

Abstract
Infections by fungal pathogens such as Candida albicans and Aspergillus fumigatus and their resistance to triazole drugs are major concerns. Fungal lanosterol 14α-demethylase belongs to the CYP51 class in the cytochrome P450 superfamily of enzymes. This monospanning bitopic membrane protein is involved in ergosterol biosynthesis and is the primary target of azole antifungal drugs, including fluconazole. The lack of high-resolution structural information for this drug target from fungal pathogens has been a limiting factor for the design of modified triazole drugs that will overcome resistance. Here we report the X-ray structure of full-length Saccharomyces cerevisiae lanosterol 14α-demethylase in complex with fluconazole at a resolution of 2.05 Å. This structure shows the key interactions involved in fluconazole binding and provides insight into resistance mechanisms by revealing a water-mediated hydrogen bonding network between the drug and tyrosine 140, a residue frequently found mutated to histidine or phenylalanine in resistant clinical isolates.
INTRODUCTION
Fungal pathogens are estimated to be responsible for ∼1.5 million deaths per year worldwide, with members of the genera Candida, Cryptococcus, and Aspergillus being those most often associated with life-threatening disseminated disease (1). Comorbidity with infectious diseases such as malaria, tuberculosis, and AIDS, the use of therapies that leave individuals immunocompromised, and the emergence of resistant clinical isolates due to antifungal prophylaxis and antifungal agrochemicals all contribute to this problem (2). In sub-Saharan Africa, Cryptococcus species are most often associated with comorbidity and cause about 400,000 deaths per annum. In the United States, Candida species are the fourth-most-common cause of nosocomial bloodstream infections, accounting for 8% to 10% of all hospital-acquired infections and resulting in an attributed mortality rate of 40% (3). Fluconazole (FLC)-resistant Candida species account for 7% of bloodstream isolates of this organism. This results in significant additional hospitalization costs and about 220 deaths per annum in the United States (4). Chemotherapy and radiotherapy patients, as well as those undergoing immunosuppressive therapies associated with organ transplants, are particularly susceptible to life-threatening invasive mycoses, mainly those caused by Candida and Aspergillus species (5).
Despite the introduction and use of the echinocandin antifungals since 2001, azole drugs such as FLC remain the most widely used class of antifungals due to their efficacy and low cost. The azole drugs used in the clinic are categorized into imidazoles and triazoles, depending on the nature of the iron-coordinating heterocycle in the drug. FLC is a first-generation triazole but has a half-life longer than and bioavailability superior to those of the second- and third-generation triazoles such as itraconazole (ITC), voriconazole (VCZ; Fig. 1), and posaconazole (PCZ) (6). Despite favorable pharmacokinetic properties and limited drug interactions, FLC is less potent than other triazoles and exhibits no activity against invasive molds (7). VCZ is structurally related to FLC but has the disadvantage of showing significant toxicity and many major drug interactions and requires therapeutic drug monitoring (8). Even so, VCZ remains the drug of choice for treatment of disseminated aspergillosis (2).
FIG 1.
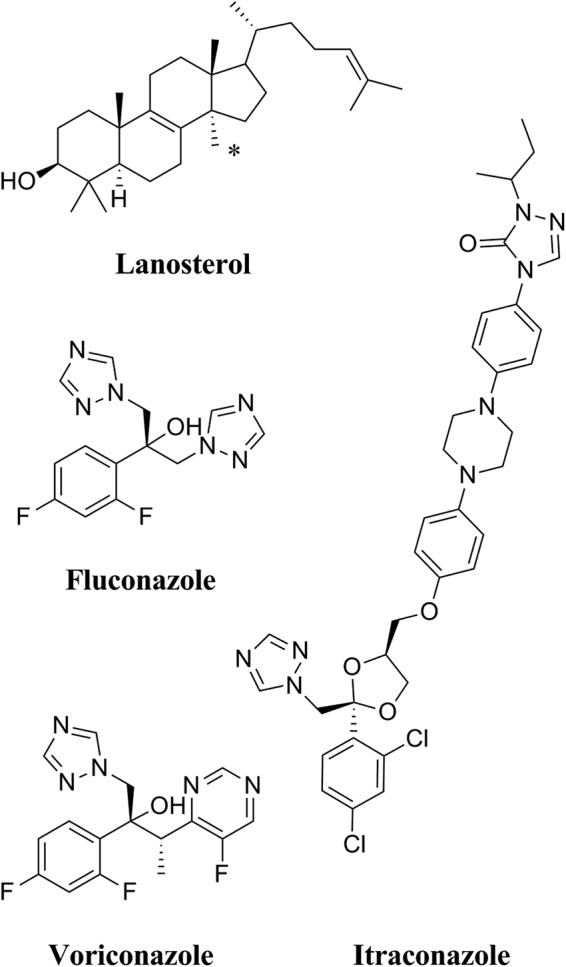
The chemical structures of the substrate lanosterol, the short-tailed triazole antifungal agents FLC and VCZ, and the long-tailed azole antifungal ITC. An asterisk (*) indicates the 14α-methyl of the lanosterol.
The antifungal activity of the azoles depends on their ability to inhibit the activity of the enzyme lanosterol 14α-demethylase (CYP51 [or Erg11p]), a member of the CYP51 class of cytochrome P450 enzymes (9). The active site of lanosterol 14α-demethylase contains a heme cofactor where the sixth position of the octahedral coordinate geometry of the iron can be occupied by a nitrogen in either the imidazole ring or the triazole ring of the azole drugs. The 14α demethylation of lanosterol (Fig. 1) is the rate-limiting step in ergosterol biosynthesis in fungi (10). Inhibition of Erg11p leads to a depletion of ergosterol from cell membranes. This affects the fluidity of the lipid bilayer and slows fungal growth. Inhibition of Erg11p also results in the accumulation of toxic metabolites such as the 14α-methyl-3,6-diol formed from 14-methyl-fecosterol (11). The combined effects of ergosterol depletion and toxic metabolite production are fungistatic for many pathogenic fungi, including Candida albicans, Candida glabrata, and Aspergillus species.
Fungi have evolved several mechanisms to circumvent the action of azoles, including mutations in the drug-binding site of Erg11p that reduce the binding affinity of triazoles and thus confer resistance (12–14). The innate resistance of Aspergillus fumigatus to FLC is thought to be due to the presence of two homologues of the drug target, with CYP51B appearing sensitive to fluconazole whereas CYP51A confers significantly reduced susceptibility (15).
