

Background: There is limited knowledge of Klebsiella pneumoniae determinants implicated in the blocking of the NF-κB signaling pathway.
Results: A high-throughput genetic screen led to the identification of 114 putative K. pneumoniae genes that are associated with suppression of NF-κB activation.
Conclusion: CPS, LPS, and the T2SS-secreted PulA are needed for immune evasion.
Significance: A new therapeutic approach to treat Klebsiella infections will be the prevention of immune evasion.
Keywords: iron, Klebsiella pneumonia, lipopolysaccharide (LPS), NF-κ B (NF-κB), toll-like receptor (TLR), pullulanase
Abstract
Klebsiella pneumoniae is an etiologic agent of community-acquired and nosocomial pneumonia. It has been shown that K. pneumoniae infections are characterized by reduced early inflammatory response. Recently our group has shown that K. pneumoniae dampens the activation of inflammatory responses by antagonizing the activation of the NF-κB canonical pathway. Our results revealed that K. pneumoniae capsule polysaccharide (CPS) was necessary but not sufficient to attenuate inflammation. To identify additional Klebsiella factors required to dampen inflammation, we standardized and applied a high-throughput gain-of-function screen to examine a Klebsiella transposon mutant library. We identified 114 mutants that triggered the activation of NF-κB. Two gene ontology categories accounted for half of the loci identified in the screening: metabolism and transport genes (32% of the mutants) and envelope-related genes (17%). Characterization of the mutants revealed that the lack of the enterobactin siderophore was linked to a reduced CPS expression, which in turn underlined the NF-κB activation induced by the mutant. The lipopolysaccharide (LPS) O-polysaccharide and the pullulanase (PulA) type 2 secretion system (T2SS) are required for full effectiveness of the immune evasion. Importantly, these factors do not play a redundant role. The fact that LPS O-polysaccharide and T2SS mutant-induced responses were dependent on TLR2-TLR4-MyD88 activation suggested that LPS O-polysaccharide and PulA perturbed Toll-like receptor (TLR)-dependent recognition of K. pneumoniae. Finally, we demonstrate that LPS O-polysaccharide and pulA mutants are attenuated in the pneumonia mouse model. We propose that LPS O-polysaccharide and PulA T2SS could be new targets for the design of new antimicrobials. Increasing TLR-governed defense responses might provide also selective alternatives for the management of K. pneumoniae pneumonia.
Introduction
Klebsiella pneumoniae is a Gram-negative pathogen causing a wide range of infections from urinary tract infections to pneumonia. Klebsiella-triggered pneumonia is particularly devastating among immunocompromised patients with mortality rates between 25 and 60% (1). K. pneumoniae is a member of the so-called ESKAPE group of microorganisms to emphasize that they effectively “escape” the effects of antibacterial drugs (2). Despite the clinical relevance of K. pneumoniae, the infection biology of this pathogen is poorly understood, and therefore it is both urgent and necessary to better understand its pathophysiology to be able to design new strategies to treat K. pneumoniae infections.
The best characterized virulence factor of this pathogen is the capsule polysaccharide (CPS).3 Isogenic CPS mutant strains are avirulent and unable to cause pneumonia and urinary tract infections (3–5). Lipid A of lipopolysaccharide (LPS), the outer membrane proteins OmpA and OmpK36, iron-scavenging systems, and several adhesins are other virulence determinants that have been characterized (6–10). Nonetheless, there is still limited knowledge of the exact role of individual virulence factors in Klebsiella infections.
A wealth of evidence indicates that the activation of early inflammatory responses is essential to clear Klebsiella infections (11–14). Any interference with this response leads to a more severe infection (15), thus in turn, augmenting the immune response with exogenous inflammatory mediators decreases the mortality associated with K. pneumoniae infection (16–19). Collectively, this evidence suggests that Klebsiella tries to counteract the induction of these host defense responses. Indeed, we (8, 20–22) and others (23) have provided compelling evidence for this notion.
At the molecular level, we have demonstrated that K. pneumoniae abrogates the activation of inflammatory responses by targeting NF-κB and MAPK signaling pathways (22, 24). Klebsiella antagonizes the activation of NF-κB via the deubiquitinase CYLD and blocks the phosphorylation of MAPKs via the MAPK phosphatase MKP-1 (22). CYLD and MKP-1 are normally regulated to return to homeostasis after inflammation to protect the host from an overwhelming inflammatory response (25, 26). To exert this anti-inflammatory effect, K. pneumoniae affects the membrane association of the receptor NOD1 (22). This is dependent on Klebsiella-triggered Rho GTPase Rac1 inhibition (22). To identify additional host factors involved in the anti-inflammatory effect, we applied an unbiased high-throughput siRNA gain-of-function screen to interrogate the human kinome (24). Follow-up validation revealed that K. pneumoniae exploits an EGF receptor (EGFR)-PI3K-AKT-PAK4-ERK-GSK3β signaling pathway to induce the expression of the deubiquitinase CYLD to attenuate the cytokine-dependent nuclear translocation of NF-κB (24). Our group uncovered a role for CPS in the activation of EGFR and EGFR-dependent signaling (24). However, because CPS does not activate NOD1-dependent responses (22), K. pneumoniae may employ additional factors to attenuate NF-κB activation.
This study was designed to identify K. pneumoniae determinants implicated in blocking the activation of the NF-κB signaling pathway. To take a systematic approach toward the identification of these bacterial factors, we performed a high-throughput genetic screen to mine a transposon mutant library of K. pneumonia strain 52145. This is a reference strain of serotype K2 highly virulent Klebsiella strains from which important virulence factors, including the large virulence plasmid harboring the regulator of mucoid phenotype (rmpA) and the aerobactin cluster, have been identified (27). We found 114 mutants that, in sharp contrast to the wild-type strain, activated the NF-κB signaling pathway. Further characterization confirmed the critical role of K. pneumoniae CPS in blocking NF-κB activation and uncovered the role of the LPS polysaccharide section and the pullulanase type II secretion system (T2SS) in immune evasion.
Experimental Procedures
Bacterial Strains, Growth Conditions, and Reagents
Strains and plasmids used in this study are listed in Table 1. K. pneumoniae 52145 (hereafter Kp52145) is a clinical isolate (serotype O1:K2) described previously (3, 28). Bacteria were grown in Luria-Bertani (LB) medium at 37 °C unless indicated otherwise. When appropriate, antibiotics were added to the growth medium at the following concentrations: ampicillin, 100 μg/ml; rifampicin, 50 μg/ml; kanamycin, 50 μg/m; cloramphenicol (Cm), 25 μg/ml; and trimethoprim (Tp), 100 μg/ml. FeSO4, and FeCl3 were used at a concentration of 10 μm, and 2-2′-dipyridyl (Sigma) was used at a concentration of 200 μm. The sequences of the primers used in this study are available from the authors upon request.
TABLE 1.
Bacterial strains and plasmids used in this study
| Bacterial strains and plasmids | Genotype or comments | Source or reference |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| CC118-λpir | Δ(ara-leu)7967 araD139 ΔlacX74 galE galK ΔphoA20 thi-1 rpsE rpoB argE(Am) recA1 | Laboratory collection |
| DH5a-λpir | DlacU169 (F80lacZDM15) recA1 endA1 hsdR17 thi-1 gyrA96 relA1 λpir phage lysogen | Laboratory collection |
| SY327-λpir | (lac pro) argE (Am) rif nalA recA56 (λpir) | Laboratory collection |
| Klebsiella pneumoniae | ||
| Kp52145 | Clinical isolate; serotype O1:K2; RifR | (3, 28) |
| 52145-ΔwcaK2 | Kp52145, ΔwcaK2, capsule mutant; RifR | (32) |
| 52OmpA2 | Kp52145, ΔompA::Cm, ompA gene is inactivated; RifR, CmR | (37) |
| 52ΔwcaK2OmpA | 52145-ΔwcaK2, ΔompA::Cm, capsule mutant in which ompA is inactivated; RifR, CmR | (37) |
| 52ΔwaaL | Kp52145, ΔwaaL, waaL is inactivated; nonpolar mutant; RifR | (68) |
| 52ΔwabM | Kp52145, ΔwabM, wabM is inactivated; nonpolar mutant; RifR | (50) |
| 52ΔwabH | Kp52145, ΔwabH, wabH is inactivated; nonpolar mutant; RifR | (50) |
| 52ΔwabK | Kp52145, ΔwabK, wabK is inactivated; non polar mutant; RifR | (50) |
| 52145-ΔwcaK2ΔwaaL | 52145-ΔwcaK2; ΔwaaL, waaL is inactivated; non polar mutant; RifR | (6) |
| 52145-ΔwcaK2ΔwabM | 52145-ΔwcaK2; ΔwabM, wabM is inactivated; non polar mutant; RifR | (6) |
| 52145-ΔwcaK2ΔwabK | 52145-ΔwcaK2; ΔwabK, wabK is inactivated; non polar mutant; RifR | (6) |
| 52145-ΔwcaK2ΔwabH | 52145-ΔwcaK2; ΔwabH, wabH is inactivated; non polar mutant; RifR | (6) |
| 52waaL::tn5 | Kp52145; ΔwaaL::tn5, waaL is inactivated; RifR, KmR | This study |
| 52entF::tn5 | Kp52145; entF::tn5, entF is inactivated; RifR, KmR | This study |
| 52145-ΔwcaK2entF | 52145-ΔwcaK2, entF:: pKNOCK-Cm_entF, capsule mutant in which entF is inactivated; RifR, CmR | This study |
| 52pulC::tn5 | Kp52145; ΔpulC::tn5, pulC is inactivated; RifR, KmR | This study |
| 52irp1 | Kp52145, irp1:: pKNOCK-Cm_IRP1, irp1 is inactivated; RifR, CmR | This study |
| 52iutA | Kp52145, iutA:: pKNOCK-Cm_iutA, iutA is inactivated; RifR, CmR | This study |
| 52glf | Kp52145, glf:: pSFglf, glf is inactivated; RifR, KmR | This study |
| 52ΔpulA | Kp52145, pulA:: pKNOCK-Cm_pulA, pulA is inactivated; RifR, CmR | This study |
| 52pulACom | 52ΔpulA harboring mini-Tn7TKmKpnpulA; PulA levels restored; RifR, CmR, KmR | This study |
| 52145-ΔwcaK2pulA | 52145-ΔwcaK2, pulA:: pKNOCK-Cm_pulA, capsule mutant in which pulA is inactivated; RifR, CmR | This study |
| 52145-ΔwcaK2pulACom | 52145-ΔwcaK2pulA mutant harboring mini-Tn7TKmKpnpulA; PulA levels restored; RifR, CmR, KmR | This study |
| 52ΔwaaLpulA | 52ΔwaaL, pulA:: pKNOCK-Cm_pulA, waaL mutant in which pulA is inactivated; RifR, CmR | This study |
| 52145-ΔwcaK2ΔwaaL pulA | 52145-ΔwcaK2ΔwaaL, pulA:: pKNOCK-Cm_pulA, capsule and waaL mutant in which pulA is inactivated; RifR, CmR | This study |
| Plasmids | ||
| pRL27 | Tn5 transposon delivery vector, KmR | (30) |
| pGEM-T Easy | Cloning plasmid, AmpR | Promega |
| pKNOCK-Cm | Suicide vector, R6K replication origin, CmR | (35) |
| pKNOCK-Cm_entF | pKNOCK-Cm containing an internal fragment of entF gene for mutant construction, CmR | This study |
| pKNOCK-Cm_IRP1 | pKNOCK-Cm containing an internal fragment of irp1 gene for mutant construction, CmR | This study |
| pKNOCK-Cm_iutA | pKNOCK-Cm containing an internal fragment of iutA gene for mutant construction, CmR | This study |
| pKNOCK-Cm_pulA | pKNOCK-Cm containing an internal fragment of pulA gene for mutant construction, CmR | This study |
| pSF100 | Suicide vector, R6K replication origin, KmR | (36) |
| pSFglf | pSF100 containing an internal fragment of glf gene for mutant construction, KmR | This study |
| pUC18R6KT-mini-Tn7TKm | pUC18R6KT-mini-Tn7TKm containing a Km cassette, AmpR, KmR | (37) |
| pTSNSK-Tp | pTSNSK-Tp containing a transposase for Tn7 insertion, KmR, TpR | (38) |
| pUC18R6KT-mini-Tn7TKm_pulACom | pUC18R6KT-mini-Tn7TKm containing pulA gene for complementation, AmpR, KmR | This study |
| p34S-Tp | Source of Tp cassette; AmpR, TpR | (40) |
| pPROBE′-gfp[LVA] | Plasmid containing gfp without promoter as reporter gene, KmR | (41) |
| pPROBE′-gfp[LVA]Tp | Trimethoprim resistance cassette cloned into SphI site of pPROBE′-gfp[LVA], TpR | This study |
| pPROBE′-gfp[LVA]Tp_entC | pPROBE′-gfp[LVA]Tp containing entC promoter region, TpR | This study |
| pPROBE′-gfp[LVA]Tp_ybtA | pPROBE′-gfp[LVA]Tp containing ybtA promoter region, TpR | This study |
| pPROBE′-gfp[LVA]Tp_psn | pPROBE′-gfp[LVA]Tp containing psn promoter region, TpR | This study |
| pPROBE′-gfp[LVA]Tp_iucA | pPROBE′-gfp[LVA]Tp containing iucA promoter region, TpR | This study |
| pGPLKpnCps | pGPL01 containing cps promoter region; AmpR | (37) |
Cell Culture and Infection
Monolayers of A549 (ATCC CCL185) were grown as described previously (22). For infections, A549 cells were seeded to 90% confluence (3 × 105 cells/well) in 24-well tissue culture plates. Cells were serum-starved for 16 h before infection. Bacteria were prepared as described (22), and infections were performed using a multiplicity of infection of 100 bacteria/cell unless indicated otherwise. For incubation times longer than 120 min, bacteria were killed by the addition of gentamicin (100 μg/ml), which was not removed until the end of the experiment. Cell viability, assessed by trypan blue dye exclusion, was >95% even after 4 h of infection.
