

Background: Axon growth and regeneration depend on kinesin-1-dependent transport.
Results: JIP3 binding to KHC promotes kinesin-1 motility along microtubules and is essential for axon elongation and regeneration.
Conclusion: JIP3 regulation of kinesin-1 motility is critical for axon elongation and regeneration.
Significance: Regulation of intracellular transport is important for proper neuronal development.
Keywords: axon, kinesin, molecular motor, neurite outgrowth, regeneration, JIP3, KHC, motility
Abstract
Kinesin-1 is a molecular motor responsible for cargo transport along microtubules and plays critical roles in polarized cells, such as neurons. Kinesin-1 can function as a dimer of two kinesin heavy chains (KHC), which harbor the motor domain, or as a tetramer in combination with two accessory light chains (KLC). To ensure proper cargo distribution, kinesin-1 activity is precisely regulated. Both KLC and KHC subunits bind cargoes or regulatory proteins to engage the motor for movement along microtubules. We previously showed that the scaffolding protein JIP3 interacts directly with KHC in addition to its interaction with KLC and positively regulates dimeric KHC motility. Here we determined the stoichiometry of JIP3-KHC complexes and observed approximately four JIP3 molecules binding per KHC dimer. We then determined whether JIP3 activates tetrameric kinesin-1 motility. Using an in vitro motility assay, we show that JIP3 binding to KLC engages kinesin-1 with microtubules and that JIP3 binding to KHC promotes kinesin-1 motility along microtubules. We tested the in vivo relevance of these findings using axon elongation as a model for kinesin-1-dependent cellular function. We demonstrate that JIP3 binding to KHC, but not KLC, is essential for axon elongation in hippocampal neurons as well as axon regeneration in sensory neurons. These findings reveal that JIP3 regulation of kinesin-1 motility is critical for axon elongation and regeneration.
Introduction
The kinesin-1 molecular motor is essential for cellular function, transporting cargoes along microtubules to distinct locales with precision timing. Kinesin-1 conventionally refers to a heterotetramer consisting of two kinesin heavy chains (KHC),2 which harbor the motor domain, and two non-motile light chains (KLC) (1–3). However, kinesin-1 can also function as a dimer of two motile KHC (4–13). Although KHC contains the force-generating component and provides motility for the kinesin-1 molecule, KHC also inhibits its own motility in the absence of cargo or regulatory proteins (14–17). The KHC tail can bind to the motor head domain and microtubules, creating a paused state for kinesin-1 (14). The KLC subunits act both as a cargo adapter for KHC and as an inhibitor of microtubule binding and motility of KHC in the absence of cargo (18, 19). Because the kinesin-1 tetramer is dually autoinhibited in the absence of binding partners, regulatory proteins must bind both KHC and KLC to relieve inhibition and promote binding and movement along microtubules (20).
Neurons depend on kinesin-1 to transport cargoes along the extreme length of axons for efficient long distance intracellular communication. Kinesin-1 plays critical roles during axon specification (21–23), growth (24), maintenance (25–27), organelle positioning (28), and regeneration (29–31). Defects in kinesin-1 cargo delivery or motility lead to neurological disorders (32–40), underscoring the importance of regulating kinesin-1 motility for neuronal function. However, the cellular mechanisms that control kinesin-1 motile properties remain poorly understood.
We previously showed that the c-Jun N-terminal kinase (JNK)-interacting protein 3, JIP3 (also known as JSAP1 or Sunday Driver) interacts with both KHC and KLC and promotes motility of dimeric KHC in vitro (5), suggesting that JIP3 regulates kinesin-1 motility in axons. In agreement with this notion, previous studies have shown that JIP3 mutation leads to axonal transport defects in Caenorhabditis elegans (41–43) and Drosophila melanogaster (44). Both JIP3 null mice and neuronal specific conditional JIP3 knock-out mice are perinatal lethal and display severe axon guidance defects of the telencephalic commissure (45–47). At the cellular level, an absence of JIP3 in cerebellar granule neurons reduces axon length but increases axon branching (48). In cortical neurons, JIP3 absence stimulates neurite elongation and branching (49). Although the localization of JIP3 to axon tips by kinesin-1 correlates with axon elongation in hippocampal neurons (50), whether modulation of kinesin-1 motile properties by JIP3 contributes to axon elongation remains unknown. We therefore sought to directly determine whether JIP3 regulates tetrameric kinesin-1 in vitro and how JIP3 interaction with kinesin-1 subunits regulates axon elongation.
Experimental Procedures
Antibodies and Reagents
Rhodamine- and biotin-labeled microtubules were generated using porcine rhodamine- and biotin-tubulin and bovine unlabeled tubulin, all obtained from Cytoskeleton (Denver, CO). The antibodies used were as follows: anti-MAP2 (Millipore, Temecula, CA), anti-Tau clone 5E2 (Millipore), anti-SCG10 (Novus, Littleton, CO), anti-JIP3 (44, 63), anti-GFP (Life Technologies, Inc.), anti-FLAG (Sigma-Aldrich), anti-α-tubulin (Abcam, Cambridge, MA), phospho-c-Jun (Cell Signaling, Danvers, MA), and anti-βIII-tubulin (Sigma-Aldrich). JIP3 knock-out mice were described previously (45). CD10 timed pregnant mice were obtained from Charles River Laboratories (O'Fallon, MO) for isolation of embryonic dorsal root ganglia (DRGs). Sprague-Dawley timed pregnant rats were obtained from Charles River Laboratories for isolation of embryonic hippocampal neurons.
Plasmid Construction
KLC-mCit was a generous gift from Dr. Kristen Verhey. FLAG-KHC and GFP-JIP3 mutants were described previously (5). Briefly, the cDNA coding sequences were cloned in-frame downstream of their respective tags in the multicloning sites of pcDNA3 and GFP-C1 vectors, respectively. Lentiviral JIP3 constructs were generated by excising the GFP-JIP3 fusion construct from their C1 vectors and inserting them into the FUGW vector using AgeI/EcoRI cloning sites.
Lentiviral Production
Lentiviral particles were concentrated from HEK293T culture medium as described previously (51). Briefly, HEK293T cells were transfected using Lipofectamine 2000 with the FUGW-JIP3 construct of interest, pVSV-G, and dR8.91 in a 1:1:3 ratio according to the manufacturer's specifications (Invitrogen). After 2 days, culture medium was collected and filtered through a 0.45-μm pore. Filtered culture medium was incubated overnight with Lenti-X lentiviral concentrator according to the manufacturer's instructions (Clontech) and centrifuged at 1,000 × g for 45 min to pellet lentiviral particles. Pellets were resuspended in Opti-MEM before adding to cultures.
In Vitro Motility Assay
An in vitro motility assay was performed as described (5, 20) with minor modifications. COS7 cells were obtained from ATCC (Manassas, VA) and cultured as recommended. Cells were transfected using Lipofectamine 2000 (KLC-mCit, all FLAG-JIP3 and GFP-JIP3 constructs) or Lipofectamine 3000 (FLAG-KHC), both from Invitrogen, according to the manufacturer's instructions. Twenty-four hours after transfection, COS7 cells were lysed in ice-cold buffer (500 mm PIPES, 1 mm EGTA, 1 mm MgSO4, 0.1% Triton X-100, Roche protease inhibitor tablet, pH 6.9), and supernatant was collected by centrifugation at 16,000 × g for 10 min at 4 °C. Flow chambers of ∼20-μl volume were assembled by adhering silanized coverslips to glass slides using double-sided tape. 250 nm rhodamine- and biotin-labeled microtubules were adhered to the coverslips using 0.01 μg/μl streptavidin (Sigma-Aldrich). The coverslip surface was then blocked with 5% Pluronic (Sigma-Aldrich). Motility mix was generated by adding 5 μl of FLAG-JIP3 (or untransfected control lysate), 1 μl of FLAG-KHC, 1 μl of KLC-mCit, and 1 μl of 40 mm Na-GTP to 8 μl of motility assay buffer (10 mm PIPES, 50 mm KOAc, 4 mm MgSO4, 1 mm EGTA) supplemented with 1 μl of 1 m DTT, 1 μl of oxygen scavenging system (250 μg/μl glucose oxidase, 35 μg/μl catalase, and 1 μl of 4.5 mg/ml glucose), and 1 μl of 0.5 mm taxol. KLC-mCit molecules were subsequently visualized at 25 °C using total internal reflection fluorescence (TIRF) microscopy on an inverted microscope (Olympus IX81). 488- and 532-nm diode-pumped solid-state lasers (Melles Griot) were used at 5-milliwatt power to visualize mCit and rhodamine, respectively. Images were captured at 1-s intervals on a back-thinned electron multiplier-CCD camera (ImagEM, Hamamatsu). KLC-mCit motility was analyzed by kymographs generated and measured in Slidebook (Intelligent Imaging Innovations). Vertical lines in the kymographs were scored as nonmotile events, whereas diagonal lines that lasted longer than two frames were scored as motile events. Only lines with a clear on and off event during the 100-s movie were analyzed to avoid inclusion of aggregates bound to microtubules. We also did not score landing events for puncta that were unusually bright, because these most likely represent aggregates.
