

Background: HSV-1 inhibits T cell functions, but the underlying mechanism is unclear.
Results: The HSV-1 Us3 protein disrupts T cell signal transduction by inhibiting the ubiquitination of LAT.
Conclusion: The Us3 protein is a crucial factor to suppress T cell activation in HSV-1 infection.
Significance: Inhibition of T cell immunity may promote HSV-1 replication or persistence.
Keywords: HSV-1, TCR, LAT, signaling, Us3, TRAF6, ubiquitination, interleukin
Abstract
Herpes simplex virus 1 (HSV-1) is the most prevalent human virus and causes global morbidity because the virus is able to infect multiple cell types. Remarkably, HSV infection switches between lytic and latent cycles, where T cells play a critical role. However, the precise way of virus-host interactions is incompletely understood. Here we report that HSV-1 productively infected Jurkat T-cells and inhibited antigen-induced T cell receptor activation. We discovered that HSV-1-encoded Us3 protein interrupted TCR signaling and interleukin-2 production by inactivation of the linker for activation of T cells. This study unveils a mechanism by which HSV-1 intrudes into early events of TCR-mediated cell signaling and may provide novel insights into HSV infection, during which the virus escapes from host immune surveillance.
Introduction
Herpes simplex virus 1 (HSV-1) belongs to the Alphaherpesviridae subfamily and is a prevalent human pathogen responsible for encephalitis and localized mucocutaneous lesions such as herpes labialis, keratitis, and genital herpes (1). Upon primary infection, HSV replicates in the mucosal epithelium and, subsequently, establishes latency. Viral reactivation occurs intermittently, which induces host immune responses.
Several lines of evidence suggest that HSV-1 adopts a variety of strategies to perturb pathogen pattern recognition receptor signaling pathways. For example, HSV-1 ICP0 inhibits the production of IFN-Ι by targeting IFN regulatory factors 3 and 7 (IRF3/7) (2–4). ICP34.5 suppresses the IFN pathway via TANK-binding kinase 1 (TBK1) and inhibitor of NF-κB kinase (5–7). Us11 inhibits double-stranded RNA-dependent protein kinase, which phosphorylates eIF-2α and inactivates protein translation (8). Us3 interrupts IFN-β production by phosphorylation of IRF3 (9). Our previous studies also discovered that the virulent factor ICP34.5 binds with both protein phosphatase 1 and eIF-2α, facilitating viral protein synthesis (10, 11).
HSV-1 also has evolved to evade adaptive immune responses. Dendritic cells and monocytes are required to initiate the pathogen-specific responses by antigen presentation and providing costimulatory factors for T cell activation. It has been demonstrated that ICP47 blocks antigen presentation via MHC-I (12, 13). ICP22 and gB of HSV-1 interrupt MHC-II-dependent antigen presentation by B cells (14, 15). We also observed that ICP34.5 interferes with dendritic cell maturation, resulting in the inhibition of T cell activation (16).
The activation of T cells is triggered by the interaction of the T cell receptor (TCR)4 with antigen epitopes on MHC concurrent with the ligation of costimulatory receptors, i.e. CD28 (17). The early events involve the formation of the immune synapse, namely, a cluster of kinases, adaptor proteins, and effector molecules, followed by a sequence of signaling (18). This ultimately leads to the transcription and expression of interleukins such as IL-2. The early event in this process is the activation of Src and Syk family tyrosine kinases, including the ζ chain-associated protein kinase of 70 kDa (ZAP-70) (19). ZAP-70 phosphorylates the linker for activation of T cells (LAT), which provides docking sites for downstream signaling molecules such as phospholipase C γ1 (PLCγ1), growth factor receptor-bound protein 2 (Grb2), and Vav1 (20). Therefore, the phosphorylation of LAT is a pivotal step in T cell activation. Recently, ubiquitination of LAT by TRAF6 has been suggested to coordinate with LAT tyrosine phosphorylation and, therefore, T cell activation (21).