Mutant Library Construction
Kp52145 was made electrocompetent following the method described by Sharma and Schimke (29). Transposon mutagenesis was performed by electroporating ∼1 μg of the pRL27 mini Tn5 transposon (30) to 50 μl of Kp52145 electrocompetent cells using a Gene Pulser Xcell (Bio-Rad) followed by a 60-min recovery in SOC (super optimal broth) medium at 37 °C. The transposon carries a kanamycin resistance cassette and an R6K origin of replication (30). Transposants were plated on LB-rifampicin-kanamycin plates, and the transposon mutant library was generated from six independent electroporation rounds. A total of 5320 mutants were arrayed in a 96-well plate format and stored at −80 °C in 20% (v/v) glycerol (master plate). One well without bacteria was kept as a negative control in all plates (typically well H12). Random transposon insertion was checked in ∼200 clones by direct genomic sequencing. Genomic DNA was extracted using the Realpure spin kit (Real). Sequencing reaction was performed in an iCycler (Bio-Rad) containing 2 μl of BigDye Master Mix, 2 μl of 5× buffer, 1 μl of betaine (5 m), 1 μm primer (tpnRL17), and 2–5 μg of genomic DNA. The thermal profile consisted of 5 min of a denaturalization step at 95 °C followed by 100 cycles of 95 °C for 30 s, 50 °C for 20 s, and 60 °C for 4 min. Sequencing reactions were purified following the manufacturer's instructions, resuspended in 10 μl of Hi-Di formamide, and run in an ABI 3730 genome sequencer (Applied Biosystems).
Construction of a NF-κB Reporter Cell Line
A549 cells were seeded into 24-well plates at a density of 4 × 104 cells/well and transfected 24 h later with 5 μg of pNifty2-SEAP (InvivoGen) using Lipofectamine 2000 (Invitrogen) lipofection reagent according to the manufacturer's instructions. In the pNifty2-SEAP vector, the proximal promoter of the endothelial cell-leukocyte adhesion molecule (ELAM-1; E-selectin), containing three NF-κB sites and lacking an AP1/CREB (cAMP-response element-binding protein) site found in the full-length promoter, controls the reporter gene, SEAP (secreted form of the human embryonic alkaline phosphatase). Twenty-four hours after transfection, cells were selected with 500 μg/ml Zeocin for 2 weeks (InvivoGen). Cells were singularized by serial dilutions in 96-well plates, and monoclonal cell lines were propagated for 2 weeks. Stably transfected A549-SEAP cells were grown in complete RPMI medium supplemented with 100 μg/ml Zeocin. A2 was the clone selected for screening purposes.
High-throughput Screening
Cell Seeding and Bacterial Culture
A549-SEAP A2 cells from the same passage number were seeded into 96-well plates at 1–2 × 104 cells/well in 100 μl of RPMI 1640. Transposon mutants and controls were grown in 96-well plates. 5 μl from the bacteria master plate were used to inoculate 150 μl of LB supplemented with the appropriate antibiotics. Plates were incubated overnight at 37 °C without shaking. Bacterial growth was determined by measuring absorbance at 600 nm (A600) using a microplate reader (Biotek PowerWave HT).
Infection
A549-SEAP A2 cells were washed twice with 200 μl of PBS, and then 150 μl of RPMI 1640 supplemented with 10% fetal calf serum (FCS) was added. Five μl of the bacterial overnight culture was used to infect the cells (multiplicity of infection, ∼100:1). Only the inner 60 wells of the plate were used. Two replicates of each control (negative/positive (IL-1β, 10 ng/ml) cells infected with Kp52145, 52OmpA, 52145ΔwcaK2, and 52145ΔwcaK2-OmpA) and 48 mutants were run in each plate. After 3 h, wells were washed twice with 200 μl of PBS, and then 100 μl of RPMI 1640 containing 100 μg/ml of gentamicin was added to each well. Plates were incubated overnight in a humidified CO2 incubator.
Colorimetric Assay
30 μl of the supernatant was mixed with 200 μl of QUANTI-Blue reagent (InvivoGen), and the mixture was incubated at 37 °C for 24 h. The levels of SEAP were measured at an absorbance at 625 nm (A625).
Statistical Analysis
The Z score was calculated for each of the 48 wells of a plate as described previously (31). For a given well, the Z score was calculated by subtracting the mean value of the wells on that plate from the value of the well and dividing by the standard deviation value for all the plate wells. A Z score ≥ 2 was considered significant. As means and standard deviations are greatly influenced by statistical outliers (31), which in the context of a screening are putative hits, we also selected mutants that induced SEAP levels higher than A625 > 0.5, which represents a 2-fold A625 value induced by the wild-type strain.
Hit Validation
Four independent validation rounds were run for each selected hit. A hit was considered validated if the SEAP levels were A625 > 0.5 in at least two of four validation runs.
Transposon Insertion Site Identification
Genomic DNA was purified using the Realpure spin kit (Real). Direct genomic sequencing was carried out as described above (see “Mutant Library Construction”).
Growth Deficiency Determination
To identify those hits with growth deficiencies, transposon mutants were plated on LB plates or M9 plates supplemented with either 0.2% glucose or 10 mm citrate and incubated at 37 °C for 48 h.
CPS Purification and Quantification
Cell-associated CPS from K. pneumoniae strains were purified using the hot phenol-water method exactly as described previously (32). CPS was quantified by determining the concentration of uronic acid in the samples using a modified carbazole assay (33).
LPS Extraction and Analysis by SDS-PAGE
The LPS from K. pneumoniae strains was extracted by a modified phenol-water purification method (34). The LPS was run on a 12% SDS-PAGE and visualized using a Pro-Q Emerald 300 lipopolysaccharide gel stain Kit (Invitrogen).
K. pneumoniae Mutant Construction
The entF, irp1, iutA, and pulA mutants were constructed by insertion mutagenesis using the pKNOCK-Cm suicide vector (35). An internal fragment of each gene was amplified using genomic DNA from wild-type K. pneumoniae 52145, Vent polymerase (New England Biolabs), and the corresponding primer pairs. PCR products were phosphorylated and cloned into the SmaI-digested and dephosphorylated pKNOCK-Cm vector to obtain pKNOCK-Cm_entF, pKNOCK-Cm_irp1, pKNOCK-Cm_iutA, and pKNOCK-Cm_pulA, respectively. The glf mutant was also constructed by insertion mutagenesis using the pir replication-dependent plasmid pSF100 (36). Plasmids were introduced into Klebsiella by conjugation and PCR, or Southern blot was used to identify strains in which plasmids were inserted into the genome by homologous recombination.
Complementation of pulA Mutant
To complement the pulA mutant, a DNA fragment of 4,008 bp containing the putative promoter region and coding region of pulA was PCR-amplified (primer pair pulACom.F-pulACom.R) using Vent polymerase, gel-purified, and cloned into SmaI-digested pUC18R6KT-mini-Tn7TKm (37) to obtain pUC18R6KT-mini-Tn7TKm_pulACom. The pTSNSK-Tp plasmid, which contains the transposase tnsABCD, necessary for Tn7 transposon mobilization (38), was electroporated into the pulA mutant. Then, the pUC18R6KT-mini-Tn7TKm_pulACom plasmid was mobilized into this strain by triparental conjugation using the helper strain Escherichia coli HB101/pRK2013. Colonies were screened for resistance to kanamycin and sensitivity to ampicillin. Because the ampicillin cassette is located outside of the Tn7 region on the vector, sensitivity to ampicillin denotes the integration of the Tn7 derivative at the attTn7 site instead of incorporation of the vector into the chromosome. Confirmation of integration of the Tn7 transposon at the established attTn7 site located downstream of the glmS gene was verified by a multiplex PCR using primers glmS.UP, glmS.DW, Tn7.L, and Tn7.R. PCR reactions were performed in a final volume of 25 μl containing 50 ng of DNA, 1.5 mm MgCl2, 0.2 mm dNTPs, each primer at 0.2 μm, and 1 unit of GoTaq polymerase (Promega). The thermal profile was as follows: an initial denaturation step at 94 °C for 5 min followed by 5 cycles of 94 °C for 45 s, a touchdown from 62 to 58 °C for 45 s and 72 °C for 45 s, and then 25 cycles of 94 °C for 45 s, 58 °C for 45 s, and 72 °C for 45 s, and a final extension step at 72 °C for 5 min. Correct insertion of the Tn7 transposon yielded two amplicons of 462 and 216 bp amplified with primer pairs glmS.UP-Tn7.L and glmS.Dw-Tn7.R, respectively. Only one amplicon of 455 bp, corresponding to an internal fragment of the glmS gene, was amplified when used as the template DNA from wild-type Klebsiella. pTSNSK-Tp from the recipient strain was cured by growing bacteria at 37 °C due to the plasmid-thermosensitive origin of replicationpSC101. Plasmid removal was confirmed by susceptibility to trimethoprim.
PulA levels on the outer membrane fraction, purified as described previously (39), were detected by immunoblotting using rabbit antibody anti-PulA (1:1000), kindly donated by A. Pugsley. Proteins were resolved using 12% SDS-PAGE and electroblotted to nitrocellulose membrane. Membranes were blocked with 4% skim milk in TBST (137 mm NaCl, 2.7 mm KCl, 19 mm Tris base (pH 7.4)).
Construction of gfp Reporter Fusions
A trimethoprim resistance cassette was obtained as a SmaI fragment from plasmid p34S-Tp (40). The cassette wascloned into SphI-digested and blunt-ended plasmid pPROBE′-gfp[LVA] to obtain pPROBE′-gfp[LVA]Tp. This vector contains a promoterless gfp gene (41). DNA fragments containing the promoter regions of entC, ybtA, psn, and iucA were amplified by PCR using Vent polymerase, gel-purified, and cloned into SmaI-digested pPROBE′-gfp[LVA]Tp plasmid. Plasmids were introduced in E. coli DH5α-λpir and then mobilized into Klebsiella by triparental conjugation using the helper strain E. coli HB101/pRK2013.
Luciferase Activity and GFP Fluorescence Measurements
Overnight cultures of the reporter strains were diluted 1/10 in fresh LB medium and incubated for 3 h at 37 °C on an orbital incubator shaker (180 rpm). The cultures were harvested(4000 × g for 20 min) and resuspended to an A600 of 1.0 in PBS. Luciferase activity and GFP fluorescence were determined as described previously and expressed as relative light units (RLU) or relative fluorescence units (RFU) (42). All measurements were performed in triplicate on at least three independent occasions.
Adhesion and Internalization Assays
Bacteria adhesion and internalization to A549 cells were determined as our group described previously (8). The results are expressed as cfu/well. Experiments were carried out in duplicate on at least three independent occasions.
IL-8 Stimulation Assay
A549 cells, seeded in 24-well plates, were infected for 2 h and then washed twice with PBS, and fresh medium plus gentamicin (100 μg/ml) was added. Supernatants were recovered after 12 h, cell debris was removed by centrifugation, and samples were frozen at −80 °C. IL-8 levels in the supernatants were determined using a commercial ELISA (eBioscience) with a sensitivity of <2 pg/ml. Experiments were run in duplicate and repeated at least three independent times.
Inmunoblotting
Eukaryotic proteins were resolved by standard 10% SDS-PAGE and electroblotted onto nitrocellulose membranes. Membranes were blocked with 4% skim milk in TBST, and protein bands were detected with specific antibodies using chemiluminescence reagents and a GeneGnome chemiluminescence imager (Syngene). Immunostainings for IκBα were performed using polyclonal rabbit anti- IκBα (1:1,000, Cell Signaling) antibodies. Immunostainings to assess phosphorylation of p38, p44/42, and JNK MAPKs were performed using polyclonal rabbit, anti-phospho-p38, anti-phospho-p44/42, and anti-phospho-JNK antibodies, respectively (all used at 1:1000; Cell Signaling). Blots were reprobed with the polyclonal antibody anti-human tubulin (1:3000, Sigma) to control that equal amounts of proteins were loaded in each lane.
Small Interfering RNA (siRNA)
Transfection of siRNAs was performed at the time of cell seeding in a 96-well plate format (2 × 104 cells/well). LipofectamineTM 2000 transfection reagent (Invitrogen) was used following the manufacturer's instructions. Transfection experiments were carried out in Opti-MEM reduced serum medium (Invitrogen). siRNAs were used at a concentration of 20 nm, and experiments were carried out 48 h after transfection. The knockdown efficiency of the siRNAs targeting NOD1, TLR2, TLR4, and EGFR was verified previously (22, 24).
Murine Infection Model
Mice were treated in accordance with the Directive of the European Parliament and the Council on the Protection of Animals Used for Scientific Purposes (Directive 2010/63/EU) and in agreement with the Bioethical Committee of the University of the Balearic Islands. This study was approved by the Bioethical Committee of the University of the Balearic Islands (authorization number 1748).
Five-to-seven-week-old female CD-1 mice (Harlan) were infected as described previously by our group (8). Non-infected mice were inoculated with 20 μl of PBS. At 24 h post-infection, mice were euthanized by cervical dislocation. Tissues were rapidly dissected for bacterial load determination, and half of the lungs were immediately frozen in liquid nitrogen and stored at −80 °C until purification of RNA was carried out.
RNA Purification and Real Time Quantitative PCR (RT-qPCR)
RNA from lungs was purified as described by us (8). cDNA was obtained by retrotranscription of 1 μg of total RNA using the M-MLV reverse transcriptase (Sigma). The reaction included one step to eliminate traces of genomic DNA. RT-qPCR analyses were performed using an iCycler real-time PCR instrument (Bio-Rad). Reactions were performed in 25 μl containing 12.5 μl of KAPA SYBR® FAST qPCR kit master mix (KapaBiosystems), each primer at 0.2 μm, and 50 ng of cDNA. Relative quantities of mRNAs were obtained using the comparative threshold cycle (ΔΔCT) method by normalizing to hprt1.
Bacteria were grown at 37 °C in 5 ml of LB medium on an orbital incubator shaker (180 rpm) until an A600 of 0.3 was reached. RNA was purified following an established protocol (43). cDNA was obtained by retrotranscription of 2 μg of total RNA using commercial M-MLV reverse transcriptase (Sigma) and random primers mixture (SABiosciences, a Qiagen company). 50 ng of cDNA was used as a template in a 25-μl reaction. RT-PCR analyses were performed with a Smart Cycler real-time PCR instrument (Cepheid, Sunnyvale, CA) using a KapaSYBR Fast qPCR kit as recommended by the manufacturer (Cultek). SYBR Green dye fluorescence was measured at 521 nm. cDNAs were obtained from two independent extractions of mRNA, and each one was amplified by RT-qPCR on two independent occasions. Relative quantities of wabM mRNAs were obtained using the comparative threshold cycle (ΔΔCT) method by normalizing to the rpoD gene.