Prokaryotic P450s are soluble cytoplasmic proteins, whereas the eukaryotic enzymes, excluding mitochondrial P450s, possess a single transmembrane helix that tethers the enzyme to the lipid bilayer. Until 2014, X-ray structures of eukaryotic CYP51s were obtained by deletion of the transmembrane helix to improve expression and crystallization (16). These CYP51s included the human CYP51 enzyme (PDB identifiers [IDs] 3LD6, 3JUV, and 3JUS) (16) and CYP51 enzymes from the protozoa Trypanosoma cruzi and Trypanosoma brucei (PDB IDs 2WX2, 2WV2, and 2X2N) (17). Other available CYP51 structures include those of Mycobacterium tuberculosis CYP51 (e.g., PDB ID 1EA1) (19) and Leishmania infantum CYP51 (PDB ID 3L4D) (20). The only structural information available on fungal CYP51s was from homology models based on the truncated structures of other P450s and, more recently, on human CYP51 and Mycobacterium tuberculosis CYP51 (21–23).
The first full-length structure of a fungal CYP51 was determined by X-ray crystallography of Erg11p from the yeast Saccharomyces cerevisiae (24). Two structures were deposited in the PDB, one with the native substrate lanosterol (PDB ID 4LXJ) bound in the active site in a precatalytic state and the other with the long-tailed triazole drug ITC (PDB ID 4K0F), at resolutions of 2.19 Å and 1.90 Å, respectively. Structures of Erg11p complexed with FLC or VCZ could be refined only at a significantly lower resolution (≥2.5 Å) and were not deposited in the PDB (24). Elucidation of the structure of the complete enzyme identified the likely orientation of the enzyme in relation to the lipid bilayer. Amphipathic N-terminal membrane helix 1 (MH1) and the adjacent transmembrane helix (TMH1) are oriented at an angle of about 60° to each other, and contacts between TMH1 and the catalytic domain help position the catalytic domain such that it is partly in the lipid bilayer (Fig. 2). The enzyme has a common P450 fold with α-helices (A to L) and β-sheets (1 to 4) constituting the catalytic domain, with the heme cofactor forming the active site. The entrance to the substrate channel is in direct contact with the surface of the lipid bilayer. This ensures that the entry of the lipidic substrate is performed directly from the surface of the membrane. In this report, we present the high-resolution crystal structure of full-length S. cerevisiae Erg11p in complex with the short-tailed triazole FLC that reveals key hydrogen bond networks in the active site.
FIG 2.
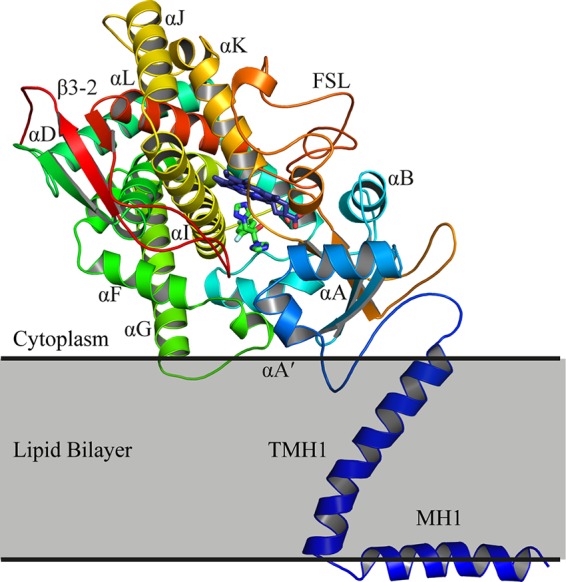
The structure of ScErg11p6×His. The protein is colored from the N terminus to the C terminus with a gradient from blue to red. The heme (purple carbons) and FLC (green carbons) are shown in sticks. For clarity, only some of the α-helices and β-sheets are labeled. FSL, fungus-specific loop.
MATERIALS AND METHODS
Yeast strains.
The yeast strains used in this study are described in Table 1. The AD2Δ strain was used for heterologous overexpression of the full-length S. cerevisiae ERG11 gene containing a sequence encoding a C-terminal hexahistidine tag (S. cerevisiae Erg11p6×His [ScErg11p6×His]). This strain is a derivative of the ADΔ strain previously described by Lamping et al. (25, 52) but with the HIS1 gene deleted. Both strains have the major ATP-binding cassette transporter pumps deleted and are therefore hypersensitive to azole drugs. The ScErg11p6×His strain was constitutively overexpressed from the PDR5 locus due to a gain-of-function pdr1–3 mutation in the Pdr1p transcriptional regulator (26). The endogenous ScERG11 gene was then replaced via homologous recombination using a histidine (His1) marker to create strain AD3ΔScErg11p (Table 1) using a previously described method (25).
TABLE 1.
Yeast strains used in this study
| Strain | Strain of origin and genotype | Reference or source |
|---|---|---|
| ADΔ | AD124567, Δpdr5::hisG Δpdr15::hisG Δura3 | Lamping et al. (25) |
| AD2Δ | ADΔ, Δhis1 | This study |
| AD2ΔScErg11p | AD2Δ, Δpdr5::ScERG11-URA3 | This study |
| AD3ΔScErg11p | AD2Δ ScErg11p, Δerg11::HIS1 | This study |
MIC80 determinations.
Triazole drugs are fungistatic and often give trailing growth even at high drug concentrations. Therefore, MICs were determined at 80% growth inhibition compared to the level seen with the nondrug controls. In addition, the RPMI medium used for standard MIC determinations with Candida albicans supports poor growth in S. cerevisiae (27) and so was not used. Instead, the MIC80s of FLC, ITC, and VCZ for yeast cells were determined using 0.079% complete synthetic media (Formedium, Norfolk, United Kingdom) containing 0.67% (wt/vol) yeast nitrogen base without amino acids (BD Difco Laboratories Inc., Franklin Lakes, NJ), 2% (wt/vol) glucose, 10 mM MES (4-morpholineethanesulfonic acid), and 20 mM HEPES (buffered with Tris to pH 6.8). The 96-well microtiter plates were seeded with cells to an optical density at 600 nm (OD600) of 0.005. The microtiter plates were incubated at 30°C with shaking at 200 rpm for 48 h, and OD600 measurements were made using a BioTek Synergy 2 multimode plate reader (BioTek Instruments, VT, USA). MIC80 determinations were done in triplicate for each strain and in 3 separate experiments.
Protein purification.
