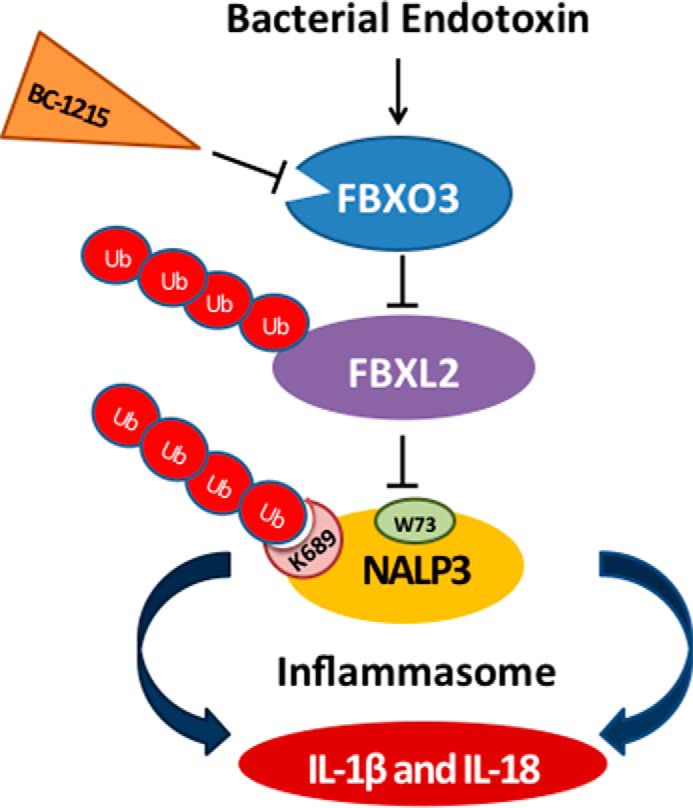
Molecular regulation of NALP3 inflammasome protein stability mediated by F-box proteins. Endotoxin robustly increases NALP3 protein levels in human inflammatory cells and subsequently induces cytokine synthesis and release. The F-box protein FBXL2 serves as a sentinel inhibitor of NALP3 by mediating its polyubiquitination (Ub) through docking at Trp-73 and ubiquitin ligation at Lys-689, resulting in proteasomal degradation in cells. Another E3 ligase component, FBXO3, targets FBXL2 for its ubiquitination and degradation (8). A small-molecule FBXO3 inhibitor, BC-1215, reduces the levels of NALP3 protein after LPS priming by restoring FBXL2 levels. BC-1215 subsequently decreases proinflammatory cytokine release after the NALP3 inflammasome is activated by a second stimulus.
Discussion
These studies demonstrate a unique mechanism whereby LPS priming prolongs the lifespan of NALP3 protein in cells. LPS stabilizes the NALP3 protein by inhibiting its FBXL2-mediated polyubiquitination and degradation, presumably by limiting its interaction with the inflammasome-SCFFBXL2 complex. We also identified Lys-689 as a putative ubiquitin acceptor site and Trp-73 as an FBXL2 binding site within the NALP3 protein. The expression of plasmids encoding mutation at these molecular sites in cells resulted in an extended half-life of NALP3 protein despite coexpression of FBXL2 or treatment with a potent FBXO3 inhibitor. Therefore, our data support an FBXO3 ⊣ FBXL2 ⊣ NALP3 inflammasome → cytokine pathway that was validated by observing decreased NALP3 protein and associated cytokines with treatment of an FBXO3 inhibitor. The results raise opportunities for the therapeutic targeting of upstream inflammasome regulators to reduce inflammation in a variety of cytokine-driven disorders, such as acute respiratory distress syndrome.