Statistical Analysis
Results were analyzed by analysis of variance (ANOVA) or with a one-tailed t test followed by a Bonferroni contrast correction for multiple testing using GraphPad Prism software (GraphPad Software Inc.). Results are given as the means ± S.D. A p value of <0.05 was considered statistically significant.
Results
High-throughput Screening to Identify K. pneumoniae Loci Involved in Attenuating NF-κB Activation
We developed an assay suitable for identifying the K. pneumoniae loci required to block activation of the NF-κB pathway. A new indicator cell line was engineered by transfection of the human epithelial cell line A549 with the reporter vector pNIFTy2-SEAP (InvivoGen). Three clones were selected, A549-SEAP A2, D2, and D4, and control experiments were carried out to determine whether different stimuli known to activate NF-κB induce the secretion of SEAP (Fig. 1A). Clone A2 secreted higher amounts of SEAP than the other two clones after cells were challenged with different stimuli (Fig. 1A). Time course experiments showed that clone A2 secreted higher levels of SEAP over time than the other two clones upon challenge with IL-1β (10 ng/ml) (Fig. 1B). Western blot analysis of IκBα levels showed no differences between A549 cells and A549-SEAP A2 after stimulation with different agonists (Fig. 1C). Clone A2 was selected for screening purposes.
FIGURE 1.
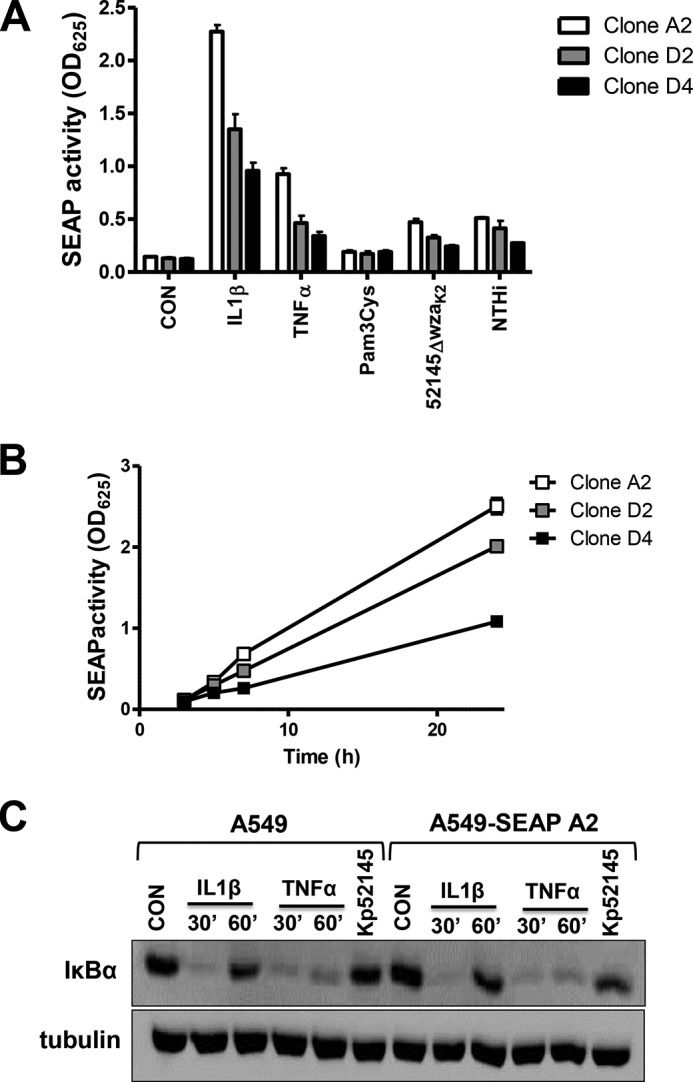
Construction of a NF-κB reporter cell line. A, SEAP levels released by three clones of A549 cells stably transfected with NF-κB reporter pNifty2-SEAP vector. Cells were left untreated (control (CON), white bars), treated with IL-1β (10 ng/ml), TNFα (10 ng/ml), or TLR2 agonist Pam3Cys (100 ng/ml), or infected with Klebsiella capsule mutant (52145-ΔwzaK2) or nontypable Haemophilus influenzae 398 (NTHi). Scale bars represent mean ± S.E. (n = 3). B, time course of SEAP levels released by the selected clones treated with IL-1β (10 ng/ml) (n = 3). C, immunoblot analysis of IκBα and tubulin levels in lysates of A549 and clone A549-SEAP A2 cells were left untreated (control), treated with IL-1β (10 ng/ml) or TNFα (50 ng/ml) for the indicated time, or infected for 3 h with Kp52145. Data are representative of three independent experiments.
SEAP levels induced by Kp52145 were not significantly different than those observed in non-infected cells (Fig. 2A). In contrast, the cps, ompA mutants, and the cps-ompA double mutant did trigger the secretion of SEAP. We have already reported that these strains elicit the activation of NF-κB (8). The differences observed between strains are consistent with data from our laboratory showing the relative importance of CPS and OmpA to attenuate NF-κB activation (8).
FIGURE 2.
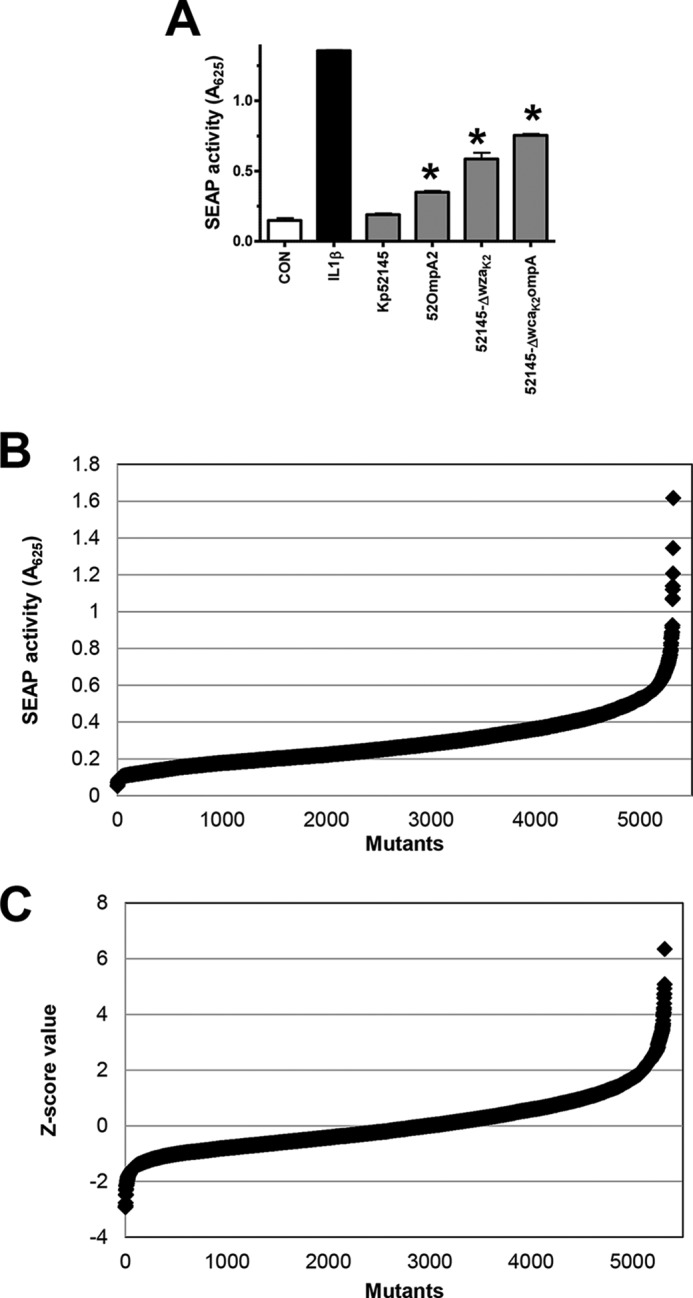
High-throughput screening for the identification of K. pneumoniae factors implicated in attenuation of NF-κB activation. A, SEAP levels released by A549-SEAP A2 cells left untreated (control (CON), white bars), treated with IL-1β (10 ng/ml) (black bars), or infected with Kp52145, 52OmpA2, 52145-ΔwzaK2, or 52145-ΔwcaK2ompA (gray bars). Scale bars represent mean ± S.E. (n = 3) *, p < 0.05 (results are significantly different from the results for Kp52145; one-tailed t test). B, distribution of SEAP levels released by A549-SEAP A2 cells infected with the mutants. C, distribution of Z score data of SEAP levels.
The assay was then transferred from a 24-well format to a 96-well format by optimizing culture conditions, cell number/well, multiplicity of infection, time of contact, and time of detection of secreted SEAP. To determine the quality of the assay, the Z′ factor (31) was calculated taking as a negative control SEAP levels secreted by cells infected with the wild-type strain and as a positive control SEAP levels secreted by cells infected with the ompA mutant or cells stimulated with IL-1β. In our case, the resulting Z′ factors were 0.73 and 0.86 for the respective comparisons. The Z′ factor is a screening window coefficient indicating the capability of a hit identification for a given assay at the defined screening conditions. Z′ factors higher than 0.5 indicate that an assay is robust enough for a high-throughput screening (31).
To identify Klebsiella loci involved in attenuating NF-κB activation, we examined a bank of 5320 transposon mutants. Each mutant was tested individually. For candidate selection the standard Z score normalization procedure was applied and mutants with a Z score ≥ 2 were selected. We also selected those mutants that induce SEAP levels higher than A625 > 0.5, which represents a 2-fold increase in the SEAP levels induced by the wild type. The distribution of A625 values and the Z scores of the mutant library are shown in Fig. 2, B and C. Following the first round of screening, 522 mutants (10% of the transposon library) were considered candidates for validation.
To validate the selected candidates, each one was tested individually in four independent screening rounds. Candidates were considered validated if they induced SEAP levels higher than A625 > 0.5 in at least two of four independent experiments upon infection of the A549-SEAP A2 cells. Using this validation criterion, 114 Kp52145 mutants were considered validated (Table 2). Control experiments showed that all mutants grew in LB agar plates, LB broth, and RPMI 1640 complete medium, whereas five and three mutants displayed growth deficiencies when cultivated in M9 agar plates supplemented with glucose and citrate, respectively (Table 2).
TABLE 2.