The purification of ScErg11p6×His was carried out according to methods described previously (24). In brief, crude membranes were prepared from liquid cultures grown overnight in YPD medium (1% [wt/vol] yeast extract [BD Difco], 2% [wt/vol] peptone [BD Difco], 2% [wt/vol] dextrose) at 30°C with shaking at 200 rpm. Cells were broken by bead beating, and crude membranes were obtained by differential centrifugation. Crude membranes were solubilized with 10× critical micelle concentration (CMC) n-decyl-β-d-maltoside (DM) (Affymetrix Inc., Santa Clara, US). ScErg11p6×His was purified from the solubilized crude membrane fraction by affinity chromatography using 2 ml of packed nickel-nitrilotriacetic acid (Ni-NTA)-agarose matrix (Qiagen) per gram of protein. FLC was added at a final concentration of 40 μM to 5 ml of pooled eluate obtained with 200 mM imidazole. The affinity-purified ScErg11p6×His was concentrated by centrifugal filtration using a 50-kDa-molecular-mass-cutoff Amicon Ultra-4 centrifugal filter (Millipore) and further purified by size exclusion chromatography (SEC) using a Superdex 200 10/300 GL column (GE Healthcare Life Sciences, United Kingdom). The column was equilibrated with SEC buffer containing 10% (wt/vol) glycerol, 150 mM NaCl, 20 mM HEPES, 0.5 mM phenylmethanesulfonyl fluoride, 6.4 mM DM (4× CMC), 10 μM FLC, and 1 Roche EDTA-free protease inhibitor pill per 400 ml. The pooled, colored fractions containing the 62-kDa ScErg11p6×His were concentrated using a 50-kDa-molecular-mass-cutoff Amicon Ultra-4 centrifugal filter.
Cytochrome P450 concentration.
Carbon monoxide-binding spectra were used to determine the concentrations of functional and nonfunctional cytochrome P450 according to the protocol described by Guengerich et al. (28). The cytochrome P450 concentration was determined using 1.5 μM ScErg11p6×His in affinity chromatography medium (10% [wt/vol] glycerol, 250 mM NaCl, 20 mM Tris, 0.5 mM phenylmethanesulfonyl fluoride, 16 mM DM [10×CMC], and 1 Roche EDTA-free protease inhibitor pill, pH 7.5). Protein spectra were measured at between 400 and 700 nm for oxidized protein and for the protein reduced with ∼6 mM sodium dithionite. The CO-P450 complex was obtained by bubbling CO through the sample in 10-mm-path-length UV transparent plastic cuvettes (GE Healthcare Life Sciences, United Kingdom) followed by addition of sodium dithionite (28). Absorption spectra were recorded using a Cary 1 Bio UV-visible spectrophotometer. The P450 concentration was determined using an extinction coefficient of 91 mM−1 cm−1 for the difference in absorbance between 445 and 490 nm (29).
Binding of triazole drugs to ScErg11p6×His.
The affinity purification for drug-binding studies used affinity chromatography medium containing 2 mM l-histidine for washing the enzyme-bound column and 50 mM l-histidine for elution of ScErg11p×His. l-Histidine was removed from the preparation by washing the enzyme with affinity chromatography medium using a 50-kDa-molecular-mass-cutoff Amicon Ultra-4 centrifugal filter (Millipore). Difference spectra were obtained by titrating affinity-purified ScErg11p6×His and used to determine dissociation constant (Kd) values for the binding to the triazole drugs FLC and VCZ (30, 31). Incremental additions of up to 7 μl of triazole drug dissolved in dimethyl sulfoxide (DMSO) were added to a 0.5-ml sample of 1 μM ScErg11p6×His in affinity chromatography buffer in a 10-mm-path-length cuvette. The same incremental volumes of DMSO were added to the reference cuvette, which contained the same amount of enzyme. Difference spectra (350 to 500 nm) were recorded, and the trough-peak absorbance changes due to type II binding were used to generate drug-binding curves. The Hill equation ΔA = ΔAmax [azole]n/([azole]n + Kdn) (where ΔA is the change in absorbance, [azole] is the azole concentration, and n is the apparent Hill number) was fitted to the curve and used to calculate Kd values. The 50% inhibitory concentration (IC50) values for the binding curves are denoted here as [azole]0.5. All calculations were carried out using GraphPad Prism 6 software (GraphPad Prism, San Diego, CA).
Crystallization and X-ray data collection.
Affinity-purified ScErg11p6×His copurified with FLC by SEC was crystallized using a hanging-drop vapor diffusion method. The crystals formed within 1 week at 18°C. The reservoir solution contained 45% polyethylene glycol 400–100 mM glycine at a pH range of 9.3 to 9.5. The drops were 4 μl in a 1:1 ratio of reservoir solution and ∼20 mg/ml of the protein in SEC buffer. The crystals were flash-cooled in liquid nitrogen prior to data collection. A complete data set was collected on the MX1 beamline at the Australian Synchrotron using an ADSC Quantum 210r detector. The data were indexed and integrated using iMosflm (32) and scaled with SCALA (33). Molecular replacement was carried out using Phaser-MR (34) from Phenix (35) and ScErg11p6×His cocrystallized with lanosterol (PDB ID 4LXJ) as the template (24). Refinement and modeling were performed using phenix.refine (35) and Coot (36), respectively. The inhibitor was modeled into the appropriate density in the active site, and quantities of water were added if at least one hydrogen bond was detected (2.5 to 3.3 Å). Iron-to-nitrogen and iron-to-sulfur distances were constrained during refinement to 2.15 Å and 2.33 Å based on the average coordinate bond distance for more than 80 known heme Fe-N (triazole) complexes and 4 heme Fe-S complexes in the Cambridge Structural Database (see Table S2 in the supplemental material) (37).
Sequence accession number.
The coordinates and structure factors have been deposited in the Protein Data Bank under accession number 4WMZ.
RESULTS AND DISCUSSION
Drug susceptibility of S. cerevisiae strains expressing Erg11p.
As expected, deletion of the HIS1 ORF in the ADΔ strain did not alter the azole susceptibility pattern in the AD2Δ derivative strain. The parent and derivative strain were equally highly sensitive to the triazoles tested (Table 2). Constitutive hyperexpression of ScErg11p6×His from the PDR5 locus under the control of the gain-of-function pdr1–3 mutation increased the resistance of the AD2Δ strain to FLC 6-fold, to ITC 3.5-fold, and to VCZ 5-fold. Removal of the endogenous ScERG11 in the AD3ΔScErg11p strain did not significantly increase susceptibility to these triazoles. This showed that the azole resistance was dependent on the presence of recombinant ScErg11p6×His and not that of the native ScErg11p.
TABLE 2.