The inflammasome has emerged as a critical intracellular multiprotein complex involved in a variety of inflammatory diseases, such as acute respiratory distress syndrome, cryopyrin-associated periodic syndrome, gout, and type II diabetes (13). To initiate the release of proinflammatory cytokines, NALP3 must first be primed by interacting with pathogen-associated molecular patterns or host-derived danger signals (danger-associated molecular patterns). When NALP3 senses second stimuli, such as pore-forming toxins or crystals, the NALP3 scaffold recruits ASC adaptor and pro-caspase 1 and assembles into a functional inflammasome. This assembled complex then serves as a platform to activate pro-caspase 1 to caspase 1, which then cleaves pro-IL-1β and pro-IL-18 to their mature forms of IL-1β and IL-18, respectively. The molecular mechanisms whereby NALP3 inflammasomes are activated to release cytokines has been the focus of significant attention because it can provide insights for understanding disease pathogenesis and may ultimately uncover potential therapeutic targets. Initially, NF-κB activation, the traditional priming signal, was thought to induce gene transcription of NALP3 and pro-IL-1β, leading to NALP3 inflammasome activation (4). However, recent studies suggest that assembly of the NALP3 inflammasome to activate caspase 1 is regulated mainly through non-transcriptional processes (14, 15). Furthermore, the final products of the activated NALP3 inflammasome, IL-1β and IL-18, are possibly regulated at posttranslational levels, suggesting concerted actions of modulators through this mode of regulatory control (16, 17).
Ubiquitination is a universal posttranslational modification process in eukaryotic organisms because it serves as a distinct signal for protein disposal or sorting in cells. Furthermore, some E3 ligases and ubiquitin components are activated by endotoxin (8, 18). Nod-like receptors (NLRs) interact with ubiquitin ligase-associated components, such as suppressor of the glycolysis regulation 2 gene, that modulate inflammasome activity (19). Studies suggest that the ubiquitination system may play an important role in NALP3 inflammasome activation to release cytokines. For example, deubiquitinases may regulate the activity of caspase 1 and IL-1β secretion via the assembly of inflammasomes (20), although the level of BRCC3, one of the deubiquitinases targeting NALP3, does not correlate with the abundance of NALP3 protein after LPS exposure (21). An E3 ligase, tripartite motif 33, and the linear ubiquitin assembly complex also seem to be involved with NALP3 inflammasome activation (22, 23). While preparing this manuscript, dopamine was reported to inhibit NALP3 inflammasome activation via the dopamine D1 receptor by promoting actions of the E3 ubiquitin ligase, MARCH7 (24). However, to date, studies have only focused on the link between ubiquitination and the terminal steps of NALP3 inflammasome activation: assembly of the inflammasome complex and subsequent release of cytokines. Despite evidence that LPS priming increases NALP3 protein levels, the precise molecular mechanisms elucidating how LPS priming up-regulates NALP3 protein levels and whether cytokine release can be altered by manipulating this priming process remain unclear. Here we describe a mechanism by which LPS primes the NALP3 inflammasome by impairing its association with a previously unrecognized NALP3 inhibitor, FBXL2, which, in turn, extends the inflammasome lifespan in cells. We identified that FBXL2 interacts with Trp-73, facilitating ubiquitin ligation to Lys-689 within the NALP3 protein. Interestingly, a tryptophan within TRAFs 1–6 also serves as a critical docking site for FBXL2 interaction (8). Therefore, tryptophan may be a common recognition site, along with other motifs, for FBXL2 recruitment to its substrates.