For the KHC dimer assay, the experimental setup was as above except 1 μl each of WT-GFP JIP3 and FLAG-KHC were used. For the stoichiometry experiments, we immobilized FLAG-KHC to microtubules using AMP-PNP and imaged WT GFP-JIP3 continuously (i.e. without any interval between frames) to photobleach GFP. The fluorescence intensity of individual GFP-JIP3 puncta was then measured over time, and clearly detectable unitary bleach steps were used to estimate the fluorescence intensity of a single GFP molecule. We then calculated the number of GFP molecules in individual puncta as (initial fluorescence intensity of a punctum − background fluorescence intensity)/fluorescence intensity of a single GFP molecule.
Hippocampal Electroporation and Culture
Hippocampuses were isolated from pups and dissociated with papain (Worthington) and DNase for 30 min and triturated in neurobasal culture medium supplemented with B-27 (1%), penicillin/streptomycin (1%), and l-glutamate (0.25%). Neurons were isolated by centrifugation and resuspended in electroporation solution. Electroporation was carried out using 5 μg of DNA in 100 μl of isotonic solution (2% Buffer I (2 g of ATP-disodium salt, 1.2 g of MgCl2·6H2O into 10 ml of H2O) into Buffer II (6 g of KH2PO4, 0.6 g of NaHCO3, 0.2 g of glucose into 300 ml of H2O, pH 7.4)) using an Amaxa Nucleofector II system (Lonza, Basel, Switzerland) factory-preset for mouse hippocampal neurons. Neurons were rapidly centrifuged after electroporation and resuspended in culture medium for plating. Neurons were cultured on poly-d-lysine-coated glass coverslips that had been etched for 3 h in nitric acid. Using this method, less than 20% of neurons are transfected. Neurons were allowed to grow for 5 days before fixing and staining. Neurons were fixed with 4% phosphate-buffered paraformaldehyde and stained with the indicated antibodies in blocking buffer containing 10% goat serum, 0.1% Triton X-100 in PBS. Cultures were mounted with Prolong mounting medium (Life Technologies, Inc.).
DRG Infection and Culture
DRG neurons were isolated from e13 mouse pups and dissociated with 0.05% trypsin-EDTA for 20 min. The trypsin was replaced with neurobasal culture medium supplemented with B27 (2%), penicillin/streptomycin (1%), Glutamax (1%), 5-fluorodeoxyuridine (0.5%), and NGF (0.1%), and neurons were triturated 25 times with a p1000 pipette tip. Medium was removed, and neurons were triturated an additional two cycles. Neurons were resuspended in enough medium to plate 5 μl/10,000 cell spots and allowed to sit on the poly-d-lysine- and laminin-coated culture surface for 5 min before a full volume of culture medium was added to the culture surface. For growth assays, neurons were infected at DIV1 and fixed and stained at DIV3 for α-tubulin. For injury assays, neurons were infected at DIV3 with lentiviral JIP3 constructs. Lentiviral infection typically provides >90% infection efficiency. At DIV7, neurons were lysed for Western blots or injured with a metal probe. 12 h after injury, neurons were fixed and stained for the axonal regeneration marker SCG10 or βIII-tubulin.
Image Acquisition and Analysis
Images were acquired with fluorescence microscopy (Nikon, Eclipse TE 200-E) and analyzed using ImageJ. For hippocampal axon morphology, neurites were traced, and length was measured using the NeuronJ plugin. NeuronJ-produced tracings were then used as input for the Sholl Analysis plugin. For DRG axon regeneration, the regeneration index was measured as described previously (51). Briefly, SCG10 images were thresholded to create a binary image that eliminated background fluorescence. Then fluorescence intensity distal to the site of injury was measured as a percentage of fluorescence intensity proximal to the site of injury for a given area (i.e. fluorescence intensity was measured over an equal sized area for proximal/distal comparisons).
Statistical Analysis
Western blots were scanned and quantified by ImageJ, and Student's t test was used for statistical analysis of band intensities. NeuronJ and Sholl Analysis plugins for ImageJ were used to measure fluorescence intensities in images of neurons. ANOVA followed by Tukey's test and Student's t test were used for statistical analysis between experimental sets as stated throughout.
Results
JIP3 Binds to KHC Dimers in at Least a 2:1 Relationship
We previously showed that the scaffolding protein JIP3 interacts directly with KHC in addition to its interaction with KLC and positively regulates dimeric KHC motility (5). However, the stoichiometry of JIP3-KHC complexes remains unknown. To determine the stoichiometry of JIP3 and KHC complexes in vitro, we first established that JIP3 movement depends on its interaction with KHC. To this end, we used a TIRF-based motility assay to measure dimeric KHC velocity by imaging GFP-JIP3 puncta in the presence of FLAG-KHC, as described previously (5, 20, 52) (Fig. 1A). COS7 cells were used to express WT GFP-JIP3 and a mutant lacking the KHC binding site (Fig. 1B). We observed that WT GFP-JIP3 puncta moved at 0.25 ± 0.02 μm/s, which was significantly faster than the speed of GFP-JIP3ΔKHC puncta, which moved at 0.16 ± 0.01 μm/s (Fig. 1, C and D). The run speed for WT GFP-JIP3 is ∼50% lower than our previously reported values (5). Although the reason for this difference is not clear, it might result from differences in the COS7 cell batches used between the experiments. Nevertheless, we found that JIP3 activates dimeric KHC for motility via its KHC-binding domain, which replicates our primary findings (5). The motile fraction of WT GFP-JIP3 was also higher compared with GFP-JIP3ΔKHC (Fig. 1F), whereas the run length was not statistically different (Fig. 1E). Together, these results indicate that the movement of GFP-JIP3 observed in these conditions is mediated by its interaction with FLAG-KHC.
FIGURE 1.

JIP3 interacts with dimeric KHC in at least a 2:1 ratio to promote KHC motility. A, schematic representation of the TIRF-based dimeric KHC motility assay. Rhodamine- and biotin-labeled microtubules (red line) are adhered to coverslips (black line) with streptavidin. A motility mix with GFP-JIP3 and FLAG-KHC protein lysates is added to the chamber. B, schematic representation of JIP3 mutants used in this study. JIP3ΔKLC removes amino acids 429–459, corresponding to the KLC binding domain. JIP3ΔKHC removes amino acids 5–81 corresponding to the KHC binding domain. JIP3ΔΔ removes both the KLC and KHC binding domains. KBD, KHC binding domain; JBD, JNK binding domain; LZ, leucine zipper (KLC binding domain). C, representative kymographs of motile FLAG-KHC events in the presence of the indicated JIP3 construct. Motile events are less frequent and shorter when JIP3 cannot bind KHC, as with JIP3ΔKHC. D, velocity of motile events. Activation of the KHC dimer by WT GFP-JIP3 in this replication was 0.25 μm/s, compared with JIP3ΔKHC, which cannot bind KHC and significantly reduces run velocity. #, p < 0.001; Student's t test. E, run length of motile events. No significant difference was detected in run length of motile events; p = 0.07, Student's t test. F, motile fraction was calculated as described by Sun et al. (5). Inability to bind KHC significantly reduces motile fraction compared with WT JIP3. #, p < 0.001, Student's t test. G, representative trace of the fluorescence intensity over time of a GFP-JIP3 punctum. Fluorescence intensity decreases in distinct steps (red lines), representing photobleaching of single GFP molecules. The total number of GFP-JIP3 molecules can then be calculated by dividing the overall drop in fluorescence intensity (blue line) by the intensity of a single GFP molecule. This example shows a punctum that is calculated to contain four GFP molecules. H, histogram of the number of GFP molecules in GFP-JIP3 bound to FLAG-KHC. Counts below 4 were rarely observed, suggesting that JIP3 associates with KHC with at least 2:1 stoichiometry. AU, arbitrary units; error bars, S.E.