FLC, ITC, and VCZ MIC80 values for S. cerevisiae strains
| Strain | MIC80 (μg/ml)a |
||
|---|---|---|---|
| FLC | ITC | VCZ | |
| ADΔ | 0.4 (± 0.04) | 0.03 | 0.05 (± 0.001) |
| AD2Δ | 0.4 (± 0.04) | 0.03 | 0.05 (± 0.001) |
| AD2Δ ScErg11p | 2.4 (± 0.13) | 0.106 (± 0.001) | 0.24 (± 0.02) |
| AD3Δ ScErg11p | 2.1 (± 0.02) | 0.105 (± 0.004) | 0.25 (± 0.05) |
MIC80 values were determined as described in Materials and Methods. The table shows the means for 3 separate clones of each strain calculated using data obtained in triplicate measurements from at least 3 different experiments (a total of 9 determinations per strain). Values for standard errors of the means (SEM) are indicated in brackets.
Spectral characteristics of purified ScErg11p.
The absorbance spectrum for ScErg11p6×His in its low-spin ferric state showed a heme Soret (γ) band at 417 nm and δ, β, and α peaks at 350, 535, and 571 nm, respectively (Fig. 3). One-electron-reduced spectra obtained using sodium dithionite gave a blue shift of the Soret peak from 417 nm to 414 nm. CO binding to the ferrous ScErg11p6×His produced a characteristic red shift to 445 nm. The minor shoulder at 420 nm indicated the presence of a small amount of inactive P420 complex in the purified enzyme preparation. The absolute, reduced, and CO spectra for purified ScErg11p6×His were comparable to those obtained previously for S. cerevisiae microsomal preparations by Yoshida and Aoyama (38) and indicated that the ScErg11p6×His preparation was almost completely active.
FIG 3.
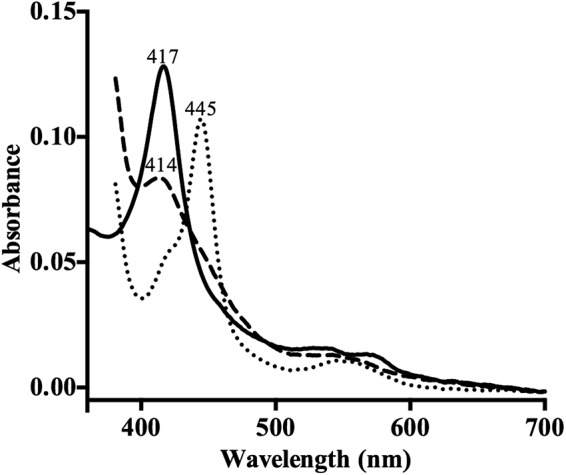
Spectral characterization of ScErg11p6×His. Data represent the absolute spectra of the ferric protein (solid trace), the ferrous protein reduced with sodium dithionite (dashed trace), and the reduced protein with bound carbon monoxide (dotted trace). Spectra were recorded using 1.5 μM ScErg11p as described in Materials and Methods.
Binding of triazole drugs to ScErg11p6×His.
The binding of FLC (Fig. 4a) and VCZ (data not shown) to ScErg11p6×His gave type II difference spectra caused by the coordination of the nitrogen atom in the triazole ring with the heme iron, replacing the water ligand and stabilizing the low-spin form (39). The absorbance spectra of ScErg11p6×His in the presence of the triazoles caused a red shift of the heme Soret peak from 417 nm to 421 nm for both VCZ and FLC. The difference spectra obtained with 1 μM ScErg11p6×His enzyme in the presence of excess FLC and VCZ were very similar, with a peak at 428 nm and a trough at 410 nm (Table 3). The binding is normally characterized as tight if the Kd values are lower than or equal to those of the enzyme concentration, and the Morrison equation is generally used to fit the data (40). However, using the Hill equation for analysis of the change in absorbance caused by drug binding versus the change in the concentration of triazole drug added, and not the rearrangement of the Morrison equation, gave the best fit (Fig. 4b and Table 3). The [azole]0.5 values were similar for the two drugs, as were the Kd values (Table 3). Similarly to previous studies performed with C. albicans CYP51, the [azole]0.5 values were found to be about half the P450 concentration values, indicating tight binding for VCZ and FLC (31, 41). The Kd value for FLC of 74 (± 15 [standard error]) nM is comparable to those previously obtained using the Morrison equation for CaCYP51 in two different studies, i.e., 46.6 (± 10.6) nM (31) and 60 (± 10) nM (42). The apparent Hill numbers of 3 for FLC and 2.7 for VCZ are unlikely to be due to positive cooperativity between ScErg11p monomers. They are more likely to result from multiple interactions between the ligand and the internal active site of an enzyme (43) that behaves as a monomer within its own detergent vesicle. Consistent with this hypothesis, size exclusion chromatography analysis of the affinity-purified enzyme gives enzyme-containing detergent vesicles that migrate with an apparent molecular mass of <100 kDa (24).
FIG 4.
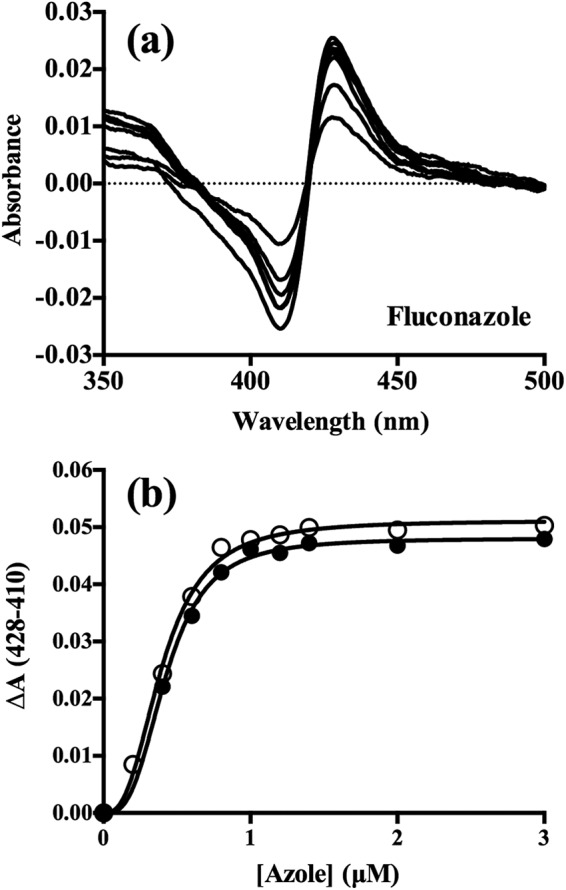
Binding of FLC and VCZ to ScErg11p6×His. (a) Difference spectra demonstrating type II binding of FLC obtained by incremental additions of the drug to 1 μM ScErg11p×His. (b) Saturation curves for FLC (filled circles) and VCZ (hollow circles) fitted to the Hill equation, with ΔA plotted against azole concentration.
TABLE 3.