Upon infection, HSV-1 is detected in mitogen-stimulated T cells (22, 23) as well as clinically isolated human CD4+ and CD8+ cells (24). In vitro infection of T cells by HSV appears to be facilitated by cell-to-cell spread (25, 26). Furthermore, HSV entry is reported to attenuate T cell activation (27, 28). However, it remains unsolved whether HSV-1 has a direct impact on the intracellular events in T cell activation.
In this study, we report that HSV-1 infection significantly inhibited TCR-activated signal transduction. The Us3 protein of HSV-1 was required to block TCR-stimulated IL-2 transcription. Similar to HSV-1 infection, ectopic expression of Us3 also suppressed TCR-mediated events such as the tyrosine phosphorylation of LAT and PLCγ1 and calcium mobilization, resulting in reduced IL-2 production. Finally, we showed that Us3 interrupted TCR signaling by reducing the ubiquitination of LAT and TRAF6, leading to a suboptimal activation of LAT. These results suggest that inhibition of T cell activation may be a mechanism that favors HSV replication or persistency.
Experimental Procedures
Cell Lines and Viruses
The Jurkat T-cell line E6.1 was obtained from the ATCC and cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS and 1% (v/v) penicillin/streptomycin at 37 °C with 5% CO2. Human peripheral blood mononuclear cells were isolated from the blood of healthy donors from the Tianjin Blood Center (Tianjin, China), and primary CD3+ T cells were sorted by magnetic beads conjugated with anti-human CD3 antibody. HEK293T and Vero cells were originally from the ATCC and cultured in DMEM with 10% FBS and 1% (v/v) penicillin/streptomycin. HSV-1(F strain) was from Dr. B. He (University of Illinois at Chicago, Chicago, IL) and propagated in Vero cells as described previously (6). Us3-null HSV-1(F) was provided by Dr. C. Zheng (Soochow University, China).
Plasmid DNA Constructs
The plasmids expressing HSV-1 proteins were constructed by PCR amplification of individual HSV-1 ORFs and insertion into the pCDH-FLAG vector (System Biosciences, Mountain View, CA) at the EcoRI/NotI cloning site. All constructs were verified by sequencing, and protein expression was verified by Western blot analysis.
Antibodies and Reagents
Antibody against total HSV-1 proteins was purchased from Dako Inc. (Glostrup, Denmark). Anti-human CD3 antibody (OKT3) and anti-human CD28 antibody (CD28.2) were purchased from BioLegend (San Diego, CA) and used as agonists to stimulate T cells. Antibodies against ERK1/2 (catalog no. 9102), phospho-ERK1/2 (catalog no. 9101), ZAP-70 (catalog no. 2705), p-ZAP-70 (catalog no. 2701), p-LAT (catalog no. 3584), PLCγ1 (catalog no. 2822), p-PLCγ1 (catalog no. 2821), and ubiquitin (catalog no. 5621) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies for TRAF6 (catalog no. sc-7221), LAT (catalog no. sc-7948, and catalog no. sc-53550), goat anti-mouse IgG, and HRP-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology. The calcium-binding dyes Fluo-3 AM and Fura Red were purchased from Enzo Life Sciences (Farmingdale, NY) and Molecular Probes (Eugene, OR), respectively.
PCR Analyses
Total RNA was isolated (using TRIzol reagent, Invitrogen) from cells and reverse-transcribed into cDNA following the instructions of the manufacturer (Promega). For HSV-1 gene expression analysis, the cDNA templates were amplified by PCR with primers of ICP27, UL30, UL44, and 18S rRNA as described previously (16). For quantitation of IL-2 mRNA, real-time PCR was performed with 100 ng of RNA from each sample in the presence of SYBR Green solution by Eppendorf Mastercycler Realplex2 and quantitated by software (realplex2.2). The relative gene expression level was calculated with 18S rRNA as a reference (29). The primers for RT-PCR analysis were as follows: IL-2, 5′-CAACTGGAGCATTTACTGCTGGA-3′ (sense) and 5′-TCAGTTCTGTGGCCTTCTTGG-3′ (antisense); 18S rRNA, 5′-CCAGTAAGTGCGGGTCATAAGC-3′ (sense) and 5′-GCCTCACTAAACCATCCAATCGG-3′ (antisense).