K. pneumoniae transposon mutants validated in the screening
| Function | Mutant code | Gene | Kp52145 locus tag | MGH78578 locus tag | Growtha |
|---|---|---|---|---|---|
| Metabolism | |||||
| Oxydoreductases | P017C4 | Pyrroline-5-carboxylate reductase | KpST66_1303 | KPN_00329 | N/N |
| P019E9 | Putative oxidoreductase | KpST66_1547 | KPN_00505 | N/Y | |
| P033F6 | Cytochrome bd-I oxidase subunit I | KpST66_2291 | KPN_01153 | Y/Y | |
| P037H10 | Putative alcohol dehydrogenase groES | KpST66_2527 | KPN_01466 | Y/Y | |
| P037H6 | γ-Aminobutyraldehyde dehydrogenase | KpST66_3019 | KPN_01924 | Y/Y | |
| P035C5 | Uncharacterized protein conserved | KpST66_3673 | KPN_02456 | Y/Y | |
| P018C10 | NADH-quinone oxidoreductase subunit F | KpST66_3892 | KPN_02674 | Y/Y | |
| P006E12 | Putative uncharacterized protein | KpST66_0795 | KPN_03309 | Y/Y | |
| P026H10 | cueO | KpST66_4209 | KPN_00131 | Y/Y | |
| Carbohydrates | P036D12 | NADPH-dependent preQ0 reductase | KpST66_3564 | Unknown function | Y/Y |
| P043B5 | N-Acetylmuramic acid 6-phosphate etherase | KpST66_2662 | KPN_01553 | Y/Y | |
| P037E12 | Deoxyribose-phosphate aldolase | KpST66_2812 | KPN_01702 | Y/Y | |
| P029H4 | Putative Carboxymuconolactone decarboxylase family protein | KpST66_1120 | KPN_03003 | Y/Y | |
| P009G9 | Propanediol dehydratase medium subunit pduD | KpST66_0902 | KPN_03206 | Y/Y | |
| P025D5 | Altronate hydrolase | KpST66_0586 | KPN_03519 | Y/Y | |
| P006H10 | L(+)-tartrate dehydratase subunit β (l-TTD β) | KpST66_0481 | KPN_03639 | Y/Y | |
| P040B5 | C-terminal of putative uncharacterized protein yhjS | KpST66_0231 | KPN_03886 | Y/Y | |
| Lipids | P025C1 | Putative acetyltransferase | KpST66_0118 | KPN_03989 | Y/Y |
| P026E12 | Putative acetyltransferase | KpST66_0118 | KPN_03989 | Y/Y | |
| P035E6 | acyl carrier protein | KpST66_3624 | A79E_1666 | Y/Y | |
| P035D1 | Glycerol dehydratase, small subunit | KpST66_0621 | KPN_03488 | Y/Y | |
| P038H3 | Putative uncharacterized protein | KpST66_4890 | Unknown function | Y/Y | |
| P007C12 | Triacylglycerol lipase | pKpST66–1_0006 | Unknown function | Y/Y | |
| P038C2 | C-terminal region of triacylglycerol lipase | pKpST66–1_0006 | Unknown function | Y/Y | |
| Amino acids | P019F6 | Dihydroxy-acid dehydratase | KpST66_4589 | KPN_04270 | N/N |
| P037B8 | Ketol-acid reductoisomerase | KpST66_4586 | KPN_04273 | Y/N | |
| Others | P044A12 | Uncharacterized protein ynjA | KpST66_2352 | KPN_01217 | Y/Y |
| P040B2 | Enterobactin synthase component F, entF | KpST66_1661 | KPN_00605 | Y/Y | |
| P005E9 | Cyclic diguanylate phosphodiesterase | KpST66_4039 | KPN_02828 | Y/Y | |
| P037G9 | Putative amidohydrolase | KpST66_1569 | KPN_00523 | Y/Y | |
| Envelope | |||||
| Protein secretion | P044G9 | Putative member of ShlA/HecA/FhaA exoprotein family | KpST66_1736 | KPN_00676 | Y/Y |
| P035G10 | C-terminal of putative uncharacterized protein ECs0126 | KpST66_4210 | KPN_00130 | Y/Y | |
| P038F11 | Pullulanase secretion envelope pulC | KpST66_4179 | KPN_00161 | Y/Y | |
| P044E5 | Putative hemolysin activator protein | KpST66_1737 | KPN_00677 | Y/Y | |
| P020H1 | Acriflavine resistance protein A, acrA | KpST66_0069 | KPN_04039 | Y/Y | |
| P018H5 | Auxiliary transport protein, membrane fusion protein (MFP) family | KpST66_3658 | Unknown function | Y/Y | |
| P029G11 | Putative plasmid transfer protein | pKpST66–2_0082 | Unknown function | Y/Y | |
| Peptidoglycan | P021E7 | Penicillin-binding protein 1B | KpST66_4173 | KPN_00164 | Y/Y |
| P035C6 | ampG muropeptide MFS transporter | KpST66_1369 | KPN_00395 | Y/Y | |
| P017C10 | Putative β-lactamase-like | KpST66_2943 | KPN_01845 | Y/Y | |
| Adhesion | P043A12 | TPR repeat lipoprotein | KpST66_0546 | KPN_03571 | Y/Y |
| P018G12 | Putative fimbrial chaperone protein | KpST66_4961 | KPN_04471 | Y/Y | |
| P043D6 | traH | pKpST66–1_0057 | KPN_pKPN4p07153 | Y/Y | |
| LPS | P020C10 | O-antigen ligase, waaL | KpST66_0145 | KPN_03966 | Y/Y |
| P025F5 | O-antigen ligase, waaL | KpST66_0144 | KPN_03966 | Y/Y | |
| Others | P050B3 | putative OmpA domain protein | KpST66_0945 | KPN_03169 | Y/Y |
| P018C9 | Putative outer membrane protein | pKpST66–1_0065 | AY703481 | Y/Y | |
| P018F7 | Putative outer membrane protein | pKpST66–1_0065 | AY703481 | Y/Y | |
| P045A8 | Phage tail fiber protein | KpST66_2280 | A79E_3050 | Y/Y | |
| P021E9 | Putative fusaric acid resistance domain protein | KpST66_3659 | KPN2242_15265 | Y/Y | |
| Transport | P025C6 | Putative extracellular solute-binding protein, family 3 | KpST66_1626 | KPN_00572 | Y/Y |
| P025D4 | Putative extracellular solute-binding protein, family 3 | KpST66_1626 | KPN_00572 | Y/Y | |
| P007C1 | Putative ABC transporter | KpST66_2950 | KPN_01850 | Y/Y | |
| P007H1 | ABC-type dipeptide/oligopeptide/nickel transport systems, permease component | KpST66_3752 | KPN_02536 | Y/Y | |
| P051H4 | Putative high-affinity nickel-transporter | KpST66_1081 | KPN_03047 | Y/Y | |
| P038C9 | d-Galactonate transporter | KpST66_0012 | KPN_04094 | Y/Y | |
| P018G8 | Spermidine/putrescine import ATP-binding protein potA | KpST66_4976 | KPN_04456 | Y/Y | |
| P008H2 | Putative ascorbate-specific PTS system enzyme IIC | KpST66_4846 | KPN_04586 | Y/Y | |
| P034F5 | Putative periplasmic-binding protein | KpST66_1095 | KPN_03033 | Y/Y | |
| Transcription | P034C6 | Threonyl-tRNA synthetase | KpST66_3559 | Unknown function | Y/Y |
| P006G10 | Transcriptional activator cadC | KpST66_1540 | KPN_00498 | Y/Y | |
| P019F8 | HTH-type transcriptional regulator YcgE | KpST66_1826 | KPN_00783 | Y/Y | |
| P009C4 | Putative transcriptional regulatory protein TyrR | KpST66_2445 | KPN_01305 | Y/Y | |
| P012G4 | Putative Mgl repressor | KpST66_4978 | KPN_04454 | Y/Y | |
| P041F4 | Putative protein YtfJ | KpST66_4824 | KPN_04609 | Y/Y | |
| P019G5 | Putative TetR family transcriptional regulator | KpST66_3249 | A79E_2099 | Y/Y | |
| P038F5 | Formylmethionylaminoacyl-tRNA deformylase | pKpST66–2_0076 | LV116 (pLVPK) | Y/Y | |
| Stress | P049G10 | Chaperone surA | KpST66_4290 | KPN_00050 | N/Y |
| P046C4 | Putative 2-component transcriptional regulator | KpST66_3517 | KPN_02412 | Y/Y | |
| P046H11 | Protease III | KpST66_0878 | KPN_03230 | Y/Y | |
| P025H2 | Putative uncharacterized protein yhbO | KpST66_0556 | KPN_03561 | Y/Y | |
| P024H5 | 33-kDa chaperonin | KpST66_0345 | KPN_03772 | Y/Y | |
| P007C4 | putative α-helix | KpST66_0117 | KPN_03990 | N/Y | |
| P044A10 | DNA helicase II | KpST66_4551 | KPN_04312 | Y/Y | |
| DNA | P012H7 | Cytoskeleton protein rodZ | KpST66_4055 | KPN_02846 | Y/Y |
| P029G3 | Putative DNA-processing protein | KpST66_0252 | KPN_03865 | Y/Y | |
| P043D4 | Putative integrase | KpST66_0789 | Unknown function | Y/Y | |
| P027A12 | Putative Isrso16-transposase orfb | pKpST66–2_0103 | Unknown function | Y/Y | |
| Hypotheticals | P018D10 | Putative lipoprotein | KpST66_3367 | Unknown functionb | Y/Y |
| P044H7 | Hypothetical CDS | KpST66_0778 | Unknown function b | Y/Y | |
| P024E6 | Putative uncharacterized protein | KpST66_0788 | Unknown functionb | Y/Y | |
| P034C10 | Putative uncharacterized protein | KpST66_4517 | Unknown functionb | Y/Y | |
| P037F10 | Hypothetical CDS | pKpST66–1_0077 | Unknown functionb | Y/Y | |
| P001G5 | Subtilisin-related serine protease | pKpST66–1_0111 | Unknown functionb | Y/Y | |
| P054G10 | Putative uncharacterized protein | KpST66_0786 | Unknown function | Y/Y | |
| P043E4 | Hypothetical protein | KpST66_3558 | Unknown function | Y/Y | |
| P053H2 | Putative uncharacterized protein | KpST66_2293 | KPN_01157 | Y/Y | |
| P013G3 | Putative uncharacterized protein | KpST66_2888 | KPN_01793 | Y/Y | |
| P020B12 | Uncharacterized protein conserved | KpST66_3673 | KPN_02457 | Y/Y | |
| P043G3 | Cyclic diguanylate phosphodiesterase domain protein | KpST66_0830 | KPN_03274 | Y/Y | |
| P043G2 | Putative uncharacterized protein | KpST66_0943 | KPN_03171 | Y/Y | |
| P025G5 | Putative uncharacterized protein | KpST66_0942 | KPN_03172 | Y/Y | |
| P045H10 | Hypothetical CDS | pKpST66–1_0020 | KPN_pKPN3p05993 | Y/Y | |
| P034F4 | Hypothetical CDS | pKpST66–2_0092 | KP1_p117 | Y/Y | |
| P043D5 | Putative uncharacterized protein | pKpST66–2_0058 | LV102a (pLVPK) | Y/Y | |
| Non-coding regions | P007G6 | Between C4-type zinc finger protein, DksA/TraR family and GTP cyclohydrolase I | KpST66_3567 and KpST66_3568 | Unknown function | Y/Y |
| P009H6 | Between LuxR-transcriptional regulator and a hypothetical protein | KpST66_1827 and KpST66_1828 | Between KPN_00784 and KPN_00785 | Y/Y | |
| P048H7 | Between putative glutathione S-transferase and d-alanyl-d-alanine carboxypeptidase; penicillin-binding protein | KpST66_1922 and KpST66_1923 | Between KPN_00870 and KPN_00871 | Y/Y | |
| P043E6 | Between deoR and deoC | KpST66_2811 and KpST66_2812 | Between KPN_01701 and KPN_01702 | Y/Y | |
| P038G8 | Between aspartate-semialdehyde dehydrogenase and a putative transcriptional regulator | KpST66_2862 and KpST66_2863 | Between KPN_01773 and KPN_01774 | Y/Y | |
| P019E6 | Between putative transposase IS4 and a putative ARAC-type regulatory protein | KpST66_2869 and KpST66_2870 | Between KPN_01779 and KPN_01780 | Y/Y | |
| P037E9 | Between putative FAD/FMN-containing dehydrogenases and a hypothetical protein | KpST66_3144 and KpST66_3145 | Between KPN_02042 and KPN_02043 | Y/Y | |
| P022H9 | Between putative enzyme and hypothetical protein | KpST66_3460 and KpST66_3461 | Between KPN_02355 and KPN_02356 | Y/Y | |
| P025H3 | Between putative transcriptional regulator and putative arylsulfatase regulator | KpST66_4042 | Between KPN_02830 and KPN_02831 | Y/Y | |
| P050B12 | Between iap and cysH | KpST66_0997 and KpST66_0998 | Between KPN_03116 and KPN_03117 | Y/Y | |
| P005D9 | Between uppP and folB | KpST66_0645 and KpST66_0646 | Between KPN_03461 and KPN_03462 | Y/Y | |
| P015G5 | Between two hypothetical proteins | KpST66_0630 and KpST66_0631 | Between KPN_03477 and KPN_03478 | Y/Y | |
| P006F8 | Between yhdP and rng | KpST66_0465 and KpST66_0466 | Between KPN_03654 and KPN_03655 | Y/Y | |
| P028H3 | Between ibpA and yidQ | KpST66_0014 and KpST66_0015 | Between KPN_04091 and KPN_04092 | Y/Y | |
| P041H3 | Between rimI and ygjF | KpST66_5016 and KpST66_5017 | Between KPN_04415 and KPN_04416 | Y/Y | |
| P024D12 | Between yhaM and psiE | KpST66_5012 and KpST66_5013 | Between KPN_04419 and KPN_04420 | Y/Y | |
| P051G11 | Between phnA and proP | KpST66_4928 and KpST66_4929 | Between KPN_04502 and KPN_04503 | Y/Y | |
| P014F6 | Between YgfA and traM | pKpST66–1_0029 and pKpST66–1_0030 | Between KPN_pKPN3p06000 and KPN_pKPN3p06001 | Y/Y | |
| P014E6 | Between hypothetical protein and iutA | pKpST66–2_0063 and pKpST66–2_0064 | Between LV106-LV107 | Y/Y | |
a Growth in M9 agar plates was supplemented with either glucose or citrate as the carbon source. N, no growth; Y, growth.
b Present only in the Kp52145 genome.
Transposon insertion sites were determined by genomic sequencing using primer tpnRL17. The sequences obtained were compared with the Kp52145 genome and plasmid sequences (GenBankTM accession numbers FO834904, FO834905, and FO834906) and to the reference sequence of K. pneumoniae strain MGH78578 (GenBankTM accession number CP000647.1). Ninety-nine insertions were found in the chromosome, whereas nine and six were located in plasmid 1 and plasmid 2, respectively. The MicrobesOnline and STRING databases were interrogated to annotate the loci. Thirty of them were related to metabolism, twenty were included in the category outer membrane and envelope-related genes, nine in transport, eight in regulation of transcription, seven in the stress response category, and four in the category of DNA-related processes (Table 2). Interestingly, six of the insertions were located in regions of the Kp52145 genome not present in other Klebsiella genomes (27). Nineteen mutants had the transposon inserted in non-coding regions of the genome, and 17 were located within the coding region of hypothetical proteins.
In summary, our high-throughput genetic screen mining a K. pneumoniae transposon library led to the identification of 114 mutants that triggered the activation of NF-κB. In the following sections we describe the characterization of the mutants on the enterobactin siderophore, WaaL encoding the O-antigen ligase and pullulanase T2SS. We selected the enterobactin and ligase mutants because iron-scavenging systems and LPS are two well characterized Klebsiella virulence determinants, although their contributions to attenuating inflammation have not been evaluated previously. On the other hand, we chose to investigate the Klebsiella pullunase T2SS because its role in Klebsiella virulence had not been established.
Analysis of the Enterobactin Mutant
One transposon insertion was identified in the enzyme EntF, a component of the biosynthetic machinery of the enterobactin siderophore (44). Enterobactin is one of the three siderophores expressed by Kp52145 (27). Enterobactin is expressed in the lungs of infected mice, and an enterobactin mutant is attenuated (9). As anticipated (9), the doubling times of the entF mutant slowed when grown in LB supplemented with the iron chelator 2-2′-dipyridyl (Fig. 3A). Similar rates of growth were observed when the wild type and the mutant were grown in LB or RPMI 1640 (data not shown).
FIGURE 3.

Indirect role of K. pneumoniae enterobactin in attenuation of NF-κB activation. A, growth of Kp52145 (black symbols) and enterobactin mutant (52entF::tn5, white symbols) in LB supplement with 2-2′-dipyridyl (DIP). B, SEAP levels released by A549-SEAP A2 cells left untreated (control (CON)) or infected with different Klebsiella strains. Scale bars represent mean ± S.E. (n = 3). C, immunoblot analysis of IκBα and tubulin levels in A549 cells left uninfected (control) or infected with Kp52145 or enterobactin mutant (52entF::tn5) for 3 h. Data are representative of three independent experiments. D, analysis of the expression of iutA, psn, ybtA, and entC by Kp52145 carrying the transcriptional fusions iutA::gfp, psn::gfp, ybtA:;gfp, and entC::gfp. Bacteria were grown in LB (white bars), RPMI 1640 (gray bars), or LB supplement with 2-2′-dipyridyl (DIP, black bars). Data are presented as mean ± S.D. (n = 3). E, adhesion (left bars) and internalization (right bars) of Klebsiella strains to A549 cells (n = 3). F, SEAP levels released by A549-SEAP A2 cells left untreated (control, white bar) or infected with Kp52145, capsule mutant (52145-ΔwzaK2), enterobactin mutant (52entF::tn5), or capsule and enterobactin double mutant (52145-ΔwzaK2entF). Scale bars represent mean ± S.E. (n = 3). ns (not significant), p > 0.05; *, p < 0.05; **, p < 0.01 (one-tailed t test).