Previously, the unique, selective, small-molecule FBXO3 inhibitor BC-1215 has been shown to block IL-1β secretion triggered by LPS (11). The inhibition of FBXO3 resulted in an increase in FBXL2 cellular mass, promoting the degradation of NALP3 protein. Here the inhibition of NALP3 protein by BC-1215 did not occur at the transcriptional level but, rather, at the posttranslational level by decreasing NALP3 protein half-life. As the amount of NALP3 protein after LPS priming is reduced with BC-1215 treatment, the absolute amount of activated NALP3 inflammasome complexes is decreased coordinately in cells, which led to a profound reduction of IL-1β and IL-18 release. One unexpected finding here is that prolonged exposure to the FBXO3 inhibitor also inhibited the LPS induction of pro-IL-1β and, therefore, reduced the release of mature IL-1β because limited substrate is available for conversion to mature cytokine. This is probably due to the ability of BC-1215 to inhibit TRAF adaptor proteins (8), leading to the interruption of NF-κB activation, which normally induces the pro-IL-1β gene (25). Indeed, shorter exposure to BC-1215 effectively decreased mature IL-1β without significant changes in pro-IL-1β, indicating reduced conversion from premature to mature IL-1β by the FBXO3 inhibitor (Fig. 7H). On the other hand, the reduction of IL-18 secretion by BC-1215 appears to be mediated simply through the inhibited conversion to mature cytokines. Pro-IL-18 is known to be expressed constitutively in cells in the absence of stimuli (17). The change in levels of pro-IL-18 protein after prolonged BC-1215 treatment was relatively small compared with that of pro-IL-1β. However, the reduction of mature IL-18 secretion was much more robust, similar to that of IL-1β, indicating that the mechanism of decreased IL-18 secretion by an FBXO3 inhibitor is mainly achieved by reducing the conversion of pro-IL-18 to mature IL-18 (Fig. 7G). These results are likely secondary to decreased levels of an activated NALP3 inflammasome complex by BC-1215.
In conclusion, our study shows that LPS prolongs the lifespan of the NALP3 protein, likely via reduced ubiquitin-mediated proteasomal processing. We found that SCFFBXL2 constitutively mediates the ubiquitination and degradation of NALP3, an effect disrupted by LPS. Through pharmacological inhibition of FBXO3, which targets the disposal of FBXL2 in cells, NALP3 protein levels are reduced profoundly, subsequently decreasing the release of IL-1β and IL-18 in human inflammatory cell lines. Our findings provide valuable mechanistic insights into inflammasome priming, and this may uncover unique opportunities to develop therapeutic strategies to modulate cytokine signaling during inflammatory illness.
Acknowledgment
We thank Sarah Dunn for assistance with the experiments.
This work was performed on the basis of work supported, in part, by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. This work was supported, in whole or in part, by National Institutes of Health R01 Grants HL096376, HL097376, HL098174, HL081784, 1UH2HL123502, P01HL114453 (to R. K. M.), and HL116472 (to B. B. C.). This work was also supported by a merit review award from the Department of Veterans Affairs, the Flight Attendant Medical Research Institute, and American Heart Association Award 12SDG12040330 (to C. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
- NALP
- NACHT, LRR and PYD domain-containing protein
- ASC
- apoptosis-associated speck-like protein containing a carboxy-terminal caspase recruitment domain
- SCF
- Skp-Cullin-F box
- TRAF
- TNF receptor-associated factor
- CHX
- cycloheximide
- MLE
- mouse lung epithelial
- NLR
- Nod-like receptor.
References
- 1. Franchi L., Muñoz-Planillo R., Núñez G. (2012) Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Makabe H., Kojika M., Takahashi G., Matsumoto N., Shibata S., Suzuki Y., Inoue Y., Endo S. (2012) Interleukin-18 levels reflect the long-term prognosis of acute lung injury and acute respiratory distress syndrome. J. Anesth. 26, 658–663 [DOI] [PubMed] [Google Scholar]