Lentivirus-based Transduction of Jurkat T Cells
The lentiviral particles were prepared with HEK293T cells as described previously (30). Basically, 293T cells were transfected with pCDH-FLAG-based constructs encoding Us3 or TRAF6 together with pCMV-VSV-G, pMDLg/pRRE, and pRSV-REV (Dr. Rong Xiang, Nankai University, China). The viral particles were collected and incubated with Jurkat T cells at 37 °C for 24 h. Jurkat T cells stably expressing Us3 or TRAF6 were selected with 0.25 μg/ml Puromycin for 7 days.
T Cell Stimulation
Jurkat T cells were collected, washed with ice-cold PBS, and mixed with 1 μg/ml OKT3 on ice. The stimulation was initiated by adding goat anti-mouse IgG to 1 μg/ml in a 37 °C water bath and stopped with an equal volume of 2× lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm EDTA, 1.0% Triton X-100, 25 mm NaF, and 1 mm Na3VO4) supplemented with protease inhibitor mixture. The samples were then prepared for SDS-PAGE and Western blot analysis.
Intracellular Calcium Measurement
1 × 106 Jurkat T cells were loaded with Fluo-3 AM (2 μm) and Fura Red (4 μm) in 1 ml of Hanks' balanced salt solution, washed, and subjected to FACS analysis on a BD FACSCalibur with an excitation wavelength of 488 nm. For each measurement, the cells were prewarmed for 5 min at 37 °C, and a baseline was recorded before stimulation with OKT3. The intracellular free calcium concentration was presented as the intensity ratio of Fluo-3 AM to Fura Red.
Luciferase Reporter Assay
1 × 107 Jurkat T cells were transfected with 15 μg of the reporter plasmid pIL2-Luc and 1 μg of pRL-TK by electroporation with an electro-square porator (ECM830, BTX Inc., San Diego, CA) at 310 V for 10 ms. After overnight recovery, cells were aliquoted and infected with viruses before stimulation by OKT3 or OKT3 plus anti-CD28 antibody. The harvested cells were lysed, and luciferase activity was determined as instructed by the manufacturer (Promega) with the Dual-Luciferase assay kit. The reporter activity was presented as the ratio of firefly luciferase activity to Renilla luciferase activity to normalize the transfection efficiency between samples, and the values are represented as the mean ± S.D. of at least three experiments.
Measurement of IL-2 Production
Jurkat or primary T-cells were cultured in a 24-well plate precoated with anti-CD3 antibody (OKT3) or together with anti-CD28 antibody for 16 h. The culture media were then collected, and IL-2 concentration was determined by ELISA kit (catalog no. EHC003, Neobioscience Technology, China), according to the instructions of the manufacturer.
Immunoprecipitation and Western Blot Analysis
Cell lysates normalized to the total amount of protein were incubated with the indicated antibodies plus 50 μl of protein G-agarose (50% slurry) rotated end over end at 4 °C. After washing with lysis buffer four times, the immobilized proteins were resolved by SDS-PAGE, blotted to a PVDF membrane, and detected with the indicated antibodies. In protein ubiquitination experiments, the lysis buffer was supplemented with SDS to 0.1% and N-ethylmaleimide to 10 mm, respectively. The quantitation of the protein bands was obtained by densitometric analysis with Quantity One software (Bio-Rad).
Virus Growth Assay
To quantitate the virions, the infected cells were collected at the indicated hours post infection (h.p.i.), freeze-thawed three times, serially diluted in medium 199v, and then laid on Vero lawn cells. The plaques were counted, and the titer of virus was expressed as plaque-forming units per milliliter.
Statistical Analysis
The quantitative data were expressed as mean ± S.D. of three independent experiments. Data were analyzed using Student's t test. p < 0.05 was considered to be significant.