Binding of triazole drugs to affinity-purified ScErg11p6×Hisa
| Triazole | ΔA max | λtrough | λpeak | Kd (μM) | Hill no. | [Azole]0.5 (μM) |
|---|---|---|---|---|---|---|
| FLC | 0.048 | 410 | 428 | 0.074 (± 0.015) | 3.0 | 0.43 |
| VCZ | 0.051 | 410 | 428 | 0.082 (± 0.018) | 2.7 | 0.4 |
The binding of azole drugs to 1 μM Ni-NTA affinity-purified ScErg11p6×His was determined as described in Materials and Methods. max, maximum. Standard errors are shown in parentheses.
X-ray crystal structure of S. cerevisiae Erg11p in complex with FLC.
Full-length S. cerevisiae lanosterol 14α-demethylase (ScErg11p6×His) copurified with FLC during SEC was used for crystallization. Data collection parameters and refinement statistics are presented in Table S1 in the supplemental material. Molecular replacement was carried out using full-length ScErg11p6×His (PDB ID 4LXJ) and revealed a single monomer in the asymmetric unit (PDB ID 4WMZ) comprising MH1, TM1, and the catalytic domain as seen previously (24) (Fig. 2). The enzyme appears to be a rigid molecule, as the tertiary structure and the shape of the substrate channel are unchanged, regardless of the size of the ligand (lanosterol, ITC, or FLC) bound in the active site (24). Some differences in ScErg11p6×His in complex with FLC, compared with the two previously deposited structures, were detected in side chains of residues of the transmembrane helix, in side chains of residues of helix-F′ and helix-G and the F′/G loop that connects them, and in some parts of the enzyme exposed to the solvent. The F/G loop is buried in the membrane, and this region has the highest β-factor values together with the transmembrane helix and the three residues Y439, S440, and V441 in all three structures, as previously discussed (24). Residues Y439 and V441 are within the N-terminal portion of a fungus-specific loop and are located at the surface of the protein.
The proposed egress channel that bifurcates from the substrate channel shows some density in the present structure comparable to that seen in the two previously published structures (24). The density has been proposed to represent zymosterol rather than the immediate product 4,4-dimethylcholesta-8,14,24-trienol, based on mass spectrometry analysis of the purified enzyme (24). The side chains of residues around this secondary vestibule are positioned similarly among all three structures except H128, which has been modeled into different positions due to the limited density. This flexibility may be required to accommodate the exit of the product or interaction with subsequent enzymes in the ergosterol biosynthesis pathway, e.g., Erg24 and the Erg25-Erg28 complex (44).
FLC binding.
FLC binds within the active site of the enzyme, with clear evidence of the ligand apparent from the molecular replacement seen immediately following the solution step of the experiment (Fig. 5a). The triazole ring of FLC is coordinated to the iron within the heme cofactor (Fe-N distance, 2.13 Å), corroborating the spectrophotometric data showing type II binding. The immediate environment surrounding FLC is hydrophobic, as would be expected from the nature of the lanosterol substrate. Residues within 4 Å of FLC are illustrated in Fig. 5b and include Y126, F134, I139, Y140, F236, G310, V311, G314, G315, T318, L380, and M509. The 2,4-difluorophenyl ring of FLC lies in a position adjacent to G310, and the coordinated triazole ring abuts G314 on the GXXXG consensus motif for sterol binding on helix-I. Consistent with other eukaryotic CYP51 structures, such as the Trypanosoma CYP51 structure, the bend in helix-I of ScErg11p at this motif is less acute than in the M. tuberculosis CYP51 structure (19). Two water molecules form hydrogen bond networks with FLC (Fig. 5c). The first water (molecule 743) mediates hydrogen bonds between the hydroxyl groups of FLC and Y140 as well as a propionate of the heme cofactor. The second water (molecule 790) forms hydrogen bonds with the carbonyl oxygen of S382, the hydroxyl of Y126, and N4 of the second triazole ring which points away from the heme (Fig. 5c). In the previously published structures of ScErg11p6×His complexed with lanosterol and ITC, neither water 743 nor water 790 was observed. Previously reported structures of other CYP51s complexed with FLC have the inhibitor bound in a similar manner. The orientation of the 2,4-difluorophenyl ring matches that seen in the M. tuberculosis (PDB ID 1EA1) and T. cruzi (PDB ID 2WX2) CYP51 structures (17). The 2,4-difluorophenyl ring is rotated 180° in several other structures, but this orientation would appear to be potentially unfavorable, as the 2-fluoro substituent and the hydroxyl group of FLC are in close proximity (PDB ID 2WV2 and 2WUZ) (17). The latter alternate orientation (2WUZ) is stabilized by a hydrogen bond between the fluorine substituent and the hydroxyl of Y103 (corresponding to Y126 in ScErg11p6×His) (17). This residue (Y103) shows a great deal of flexibility among the trypanosomal structures, including occupying the site for key water molecule 743.
FIG 5.