To determine the stoichiometry of JIP3 and KHC complexes in vitro, we immobilized dimeric KHC and its associated WT GFP-JIP3 molecules on rhodamine-labeled microtubules by using AMP-PNP and conducted single-molecule photobleaching assays as described previously (53). This TIRF-based photobleaching experiment is an extremely well established technique (54–57), and it offers the great advantage of determining stoichiometry specifically for motile, microtubule-bound KHC-JIP3 complexes, as opposed to non-motile complexes that might exist in the cytoplasm. In these assays, the most frequent number of photobleaching steps was four, indicating four JIP3 molecules per KHC dimer (Fig. 1, G and H). We also measured up to eight photobleaching steps, which may result from unresolved KHC dimers bound to a microtubule and photobleaching of all or some of the associated GFP-JIP3 molecules. We rarely measured photobleaching steps fewer than four. Based on this analysis, we conclude that at least four JIP3 molecules bind each KHC dimer, and therefore the stoichiometry for JIP3/KHC is likely to be at least 2:1.
JIP3 Differentially Regulates Kinesin-1 for Microtubule Binding and Motility
By virtue of its non-overlapping KLC and KHC binding domains, JIP3 has the potential to activate tetrameric kinesin-1 for microtubule binding and motility (5, 20). To test this possibility, we modified our TIRF assay by expressing FLAG-tagged WT JIP3 or JIP3 mutants (5) (Fig. 1B) as well as KLC-mCit and FLAG-KHC (Fig. 2, A–C). Cell lysates were mixed before the motility assay, and an excess of FLAG-JIP3 (WT or deletion mutants) was used. Because KLC has no intrinsic motile activity, mCit-positive puncta moving along rhodamine-labeled microtubules represent KLC-KHC complexes (i.e. tetrameric kinesin-1) (20).
FIGURE 2.

JIP3 regulates distinct aspects of kinesin-1 motility via differential binding to KLC and KHC subunits. A, schematic representation of the tetrameric kinesin-1 motility assay. FLAG-KHC and KLC-mCit are used with FLAG-JIP3 mutants as described in the legend to Fig. 1B. Because KLC has no intrinsic motility along microtubules, mCit puncta move along microtubules only when kinesin-1 is functioning as a heterotetramer. B, representative Western blot of KLC-mCit and FLAG-KHC lysates. FLAG-KHC and KLC-mCit were both present in their expected size ranges (n = 3). C, representative Western blot of FLAG-JIP3 lysates, including a control lysate lacking JIP3 and FLAG-KHC for comparison. 1 μl of each of the JIP3 mutants and control lysate and 3 μl of the FLAG-KHC were loaded in the gel. All JIP3 constructs were expressed at much greater levels than FLAG-KHC (n = 3). D, representative kymographs from TIRF experiments. Each y axis represents 100 s of imaging time. Scale bar, 2 μm. E, binding frequency was measured as the number of motile and nonmotile binding events per micron of microtubule. WT JIP3 and JIP3ΔKHC significantly increased the microtubule binding frequency of kinesin-1 compared with untransfected controls, but JIP3ΔKLC did not. F and G, n = 74 (control), 93 (WT JIP3), 85 (JIP3ΔKLC), and 92 (JIP3ΔKHC) microtubules analyzed in four replicates (ANOVA, p = 1.65 × 10−5; #, p ≪ 0.001 compared with control; Tukey's HSD). F, motile efficiency was measured as the number of motile events per 10 μm of microtubule. All JIP3 mutants increased motile efficiency compared with control, but JIP3ΔKHC produced a significantly lower motile efficiency than WT JIP3 (ANOVA, p = 0.01; *, p < 0.05; ***, p < 0.001; n.s., not significant; Tukey's HSD). G, velocity of motile events. WT JIP3 and JIP3ΔKLC significantly increase run speed compared with control, but JIP3ΔKHC did not. G–J, n = 41 (control), 108 (WT JIP3), 80 (JIP3ΔKLC), or 86 (JIP3ΔKHC) motile events in four replicates (ANOVA, p = 9.65 × 10−5; **, p < 0.01; ***, p < 0.001 compared with control; Tukey's HSD). H, run length of motile events. WT JIP3, JIP3ΔKLC, and JIP3ΔKHC all significantly increase run length compared with controls, but JIPΔKHC does not increase run length to the extent that WT JIP3 does (Kruskal-Wallis, p = 4.2 × 10−5; *, p < 0.05; **, p < 0.01; #, p ≪ 0.001; Mann-Whitney U test). I, histogram of velocity of motile events. J, histogram of run length of motile events. Error bars, S.E.
When mixed with control lysates lacking JIP3, KLC-mCit rarely displayed motile events (supplemental Video S1), in agreement with previous findings (20). This was expected because KLC inhibits the microtubule binding and motility of KHC (19, 20, 58). Under control conditions, the binding frequency of KLC-mCit is 0.11 ± 0.01 events/μm, and the motile efficiency is 0.22 ± 0.04 events/10 μm of microtubule (Fig. 2, D–F). The presence of WT JIP3 lysate doubled both the binding frequency (0.22 ± 0.02 events/μm) and motile efficiency (0.50 ± 0.07 events/10 μm) of KLC-mCit (Fig. 2, D–F, and supplemental Video S2), indicating that WT JIP3 activates kinesin-1 for microtubule binding and motility. JIP3ΔKLC lysates were unable to significantly increase the binding frequency of KLC-mCit (0.15 ± 0.02 events/μm) compared with control lysates, but they increased the motile efficiency of KLC-mCit close to the levels obtained using WT JIP3 lysates (0.42 ± 0.08 events/10 μm) (Fig. 2, D–F, and supplemental Video S3). In contrast, JIP3ΔKHC lysates resulted in a high binding frequency that is similar to WT JIP3 lysates (0.21 ± 0.02 events/μm) but only modestly increased the motile efficiency compared with control lysates (0.34 ± 0.05 events/10 μm) (Fig. 2, D–F, and supplemental Video S4). Together, these data suggest that JIP3 interaction with KLC promotes binding of kinesin-1 to microtubules and that JIP3 interaction with KHC binding promotes motility of kinesin-1 along microtubules.
To explore further how JIP3 regulates kinesin-1 motility, we measured the velocity and run length of motile events (Fig. 1, G–J, and supplemental Videos S1–S4). Rare motile events observed in the presence of control lysates moved at an average velocity of 0.17 ± 0.01 μm/s and an average run length of 0.44 ± 0.02 μm. In the presence of WT JIP3, we observed a significant increase in the velocity (0.21 ± 0.01 μm/s) and run length (0.64 ± 0.03 μm) of motile events compared with control lysates. The velocities of KLC-mCit puncta were comparable with those reported using a similar in vitro assay (20) and similar to those measured in our dimeric kinesin assay (Fig. 1D). Importantly, we observed statistically significant differences in kinesin-1 velocity and run length in the presence of the different JIP3 constructs. JIP3ΔKLC lysate increased the velocity of KLC-mCit puncta similar to WT JIP3 lysate, whereas JIP3ΔKHC lysate did not (Fig. 1, G and I). These data indicate that JIP3 binding to KHC, but not KLC, contributes to regulation of kinesin-1 velocity. Run lengths of KLC-mCit puncta were highest in the presence of WT JIP3 lysate and were modestly lower in the presence of JIP3ΔKHC lysates (Fig. 1, H and J), indicating that the binding of JIP3 to KHC regulates the off-rate of kinesin-1. In the presence of JIP3ΔKLC lysate, we measured slightly but not significantly lower run lengths than with WT JIP3 lysate (p = 0.08), indicating that JIP3 binding to KLC might also be important for kinesin-1 off-rates. Importantly, all JIP3 constructs significantly increased run lengths compared with controls.