Results
HSV-1 Can Productively Infect Jurkat T Cells
It has been documented by Hammer et al. (22) and Braun et al. (23) that HSV-1 can be detected clinically in T lymphocytes. Because Jurkat T-cells are used widely to study T cell activation and signal transduction, we verified HSV replication in Jurkat T-cells. First we examined the transcription of HSV genes. As shown in Fig. 1A, viral genes representing the immediate early, early, and late kinetic classes were detected at different hours post-infection. The production of viral proteins was increased over time, as determined by Western blot analysis using antibody against total HSV-1 proteins (Fig. 1B). We further determined viral production by plaque assay, and the virus titer showed a 100-fold increase at 48 h.p.i (Fig. 1C). By flow cytometry analysis, we observed 45–50% Jurkat T cells in the lytic phase (Fig. 1D). The above data indicate that HSV-1 can directly infect and replicate in Jurkat T cells.
FIGURE 1.

Productive infection of Jurkat T-cells by HSV-1. A, Jurkat T-cells were infected with HSV-1 at 1 pfu/cell. At the indicated hours post-infection, cells were collected, and total RNA was extracted for RT-PCR amplification of viral genes: ICP27 (IE gene), UL30 (E gene), UL44 (L gene), and cellular 18S rRNA as an internal control. B, whole cell lysates were prepared for Western blot analysis using polyclonal antibody against HSV-1 or tubulin. C, virus was titrated at the indicated hours post-infection, and the growth curve was plotted with the log (10) plaque-forming units as the vertical axis and the time as the horizontal axis. Data are mean ± S.D. of three independent experiments. D, Jurkat T cells were mock-infected or infected with HSV-1 at 1 pfu/cell for 8 h and collected for flow cytometry analysis using anti-HSV-1 antibody. The plot is representative of three experiments, and the percentage of HSV-1 positive cells is expressed as mean ± S.D.
HSV-1 Suppresses TCR-induced IL-2 Production
The hallmark of T cell activation is the transcription and expression of IL-2. To determine the effect of HSV-1 infection, we used OKT3, an agonist antibody against CD3, to mimic the ligation and stimulation of TCR and measured the transcription of IL-2 by luciferase reporter assay. As shown in Fig. 2A, in mock-infected cells, the stimulated IL-2 promoter activity was 45-fold over the basal level, whereas, with HSV-1 infection, only 5-fold activation was achieved. A similar trend of effects was seen in IL-2 mRNA (Fig. 2B). Furthermore, we analyzed the secretion of IL-2 by infected cells treated with OKT3 alone (Fig. 2C, filled columns) or OKT3 plus anti-CD28 antibody, which provides the secondary signal for T cell activation (Fig. 2C, hatched columns). TCR-mediated IL-2 production was reduced at least 50% in HSV-1-infected cells compared with mock-infected cells. These observations demonstrate that HSV-1 infection negatively affects the signal transduction induced by TCR engagement.
FIGURE 2.
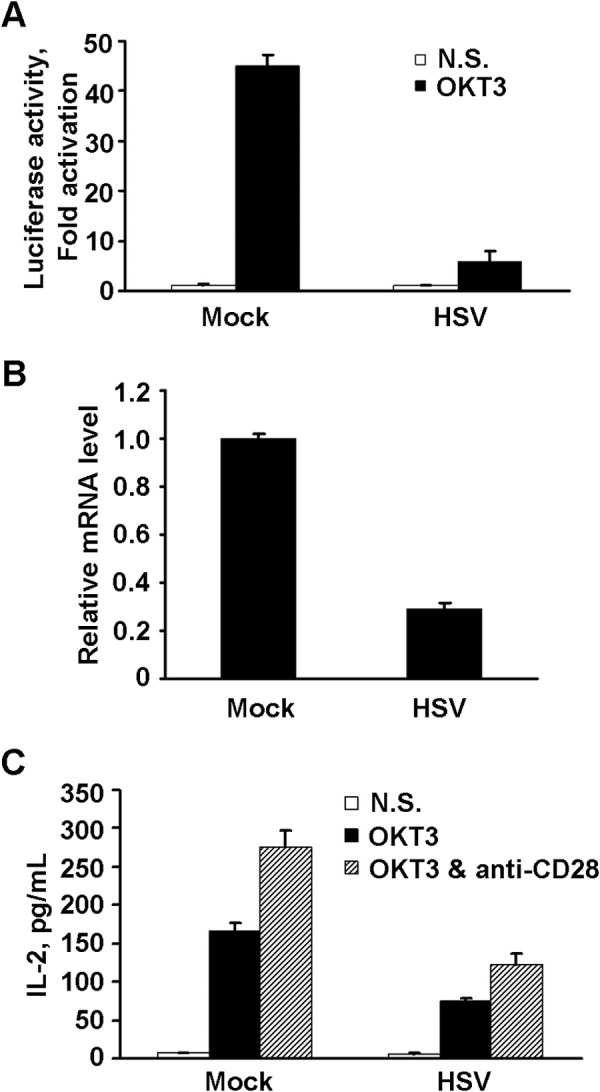