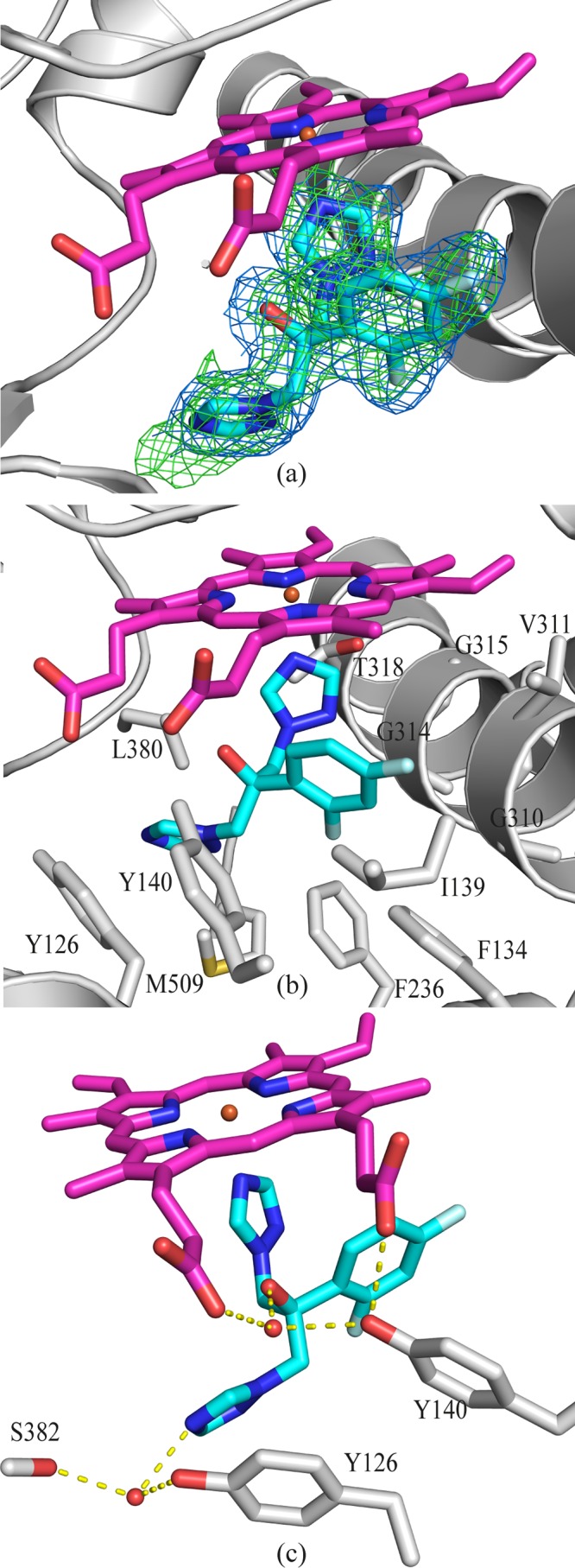
FLC binding in the active site of ScErg11p. (a) OMIT map for FLC (Fo-Fc map [green mesh] contoured at 3σ; 2Fo-Fc map [blue mesh] contoured at 1σ). The Fo-Fc map was calculated using Fcalc refined from coordinates with no ligand at the active site. The 2Fo-Fc map was calculated following the final refinement. The main chain is indicated in gray, and fluconazole is indicated as sticks, with C atoms in cyan, N atoms blue, O atoms in red, and F atoms in pale blue. The heme is shown as sticks, with C atoms colored magenta. (b) Side chains of amino acid residues within 4 Å of FLC are indicated in gray. The main chain atoms are shown for G310, G314, and G315. (c) Water-mediated hydrogen bonding (yellow dashed lines) between HOH743, FLC, heme, and Y140, as well as between HOH790, FLC, and S382.
VCZ, another short-tailed antifungal agent, is similar to FLC and differs only in that the second noncoordinating triazole is replaced with a fluoropyrimidine group along with the addition of a methyl group. In the previous modeled structure obtained with a lower-resolution data set (2.8 Å), the fluoropyrimidine ring is positioned in the same plane as the triazole ring, with the fluoro substituent positioned in the direction opposite to that seen with the hydroxyl of the inhibitor and the methyl group (24). The presence of the key water (743) would also provide the potential for a hydrogen bond with N3 of the fluoropyrimidine in this orientation in preference to the flipped conformation.
Antifungal resistance.
Mutations which reduce the effectiveness of azole drugs are frequently found in resistant pathogenic fungi. Of the residues within 4 Å of FLC in ScErg11p6×His that have residues equivalent to those found to be mutated in C. albicans Erg11p, only Y132F/H (Y140F/H in ScErg11p) is known to confer resistance to FLC as a single mutation (13, 45). The others confer resistance in combination with additional mutations or confer susceptible phenotypes alone (46). The single-amino-acid substitutions Y132F, K143R, F145L, S405F, D446E, G448E, F449V, G450E, and G464S in C. albicans Erg11p have been found to confer resistance to FLC (45). The FLC MICs for those mutants were increased ∼4-fold compared to those for the susceptible strain. Our structure shows that only Y132F, K143R, and G464S are located close enough to the active site and the heme cofactor to directly affect the binding of FLC. Residues D446, G448, F449, and G450 (fungus-specific loop) as well as F145 and S405 are too far away to directly affect the heme or interfere with FLC binding.
The Y132F/H mutation in C. albicans is commonly found in clinical isolates of other fungal pathogens of humans and plants that have reduced susceptibility to FLC and other azoles (12, 47). As described above, a key water molecule (743) mediates hydrogen bonds between the hydroxyl group of the inhibitor, the hydroxyl group of Y140, and the propionate group of the heme (Fig. 5c). Homologous mutations at Y140 to either phenylalanine or histidine could disrupt this hydrogen bonding network and therefore reduce susceptibility to FLC.
Mutations equivalent to G464S (S. cerevisiae and C. albicans numbering) have been found in both C. albicans and A. fumigatus (see Fig. S2 in the supplemental material). The C. albicans G464S mutation has been found in clinical isolates and gives rise to FLC resistance (13, 45, 48), while the equivalent G448S mutation in A. fumigatus CYP51B is thought to confer resistance to the long-tailed drugs ITC and PCZ but not resistance to the short-tailed VCZ drug (49). It has been suggested that this substitution would disrupt the positioning of the heme, reduce the binding of the azole, and increase resistance (22). However, its differential effect on short-tailed azoles compared to long-tailed azoles is not understood. G464 is located on the same face of the heme as the coordinating cysteine residue and is close to a propionate group of the cofactor that makes a hydrogen bond to a water molecule (see Fig. S2). The G464S mutation may replace this water and alter the interaction with the heme carboxylate, thus potentially tilting the heme.
The K143R mutation in C. albicans CYP51 (K151 S. cerevisiae numbering) confers resistance to FLC (50). This residue is also located on the same side of the heme cofactor as the coordinating cysteine (see Fig. S2 in the supplemental material). In the current crystal structure, the side chain of K151 forms an ionic interaction with the carboxylate group of the heme. The change from lysine to arginine, while being functionally conservative, results in the addition of a large guanidinium group in an environment optimized for lysine. The change causes local disruption, but its extent is unknown.
A. fumigatus has innate resistance to FLC, which may be a result of sequence differences between the two homologues of CYP51 present in this species (see Fig. S1 in the supplemental material). Most fungal species have a threonine at the position corresponding to T322 on helix I (S. cerevisiae numbering). A. fumigatus CYP51A has an isoleucine at this position (A. fumigatus CYP51A numbering, I301) whereas A. fumigatus CYP51B maintains the threonine (A. fumigatus CYP51B numbering, T315). It has been hypothesized that this difference confers resistance to FLC (51). Alanine mutagenesis at the homologous T315 position in C. albicans has been shown to result in increased resistance to FLC CYP51 (41). The current structure of ScErg11p shows that T322 is located adjacent to the active site on helix-I and close to an ethylene group of the heme but about 10 Å from the iron-coordinated triazole group. G310 (S. cerevisiae numbering) is within a distance of 4 Å of the drug. Glycine or alanine is accommodated in this position in fungal CYP51s, apart from A. fumigatus CYP51A, which has threonine (T289), and in one of two sequences available for Coccidioides immitis RS CYP51 (T303). This residue appears to be in better position than T322 to alter the binding of FLC (Fig. 5c).
Future prospects.