We sought to determine whether the other two siderophores encoded by Kp52145, aerobactin and yersiniabactin, are implicated in attenuation of inflammation. However, the SEAP levels induced by the aerobactin mutant, strain 52iutA, and the yersiniabactin mutant, strain 52irp1, were not significantly different than those induced by the wild-type strain (Fig. 3B). In contrast, SEAP levels induced by the entF mutant were higher than those induced by Kp52145, which in turn, were not significantly different than those found in the supernatant of non-infected cells (Fig. 3B). In the canonical NF-κB activation pathway, nuclear translocation of NF-κB is preceded by phosphorylation and subsequent degradation of IκBα (45). Immunoblot analysis demonstrated that the entF mutant induced the degradation of IκBα in A549 cells (Fig. 3C).
These findings suggested that enterobactin, but not the other siderophores, was expressed under our infection conditions. To monitor the transcription of the siderophores quantitatively, transcriptional fusions were constructed in which a promoterless gfp gene was under the control of the iutA, psn, ybtA, and entC promoter regions (see “Experimental Procedures”). iutA has been used previously to study aerobactin expression and psn and ybtA to analyze yersiniabactin expression, whereas entC has been used to determine enterobactin expression (9). gfp fusions were introduced into Kp52145, and fluorescence was determined after growing the reporter strains in LB, LB supplemented with iron chelator 2-2′-dipyridyl, or RPMI 1640 (Fig. 3D). The entC::gfp fusion was the only one expressed under iron-limited conditions, LB plus 2-2′-dipyridyl and RPMI 1640 (Fig. 3D). It has been reported previously that bacteria up-regulate iron-scavenging systems when grown in tissue culture medium (46). Further confirming this finding, the addition of FeCl3 to RPMI 1640 decreased the expression of the entC::gfp fusion by 75% (9106 ± 850 RFU and 2190 ± 323 RFU, respectively; p < 0.05, one-tailed t test).
Collectively, our data are consistent with a model in which K. pneumoniae experiences a poor iron environment when infecting A549 cells, hence leading to the activation of the siderophore enterobactin. There are reports suggesting that siderophores may inhibit immune responses (47), hence making it possible that enterobactin may directly attenuate NF-κB activation. However, it has been also shown that K. pneumoniae CPS expression is down-regulated in iron-poor media (48). Therefore, the involvement of enterobactin in attenuating NF-κB activation would be indirect considering that CPS is known to prevent NF-κB activation (20, 21). Indirectly supporting the latter evidence, the entF mutant expressed 50% less cell-bound CPS than the wild-type strain (6.11 ± 2.99 mg/107 cells and 11.49 ± 1.91 mg/107 cells, respectively; p < 0.05, one-tailed t test), and the activity of the transcriptional cps::lucFF fusion was 80% lower in the entF mutant background than in the wild-type one (3.1 × 106 ± 16,000 RLU and 1.7 × 107 ± 34,000 RLU, respectively; p < 0.01, one-tailed t test). Further confirming the inverse correlation between the CPS levels and the adhesion and internalization to epithelial cells (49), the entF mutant adhered to and was internalized by A549 cells in higher numbers than the wild type (Fig. 3E). Altogether, this evidence suggests that entF-induced NF-κB activation could be explained by the reduced CPS levels expressed by the mutant. To validate this hypothesis, we asked whether the SEAP levels induced by the entF mutant were significantly different than those triggered by the cps mutant or the double cps-entF mutant. The data shown in Fig. 3F demonstrate that indeed there were no differences in the SEAP levels triggered by any of the three strains.
In aggregate, our findings revealed that enterobactin was activated under our tissue culture infection conditions and that the lack of the siderophore was linked to a reduced CPS expression. In turn, the low CPS levels underlined the NF-κB activation induced by the entF mutant.
Analysis of the LPS Polysaccharide Mutants
Two transposon insertions were identified in the waaL gene. WaaL is the O-antigen ligase responsible for ligating the LPS O-polysaccharide (OPS) to the core (50). LPS analysis confirmed that neither of the mutants express OPS, and the LPS pattern was similar to that displayed by a nonpolar waaL mutant (50) (Fig. 4A). waaL is encoded within the operon responsible for the synthesis of the LPS core (50), hence making it possible that the transposon insertion would affect the expression of downstream genes. However, RT-qPCR experiments showed that the expression of the downstream gene wabM was not significantly different between the wild type and the transposon mutants (data not shown). WabM encodes the glycosyltransferase, adding the first glucose residue of the core where the OPS is attached (50). The SEAP levels induced by the ligase mutants were significantly higher than those induced by the wild type (Fig. 4B). No significant differences were found in the SEAP levels induced by any of the mutants (Fig. 4B).
FIGURE 4.

K. pneumoniae LPS O-polysaccharide is implicated in attenuation of NF-κB activation. A, LPS analysis by SDS-PAGE. Data are representative of three independent experiments. B, SEAP levels released by A549 cells left untreated (control (CON), white bar) or infected with Kp52145 or waaL mutants (52waaL::tn5 and 52ΔwaaL). Scale bars represent mean ± S.E. (n = 3). C, immunoblot analysis of IκBα and tubulin levels in A549 cells left uninfected (control) or infected with the indicated strains for 3 h. Data are representative of three independent experiments. D, immunoblots showing phosphorylated MAPKs and tubulin levels in cell extracts of A549 cells left uninfected (control, time 0) or infected with K. pneumoniae strains for different times. The results are representative of three independent experiments. E, SEAP levels released by A549 cells left untreated (control, white bar) or infected with the indicated strains (n = 3). F, ELISA of IL-8 released by A549 cells left untreated (control, white bar) or infected for 12 h with the indicated strains (n = 3). G and H, immunoblot analysis of IκBα and tubulin levels in lysates of A549 cells infected with the indicated strains. The cells were left untreated, stimulated with IL-1β (1 ng/ml, 10 min), infected for 3 h, or infected for 3 h and then stimulated with IL-1β. In H, the siRNA targets are indicated in the top row of each panel. (AS−, AllStars control siRNA). Data are representative of three independent experiments. I, adhesion (left panel) and internalization (right panel) of Klebsiella strains to A549 cells (n = 3). *, p < 0.05 (one-tailed t test); ns (not significant), p > 0.05 (one-tailed t test). In E and F, CPS-expressing strains, gray bars; CPS negative strains, black bars.
To rule out the possibility that the non-ligated OPS exerted an unanticipated effect on Klebsiella physiology, we tested the glf glycosyltransferase mutant, strain 52glf. Glf is a UDP-galactopyranose mutase essential for OPS biosynthesis (51). Immunoblot analysis revealed that the ligase and OPS mutants triggered the degradation of IκBα in A549 cells in contrast to the wild-type strain (Fig. 4C). Many cellular stimuli also activate MAPK pathways. The activation of the three MAPKs (p38, JNK, and p44/42) occurred through phosphorylation of serine and threonine residues. All strains induced the phosphorylation of the three MAPKs; however the phosphorylation of p38 and p44/42 was more apparent in cells infected with the ligase and OPS mutants than in those infected with the wild type (Fig. 4D). No differences were observed between both mutants (Fig. 4, C and D). Altogether, these results support the view that Kp52145 OPS is required to attenuate inflammation.
waaL mutants express similar levels of CPS as the wild type (Ref. 50 and data not shown). To assess the relative contribution of CPS and OPS to attenuate NF-κB activation, we investigated whether the absence of OPS increases SEAP levels induced by the cps mutant. Results shown in Fig. 4E confirmed that this was the case.
To further analyze the contribution of the LPS polysaccharide section to the attenuation of NF-κB activation, we sought to determine whether the LPS core section plays any role in Klebsiella attenuation of NF-κB activation. wabM, wabK, and wabH mutants lack, in addition to the OPS, the first, second, and third sugars of the LPS core, respectively (50), but they express similar levels of CPS as the wild type (50). The three mutants triggered the secretion of SEAP by infected cells (Fig. 4E). SEAP levels were higher than those induced by the wild type; the levels induced by the wabM and wabK mutants were also significantly higher than those induced by the waaL mutant (Fig. 4E). No significant differences were found between the wabM and wabK mutants (Fig. 4E). In the genetic background of the cps mutant, the wabM and wabK mutants induced similar levels of SEAP, which were higher than those triggered by the waaL mutant (Fig. 4E). SEAP levels induced by the wabH mutant were not significantly different than those induced by the waaL mutant (Fig. 4E). Comparatively similar results were found when the secretion of IL-8 was evaluated (Fig. 4F). Collectively, these data show that the CPS, the OPS, and the first glucose of the core are bacterial factors required to attenuate the activation of NF-κB by epithelial cells upon K. pneumoniae infection.
Recently, we have shown that Kp52145 attenuates proinflammatory mediator-induced NF-κB activation (22). This process requires bacteria-cell contact, and removal of bacteria by washing followed by a 1-h gentamicin treatment renders the cells responsive to agonist-induced IL-8 secretion (22). To exert this anti-inflammatory effect, Kp52145 engages NOD1 and EGFR receptors (22, 24). To explore whether the OPS could account for the Kp52145 anti-inflammatory effect, we determined the effect of the waaL mutant on IL-1β-induced IκBα degradation using the assay that we described previously (22, 24). Similar to Kp52145, waaL attenuated IL-1β-induced IκBα degradation (Fig. 4G). As with the wild type, this effect was abrogated in cells in which NOD1 and EGFR were knocked down using siRNA (Fig. 4H).
Control experiments revealed that the ligase and core mutants adhered to A549 cells in higher numbers than the wild type (for each comparison between Kp52145 and the mutants, p < 0.05 (one-tailed Student's t test)) (Fig. 4I). The adhesion levels of the wabM and wabK mutants were higher than that of the waaL mutant (Fig. 4I). As reported previously (49), strains lacking CPS adhered in higher numbers than CPS-expressing strains (for each comparison between CPS-expressing and CPS-negative strains, p < 0.01 (one-tailed Student's t test)) (Fig. 4I). In the cps mutant background, no significant differences in adhesion between strains were found (Fig. 4I). waaL and core mutants were internalized in higher numbers than the wild type (for each comparison between Kp52145 and the LPS mutants, p < 0.01 (one-tailed Student's t test)) (Fig. 4I). In the cps mutant background, however, waaL, wabM, and wabK mutants were internalized in lower numbers than the cps mutant (Fig. 4I), and the levels were not significantly different than those of the wild type (for each comparison between Kp52145 and the LPS mutants, p > 0.05 (one-tailed Student's t test)). Interestingly, wabH mutant displayed internalization levels similar to those of the cps mutant (Fig. 4I).
Finally, we sought to determine the innate receptor(s) involved in the recognition of the OPS mutant by using as cellular readouts of the NF-κB-dependent secretion of SEAP and the secretion of IL-8. Almost all TLRs activate cellular signaling pathways through TIR domain-mediated interactions with the adaptor molecule MyD88 (52). To explore the involvement of TLRs in waaL mutant-induced cell activation, the function of the MyD88 adaptor molecule was interrupted by siRNA. In MyD88 knocked-down cells, waaL mutant induced neither the activation of the NF-κB reported construct nor the secretion of IL-8 (Fig. 5, A and B, respectively). To further dissect the contribution of TLR-dependent signaling to waaL mutant-induced cell activation, TLR2 and TLR4 were knocked down by siRNA. The results shown in Fig. 5 indicate that both receptors contributed to waaL-induced cell activation. On the whole, these data suggest that waaL mutant induced NF-κB activation and IL-8 secretion is mediated by TLR4-TLR2-MyD88 pathway.
FIGURE 5.

Role of MyD88, TLR2, and TLR4 in K. pneumoniae waaL mutant-induced cell activation. A, SEAP levels released by A549 cells transfected with the indicated siRNAs for different pattern recognition receptors, which were left untreated (control (CON), white bars) or infected with Kp52145 or waaL mutant (n = 3). B, ELISA of IL-8 released by A549 cells transfected with either control or the indicated siRNA for different pattern recognition receptors, which were left untreated (control, white bars) or infected (n = 3). Scale bars represent mean ± S.E. **, p < 0.01; *, p < 0.05 (results are significantly different from the results obtained by infecting control siRNA-transfected cells with waaL mutant; one-way ANOVA).
Analysis of the Pullulanase Mutants
Two transposon insertions were found in the pulC and loci. pulC is the first locus of the Klebsiella T2SS that secrets the enzyme pullulanase encoded by pulA (53). yacC encodes for a lipoprotein containing a domain related to the pulS/outS family but for which the exact function has not yet been described. To help understand the contribution of Klebsiella T2SS to NF-κB attenuation, we constructed a pulA mutant, because PulA is the only known protein secreted by K. pneumoniae T2SS (54). Immunoblot analysis confirmed the absence of the enzyme pullulanse in the outer membranes of the pulC and yacC mutants and, as expected, also in the outer membrane of the pulA mutant (Fig. 6A). The amount of cell-bound CPS expressed by the mutants was quantified, and no differences were found between the CPS expressed by the wild type (11.49 ± 1.91 mg/107 cells) and the CPS expressed by any of the mutants (52ΔpulA, 15.21 ± 3.65 mg/107 cells; 52pulC::tn5, 9.44 ± 2.91 mg/107 cells; and 52yacC::tn5, 13.26 ± 4.97 mg/107 cells; for each comparison between wild-type CPS levels and mutant levels, p > 0.05, one-tailed Student's t test). Control experiments revealed that the adhesion levels of the pulC and yacC mutants were higher than those of the pulA and Kp52145 strains, which in turn, were not significantly different (Fig. 6B). In contrast, no significant differences were found in the internalization to cells between any of the strains (Fig. 6B).
FIGURE 6.