Inhibition of TCR stimulation by HSV-1. A, Jurkat T cells were transfected with pIL2-Luc and pRL-TK and mock-infected or infected with HSV (1 pfu/cell). At 8 h.p.i, cells were stimulated with anti-CD3 antibody (OKT3) or left unstimulated for an additional 4 h, followed by a luciferase assay. Data are presented as -fold of activation, and the ratio is that of OKT3-stimulated luciferase activity (closed columns) relative to an unstimulated sample (open columns). N.S., not stimulated. B, mock-infected or infected cells were collected at 8 h.p.i, and IL-2 mRNA was quantitated by real-time PCR. The mRNA level was normalized to that of 18S rRNA, and the level of IL-2 mRNA of each sample was calculated and is presented relative to the mock sample. C, the infected cells were left unstimulated (open columns) or stimulated for 24 h with OKT3 alone (closed columns) or with OKT3 plus anti-CD28 (hatched columns). The medium of each sample was collected, and IL-2 was determined by ELISA as described under “Experimental Procedures.” The above data were obtained from three independent experiments and are expressed as mean ± S.D.
The HSV-1 Tegument Protein Us3 Inhibits TCR-stimulated IL-2 Expression
The HSV-1 genome encodes 89 proteins, and several of them, including ICP34.5 and Us3, target host cellular proteins and suppress antiviral responses (31–33). To assess whether one or more HSV proteins interfere with TCR activation, we carried out reporter assays in the presence of selected viral proteins. As illustrated in Fig. 3A, the control sample (without viral protein) or cells transfected with ICP34.5, UL13, or UL51 displayed a more than 200-fold induction upon TCR stimulation (Fig. 3, A and B). In stark contrast, Us3 dramatically inhibited IL-2 promoter activation to a marginal level (Fig. 3A). The inhibitory effect of Us3 on IL-2 transcription was confirmed to be dose-dependent (Fig. 3C). Consistently, expression of Us3 effectively inhibited endogenous IL-2 secretion in Jurkat T-cells, as measured by ELISA (Fig. 3D).
FIGURE 3.

The effect of HSV-1 proteins on TCR stimulated IL-2. A, plasmids encoding the indicated viral proteins with a FLAG tag, together with pIL2-Luc and pRL-TK, were transfected into Jurkat T cells, and the cells were stimulated with OKT3 or remained non-stimulated. IL-2 promoter activity is presented as -fold of activation; namely, the ratio of the reporter in activated cells to that of non-stimulated cells. Data are mean ± S.D. (n = 3). Ctrl, control. B, the expression of viral proteins was determined by Western blot analysis with antibodies against FLAG and tubulin. C, Jurkat T cells were transfected with increasing amounts of Us3-encoding plasmid (samples 1–4 are 0, 50, 200, and 900 ng, respectively) together with the pIL2-Luc and pRL-TK reporter constructs. IL-2 promoter activity is presented as -fold of activation as described in A and is the mean ± S.D. (n = 3). D, Jurkat T cells stably expressing FLAG-Us3 fusion protein were left unstimulated (N.S., open columns) or stimulated with OKT3 plus anti-CD28 (closed columns), and the secreted IL-2 was determined and is presented as mean ± S.D. (n = 3). The inset represented the expression of Us3 protein and the internal control, GAPDH.