The high-resolution X-ray crystal structure of full-length recombinant S. cerevisiae CYP51 (ScErg11p6×His) complexed with FLC and the resultant discovery of hydrogen bonding networks involving key water molecules within the active site provide the basis for detailed exploration of both the binding and resistance mechanisms of azole antifungal drugs, in particular, FLC and VCZ, in fungal pathogens. Due to the strong homology of fungal CYP51s, ScErg11p can be used, both structurally and experimentally, as a model to study the mutations that have been observed in resistant clinical isolates of fungal pathogens. The presence of a water-mediated hydrogen bond network between an inhibitor and Y140 in the active site of lanosterol 14α-demethylase when complexed with the short-chain FLC but not with the long-chain ITC, as well as the interactions of the long tail of ITC in the enzyme entrance channel (24), may explain the differential levels of susceptibility to FLC and ITC conferred by mutations equivalent to Y140F/H. Structural and functional analysis will enable rigorous determination of the effects of these mutations on the protein and its affinity for these drugs.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by grants from the Marsden Fund of the Royal Society of New Zealand and the Health Research Council of New Zealand awarded to B.C.M. Funding for travel to the Australian Synchrotron was provided by the New Zealand Synchrotron Group.
The mentoring from Franziska Huschmann is deeply appreciated. We thank Sigurd Wilbanks for his wealth of knowledge and sound advice.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00925-15.
REFERENCES
- 1.Brown GD, Denning DW, Levitz SM. 2012. Tackling human fungal infections. Science 336:647. doi: 10.1126/science.1222236. [DOI] [PubMed] [Google Scholar]
- 2.Denning DW, Bromley MJ. 2015. How to bolster the antifungal pipeline. Science 347:1414–1416. doi: 10.1126/science.aaa6097. [DOI] [PubMed] [Google Scholar]
- 3.Horn DL, Fishman JA, Steinbach WJ, Anaissie EJ, Marr KA, Olyaei AJ, Pfaller MA, Weiss MA, Webster KM, Neofytos D. 2007. Presentation of the PATH Alliance registry for prospective data collection and analysis of the epidemiology, therapy, and outcomes of invasive fungal infections. Diagn Microbiol Infect Dis 59:407–414. doi: 10.1016/j.diagmicrobio.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 4.US Department of Health and Human Services, Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States. US Department of Health and Human Services, Centers for Disease Control and Prevention. http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf.
- 5.Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, Booker R, Castanheira M, Messer SA, Perlin DS, Pfaller MA. 2013. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis 56:1724–1732. doi: 10.1093/cid/cit136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lipp HP. 2008. Antifungal agents—clinical pharmacokinetics and drug interactions. Mycoses 51(Suppl 1):S7–S18. doi: 10.1111/j.1439-0507.2008.01523.x. [DOI] [PubMed] [Google Scholar]
- 7.Kontoyiannis DP, Lewis RE. 2002. Antifungal drug resistance of pathogenic fungi. Lancet 359:1135–1144. doi: 10.1016/S0140-6736(02)08162-X. [DOI] [PubMed] [Google Scholar]
- 8.Ashbee HR, Barnes RA, Johnson EM, Richardson MD, Gorton R, Hope WW. 2014. Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology. J Antimicrob Chemother 69:1162–1176. doi: 10.1093/jac/dkt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lepesheva GI, Waterman MR. 2011. Structural basis for conservation in the CYP51 family. Biochim Biophys Acta 1814:88–93. doi: 10.1016/j.bbapap.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lepesheva GI, Hargrove TY, Kleshchenko Y, Nes WD, Villalta F, Waterman MR. 2008. CYP51: A major drug target in the cytochrome P450 superfamily. Lipids 43:1117–1125. doi: 10.1007/s11745-008-3225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watson PF, Rose ME, Ellis SW, England H, Kelly SL. 1989. Defective sterol C5–6 desaturation and azole resistance: a new hypothesis for the mode of action of azole antifungals. Biochem Biophys Res Commun 164:1170–1175. doi: 10.1016/0006-291X(89)91792-0. [DOI] [PubMed] [Google Scholar]
- 12.Becher R, Wirsel SG. 2012. Fungal cytochrome P450 sterol 14alpha-demethylase (CYP51) and azole resistance in plant and human pathogens. Appl Microbiol Biotechnol 95:825–840. doi: 10.1007/s00253-012-4195-9. [DOI] [PubMed] [Google Scholar]
- 13.Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob Agents Chemother 42:241–253. doi: 10.1093/jac/42.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snelders E, Karawajczyk A, Schaftenaar G, Verweij PE, Melchers WJ. 2010. Azole resistance profile of amino acid changes in Aspergillus fumigatus CYP51A based on protein homology modeling. Antimicrob Agents Chemother 54:2425–2430. doi: 10.1128/AAC.01599-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mellado E, Garcia-Effron G, Buitrago MJ, Alcazar-Fuoli L, Cuenca-Estrella M, Rodriguez-Tudela JL. 2005. Targeted gene disruption of the 14-alpha sterol demethylase (cyp51A) in Aspergillus fumigatus and its role in azole drug susceptibility. Antimicrob Agents Chemother 49:2536–2538. doi: 10.1128/AAC.49.6.2536-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strushkevich N, Usanov SA, Park HW. 2010. Structural basis of human CYP51 inhibition by antifungal azoles. J Mol Biol 397:1067–1078. doi: 10.1016/j.jmb.2010.01.075. [DOI] [PubMed] [Google Scholar]
- 17.Chen CK, Leung SS, Guilbert C, Jacobson MP, McKerrow JH, Podust LM. 2010. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl Trop Dis 4:e651. doi: 10.1371/journal.pntd.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reference deleted.