JIP3 Is Required for Axon Elongation
To test whether modulation of kinesin-1 motile properties by JIP3 contributes to axon elongation, we first evaluated axon morphology of cultured hippocampal neurons isolated from JIP3 knock-out (KO) (45) or littermate control embryonic day 18 embryos (Fig. 3, A and B). After 5 days in culture, neurons were fixed and stained for Tau, an axonal marker, and MAP2, which is enriched in dendrites (Fig. 3C). Given the possible function of JIP3 at the axon initial segment (43) and the role of the JIP1 in axon specification (59), we first determined whether absence or reduction of JIP3 affects axon specification by measuring the number of Tau-positive axons. No effect on the number of Tau-positive axons was detected between the groups, indicating that JIP3 is dispensable for axon specification (Fig. 3D). We next measured the length and branching of both axons and dendrites. Absence of JIP3 significantly reduced axon length (379 ± 18 μm for WT versus 322 ± 15 μm for KO) (Fig. 3E), in agreement with what was reported in cerebellar granule neurons (48). JIP3 heterozygous neurons also showed a reduction in axon length compared with WT (332 ± 13 μm). We observed no effect on axon branching (ANOVA, p = 0.23; Fig. 3F), in contrast to what has been reported in cerebellar granule and cortical neurons (48, 49). Importantly, neither length nor branching of MAP2-positive dendrites were affected by the absence of JIP3 (ANOVA, p = 0.81 and 0.86, respectively; Fig. 3, G and H), indicating that JIP3 contributes specifically to axon elongation in mouse hippocampal neurons.
FIGURE 3.

JIP3 is required for axon elongation. A, representative Western blots from JIP3 WT (+/+), heterozygous (+/−), and KO (−/−) littermate embryonic whole brain lysates. B, quantification of A. JIP3 heterozygosity results in an ∼50% reduction of JIP3 expression compared with WT. n = 5 pups in each group, litter-matched. C, Tau (axons) and MAP2 (dendrites) staining of littermate WT, heterozygous, and KO embryonic hippocampal neurons at DIV5. Most neurons produce one Tau-positive axon and multiple MAP2-enriched dendrites. Scale bar, 50 μm. D, quantification of axon number from C. Axons were defined as Tau-positive neurites. No difference in axon number was detected between any of the groups (Kruskal-Wallis, p = 0.22). D–H, n = 100 neurons/group over two replicates. E, quantification of total axon length from C. Heterozygous and KO axons are significantly shorter than WT (ANOVA, p = 0.018; *, p < 0.05; **, p < 0.01 compared with WT; Tukey's HSD). F, quantification of axon branching from C by Sholl analysis. No differences in axon branching were detected in any of the groups (ANOVA, p = 0.23). G, total dendrite length quantification of C. No differences in dendrite length were detected in any group (ANOVA, p = 0.81). H, dendrite branching quantification of C by Sholl analysis. No difference in dendrite branching was detected in any group (ANOVA, p = 0.87). n.s., not significant; Error bars, S.E.
JIP3 Binding to KHC Is Required to Promote Axon Elongation
To determine whether binding of JIP3 to KHC and KLC differentially affects axon elongation, we performed rescue experiments with JIP3 deletion mutants. GFP-tagged WT JIP3, JIP3ΔKLC, JIP3ΔKHC, and JIP3ΔΔ (Fig. 1B) were expressed in hippocampal neurons isolated from JIP3 KO embryonic day 18 embryos. At DIV5, neurons were fixed and stained for Tau and MAP2 to assess axon morphology (Fig. 4A). We note that electroporated JIP3 KO neurons consistently grew more robustly than non-electroporated neurons (compare Figs. 3E and 4C). Only neurons with GFP signal evident in the axon were selected for analysis. Both JIP3ΔKLC and JIP3ΔKHC localized to the axon and were enriched at the axon tip, similar to WT JIP3, whereas JIP3ΔΔ primarily localized to the cell body and initial portion of the axon (Fig. 4A), as reported previously (5, 50). Compared with the GFP vector control, neurons expressing WT JIP3 and JIP3ΔKLC extended longer axons, indicating that both constructs rescued the axon elongation defects (441 ± 40, 577 ± 53, and 636 ± 62 μm, respectively; Fig. 4, A and C). In contrast, expression of JIP3ΔKHC or JIP3ΔΔ failed to rescue axon length defects (498 ± 44 and 505 ± 54 μm, respectively). We observed no significant effect on axon branching (ANOVA, p = 0.35; Fig. 4D) or on the length and branching of MAP2-enriched dendrites (ANOVA, p = 0.25 and 0.33, respectively; Fig. 4. E and F). These results suggest that JIP3 binding to KHC, but not KLC, is sufficient to promote axon elongation.
FIGURE 4.

JIP3 binding to KHC is required to promote axon elongation. A, rescue of axon length defects in JIP3 KO mouse hippocampal neurons by JIP3 WT and mutants shown in Fig. 1B. Top, Tau (axons) and MAP2 (dendrites) staining of mouse JIP3 KO hippocampal neurons. Bottom, GFP signal produced by the GFP-JIP3 fusion constructs. As in Fig. 2C, most neurons extend a single axon. Arrowheads, cell bodies; arrows, tip of the longest axon. Scale bar, 50 μm. B, quantification of axon number from A. No differences in axon number were detected between groups (p = 0.15, Kruskal-Wallis). C, quantification of total axon length from A. WT JIP3 and JIP3ΔKLC rescued axon length defects of the JIP3 KO, but JIP3ΔKHC and JIP3ΔΔ did not. B–F, n = 59 (vector), 66 (WT JIP3), 71 (JIP3ΔKLC), 67 (JIP3ΔKHC), or 53 (JIP3ΔΔ) neurons from three replicates (ANOVA, p = 0.065; *, p < 0.05; **, p < 0.01 compared with vector; Tukey's HSD). D, quantification of axon branching from A by Sholl analysis. No changes in axon branching was detected among any of the groups (ANOVA, p = 0.35). E, total dendrite length quantification from A. No change in dendrite length was detected between any of the groups (ANOVA, p = 0.25). F, dendrite branching from A analyzed by Sholl analysis. No change in dendrite branching was measured between any groups (ANOVA, p = 0.34). n.s., not significant; error bars, S.E.
Deletion of the KHC Binding Domain in JIP3 Has a Dominant Negative Effect
Because JIP3 can homo-oligomerize (5, 60) we next determined whether JIP3 lacking the KHC binding domain had dominant negative effects on axon morphology in a WT background. For these experiments, we used rat hippocampal neurons and assessed axon and dendrite morphology at DIV5 with Tau/MAP2 staining as above (Fig. 5A). Only neurons with GFP signal present in the axon were analyzed. As observed in mouse hippocampal neurons, both JIP3ΔKLC and JIP3ΔKHC localized to the axon and were enriched at the axon tip, whereas JIP3ΔΔ primarily localized in a punctate pattern in the cell body and initial portion of the axon (Fig. 5A). Whereas expression of WT JIP3 and JIP3ΔKLC had no effect on axon length when compared with vector controls (655 ± 39, 626 ± 38, and 562 ± 36 μm, respectively; Fig. 5, A and C), expression of JIP3ΔKHC or JIP3ΔΔ essentially halved the total axon length compared with vector controls (347 ± 25 and 326 ± 24 μm, respectively). These results suggest that JIP3ΔKHC and JIP3ΔΔ act as dominant negative mutants. The dominant negative effect of JIP3ΔΔ on axon growth has been reported previously (50) and attributed to its inability to localize to axon tips. However, we observed that JIP3ΔKHC, which localized to the axon tip, also reduced axon elongation (Fig. 5, A and C). The dominant negative effect therefore seems attributable to lack of JIP3 interaction with KHC and not solely due to the mislocalization of JIP3. In this rat hippocampal assay, JIP3ΔKHC and JIP3ΔΔ reduced axon branching compared with vector controls (6.8 ± 0.4, 6.5 ± 0.5, and 9.8 ± 0.4 branches, respectively; Fig. 5D), suggesting that JIP3 can regulate axon branching in certain cases, as reported previously (48, 49). Importantly, dendrite length was not different between any of the groups (ANOVA, p = 0.42), and dendrite branching, although reduced by the expression of JIP3ΔKHC and JIP3ΔΔ compared with controls (4.9 ± 0.2, 4.6 ± 0.2, and 5.7 ± 0.3 branches, respectively), was affected to a much smaller degree than axon branching (Fig. 5, E and F). These data provide further evidence for the importance of JIP3 binding to KHC, but not KLC, for axon elongation and, in some cell types, branching.
FIGURE 5.