The Us3 Protein Is Required for HSV-1 Suppression of TCR-mediated IL-2 Production
Next we confirmed the inhibitory role of endogenous Us3 during viral infection. For this purpose, Jurkat T cells were infected with wild-type virus (HSV) or a recombinant HSV with deletion of the Us3 gene (Us3-null) (9). The cells were then treated with OKT3 and subjected to real-time PCR and ELISA analyses. As shown in Fig. 4A, wild-type HSV-1 significantly inhibited IL-2 mRNA transcription compared with mock-infected cells, whereas the Us3-null virus displayed a mild effect. A similar pattern of inhibition was seen in the secretion of IL-2 (Fig. 4B). In addition, we performed the same test on human primary T cells. As shown in Fig. 4C, TCR-mediated IL-2 production was suppressed only in cells infected with HSV-1, whereas cells infected with Us3-null virus exhibited moderate IL-2 secretion compared with that in mock-infected cells. The above data, with both the Jurkat T cell line and primary T cells, suggest that the HSV-1 Us3 protein is involved in the down-regulation of TCR-induced IL-2 expression.
FIGURE 4.
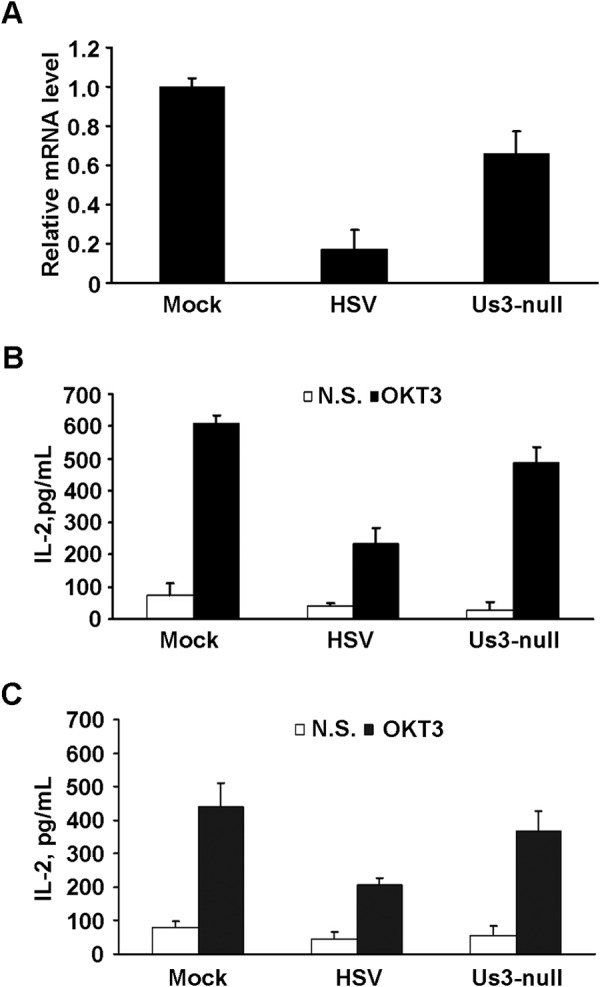
The effect of HSV-1 Us3 on T cell activation. Jurkat T cells were mock-infected or infected with wild-type or Us3-null HSV-1, followed by stimulation with OKT3. A, IL-2 mRNA was measured by real-time PCR and normalized to that of 18S rRNA. The level of IL-2 mRNA of each sample was calculated and is presented relative to the mock sample. B, the medium was collected, and secreted IL-2 was monitored by ELISA as described above. Non-stimulated sample (N.S., open columns) and OKT3-stimulated (closed columns) IL-2 measurements are presented as mean ± S.D. of three independent experiments. C, human CD3+ T cells were infected with the indicated viruses, and secretion of IL-2 was determined as described in B.
Us3 Inhibits TCR-induced Phosphorylation of LAT
The engagement of the T cell receptor evokes the proximal membrane activation of a series of tyrosine kinases (i.e. ZAP-70) and adapter proteins (i.e. LAT), which leads to the recruitment of signal molecules, including scaffold proteins and enzymes (18, 34). For example, PLCγ1 is tyrosine-phosphorylated and -activated, resulting in the production of second-messenger IP3 and enhancement of the calcium concentration. Concurrently, the PKCθ and Ras-ERK pathways are also initiated. The orchestration of these events ultimately leads to the transcription and production of IL-2 (18, 34).