- 19.Podust LM, Poulos TL, Waterman MR. 2001. Crystal structure of cytochrome P450 14 alpha-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci U S A 98:3068–3073. doi: 10.1073/pnas.061562898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hargrove TY, Wawrzak Z, Liu J, Nes WD, Waterman MR, Lepesheva GI. 2011. Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14alpha-demethylase (CYP51) from Leishmania infantum. J Biol Chem 286:26838–26848. doi: 10.1074/jbc.M111.237099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boscott PE, Grant GH. 1994. Modeling cytochrome P450 14 alpha demethylase (Candida albicans) from P450cam. J Mol Graph 12:185–192, 195. doi: 10.1016/0263-7855(94)80086-3. [DOI] [PubMed] [Google Scholar]
- 22.Fraczek MG, Bromley M, Bowyer P. 2011. An improved model of the Aspergillus fumigatus CYP51A protein. Antimicrob Agents Chemother 55:2483–2486. doi: 10.1128/AAC.01651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao L, Madison V, Chau AS, Loebenberg D, Palermo RE, McNicholas PM. 2004. Three-dimensional models of wild-type and mutated forms of cytochrome P450 14 alpha-sterol demethylases from Aspergillus fumigatus and Candida albicans provide insights into posaconazole binding. Antimicrob Agents Chemother 48:568–574. doi: 10.1128/AAC.48.2.568-574.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JD, O'Connell JD III, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM. 2014. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc Natl Acad Sci U S A 111:3865–3870. doi: 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamping E, Monk BC, Niimi K, Holmes AR, Tsao S, Tanabe K, Niimi M, Uehara Y, Cannon RD. 2007. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot Cell 6:1150–1165. doi: 10.1128/EC.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura K, Niimi M, Niimi K, Holmes AR, Yates JE, Decottignies A, Monk BC, Goffeau A, Cannon RD. 2001. Functional expression of Candida albicans drug efflux pump Cdr1p in a Saccharomyces cerevisiae strain deficient in membrane transporters. Antimicrob Agents Chemother 45:3366–3374. doi: 10.1128/AAC.45.12.3366-3374.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niimi K, Harding DR, Parshot R, King A, Lun DJ, Decottignies A, Niimi M, Lin S, Cannon RD, Goffeau A, Monk BC. 2004. Chemosensitization of fluconazole resistance in Saccharomyces cerevisiae and pathogenic fungi by a d-octapeptide derivative. Antimicrob Agents Chemother 48:1256–1271. doi: 10.1128/AAC.48.4.1256-1271.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guengerich FP, Martin MV, Sohl CD, Cheng Q. 2009. Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat Protoc 4:1245–1251. doi: 10.1038/nprot.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Omura T, Sato R. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378. [PubMed] [Google Scholar]
- 30.Warrilow AG, Melo N, Martel CM, Parker JE, Nes WD, Kelly SL, Kelly DE. 2010. Expression, purification, and characterization of Aspergillus fumigatus sterol 14-alpha demethylase (CYP51) isoenzymes A and B. Antimicrob Agents Chemother 54:4225–4234. doi: 10.1128/AAC.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warrilow AG, Martel CM, Parker JE, Melo N, Lamb DC, Nes WD, Kelly DE, Kelly SL. 2010. Azole binding properties of Candida albicans sterol 14-alpha demethylase (CaCYP51). Antimicrob Agents Chemother 54:4235–4245. doi: 10.1128/AAC.00587-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen FH. 2002. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B 58:380–388. doi: 10.1107/S0108768102003890. [DOI] [PubMed] [Google Scholar]
- 38.Yoshida Y, Aoyama Y. 1987. Interaction of azole antifungal agents with cytochrome-P-45014dm purified from Saccharomyces cerevisiae microsomes. Biochem Pharmacol 36:229–235. doi: 10.1016/0006-2952(87)90694-0. [DOI] [PubMed] [Google Scholar]
- 39.Locuson CW, Hutzler JM, Tracy TS. 2007. Visible spectra of type II cytochrome P450-drug complexes: evidence that “incomplete” heme coordination is common. Drug Metab Dispos 35:614–622. doi: 10.1124/dmd.106.012609. [DOI] [PubMed] [Google Scholar]
- 40.Morrison JF. 1969. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim Biophys Acta 185:269–286. doi: 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- 41.Lamb DC, Kelly DE, Schunck WH, Shyadehi AZ, Akhtar M, Lowe DJ, Baldwin BC, Kelly SL. 1997. The mutation T315A in Candida albicans sterol 14alpha-demethylase causes reduced enzyme activity and fluconazole resistance through reduced affinity. J Biol Chem 272:5682–5688. doi: 10.1074/jbc.272.9.5682. [DOI] [PubMed] [Google Scholar]
- 42.Park HG, Lee IS, Chun YJ, Yun CH, Johnston JB, Montellano PR, Kim D. 2011. Heterologous expression and characterization of the sterol 14alpha-demethylase CYP51F1 from Candida albicans. Arch Biochem Biophys 509:9–15. doi: 10.1016/j.abb.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prinz H, Schonichen A. 2008. Transient binding patches: a plausible concept for drug binding. J Chem Biol 1:95–104. doi: 10.1007/s12154-008-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mo C, Bard M. 2005. A systematic study of yeast sterol biosynthetic protein-protein interactions using the split-ubiquitin system. Biochim Biophys Acta 1737:152–160. doi: 10.1016/j.bbalip.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 45.Flowers SA, Colon B, Whaley SG, Schuler MA, Rogers PD. 2015. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob Agents Chemother 59:450–460. doi: 10.1128/AAC.03470-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morio F, Loge C, Besse B, Hennequin C, Le Pape P. 2010. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn Microbiol Infect Dis 66:373–384. doi: 10.1016/j.diagmicrobio.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 47.Wheat LJ, Connolly P, Smedema M, Durkin M, Brizendine E, Mann P, Patel R, McNicholas PM, Goldman M. 2006. Activity of newer triazoles against Histoplasma capsulatum from patients with AIDS who failed fluconazole. J Antimicrob Chemother 57:1235–1239. doi: 10.1093/jac/dkl133. [DOI] [PubMed] [Google Scholar]
- 48.Chau AS, Mendrick CA, Sabatelli FJ, Loebenberg D, McNicholas PM. 2004. Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azoles. Antimicrob Agents Chemother 48:2124–2131. doi: 10.1128/AAC.48.6.2124-2131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bueid A, Howard SJ, Moore CB, Richardson MD, Harrison E, Bowyer P, Denning DW. 2010. Azole antifungal resistance in Aspergillus fumigatus: 2008 and 2009. J Antimicrob Chemother 65:2116–2118. doi: 10.1093/jac/dkq279. [DOI] [PubMed] [Google Scholar]
- 50.Manavathu EK, Kallakuri S, Arganoza MT, Vazquez JA. 1999. Amino acid variations of cytochrome P-450 lanosterol 14 alpha-demethylase (CYP51A1) from fluconazoleresistant clinical isolates of Candida albicans. Rev Iberoam Micol 16:198–203. [PubMed] [Google Scholar]
- 51.Edlind TD, Henry KW, Metera KA, Katiyar SK. 2001. Aspergillus fumigatus CYP51 sequence: potential basis for fluconazole resistance. Med Mycol 39:299–302. doi: 10.1080/mmy.39.3.299.302. [DOI] [PubMed] [Google Scholar]
- 52.Monk BC, Cannon RD, Nakamura K, Niimi M, Niimi K, Holmes AR, Lamping E, Harding DRK, Goffeau A, Decottignies A. May 2014. Yeast membrane protein expression system and its application in drug screening. US patent 8728797 B2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.