JIP3 mutants unable to bind KHC are dominant negative. Analysis of JIP3 deletion mutants shown in Fig. 1B in WT rat hippocampal neurons. A, WT rat hippocampal neurons electroporated with the indicated constructs were fixed and stained for Tau/MAP3 (axons/dendrites) at DIV5 (top). Arrowheads, cell bodies; arrows, tip of the longest axon. Scale bar, 50 μm. B, axon number was not significantly different between groups (p = 0.09, Kruskal-Wallis). C, total axon length quantification of A. Expression of JIP3ΔKHC or JIP3ΔΔ, but not WT JIP3 or JIP3ΔKLC, significantly reduced axon length compared with vector controls. B–F, n = 70 (vector), 68 (WT JIP3), 72 (JIP3ΔKLC), 73 (JIP3ΔKHC), or 66 (JIP3ΔΔ) neurons from five replicates (ANOVA, p ≪0.001; #, p ≪ 0.001 compared with vector; Tukey's HSD). D, axon branching quantification by Sholl analysis of A. Expression of JIP3ΔKHC and JIP3ΔΔ reduced axon branching, but expression of WT JIP3 or JIP3ΔKLC did not (ANOVA, p ≪ 0.001; #, p ≪ 0.001 compared with vector; Tukey's HSD). E, total dendrite length quantification of A. No difference in dendrite length was measured between any groups (ANOVA, p = 0.42). F, dendrite branching of A by Sholl analysis. Expression of JIP3ΔKHC and JIP3ΔΔ significantly reduced the number of dendrite branches but to a much smaller extent than axon branches (see Fig. 5C) (ANOVA, p = 2.0 × 10−4; **, p < 0.01; Tukey's HSD). n.s., not significant; error bars, S.E.
JIP3 Interaction with KHC Is Required for Axon Regeneration
To test if JIP3-dependent regulation of kinesin-1 contributes to axon growth following injury, we used axon regeneration assays. Axon growth after injury is distinct from axon growth during development, but both processes depend on kinesin-1-driven transport (29–31). We used embryonic DRG neurons to test for axon regeneration. This neuronal type extends only axons (61) and can be cultured in a “spot,” which leads to large numbers of radially growing axons that are amenable to measuring growth and regeneration (51). To establish a baseline for the effects of JIP3 mutants in this neuronal subtype, we used a lentiviral system to infect DRGs with GFP-tagged WT and mutant JIP3 on DIV1 and then assayed their growth on DIV3. WT JIP3 and JIP3 deletion mutants were expressed at a similar level (Fig. 6A). However, none of the JIP3 mutants had an effect on axon outgrowth during normal development of DRG neurons (Fig. 6, B and C), in contrast to our hippocampal neuron results (Figs. 3–5). This finding indicates that JIP3 function probably differs in different neuronal subtypes.
FIGURE 6.
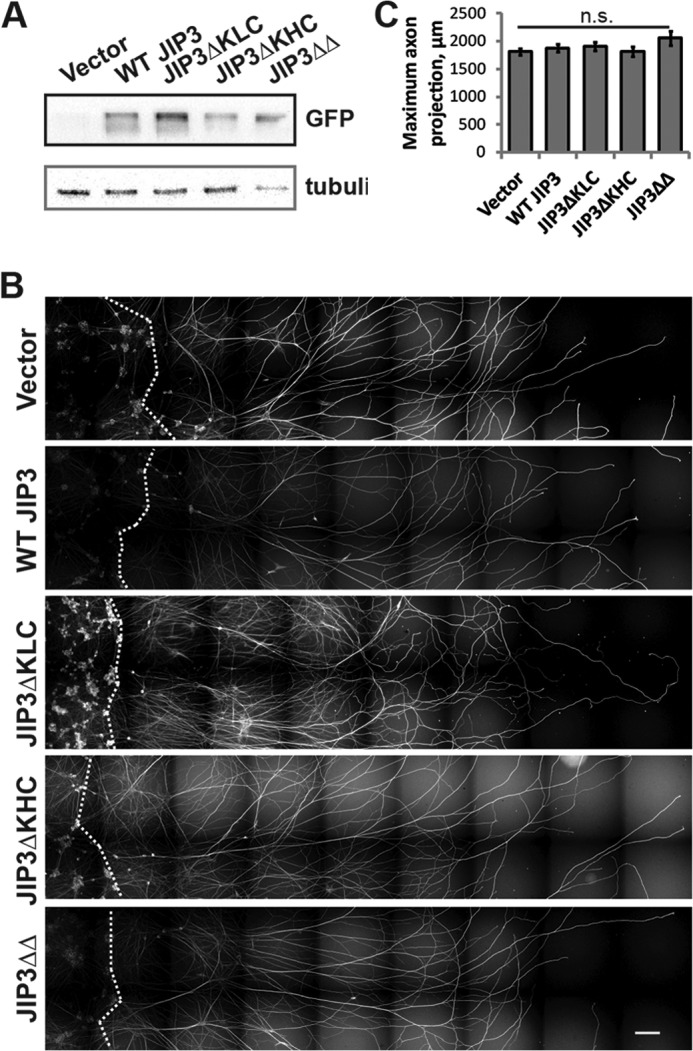
JIP3 binding to KHC is not essential for axon outgrowth in DRG neurons. A, representative Western blot of spot cultures infected with lentiviral vector, GFP-tagged WT JIP3, JIP3ΔKLC, JIP3ΔKHC, or JIP3ΔΔ. GFP was used to probe for transgenic JIP3 expression. Note that the gel used did not allow for resolution of molecular weight changes between mutants and WT. B, axon growth of DRG neurons infected at DIV1 with mutant JIP3 lentivirus and assessed for axon growth at DIV3. Dotted lines indicate the boundary of the cell body spot and the point from which axon length was assessed. Expression of JIP3 mutants did not affect growth of DRG neurons. The gridlike overlay is an artifact of stitching images together to visualize the entire spot field. C, quantification of B. No significant differences in maximum axon projection from the spot culture boundary were detected between different conditions. n = 17 (vector), 19 (WT JIP3), 24 (JIP3ΔKLC), 21 (JIP3ΔKHC), or 17 (JIP3ΔΔ) fields of view analyzed from three replicates (4 spots/group/replicate); ANOVA, p = 0.30. n.s., not significant; error bars, S.E.
Nevertheless, we tested the effect of JIP3/kinesin-1 interactions on axon regeneration, using an assay we developed previously (51). Embryonic DRG neurons were plated in a spot culture and infected with lentiviral GFP-tagged WT or mutant JIP3 at DIV3. At DIV7, axons were injured mechanically, and 12 h later, cultures were fixed and stained with the regenerating axon marker SCG10 (62). Axon regeneration was assessed by measuring the regeneration index, normalizing SCG10 intensity distal to the site of injury to SCG10 intensity proximal to the site of injury. A regeneration index of 0 would indicate a complete lack of regeneration. Vector-treated embryonic DRGs produced a regeneration index of 36 ± 4%, indicating that these neurons produce robust axon regeneration in response to injury (Fig. 7, A and B). WT JIP3 and JIP3ΔKLC produced modestly but not significantly lower regeneration indices of 28 ± 3 and 28 ± 3%, respectively, compared with vector controls. Expression of either JIP3ΔKHC or JIP3ΔΔ significantly reduced the regeneration index compared with vector controls (21 ± 2 and 22 ± 3%, respectively). Importantly, the reduction in the regeneration index was not due to mislocalization of the regeneration marker SCG10 (Fig. 7C).
FIGURE 7.

JIP3 binding to KHC is required for axon regeneration in DRG neurons. A, representative images of infected spot cultures 12 h after axon injury, stained for the regenerating axon marker SCG10. Vector-, WT JIP3-, and JIP3ΔKLC-infected neurons undergo robust regeneration after injury, which is markedly reduced with expression of JIP3ΔKHC or JIP3ΔΔ. Dashed lines, site of injury. Scale bar, 100 μm. B, quantification of A. WT JIP3 and JIP3ΔKLC slightly, but not significantly, reduce axon regeneration compared with vector controls (p = 0.077 and 0.088, respectively). In contrast, JIP3ΔKHC and JIP3ΔΔ attenuate axon regeneration after injury. n = 15 (vector), 18 (WT JIP3), 23 (JIP3ΔKLC), 30 (JIP3ΔKHC), or 31 (JIP3ΔΔ) fields of view analyzed from four replicates (4 spots/group/replicate); ANOVA, p = 0.002; **, p < 0.01; ***, p < 0.001; Tukey's HSD). C, staining of the regeneration marker SCG10 compared with total tubulin. Note that SCG10 is present in regenerating tips in all groups, indicating that transport of SCG10 was not affected by JIP3 expression. However large tubulin-positive, SCG10-negative axon debris was noted in all groups (arrowheads), indicating that SCG10 is a suitable marker for axon regeneration. Scale bar, 50 μm. Dotted line, axotomy. D, top two rows, staining of activated phosphorylated c-Jun (p-cJun) after axotomy in JIP3 mutant-expressing neurons. All groups display strong nuclear staining of phospho-c-Jun, as indicated, with the DAPI counterstain. Bottom row, uninjured controls display no phospho-c-Jun staining in DAPI-positive cell bodies. Error bars, S.E.