To explore the mechanism involved in HSV-1 blocking of TCR-mediated signal transduction, we first examined the activation and tyrosine phosphorylation of the key proteins essential in the early events of T cell activation, including ZAP-70, LAT, PLCγ1, and ERK (Fig. 5, A and B). In comparison with mock-infected cells, HSV-1 infection had no effect on Tyr phosphorylation of ZAP-70, whereas the tyrosine phosphorylation of LAT (p-LAT) and downstream effectors PLCγ1 and ERK were apparently reduced. In contrast, infection by the Us3-null variant of HSV-1 hardly affected the tyrosine phosphorylation of those molecules, except for PLCγ1, although it was significantly higher than that of wild-type HSV-1. Besides, TCR-induced calcium mobilization was severely impaired in HSV-1-infected cells, whereas it remained intact in Us3-null HSV-infected cells (Fig. 5C). Taken together, this suggests that the viral protein Us3 plays an impeditive role in TCR signaling at its proximal adaptor, LAT.
FIGURE 5.

Inhibition of TCR-mediated signaling by HSV-1. Jurkat T cells were mock-infected or infected with HSV-1 and the recombinant Us3-null virus. A, the phosphorylation of the indicated proteins was resolved by Western blot analysis with the corresponding total proteins. B, each band was quantified by densitometer, and the -fold of activation of each protein was calculated by the ratio of each phosphorylated band density to that of the non-stimulated band. Data are mean ± S.D. (n = 3). *, p < 0.05 by Student's t test. C, the infected cells were loaded with fluorescent dyes, and intracellular calcium was monitored as described under “Experimental Procedures.” The vertical axis displays the relative free calcium concentration, and the plot represents three independent experiments.
Us3 Impairs the Ubiquitination and Activation of LAT via TRAF6
The phosphorylation of LAT is triggered by the engagement of TCR (20), whereas the simultaneous activation of an E3 ubiquitin ligase, TRAF6, has been reported to affect the ubiquitination and optimal activation of LAT (21). Because Us3 has been reported to repress TRAF6 activity in HEK293T cells (32), we hypothesize that Us3 may influence the phosphorylation of LAT via ubiquitination by TRAF6. First we examined the ubiquitination of LAT in Jurkat T cells infected by HSV-1 or the Us3-null HSV-1 variant (Fig. 6A). The ubiquitination of LAT was induced by OKT3 in mock-infected cells (Fig. 6A, lanes 2 and 5) and decreased severely in HSV-1-infected cells (Fig. 6A, lane 6). Us3-null HSV-1 had a moderate effect (Fig. 6A, lane 7). Because TRAF6 is concomitantly ubiquitinated upon TCR ligation (21), we also performed a ubiquitination analysis on TRAF6. As shown in Fig. 6B, HSV-1 exhibited remarkable inhibition compared with mock and Us-null virus (lane 6 versus lanes 5 and 7). Second, as reported previously, TRAF6 is an E3 ligase, and its activity relies on the Cys-70 amino acid residue (35). Therefore, we generated Jurkat T cells stably expressing wild-type TRAF6 (T6) and a dominant inactive mutation of TRAF6 (T6(C70A)) and measured TCR-induced p-LAT. As shown in Fig. 6, C and D, HSV-1-suppressed p-LAT (Fig. 6C, lane 2) was restored by the overexpression of wild-type TRAF6 (Fig. 6C, lane 5) but not the C70A mutant (Fig. 6C, lane 8). Taking the mock infection as a reference, HSV-1 infection (Fig. 6D, open columns) resulted in a dramatically inhibited level of p-LAT (Fig. 6D, Vector) that was changed to more than 80% in the presence of overexpressed TRAF6 (Fig. 6D, T6). In contrast, cells transduced with the inactive form of TRAF6 C70A exhibited down-regulated p-LAT (Fig. 6D, T6(C70A)). As a comparison, the Us3-null virus infection scarcely impacted p-LAT regardless of the expression of wild-type or C70A TRAF6 (Fig. 6D, closed columns). The above data imply that HSV-1 hindering TCR-mediated signaling requires the E3 ligase activity of TRAF6.