K. pneumoniae PulA T2SS is required for attenuation of NF-κB activation. A, immunoblot analysis of PulA levels in the outer membranes of Klebsiella strains. An asterisk marks a protein also recognized by the anti-PulA antibody, which served as a loading control. Data are representative of three independent experiments. B, adhesion (left panel) and internalization (right panel) of Klebsiella strains to A549 cells (n = 3). C, SEAP levels released by A549 cells left untreated (control (CON), white bar) or infected with the indicated strains. Scale bars represent mean ± S.E. (n = 3). D, immunoblot analysis of IκBα and tubulin levels in A549 cells left uninfected (control) or infected for 3 h. Data are representative of three independent experiments. E, ELISA of IL-8 released by A549 cells left untreated (control, white bar) or infected for 12 h with the indicated strains (n = 3). F, immunoblots showing phosphorylated MAPKs and tubulin levels in cell extracts of A549 cells left uninfected (control, time 0) or infected with K. pneumoniae strains for different times. The results are representative of three independent experiments. G and H, immunoblot analysis of IκBα and tubulin levels in lysates of A549 cells infected with the indicated strains. The cells were left untreated, stimulated with IL-1β (1 ng/ml, 10 min), infected for 3 h, or infected for 3 h and then stimulated with IL-1β. In H, siRNA targets are indicated in the top row of each panel (AS−, AllStars control siRNA). Data are representative of three independent experiments. *, p < 0.05 (results are significantly different from the results for Kp52145; one-tailed t test). ns (not significant), p > 0.05 (for the indicated comparisons; one-tailed t test).
SEAP levels induced by the T2SS mutants were higher than those induced by the wild type (Fig. 6C), and IκBα levels were lower in those cells infected with the T2SS mutants (Fig. 6D). Complementation of the pulA mutant restored the SEAP levels to those triggered by the wild type, and we found an increase in the levels of IκBα in the lysates of infected cells (Fig. 6, C and D). Cells infected with the pulA mutants also secreted higher levels of IL-8 than cells infected with the wild type (Fig. 6E). Complementation of the mutant restored IL-8 secretion to wild-type levels. We also evaluated the activation of MAPKs in pulA mutant-infected cells. Western blot analysis shown in Fig. 6E revealed that infection with the pulA mutant triggered the phosphorylation of the three MAPKs. Phosphorylation levels of p38 and p44/42 were higher at 60 and 90 min post-infection in cells infected with the pulA mutant than in those infected with Kp52145 (Fig. 6E). Kp52145 and pulA mutant triggered similar levels of phosphorylated JNK (Fig. 6E). In aggregate, these findings indicate that K. pneumoniae that PulA contributes to limiting the activation of the NF-κB canonical pathway and MAPKs p38 and p44/42.
We next sought to clarify whether T2SS-secreted PulA is required for K. pneumoniae anti-inflammatory phenotype (22, 24). Similar to Kp52145, pulA mutant attenuated IL-1β-induced IκBα degradation (Fig. 6G). Furthermore, this effect was dependent on NOD1 and EGFR, because the pulA mutant did not block cytokine-triggered IκBα degradation in NOD1-EGFR knockdown cells (Fig. 6H).
siRNA-based experiments were carried out to investigate whether a TLR-MyD88-dependent pathway is implicated in the recognition of the pulA mutant. SEAP levels induced by the pulA mutant were reduced to those induced by the wild type only in MyD88 knocked-down cells and in TLR2-TLR4 knocked-down cells (Fig. 7A). Likewise, pulA mutant-triggered IL-8 secretion was also dependent on TLR4-TLR2-MyD88 (Fig. 7B). Altogether, our results demonstrated that K. pneumoniae PulA is required to attenuate the activation of NF-κB by limiting the activation of a TLR4-TLR2-MyD88 pathway.
FIGURE 7.
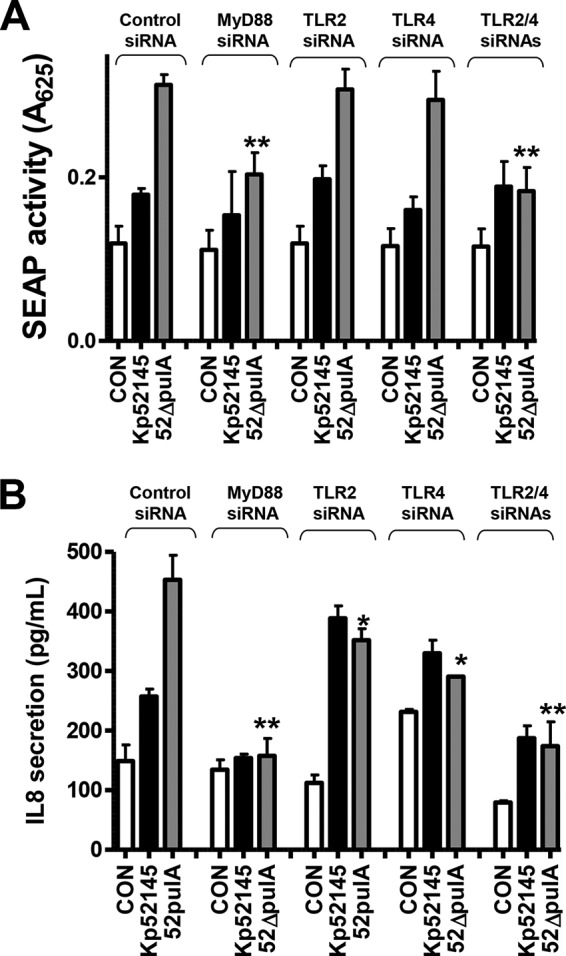
Role of MyD88, TLR2, and TLR4 in K. pneumoniae pulA mutant-induced cell activation. A, SEAP levels released by A549 cells transfected with the indicated siRNAs for different pattern recognition receptors, which were left untreated (control (CON), white bars) or infected with Kp52145 or pulA mutant (n = 3). B, ELISA of IL-8 released by A549 cells transfected with either control or the indicated siRNA for different pattern recognition receptors, which were left untreated (control, white bars) or infected (n = 3). Scale bars represent mean ± S.E. **, p < 0.01; *, p < 0.05 (results are significantly different from the results by obtained infecting control siRNA-transfected cells with pulA mutant; one-way ANOVA).
Relative Contribution of CPS, O-polysaccharide, and pulA to K. pneumoniae Attenuation of Inflammation
The fact that CPS, LPS OPS, and PulA are all involved in attenuating inflammatory responses led us to study the relative contribution of each of these factors. Therefore, we asked whether the absence of PulA further increases the SEAP levels and the secretion of IL-8 induced by the cps and waaL mutants. Data displayed in Fig. 7A show that SEAP levels induced by the double mutant waaL-pulA were higher than those induced by either the waaL or pulA single mutant. In turn, waaL-infected cells secreted higher SEAP levels than cells infected with the pulA mutant (Fig. 8A). In the cps mutant background, the strain inducing the highest SEAP levels was the double mutant waaL-pulA. No significant differences were observed between the waaL and pulA mutants. Similar picture was obtained when the IL-8 secretion was used as read out for cellular activation (Fig. 8B). Collectively, these data show that the CPS, the LPS OPS and PulA are Klebsiella factors require to limit NF-κB activation and the secretion of IL-8 by infected epithelial cells.
FIGURE 8.
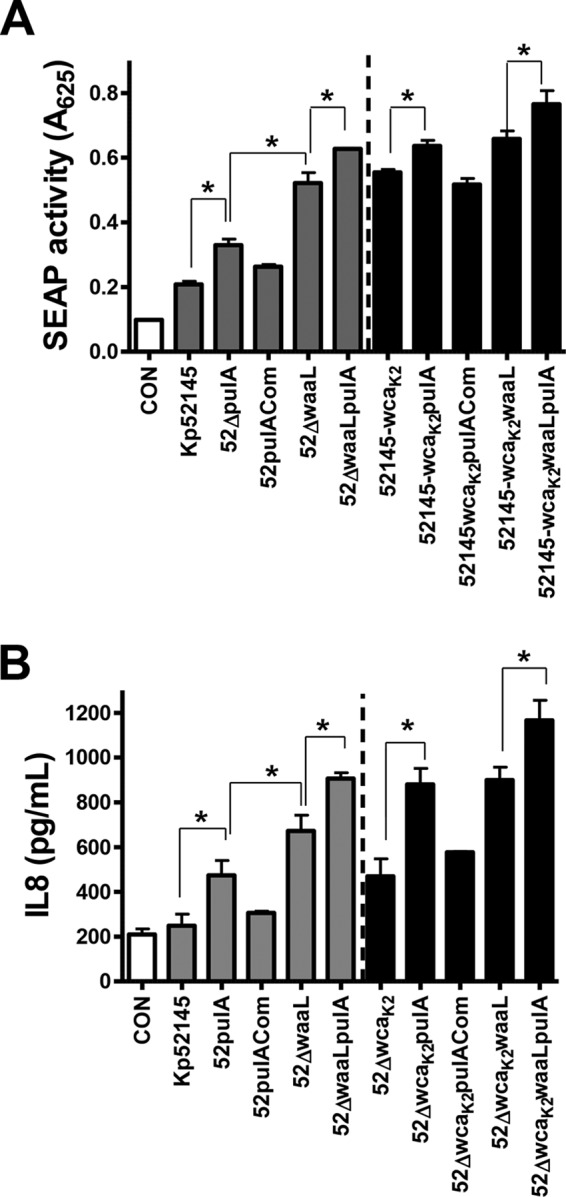
Relative contribution of CPS, LPS O-polysaccharide, and PulA T2SS to K. pneumoniae-triggered attenuation of NF-κB activation. A, SEAP levels released by A549 cells left untreated (control (CON), white bar) or infected with the indicated strains (n = 3). B, ELISA of IL-8 released by A549 cells left untreated (control, white bar) or infected for 12 h with the indicated strains (n = 3). Scale bars represent mean ± S.E. CPS-expressing strains, gray bars; CPS negative strains, black bars. *, p < 0.05 (for the indicated comparisons, one-tailed t test).
Virulence of K. pneumoniae Mutants
To determine the ability of pulA and waaL mutants to cause pneumonia, CD-1 mice were infected intranasally, and 24 h post-infection the bacterial loads in trachea, lung, spleen, and liver homogenates were determined. No differences were found in the bacterial loads in the trachea (Fig. 9A). In contrast, the bacterial loads of the pulA and waaL mutants were significantly lower in lung, spleen, and liver than those of the wild type (Fig. 9A).
FIGURE 9.

Virulence of K. pneumoniae waaL and pulA mutants. A, bacterial counts in mouse organs at 24 h post-infection. Mice were infected intranasally with the indicated strains (Kp52145, ■), pulA mutant (52ΔpulA, ○) or waaL mutant (52ΔwaaL, ▾). Results were reported as log cfu/g of tissue. **, p < 0.01; *, p < 0.05 (results are significantly different from the results obtained infecting with Kp52145; one-way ANOVA). B, kc and tnfα mRNA expressions in whole lungs were assessed by RT-qPCR. Mice were non-infected (control (CON), white bars, n = 5) or infected with Kp52145 (black bars, n = 5) or waaL and pulA mutants (gray bars; n = 5 for each mutant). Scale bars represent mean ± S.E. *, p < 0.05 (results are significantly different from the results obtained by infecting with Kp52145; one-way ANOVA).
We also assessed by RT-qPCR the expression levels of kc and tnfα cytokines in the lungs of infected mice. The levels of kc were higher in lungs of infected mice than in lungs of non-infected animals (p < 0.05 for all comparisons versus non-infected mice; one-tailed t test) (Fig. 9B). However, kc levels were significantly higher in the lungs of mice infected with pulA and waaL mutants than in those infected with the wild type (Fig. 9B). tnfα levels were higher in the lungs of mice infected with pulA mutant than in those infected with the wild type or the waaL mutant.
Discussion
We recently showed that K. pneumoniae dampens the activation of inflammatory responses in host cells by targeting the activation of the NF-κB canonical pathway (22, 24). Our results revealed that K. pneumoniae CPS is necessary but not sufficient to attenuate inflammation (22, 24). To identify additional Klebsiella factors required to dampen inflammation, we standardized and applied a high-throughput gain-of-function screen, mining a Klebsiella transposon mutant library. Using this approach, we identified 114 mutants that triggered the activation of NF-κB. Follow-up validation uncovered that, in addition to the CPS, K. pneumoniae LPS OPS and the pullulanase T2SS are required for evasion of innate immune responses. Table 3 summarizes the findings obtained in this study. This study represents the first functional genomics-driven identification of previously unknown bacterial factors required to down-regulate the NF-κB signaling pathway.
TABLE 3.
Summary of the phenotypes displayed by the K. pneumoniae mutants characterized in this study
NT, not tested; —, no capsule; ∼, similar to the wild-type strain; ↑, higher/stronger than the wild type, where the number of arrows gives an indication of relative levels compared with the wild type; ↓, less than the wild type.
| Klebsiella factor | Phenotype of the mutants |
||||||
|---|---|---|---|---|---|---|---|
| Adhesion/internalization | CPS levels | NF-κB activationa | MAPK activationb |
TLR signalingc | |||
| p38 | p44/42 | JNK | |||||
| Siderophores | |||||||
| entF | ↑ | ↓ | ↑↑ | NT | NT | ||
| cps-entF | NT | — | ↑↑ | NT | NT | ||
| iutA | NT | NT | ∼ | NT | NT | ||
| Irp1 | NT | NT | ∼ | NT | NT | ||
| LPS | |||||||
| waaL | ∼ | ∼ | ↑ | ↑ | ↑ | ∼ | Yes |
| glf | ∼ | ∼ | ↑ | ↑ | ↑ | ∼ | NT |
| wabM | ∼ | ∼ | ↑↑ | NT | NT | ||
| cps-waaL | ∼ | — | ↑↑ | NT | NT | ||
| cps-wabM | ↑ | — | ↑↑↑↑ | NT | NT | ||
| T2SS | |||||||
| pulA | ∼ | ∼ | ↑ | ↑ | ↑ | ∼ | Yes |
| cps-pulA | NT | — | ↑↑ | NT | NT | ||
| cps-waaL-pulA | NT | — | ↑↑↑↑ | NT | NT | ||
a SEAP levels as read-out.
b Phosphorylation of the MAPKs as read-out.
c IL-8 secretion and SEAP levels are dependent on the TLR4-TLR2-MyD88 signaling pathway.