Because JIP3 also plays a role in retrograde injury signaling and activation of the proregenerative program (62, 63), we tested whether JIP3ΔKHC and JIP3ΔΔ reduced axon regeneration by decreasing retrograde injury signaling. We used the phosphorylation of c-Jun, a regeneration-associated protein (64), as a readout for retrograde injury signaling, as described (65). DRG neurons were cultured and infected as described above and immunostained for phosphorylated c-Jun (p-c-Jun) (Fig. 7D). Compared with uninjured controls, injury elicited a robust phospho-c-Jun staining in all cases. These results indicate that the reduced axon regeneration observed in the presence of JIP3ΔKHC or JIP3ΔΔ does not result from defects in retrograde signaling after injury and probably involves other activities that rely on the JIP3-KHC interaction.
Discussion
Here, we show that JIP3 activates tetrameric kinesin-1 in two different ways, depending on whether it binds to KLC or KHC. Specifically, interaction of JIP3 with KLC promotes binding of kinesin-1 to microtubules, whereas interaction of JIP3 with KHC promotes motility of kinesin-1 along microtubules. Although this dual action of JIP3 might be important for maximally activating kinesin-1 motility, our in vivo experiments show that JIP3 binding to KHC, but not KLC, is sufficient to rescue axon elongation defects caused by the loss of JIP3 in cultured neurons. In addition, expression of a JIP3 mutant lacking the KHC binding domain reduces axon elongation and axon regeneration. Our data reveal that regulation of kinesin-1 motility by JIP3 is critical for axon growth. Together with the recent observation that modulation of dynein's velocity affects axon elongation (66), our study underscores the importance of regulation of intracellular transport for neuronal development.
Binding of JIP3 to KLC enhanced kinesin-1 binding to microtubules in vitro, indicating that JIP3 probably relieves KLC-based inhibition of KHC-microtubule binding. This is consistent with previous findings that KLC inhibits binding of KHC to microtubules (19) and that KLC must be bound by regulatory proteins in order for kinesin-1 to efficiently dock on microtubules (20). However, the absence of JIP3-KLC binding did not have any effect on axon elongation or regeneration. One possible reason for this observation is that cargoes associated with KLC might compensate for loss of JIP3 interaction with KLC. Binding to the tetratricopeptide domain of KLC is sufficient to activate kinesin-1 transport (67), and several neuronal proteins interact with KLC via its tetratricopeptide domain, including amyloid precursor protein (68), calsyntenin (69), alcadein (25), and caytaxin (70). Alternatively, other JIPs may compensate for loss of JIP3-KLC interactions in cells. Indeed, JNK-interacting protein 1 (JIP1), a protein functionally and structurally related to JIP3, also regulates kinesin-1 microtubule binding and motility (20), regulates cortical axon development (59), and can partially rescue the axon projection defects present in JIP3 KO mice (47). Therefore, neurons seem to have multiple means of ensuring kinesin-1 docking to microtubules.
JIP3 interaction with KHC specifically promoted kinesin-1 motility in our TIRF assay. This is in agreement with reports that the tail domain of KHC autoinhibits its motility along microtubules and must be bound by regulatory proteins for efficient motility (14–17, 20). Exactly how JIP3 regulates kinesin-1 motility remains to be studied, but our stoichiometry experiments, which show that at least four JIP3 molecules bind per KHC dimer, suggest that JIP3 oligomerization may be important. Our finding that neurons cultured from heterozygous JIP3 KO mice show significant axon elongation defects also suggests that the amount of JIP3 protein is important for its function. Our experiments in neurons further indicate that JIP3 binding to KHC is a key event for axon growth and elongation.
Previously, we reported that JIP3 lacking both KHC and KLC binding domains fails to localize to axon tips (5), and subsequently this inability was posited to be responsible for the failure of JIP3ΔΔ neurons to promote axon elongation (50). However, we show that JIP3ΔKHC, which is present in axon tips, is defective for axon growth and regeneration. These results suggest that regulation of kinesin-1 motility by JIP3 in addition to its subcellular localization is important for axonal outgrowth. Our data do not rule out the possibility that the axonal growth defects of JIP3ΔKHC may be due to compromised ability to serve as a JNK scaffolding protein, although we think this is unlikely because the KHC and JNK binding domains of JIP3 do not overlap (5, 71).
We found that JIP3 promotes axon elongation in mouse hippocampal neurons but not in mouse DRG neurons, indicating that JIP3 functions differentially in different cell types. In addition, the effect of JIP3 can also vary between species because JIP3 promotes axon elongation but not axon branching in mouse hippocampal neurons, whereas it induces both axon elongation and branching in rat hippocampal neurons. Our findings are consistent with previous work, which showed that JIP3 promotes axon length in rat hippocampal neurons (50) but reduces axon length and branching in mouse cortical neurons (49) and restricts axon branching in rat cerebellar granule neurons (48). The diversity of neuronal responses to JIP3 is probably a reflection of the multiple functions of JIP3, as a JNK scaffolding protein (60, 72–75), a transport regulator (41, 42, 44), and an axon gatekeeper (43). We propose that different neuronal subtypes in different species may be selectively sensitive to one or more functional modules of JIP3.
Why does modulation of kinesin-1 motility on microtubules affect axon growth, especially axon length? A simple explanation is that delivery of trophic cargoes to the axon tip is disrupted when kinesin-1 motility is disrupted. However, our results are interesting in light of a recent model of axon length-sensing proposed by Rishal et al. (24). This model posits a frequency-based signaling module, whereby a cell senses the length of an axon based on the frequency of an oscillating signal sent anterogradely to the axon tip and returned via retrograde transport. Given that JIP3 associates with both kinesin-1 and dynein during axon growth and after injury (63, 76–78), JIP3 may be a promising lead to elucidate possible oscillating signaling modules.
Supplementary Material
Acknowledgments
We thank Chuanmei Zhu for assistance with training D. W. in the TIRF assay. We thank Domini Montgomery for assistance with the maintenance of the JIP3 knock-out line and Doug Larson for cloning of the JIP3 lentiviral constructs.
This work was supported, in whole or in part, by National Institutes of Health, NINDS, Grant R01 NS082446 (to V. C.). This work was also supported by National Science Foundation Grant MCB-1121287 (to R. D.).

This article contains supplemental Videos 1–4.
- KHC
- kinesin heavy chain(s)
- KLC
- kinesin light chain(s)
- TIRF
- total internal reflection fluorescence
- DRG
- dorsal root ganglion
- DIVn
- day in vitro n
- ANOVA
- analysis of variance
- HSD
- honest significant difference.