- 3. Dolinay T., Kim Y. S., Howrylak J., Hunninghake G. M., An C. H., Fredenburgh L., Massaro A. F., Rogers A., Gazourian L., Nakahira K., Haspel J. A., Landazury R., Eppanapally S., Christie J. D., Meyer N. J., Ware L. B., Christiani D. C., Ryter S. W., Baron R. M., Choi A. M. (2012) Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am. J. Respir. Crit. Care Med. 185, 1225–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bauernfeind F. G., Horvath G., Stutz A., Alnemri E. S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B. G., Fitzgerald K. A., Hornung V., Latz E. (2009) Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 6. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 7. Cardozo T., Pagano M. (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5, 739–751 [DOI] [PubMed] [Google Scholar]
- 8. Chen B. B., Coon T. A., Glasser J. R., McVerry B. J., Zhao J., Zhao Y., Zou C., Ellis B., Sciurba F. C., Zhang Y., Mallampalli R. K. (2013) A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol. 14, 470–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davies J. Q., Gordon S. (2005) Isolation and culture of human macrophages. Methods Mol. Biol. 290, 105–116 [DOI] [PubMed] [Google Scholar]
- 10. Chen B. B., Mallampalli R. K. (2009) Masking of a nuclear signal motif by monoubiquitination leads to mislocalization and degradation of the regulatory enzyme cytidylyltransferase. Mol. Cell. Biol. 29, 3062–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mallampalli R. K., Coon T. A., Glasser J. R., Wang C., Dunn S. R., Weathington N. M., Zhao J., Zou C., Zhao Y., Chen B. B. (2013) Targeting F box protein Fbxo3 to control cytokine-driven inflammation. J. Immunol. 191, 5247–5255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Radivojac P., Vacic V., Haynes C., Cocklin R. R., Mohan A., Heyen J. W., Goebl M. G., Iakoucheva L. M. (2010) Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78, 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schroder K., Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 14. Juliana C., Fernandes-Alnemri T., Kang S., Farias A., Qin F., Alnemri E. S. (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 287, 36617–36622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fernandes-Alnemri T., Kang S., Anderson C., Sagara J., Fitzgerald K. A., Alnemri E. S. (2013) Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J. Immunol. 191, 3995–3999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niebler M., Qian X., Höfler D., Kogosov V., Kaewprag J., Kaufmann A. M., Ly R., Böhmer G., Zawatzky R., Rösl F., Rincon-Orozco B. (2013) Post-translational control of IL-1β via the human papillomavirus type 16 E6 oncoprotein: a novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog. 9, e1003536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghonime M. G., Shamaa O. R., Das S., Eldomany R. A., Fernandes-Alnemri T., Alnemri E. S., Gavrilin M. A., Wewers M. D. (2014) Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 192, 3881–3888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu Y., Nguyen T. T., Bui K. C., Demello D. E., Smith J. B. (2005) A novel inflammation-induced ubiquitin E3 ligase in alveolar type II cells. Biochem. Biophys. Res. Commun. 333, 253–263 [DOI] [PubMed] [Google Scholar]
- 19. Mayor A., Martinon F., De Smedt T., Pétrilli V., Tschopp J. (2007) A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 8, 497–503 [DOI] [PubMed] [Google Scholar]
- 20. Lopez-Castejon G., Luheshi N. M., Compan V., High S., Whitehead R. C., Flitsch S., Kirov A., Prudovsky I., Swanton E., Brough D. (2013) Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J. Biol. Chem. 288, 2721–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Py B. F., Kim M. S., Vakifahmetoglu-Norberg H., Yuan J. (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338 [DOI] [PubMed] [Google Scholar]
- 22. Weng L., Mitoma H., Trichot C., Tricot C., Bao M., Liu Y., Zhang Z., Liu Y. J. (2014) The E3 ubiquitin ligase tripartite motif 33 is essential for cytosolic RNA-induced NLRP3 inflammasome activation. J. Immunol. 193, 3676–3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodgers M. A., Bowman J. W., Fujita H., Orazio N., Shi M., Liang Q., Amatya R., Kelly T. J., Iwai K., Ting J., Jung J. U. (2014) The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 211, 1333–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yan Y., Jiang W., Liu L., Wang X., Ding C., Tian Z., Zhou R. (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73 [DOI] [PubMed] [Google Scholar]
- 25. Zhu L. J., Yang X., Li X. Y., Liu Q. H., Tang X. Q., Zhou S. F., Kong Q. Y., Axelsson J., Yu X. Q. (2010) Suppression of tumor necrosis factor receptor associated factor (TRAF)-2 attenuates the proinflammatory and proliferative effect of aggregated IgG on rat renal mesangial cells. Cytokine 49, 201–208 [DOI] [PubMed] [Google Scholar]