The high number of Klebsiella loci involved in the attenuation of NF-κB activation was somewhat unanticipated. To the best of our knowledge the majority of the loci identified in our screening have never been implicated in NF-κB control, and even the function of most of them is largely speculative, as it is based on in silico predictions. Nevertheless, it should be noted that the transposon mutant library examined is not saturated, hence making it possible that additional loci are needed for fully effective immune evasion. For example, in our screening we did not retrieve any cps mutant, despite the fact that CPS plays a crucial role in Klebsiella anti-immune strategies (8, 20, 24). However, this is not unprecedented in this type of studies; for example, cps mutants were not isolated in other Klebsiella screenings searching for virulence factors (4, 55, 56).
The two gene ontology categories including half of the loci identified in the screening are: metabolism and transport, 32% of the mutants, and envelope-related genes, 17% of the mutants. None of the metabolic and transport mutants displayed any growth defects under the screening conditions or in LB, hence making it unlikely that any gross growth defect underlies the activation of NF-κB in infected cells. As this will be the subject of future studies, at present we speculated that their contribution to Klebsiella immune evasion is indirect and related to their impact on other Klebsiella factors. Supporting this hypothesis, the follow-up analysis of the enterobactin mutant revealed that the lack of the siderophore is linked to a reduced CPS expression, which in turn explains the NF-κB activation induced by the mutant. The regulatory connection between iron levels and CPS expression has been already established (48). Because our wild-type strain encodes for two additional iron-scavenging systems, yersiniabactin and aerobactin, we sought to determine whether any Klebsiella siderophore mutant would induce NF-κB activation. However, and in good agreement with Lawlor et al. (9), enterobactin was the only siderophore expressed under our infection conditions. Therefore, it was not unexpected to find that the mutants in the other two scavenging systems did not activate NF-κB. Nonetheless, the evidence indicates that yersiniabactin is the Klebsiella siderophore that plays a dominant role in vivo (9), hence making possible that yersiniabactin would be the siderophore implicated in Klebsiella immune evasion during pneumonia.
The other major set of mutants were related to envelope structures, thereby adding further evidence to the critical role played by bacterial surface structures on host-pathogen interactions. Although in this work we have characterized only the role of the OPS and T2SS systems, it is worth discussing the possible contribution of the peptidoglycan and adhesion mutants to Klebsiella immune evasion. The seminal discoveries showing that NOD receptors recognize peptidoglycan motifs from Gram-negative bacteria provide a mechanistic explanation for the observation that peptidoglycan mutants induce NF-κB activation (57, 58). This is because these mutants secrete peptidoglycan fragments into the medium, which then activate NOD1 (59). Future efforts in our laboratory will confirm whether this is the case with our set of mutants. In addition, we will be eager to explore whether K. pneumoniae remodels its peptidoglycan to attenuate detection by NOD receptors.
In our previous studies we did demonstrate that Klebsiella-cell contact is essential to attenuate NF-κB activation (22). In this scenario, it can be predicted that any mutant deficient in adhesion will be affected on its ability to block NF-κB activation. Of note, in our recent siRNA-based screening we uncovered the fact that Klebsiella may manipulate the β1-integrin-ILK signaling cascade to dampen inflammatory responses (24). β1-integrins are used by many pathogens as cellular receptors for attachment (60). It is then tempting to speculate that the adhesion-related mutants found in the present study might not be able to target the β1-integrin-ILK signaling cascade.
The LPS OPS is perhaps the most widely found factor in any screen designed to identify virulence factors of Gram-negative bacteria. However, to the best of our knowledge, ours is the first screening to demonstrate its involvement in attenuating NF-κB activation. The fact that the double mutant lacking CPS and OPS induced higher responses than each of the single mutants indicates that the Klebsiella polysaccharides do not play redundant roles in host-Klebsiella interactions. Having established the role of the OPS, we next investigated whether LPS core residues may also participate in immune evasion. To investigate the contribution of core residues in an OPS-bearing strain, we used defined mutants that lack the OPS in addition to core residues, which in turn might suggest that the core residues will never be exposed in a wild-type strain. However, it should be noted that epidemiological data indicate that nearly 10% of Klebsiella clinical isolates do not express the LPS OPS (61), and therefore, core residues will not be masked by the OPS. Our results revealed that the first glucose residue of the LPS core is necessary to attenuate NF-κB activation, because the SEAP levels induced by the wabM mutant, lacking also OPS, were higher than those triggered by the OPS mutant. Elimination of additional core residues did not further increase the SEAP levels.
The invasion experiments revealed unexpected features of the contribution of CPS and LPS to Klebsiella interaction with epithelial cells. Available evidence supports the notion that Klebsiella CPS is the critical factor preventing Klebsiella internalization by epithelial cells (49). Our data further corroborated this notion but also highlighted the contribution of OPS and core residues. However, in the absence of CPS, the d-galactan OPS mediates Klebsiella internalization by epithelial cells because the internalization of those mutants lacking the OPS, mutants waaL, wabM, and wabK, was reduced to the levels of the wild-type strain. Surprisingly, the internalization levels of the wabH mutant were similar to those of the CPS mutant. Structural studies showed that a core galactose residue gets exposed in this mutant (50), which in turn could account for the mutant internalization.
The contribution of the LPS core to virulence is poorly characterized in most Gram-negative pathogens, and it has been rigorously established only for Yersinia enterocolitica and Kp52145 (50, 62). In a previous study we uncovered the contribution of the Klebsiella core in preventing macrophage phagocytosis (6), which, together with the evidence presented in this work, led us to put forward the notion that core residues are important for Klebsiella immune evasion. Interestingly, this might be a general feature of LPS core sections, as it has been reported recently that Brucella abortus core acts as a shield against immune recognition (63). Future studies testing additional pathogens should help to further validate this hypothesis.
In this work, we discovered that Klebsiella pullulanase T2SS is important for immune evasion in vitro and in vivo. The Klebsiella pullulanase secretion system is the archetype of T2SS, and most of the previous research efforts have been devoted to characterizing the molecular structure and function of the secretion system (54). However, its contribution to Klebsiella virulence, if any, was unknown. Perusal of the literature indicates that PulA is the only protein secreted by K. pneumoniae T2SS, although presently we cannot rigorously rule out that other proteins yet to be found are also secreted by the system. Nonetheless, the fact that SEAP levels induced by the pulA, pulC, and yacC mutants were not significantly different may support the idea that indeed the only protein secreted by the T2SS is PulA. Taking into account the evidence available for similar enzymes from other pathogens (64, 65), we speculate that PulA interacts with glycan(s) located in the epithelial surface, hence leading to limited activation of the TLR4-TLR2-MyD88 pathway upon Klebsiella infection. Future efforts of the laboratory will be devoted to identify these host glycan(s).
In this work, we have demonstrated for the first time, using a pneumonia mouse model, that PulA is important for bacterial survival in the lung. Research over the last 20 years has demonstrated a correlation between the activation of inflammatory responses and Klebsiella clearance from the airways (11–14). Hence the higher inflammatory response induced by the pulA mutant may contribute to mutant clearance. It should be noted that cytokines and chemokines released by epithelial cells do increase the bactericidal activity of professional phagocytes. Studies are ongoing to determine whether PulA plays any role in the interaction of K. pneumoniae with alveolar macrophages and neutrophils.
Recently, we demonstrated that K. pneumoniae exerts an anti-inflammatory effect by engaging NOD1 and EGF receptors (22, 24). Bacterial removal by gentamicin treatment renders cells again responsive to pro-inflammatory mediators (22). Considering that CPS is necessary but not sufficient to attenuate inflammation (22, 24), in this work we asked whether OPS and PulA could mediate this anti-inflammatory effect. However, our findings shown in Figs. 4 and 6 do not support this. These results do not contradict the other findings reported in this work showing that OPS and T2SS mutants activate inflammatory responses. It should be noted that in most of the experiments in this study gentamicin was used to remove bacteria after a 2-h infection; for example, IL-8 in the supernatants was measured at 12 h post-infection. In turn, the fact that waaL and pulA mutant-induced responses were dependent on TLR2-TLR4-MyD88 activation suggests that OPS and PulA perturb TLR-dependent recognition of K. pneumoniae. These results further corroborate in vitro and in vivo data showing that TLR2 and TLR4 play a dominant role in detecting K. pneumoniae infections (20, 66, 67).
On the whole, the evidence is consistent with the notion that an essential aspect of K. pneumoniae infection biology is to thwart the TLR-dependent activation of host defense responses. Our studies have revealed the concerted action of CPS, OmpA, LPS polysaccharide, and T2SS to prevent TLR-dependent responses (this work and others (8, 20, 21)). Further highlighting the importance of immune evasion for Klebsiella virulence, all of these mutants are attenuated in the pneumonia model (this work and others (3, 8)). Therefore, we put forward the notion that a new therapeutic approach to the treatment of Klebsiella infections will be to prevent this virulence strategy, either by using molecules directed to block some of these factors or by increasing TLR-governed defense responses. Supporting the feasibility of the latter approach, the administration of the TLR ligand CpG augments anti-Klebsiella immunity in pneumonia (17).
Acknowledgments
We thank Anthony Pugsley for the anti-PulA antibody as well as for insights into the pullulanase system. We are grateful to members of the Bengoechea laboratory for helpful discussions. The Centro de Investigación Biomédica en Red Enfermedades Respiratorias (CIBERES) is an initiative of the Instituto de Salud Carlos III.
This work was supported by a grant from ERA-Net Pathogenomic 2008 (to P. J. S. and J. A. B.), Marie Curie Career Integration Grant (U-KARE) PCIG13-GA-2013-618162, Biotechnology and Biological Sciences Research Council (BBSRC) Grant BB/L007223/1, and Queen's University Belfast start-up funds (to J. A. B.). The authors declare that they have no conflicts of interest with the contents of this article.
- CPS
- capsule polysaccharide
- EGFR
- EGF receptor
- Omp
- outer membrane protein
- TLR
- Toll-like receptor
- OPS
- O-polysaccharide
- T2SS
- type 2 secretion system
- SEAP
- secreted form of the human embryonic alkaline phosphatase
- RLU
- relative light unit
- RT-qPCR
- real time quantitative PCR
- ANOVA
- analysis of variance
- Cm
- cloramphenicol
- Tp
- trimethoprim
- RFU
- relative fluorescence units.