References
- 1. Johnson C. S., Buster D., Scholey J. M. (1990) Light chains of sea urchin kinesin identified by immunoadsorption. Cell Motil. Cytoskeleton 16, 204–213 [DOI] [PubMed] [Google Scholar]
- 2. Gindhart J. G. (1998) Kinesin light chains are essential for axonal transport in Drosophila. J. Cell Biol. 141, 443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rahman A., Kamal A., Roberts E. A., Goldstein L. S. B. (1999) Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J. Cell Biol. 146, 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moua P., Fullerton D., Serbus L. R., Warrior R., Saxton W. M. (2011) Kinesin-1 tail autoregulation and microtubule-binding regions function in saltatory transport but not ooplasmic streaming. Development 138, 1087–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun F., Zhu C., Dixit R., Cavalli V. (2011) Sunday Driver/JIP3 binds kinesin heavy chain directly and enhances its motility. EMBO J. 30, 3416–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cai Q., Gerwin C., Sheng Z.-H. (2005) Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 170, 959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skoufias D. A., Cole D. G., Wedaman K. P., Scholey J. M. (1994) The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J. Biol. Chem. 269, 1477–1485 [PubMed] [Google Scholar]
- 8. Loiseau P., Davies T., Williams L. S., Mishima M., Palacios I. M. (2010) Drosophila PAT1 is required for Kinesin-1 to transport cargo and to maximize its motility. Development 137, 2763–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanai Y., Dohmae N., Hirokawa N. (2004) Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525 [DOI] [PubMed] [Google Scholar]
- 10. Palacios I. M. (2002) Kinesin light chain-independent function of the Kinesin heavy chain in cytoplasmic streaming and posterior localisation in the Drosophila oocyte. Development 129, 5473–5485 [DOI] [PubMed] [Google Scholar]
- 11. Glater E. E., Megeath L. J., Stowers R. S., Schwarz T. L. (2006) Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 173, 545–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Su Q., Cai Q., Gerwin C., Smith C. L., Sheng Z.-H. (2004) Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat. Cell Biol. 6, 941–953 [DOI] [PubMed] [Google Scholar]
- 13. Williams L. S., Ganguly S., Loiseau P., Ng B. F., Palacios I. M. (2014) The auto-inhibitory domain and ATP-independent microtubule-binding region of Kinesin heavy chain are major functional domains for transport in the Drosophila germline. Development 141, 176–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dietrich K. A., Sindelar C. V., Brewer P. D., Downing K. H., Cremo C. R., Rice S. E. (2008) The kinesin-1 motor protein is regulated by a direct interaction of its head and tail. Proc. Natl. Acad. Sci. U.S.A. 105, 8938–8943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaan H. Y. K., Hackney D. D., Kozielski F. (2011) The structure of the kinesin-1 motor-tail complex reveals the mechanism of autoinhibition. Science 333, 883–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu J., Reddy B. J. N., Anand P., Shu Z., Cermelli S., Mattson M. K., Tripathy S. K., Hoss M. T., James N. S., King S. J., Huang L., Bardwell L., Gross S. P. (2012) Casein kinase 2 reverses tail-independent inactivation of kinesin-1. Nat. Commun. 3, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watanabe T. M., Yanagida T., Iwane A. H. (2010) Single molecular observation of self-regulated kinesin motility. Biochemistry 49, 4654–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Woźniak M. J., Allan V. J. (2006) Cargo selection by specific kinesin light chain 1 isoforms. EMBO J. 25, 5457–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wong Y. L., Rice S. E. (2010) Kinesin's light chains inhibit the head- and microtubule-binding activity of its tail. Proc. Natl. Acad. Sci. U.S.A. 107, 11781–11786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blasius T. L., Cai D., Jih G. T., Toret C. P., Verhey K. J. (2007) Two binding partners cooperate to activate the molecular motor Kinesin-1. J. Cell Biol. 176, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hammond J. W., Huang C.-F., Kaech S., Jacobson C., Banker G., Verhey K. J. (2010) Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell. 21, 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakata T., Niwa S., Okada Y., Perez F., Hirokawa N. (2011) Preferential binding of a kinesin-1 motor to GTP-tubulin-rich microtubules underlies polarized vesicle transport. J. Cell Biol. 194, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacobson C., Schnapp B., Banker G. A. (2006) A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron 49, 797–804 [DOI] [PubMed] [Google Scholar]
- 24. Rishal I., Kam N., Perry R. B. T., Shinder V., Fisher E. M. C., Schiavo G., Fainzilber M. (2012) A motor-driven mechanism for cell-length sensing. Cell Rep. 1, 608–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Araki Y., Kawano T., Taru H., Saito Y., Wada S., Miyamoto K., Kobayashi H., Ishikawa H. O., Ohsugi Y., Yamamoto T., Matsuno K., Kinjo M., Suzuki T. (2007) The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 26, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Terada S., Kinjo M., Aihara M., Takei Y., Hirokawa N. (2010) Kinesin-1/Hsc70-dependent mechanism of slow axonal transport and its relation to fast axonal transport. EMBO J. 29, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hoerndli F. J., Maxfield D. A., Brockie P. J., Mellem J. E., Jensen E., Wang R., Madsen D. M., Maricq A. V. (2013) Kinesin-1 regulates synaptic strength by mediating the delivery, removal, and redistribution of AMPA receptors. Neuron 80, 1421–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Campbell P. D., Marlow F. L. (2013) Temporal and tissue specific gene expression patterns of the zebrafish kinesin-1 heavy chain family, kif5s, during development. Gene Expr. Patterns 13, 271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goldberg D. J., Schacher S. (1987) Differential growth of the branches of a regenerating bifurcate axon is associated with differential axonal transport of organelles. Dev. Biol. 124, 35–40 [DOI] [PubMed] [Google Scholar]
- 30. Griffin J. W., Drachman D. B., Price D. L. (1976) Fast axonal transport in motor nerve regeneration. J. Neurobiol. 7, 355–370 [DOI] [PubMed] [Google Scholar]
- 31. Mar F. M., Simões A. R., Leite S., Morgado M. M., Santos T. E., Rodrigo I. S., Teixeira C. A., Misgeld T., Sousa M. M. (2014) CNS axons globally increase axonal transport after peripheral conditioning. J. Neurosci. 34, 5965–5970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Falzone T. L., Gunawardena S., McCleary D., Reis G. F., Goldstein L. S. B. (2010) Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum. Mol. Genet. 19, 4399–4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Falzone T. L., Stokin G. B., Lillo C., Rodrigues E. M., Westerman E. L., Williams D. S., Goldstein L. S. B. (2009) Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J. Neurosci. 29, 5758–5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goldstein L. S. (2001) Kinesin molecular motors: transport pathways, receptors, and human disease. Proc. Natl. Acad. Sci. U.S.A. 98, 6999–7003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kuribayashi J., Kitaoka Y., Munemasa Y., Ueno S. (2010) Kinesin-1 and degenerative changes in optic nerve axons in NMDA-induced neurotoxicity. Brain Res. 1362, 133–140 [DOI] [PubMed] [Google Scholar]
- 36. Morfini G. A., Bosco D. A., Brown H., Gatto R., Kaminska A., Song Y., Molla L., Baker L., Marangoni M. N., Berth S., Tavassoli E., Bagnato C., Tiwari A., Hayward L. J., Pigino G. F., Watterson D. M., Huang C.-F., Banker G., Brown R. H., Jr., Brady S. T. (2013) Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase. PLoS One 8, e65235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Willemsen M. H., Ba W., Wissink-Lindhout W. M., de Brouwer A. P. M., Haas S. A., Bienek M., Hu H., Vissers L. E. L. M., van Bokhoven H., Kalscheuer V., Nadif Kasri N., Kleefstra T. (2014) Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 51, 487–494 [DOI] [PubMed] [Google Scholar]
- 38. Yamada M., Toba S., Takitoh T., Yoshida Y., Mori D., Nakamura T., Iwane A. H., Yanagida T., Imai H., Yu-Lee L.-Y., Schroer T., Wynshaw-Boris A., Hirotsune S. (2010) mNUDC is required for plus-end-directed transport of cytoplasmic dynein and dynactins by kinesin-1. EMBO J. 29, 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poirier K., Lebrun N., Broix L., Tian G., Saillour Y., Boscheron C., Parrini E., Valence S., Pierre B. S., Oger M., Lacombe D., Geneviève D., Fontana E., Darra F., Cances C., Barth M., Bonneau D., Bernadina B. D., N'guyen S., Gitiaux C., Parent P., des Portes V., Pedespan J. M., Legrez V., Castelnau-Ptakine L., Nitschke P., Hieu T., Masson C., Zelenika D., Andrieux A., Francis F., Guerrini R., Cowan N. J., Bahi-Buisson N., Chelly J. (2013) Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 45, 639–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gunawardena S., Yang G., Goldstein L. S. B. (2013) Presenilin controls kinesin-1 and dynein function during APP-vesicle transport in vivo. Hum. Mol. Genet. 22, 3828–3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Byrd D. T., Kawasaki M., Walcoff M., Hisamoto N., Matsumoto K., Jin Y. (2001) UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 32, 787–800 [DOI] [PubMed] [Google Scholar]