References
- 1. Sahly H., Podschun R. (1997) Clinical, bacteriological, and serological aspects of Klebsiella infections and their spondylarthropathic sequelae. Clin. Diagn. Lab. Immunol. 4, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boucher H. W., Talbot G. H., Bradley J. S., Edwards J. E., Gilbert D., Rice L. B., Scheld M., Spellberg B., Bartlett J. (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12 [DOI] [PubMed] [Google Scholar]
- 3. Cortés G., Borrell N., de Astorza B., Gómez C., Sauleda J., Albertí S. (2002) Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect. Immun. 70, 2583–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lawlor M. S., Hsu J., Rick P. D., Miller V. L. (2005) Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol. Microbiol. 58, 1054–1073 [DOI] [PubMed] [Google Scholar]
- 5. Camprubí S., Merino S., Benedí V. J., Tomás J. M. (1993) The role of the O-antigen lipopolysaccharide and capsule on an experimental Klebsiella pneumoniae infection of the rat urinary tract. FEMS Microbiol. Lett. 111, 9–13 [DOI] [PubMed] [Google Scholar]
- 6. March C., Cano V., Moranta D., Llobet E., Pérez-Gutiérrez C., Tomás J. M., Suárez T., Garmendia J., Bengoechea J. A. (2013) Role of bacterial surface structures on the interaction of Klebsiella pneumoniae with phagocytes. PLoS One 8, e56847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Llobet E., Campos M. A., Giménez P., Moranta D., Bengoechea J. A. (2011) Analysis of the networks controlling the antimicrobial-peptide-dependent induction of Klebsiella pneumoniae virulence factors. Infect. Immun. 79, 3718–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. March C., Moranta D., Regueiro V., Llobet E., Tomás A., Garmendia J., Bengoechea J. A. (2011) Klebsiella pneumoniae outer membrane protein A is required to prevent the activation of airway epithelial cells. J. Biol. Chem. 286, 9956–9967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lawlor M. S., O'connor C., Miller V. L. (2007) Yersiniabactin is a virulence factor for Klebsiella pneumoniae during pulmonary infection. Infect. Immun. 75, 1463–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murphy C. N., Mortensen M. S., Krogfelt K. A., Clegg S. (2013) Role of Klebsiella pneumoniae type 1 and type 3 fimbriae in colonizing silicone tubes implanted into the bladders of mice as a model of catheter-associated urinary tract infections. Infect. Immun. 81, 3009–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greenberger M. J., Kunkel S. L., Strieter R. M., Lukacs N. W., Bramson J., Gauldie J., Graham F. L., Hitt M., Danforth J. M., Standiford T. J. (1996) IL-12 gene therapy protects mice in lethal Klebsiella pneumonia. J. Immunol. 157, 3006–3012 [PubMed] [Google Scholar]
- 12. Greenberger M. J., Strieter R. M., Kunkel S. L., Danforth J. M., Laichalk L. L., McGillicuddy D. C., Standiford T. J. (1996) Neutralization of macrophage inflammatory protein-2 attenuates neutrophil recruitment and bacterial clearance in murine Klebsiella pneumonia. J. Infect. Dis. 173, 159–165 [DOI] [PubMed] [Google Scholar]
- 13. Happel K. I., Dubin P. J., Zheng M., Ghilardi N., Lockhart C., Quinton L. J., Odden A. R., Shellito J. E., Bagby G. J., Nelson S., Kolls J. K. (2005) Divergent roles of IL-23 and IL-12 in host defense against. Klebsiella pneumoniae. J. Exp. Med. 202, 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Happel K. I., Zheng M., Young E., Quinton L. J., Lockhart E., Ramsay A. J., Shellito J. E., Schurr J. R., Bagby G. J., Nelson S., Kolls J. K. (2003) Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J. Immunol. 170, 4432–4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laichalk L. L., Bucknell K. A., Huffnagle G. B., Wilkowski J. M., Moore T. A., Romanelli R. J., Standiford T. J. (1998) Intrapulmonary delivery of tumor necrosis factor agonist peptide augments host defense in murine Gram-negative bacterial pneumonia. Infect. Immun. 66, 2822–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karaolis D. K., Newstead M. W., Zeng X., Hyodo M., Hayakawa Y., Bhan U., Liang H., Standiford T. J. (2007) Cyclic di-GMP stimulates protective innate immunity in bacterial pneumonia. Infect. Immun. 75, 4942–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng J. C., Moore T. A., Newstead M. W., Zeng X., Krieg A. M., Standiford T. J. (2004) CpG oligodeoxynucleotides stimulate protective innate immunity against pulmonary Klebsiella infection. J. Immunol. 173, 5148–5155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Standiford T. J., Wilkowski J. M., Sisson T. H., Hattori N., Mehrad B., Bucknell K. A., Moore T. A. (1999) Intrapulmonary tumor necrosis factor gene therapy increases bacterial clearance and survival in murine Gram-negative pneumonia. Hum. Gene Ther. 10, 899–909 [DOI] [PubMed] [Google Scholar]
- 19. Tsai W. C., Strieter R. M., Wilkowski J. M., Bucknell K. A., Burdick M. D., Lira S. A., Standiford T. J. (1998) Lung-specific transgenic expression of KC enhances resistance to Klebsiella pneumoniae in mice. J. Immunol. 161, 2435–2440 [PubMed] [Google Scholar]
- 20. Moranta D., Regueiro V., March C., Llobet E., Margareto J., Larrate E., Garmendia J., Bengoechea J. A. (2010) Klebsiella pneumoniae capsule polysaccharide impedes the expression of β-defensins by airway epithelial cells. Infect. Immun. 78, 1135–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Regueiro V., Campos M. A., Pons J., Albertí S., Bengoechea J. A. (2006) The uptake of a Klebsiella pneumoniae capsule polysaccharide mutant triggers an inflammatory response by human airway epithelial cells. Microbiology 152, 555–566 [DOI] [PubMed] [Google Scholar]
- 22. Regueiro V., Moranta D., Frank C. G., Larrarte E., Margareto J., March C., Garmendia J., Bengoechea J. A. (2011) Klebsiella pneumoniae subverts the activation of inflammatory responses in a NOD1-dependent manner. Cell. Microbiol. 13, 135–153 [DOI] [PubMed] [Google Scholar]
- 23. Lawlor M. S., Handley S. A., Miller V. L. (2006) Comparison of the host responses to wild-type and cpsB mutant Klebsiella pneumoniae infections. Infect. Immun. 74, 5402–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frank C. G., Reguerio V., Rother M., Moranta D., Maeurer A. P., Garmendia J., Meyer T. F., Bengoechea J. A. (2013) Klebsiella pneumoniae targets an EGF receptor-dependent pathway to subvert inflammation. Cell. Microbiol. 15, 1212–1233 [DOI] [PubMed] [Google Scholar]
- 25. Liu Y., Shepherd E. G., Nelin L. D. (2007) MAPK phosphatases: regulating the immune response. Nat. Rev. Immunol. 7, 202–212 [DOI] [PubMed] [Google Scholar]
- 26. Sun S. C. (2008) Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 8, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lery L. M., Frangeul L., Tomas A., Passet V., Almeida A. S., Bialek-Davenet S., Barbe V., Bengoechea J. A., Sansonetti P., Brisse S., Tournebize R. (2014) Comparative analysis of Klebsiella pneumoniae genomes identifies a phospholipase D family protein as a novel virulence factor. BMC Biol. 12, 41–7007-12–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nassif X., Fournier J. M., Arondel J., Sansonetti P. J. (1989) Mucoid phenotype of Klebsiella pneumoniae is a plasmid-encoded virulence factor. Infect. Immun. 57, 546–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharma R. C., Schimke R. T. (1996) Preparation of electrocompetent E. coli using salt-free growth medium. BioTechniques 20, 42–44 [DOI] [PubMed] [Google Scholar]
- 30. Larsen R. A., Wilson M. M., Guss A. M., Metcalf W. W. (2002) Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch. Microbiol. 178, 193–201 [DOI] [PubMed] [Google Scholar]
- 31. Malo N., Hanley J. A., Cerquozzi S., Pelletier J., Nadon R. (2006) Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 24, 167–175 [DOI] [PubMed] [Google Scholar]
- 32. Llobet E., Tomás J. M., Bengoechea J. A. (2008) Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 154, 3877–3886 [DOI] [PubMed] [Google Scholar]
- 33. Rahn A., Whitfield C. (2003) Transcriptional organization and regulation of the Escherichia coli K30 group 1 capsule biosynthesis (cps) gene cluster. Mol. Microbiol. 47, 1045–1060 [DOI] [PubMed] [Google Scholar]
- 34. Marolda C. L., Lahiry P., Vinés E., Saldías S., Valvano M. A. (2006) Micromethods for the characterization of lipid A-core and O-antigen lipopolysaccharide. Methods Mol. Biol. 347, 237–252 [DOI] [PubMed] [Google Scholar]
- 35. Alexeyev M. F. (1999) The pKNOCK series of broad-host-range mobilizable suicide vectors for gene knockout and targeted DNA insertion into the chromosome of Gram-negative bacteria. BioTechniques 26, 824-826 [DOI] [PubMed] [Google Scholar]
- 36. Rubirés X., Saigi F., Piqué N., Climent N., Merino S., Albertí S., Tomás J. M., Regué M. (1997) A gene (wbbL) from Serratia marcescens N28b (O4) complements the rfb-50 mutation of Escherichia coli K-12 derivatives. J. Bacteriol. 179, 7581–7586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Llobet E., March C., Giménez P., Bengoechea J. A. (2009) Klebsiella pneumoniae OmpA confers resistance to antimicrobial peptides. Antimicrob. Agents Chemother. 53, 298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crépin S., Harel J., Dozois C. M. (2012) Chromosomal complementation using Tn7 transposon vectors in Enterobacteriaceae. Appl. Environ. Microbiol. 78, 6001–6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pérez-Gutiérrez C., Llobet E., Llompart C. M., Reinés M., Bengoechea J. A. (2010) Role of lipid A acylation in Yersinia enterocolitica virulence. Infect. Immun. 78, 2768–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dennis J. J., Zylstra G. J. (1998) Plasposons: modular self-cloning minitransposon derivatives for rapid genetic analysis of Gram-negative bacterial genomes. Appl. Environ. Microbiol. 64, 2710–2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miller W. G., Leveau J. H., Lindow S. E. (2000) Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol. Plant Microbe Interact. 13, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 42. Reinés M., Llobet E., Llompart C. M., Moranta D., Pérez-Gutiérrez C., Bengoechea J. A. (2012) Molecular basis of Yersinia enterocolitica temperature-dependent resistance to antimicrobial peptides. J. Bacteriol. 194, 3173–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reinés M., Llobet E., Dahlström K. M., Pérez-Gutiérrez C., Llompart C. M., Torrecabota N., Salminen T. A., Bengoechea J. A. (2012) Deciphering the acylation pattern of Yersinia enterocolitica lipid A. PLoS Pathog. 8, e1002978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raymond K. N., Dertz E. A., Kim S. S. (2003) Enterobactin: an archetype for microbial iron transport. Proc. Natl. Acad. Sci. U.S.A. 100, 3584–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hayden M. S., Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 46. Liu Z., Reba S., Chen W. D., Porwal S. K., Boom W. H., Petersen R. B., Rojas R., Viswanathan R., Devireddy L. (2014) Regulation of mammalian siderophore 2,5-DHBA in the innate immune response to infection. J. Exp. Med. 211, 1197–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Autenrieth I., Hantke K., Heesemann J. (1991) Immunosuppression of the host and delivery of iron to the pathogen: a possible dual role of siderophores in the pathogenesis of microbial infections?. Med. Microbiol. Immunol. 180, 135–141 [DOI] [PubMed] [Google Scholar]
- 48. Lin C. T., Wu C. C., Chen Y. S., Lai Y. C., Chi C., Lin J. C., Chen Y., Peng H. L. (2011) Fur regulation of the capsular polysaccharide biosynthesis and iron-acquisition systems in Klebsiella pneumoniae CG43. Microbiology 157, 419–429 [DOI] [PubMed] [Google Scholar]
- 49. Cortés G., Alvarez D., Saus C., Albertí S. (2002) Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect. Immun. 70, 1075–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Regué M., Izquierdo L., Fresno S., Piqué N., Corsaro M. M., Naldi T., De Castro C., Waidelich D., Merino S., Tomás J. M. (2005) A second outer-core region in Klebsiella pneumoniae lipopolysaccharide. J. Bacteriol. 187, 4198–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Köplin R., Brisson J. R., Whitfield C. (1997) UDP-galactofuranose precursor required for formation of the lipopolysaccharide O antigen of Klebsiella pneumoniae serotype O1 is synthesized by the product of the rfbDKPO1 gene. J. Biol. Chem. 272, 4121–4128 [DOI] [PubMed] [Google Scholar]
- 52. Kenny E. F., O'Neill L. A. (2008) Signalling adaptors used by Toll-like receptors: an update. Cytokine 43, 342–349 [DOI] [PubMed] [Google Scholar]
- 53. Korotkov K. V., Sandkvist M., Hol W. G. (2012) The type II secretion system: biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol. 10, 336–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pugsley A. P., Francetic O., Hardie K., Possot O. M., Sauvonnet N., Seydel A. (1997) Pullulanase: model protein substrate for the general secretory pathway of Gram-negative bacteria. Folia Microbiol. (Praha) 42, 184–192 [DOI] [PubMed] [Google Scholar]
- 55. Maroncle N., Balestrino D., Rich C., Forestier C. (2002) Identification of Klebsiella pneumoniae genes involved in intestinal colonization and adhesion using signature-tagged mutagenesis. Infect. Immun. 70, 4729–4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tu Y. C., Lu M. C., Chiang M. K., Huang S. P., Peng H. L., Chang H. Y., Jan M. S., Lai Y. C. (2009) Genetic requirements for Klebsiella pneumoniae-induced liver abscess in an oral infection model. Infect. Immun. 77, 2657–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Qiu S., Saab L., Ogura Y., Kawasaki A., Fukase K., Kusumoto S., Valvano M. A., Foster S. J., Mak T. W., Nuñez G., Inohara N. (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707 [DOI] [PubMed] [Google Scholar]
- 58. Girardin S. E., Boneca I. G., Carneiro L. A., Antignac A., Jéhanno M., Viala J., Tedin K., Taha M. K., Labigne A., Zähringer U., Coyle A. J., DiStefano P. S., Bertin J., Sansonetti P. J., Philpott D. J. (2003) Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science 300, 1584–1587 [DOI] [PubMed] [Google Scholar]
- 59. Nigro G., Fazio L. L., Martino M. C., Rossi G., Tattoli I., Liparoti V., De Castro C., Molinaro A., Philpott D. J., Bernardini M. L. (2008) Muramylpeptide shedding modulates cell sensing of. Shigella flexneri. Cell. Microbiol. 10, 682–695 [DOI] [PubMed] [Google Scholar]
- 60. Boyle E. C., Finlay B. B. (2003) Bacterial pathogenesis: exploiting cellular adherence. Curr. Opin. Cell Biol. 15, 633–639 [DOI] [PubMed] [Google Scholar]
- 61. Hansen D. S., Mestre F., Alberti S., Hernández-Allés S., Alvarez D., Doménech-Sánchez A., Gil J., Merino S., Tomás J. M., Benedí V. J. (1999) Klebsiella pneumoniae lipopolysaccharide O typing: revision of prototype strains and O-group distribution among clinical isolates from different sources and countries. J. Clin. Microbiol. 37, 56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Skurnik M., Venho R., Bengoechea J. A., Moriyón I. (1999) The lipopolysaccharide outer core of Yersinia enterocolitica serotype O:3 is required for virulence and plays a role in outer membrane integrity. Mol. Microbiol. 31, 1443–1462 [DOI] [PubMed] [Google Scholar]
- 63. Conde-Álvarez R., Arce-Gorvel V., Iriarte M., Manček-Keber M., Barquero-Calvo E., Palacios-Chaves L., Chacón-Díaz C., Chaves-Olarte E., Martirosyan A., von Bargen K., Grilló M. J., Jerala R., Brandenburg K., Llobet E., Bengoechea J. A., Moreno E., Moriyón I., Gorvel J. P. (2012) The lipopolysaccharide core of Brucella abortus acts as a shield against innate immunity recognition. PLoS Pathog. 8, e1002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shelburne S. A., 3rd, Sahasrobhajane P., Suber B., Keith D. B., Davenport M. T., Horstmann N., Kumaraswami M., Olsen R. J., Brennan R. G., Musser J. M. (2011) Niche-specific contribution to streptococcal virulence of a MalR-regulated carbohydrate-binding protein. Mol. Microbiol. 81, 500–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. van Bueren A. L., Higgins M., Wang D., Burke R. D., Boraston A. B. (2007) Identification and structural basis of binding to host lung glycogen by streptococcal virulence factors. Nat. Struct. Mol. Biol. 14, 76–84 [DOI] [PubMed] [Google Scholar]
- 66. Regueiro V., Moranta D., Campos M. A., Margareto J., Garmendia J., Bengoechea J. A. (2009) Klebsiella pneumoniae increases the levels of Toll-like receptors 2 and 4 in human airway epithelial cells. Infect. Immun. 77, 714–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wieland C. W., van Lieshout M. H., Hoogendijk A. J., van der Poll T. (2011) Host defence during Klebsiella pneumonia relies on haematopoietic-expressed Toll-like receptors 4 and 2. Eur. Respir. J. 37, 848–857 [DOI] [PubMed] [Google Scholar]
- 68. Izquierdo L., Coderch N., Piqué N., Bedini E., Corsaro M. M., Merino S., Fresno S., Tomás J. M., Regué M. (2003) The Klebsiella pneumoniae wabG gene: role in biosynthesis of the core lipopolysaccharide and virulence. J. Bacteriol. 185, 7213–7221 [DOI] [PMC free article] [PubMed] [Google Scholar]