- 42. Brown H. M., Van Epps H. A., Goncharov A., Grant B. D., Jin Y. (2009) The JIP3 scaffold protein UNC-16 regulates RAB-5 dependent membrane trafficking at C. elegans synapses. Dev. Neurobiol. 69, 174–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edwards S. L., Yu S. C., Hoover C. M., Phillips B. C., Richmond J. E., Miller K. G. (2013) An organelle gatekeeper function for Caenorhabditis elegans UNC-16 (JIP3) at the axon initial segment. Genetics 194, 143–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bowman A. B., Kamal A., Ritchings B. W., Philp A. V., McGrail M., Gindhart J. G., Goldstein L. S. (2000) Kinesin-dependent axonal transport is mediated by the Sunday driver (SYD) protein. Cell 103, 583–594 [DOI] [PubMed] [Google Scholar]
- 45. Kelkar N., Delmotte M.-H., Weston C. R., Barrett T., Sheppard B. J., Flavell R. A., Davis R. J. (2003) Morphogenesis of the telencephalic commissure requires scaffold protein JNK-interacting protein 3 (JIP3). Proc. Natl. Acad. Sci. U.S.A. 100, 9843–9848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iwanaga A., Sato T., Sugihara K., Hirao A., Takakura N., Okamoto H., Asano M., Yoshioka K. (2007) Neural-specific ablation of the scaffold protein JSAP1 in mice causes neonatal death. Neurosci. Lett. 429, 43–48 [DOI] [PubMed] [Google Scholar]
- 47. Ha H.-Y., Cho I.-H., Lee K.-W., Lee K.-W., Song J.-Y., Kim K.-S., Yu Y.-M., Lee J.-K., Song J.-S., Yang S.-D., Shin H.-S., Han P.-L. (2005) The axon guidance defect of the telencephalic commissures of the JSAP1-deficient brain was partially rescued by the transgenic expression of JIP1. Dev. Biol. 277, 184–199 [DOI] [PubMed] [Google Scholar]
- 48. Bilimoria P. M., de la Torre-Ubieta L., Ikeuchi Y., Becker E. B. E., Reiner O., Bonni A. (2010) A JIP3-regulated GSK3β/DCX signaling pathway restricts axon branching. J. Neurosci. 30, 16766–16776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suzuki A., Arikawa C., Kuwahara Y., Itoh K., Watanabe M., Watanabe H., Suzuki T., Funakoshi Y., Hasegawa H., Kanaho Y. (2010) The scaffold protein JIP3 functions as a downstream effector of the small GTPase ARF6 to regulate neurite morphogenesis of cortical neurons. FEBS Lett. 584, 2801–2806 [DOI] [PubMed] [Google Scholar]
- 50. Sun T., Yu N., Zhai L.-K., Li N., Zhang C., Zhou L., Huang Z., Jiang X.-Y., Shen Y., Chen Z.-Y. (2013) c-Jun NH2-terminal kinase (JNK)-interacting protein-3 (JIP3) regulates neuronal axon elongation in a kinesin- and JNK-dependent manner. J. Biol. Chem. 288, 14531–14543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cho Y., Cavalli V. (2012) HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 31, 3063–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fu M. M., Holzbaur E. L. F. (2013) JIP1 regulates the directionality of APP axonal transport by coordinating kinesin and dynein motors. J. Cell Biol. 202, 495–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dixit R., Ross J. L., Goldman Y. E., Holzbaur E. L. F. (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cherny D., Gooding C., Eperon G. E., Coelho M. B., Bagshaw C. R., Smith C. W. J., Eperon I. C. (2010) Stoichiometry of a regulatory splicing complex revealed by single-molecule analyses. EMBO J. 29, 2161–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gordon M. P., Ha T., Selvin P. R. (2004) Single-molecule high-resolution imaging with photobleaching. Proc. Natl. Acad. Sci. U.S.A. 101, 6462–6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang H., Guo P. (2014) Single molecule photobleaching (SMPB) technology for counting of RNA, DNA, protein and other molecules in nanoparticles and biological complexes by TIRF instrumentation. Methods 67, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coffman V. C., Wu J.-Q. (2012) Counting protein molecules using quantitative fluorescence microscopy. Trends Biochem. Sci. 37, 499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cai D., Hoppe A. D., Swanson J. A., Verhey K. J. (2007) Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J. Cell Biol. 176, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dajas-Bailador F., Jones E. V., Whitmarsh A. J. (2008) The JIP1 scaffold protein regulates axonal development in cortical neurons. Curr. Biol. 18, 221–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kelkar N., Gupta S., Dickens M., Davis R. J. (2000) Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 20, 1030–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zheng J.-Q., Kelly T. K., Chang B., Ryazantsev S., Rajasekaran A. K., Martin K. C., Twiss J. L. (2001) A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J. Neurosci. 21, 9291–9303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shin J. E., Geisler S., DiAntonio A. (2014) Dynamic regulation of SCG10 in regenerating axons after injury. Exp. Neurol. 252, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cavalli V., Kujala P., Klumperman J., Goldstein L. S. B. (2005) Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 168, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Broude E., McAtee M., Kelley M. S., Bregman B. S. (1997) c-Jun expression in adult rat dorsal root ganglion neurons: differential response after central or peripheral axotomy. Exp. Neurol. 148, 367–377 [DOI] [PubMed] [Google Scholar]
- 65. Shin J. E., Cho Y., Beirowski B., Milbrandt J., Cavalli V., DiAntonio A. (2012) Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74, 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schlager M. A., Serra-Marques A., Grigoriev I., Gumy L. F., Esteves da Silva M., Wulf P. S., Akhmanova A., Hoogenraad C. C. (2014) Bicaudal d family adaptor proteins control the velocity of Dynein-based movements. Cell Rep. 8, 1248–1256 [DOI] [PubMed] [Google Scholar]
- 67. Kawano T., Araseki M., Araki Y., Kinjo M., Yamamoto T., Suzuki T. (2012) A small peptide sequence is sufficient for initiating kinesin-1 activation through part of TPR region of KLC1. Traffic 13, 834–848 [DOI] [PubMed] [Google Scholar]
- 68. Kamal A., Stokin G. B., Yang Z., Xia C. H., Goldstein L. S. (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28, 449–459 [DOI] [PubMed] [Google Scholar]
- 69. Konecna A., Frischknecht R., Kinter J., Ludwig A., Steuble M., Meskenaite V., Indermühle M., Engel M., Cen C., Mateos J.-M., Streit P., Sonderegger P. (2006) Calsyntenin-1 docks vesicular cargo to kinesin-1. Mol. Biol. Cell. 17, 3651–3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Aoyama T., Hata S., Nakao T., Tanigawa Y., Oka C., Kawaichi M. (2009) Cayman ataxia protein caytaxin is transported by kinesin along neurites through binding to kinesin light chains. J. Cell Sci. 122, 4177–4185 [DOI] [PubMed] [Google Scholar]
- 71. Ito M., Akechi M., Hirose R., Ichimura M., Takamatsu N., Xu P., Nakabeppu Y., Tadayoshi S., Yamamoto K., Yoshioka K. (2000) Isoforms of JSAP1 scaffold protein generated through alternative splicing. Gene 255, 229–234 [DOI] [PubMed] [Google Scholar]
- 72. Ito M., Yoshioka K., Akechi M., Yamashita S., Takamatsu N., Sugiyama K., Hibi M., Nakabeppu Y., Shiba T., Yamamoto K. I. (1999) JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol. Cell. Biol. 19, 7539–7548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Matsuguchi T., Masuda A., Sugimoto K., Nagai Y., Yoshikai Y. (2003) JNK-interacting protein 3 associates with Toll-like receptor 4 and is involved in LPS-mediated JNK activation. EMBO J. 22, 4455–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Matsuura H., Nishitoh H., Takeda K., Matsuzawa A., Amagasa T., Ito M., Yoshioka K., Ichijo H. (2002) Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1-JNK signaling pathway: a new mode of regulation of the MAP kinase cascade. J. Biol. Chem. 277, 40703–40709 [DOI] [PubMed] [Google Scholar]
- 75. Takino T., Nakada M., Miyamori H., Watanabe Y., Sato T., Gantulga D., Yoshioka K., Yamada K. M., Sato H. (2005) JSAP1/JIP3 cooperates with focal adhesion kinase to regulate c-Jun N-terminal kinase and cell migration. J. Biol. Chem. 280, 37772–37781 [DOI] [PubMed] [Google Scholar]
- 76. Drerup C. M., Nechiporuk A. V. (2013) JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS Genet. 9, e1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Arimoto M., Koushika S. P., Choudhary B. C., Li C., Matsumoto K., Hisamoto N. (2011) The Caenorhabditis elegans JIP3 protein UNC-16 functions as an adaptor to link kinesin-1 with cytoplasmic dynein. J. Neurosci. 31, 2216–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Abe N., Almenar-Queralt A., Lillo C., Shen Z., Lozach J., Briggs S. P., Williams D. S., Goldstein L. S. B., Cavalli V. (2009) Sunday driver interacts with two distinct classes of axonal organelles. J. Biol. Chem. 284, 34628–34639 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.