FIGURE 6.

HSV-1 inhibition of LAT by blocking the binding of TRAF6. Jurkat T-cells were mock-infected or infected with HSV-1 or Us3-null virus and treated with (+) or without (−) OKT3 as indicated. The cell lysates were subjected to immunoprecipitation (IP) using anti-LAT antibody (A and F) or anti-TRAF6 antibody (B). The immobilized proteins were resolved by Western blot analysis with the indicated antibodies. The cognate IgG was used as a negative control (lane 1 in A, B, and F). The expression of those proteins in whole cell extract (WCE) was monitored (bottom panels). C, Jurkat T cells transduced lentivirally with vector, wild-type TRAF6 (T6), or inactive mutant TRAF6 (T6 C70A) were infected as indicated and stimulated with OKT3. p-LAT, total LAT, TRAF6, and tubulin were analyzed by Western blot and quantitated by densitometry. D, the relative activation of LAT is presented by the ratio of infected samples to that of the mock-infected sample in the same transduction group. E, HEK293T cells were transiently transfected with the indicated plasmids, and cell extracts normalized to an equal amount of protein were applied to immunoprecipitation by anti-Myc antibody. The immobilized proteins (top panels) and whole cell extracts (bottom panels) were subjected to SDS-PAGE and Western blot analysis. F, Jurkat T cells were infected as indicated and stimulated with (+) or without (−) OKT3. Cell lysates were subjected to immunoprecipitation with anti-LAT antibody and Western blot analysis with the indicated antibodies.
Previous report have suggested that LAT interacts with TRAF6 during T cell activation and that, together with the ubiquitination of LAT, it contributes to the optimal activation of LAT (21). Therefore, we examined the interaction between Us3, TRAF6, and LAT in HEK293T cells by coimmunoprecipitation. As shown in Fig. 6E, lane 2, TRAF6 associated with LAT. However, the presence of Us3 or its kinase-dead mutant K220M abolished the interaction between LAT and TRAF6 (Fig. 6E, lanes 3 and 4, respectively). Intriguingly, Us3 and the mutant K220M were coimmobilized with LAT (Fig. 6E, lanes 3 and 4), implying that the LAT-TRAF6 interaction was interrupted by Us3 independent of its kinase activity. The finding was further confirmed endogenously in Jurkat T cells. As shown in Fig. 6F, TRAF6 appeared in coimmunoprecipitation fraction with LAT in the absence of Us3 (lanes 5 and 7) but was not detectable in HSV-1-infected cells (lane 6). Taken together, our data strongly suggest that Us3 hijacks TCR signaling at a pivotal effector, LAT, by blocking the recruitment and binding of TRAF6, and that results in impaired activation of LAT (Fig. 7).
FIGURE 7.

Proposed model for the inhibition of TCR signaling by HSV-1. In quiescent T cells, the signal molecules stay in their inactive form (a) until the TCR is engaged by antigen or the agonist antibody OKT3 (in context) (b). b, LAT interacts with and recruits TRAF6. The latter is required for the ubiquitination and optimal tyrosine phosphorylation of LAT. Upon HSV-1 infection (c), the viral protein Us3 associates with LAT and interferes with the interaction between LAT and TRAF6. Therefore, the ubiquitination and phosphorylation of LAT are impaired so that TCR-mediated signal transduction and IL-2 production are blocked.
Discussion
The Us3 protein of HSV-1 is a multifunctional protein that plays a crucial role in viral virulence. Several lines of evidence indicate that Us3 participates in a variety of processes, such as promoting viral egress (36, 37), enhancing viral protein synthesis (38), preventing apoptosis (39–41), regulating the epigenetic modification of chromosomes (42, 43), and suppressing innate immunity (9, 44). In this study, we showed that Us3 negatively regulated TCR-mediated signaling, resulting in diminished calcium mobilization and IL-2 secretion. This involved a functional interaction of Us3, the TRAF6 ubiquitin ligase, and LAT. Therefore, these results reveal a previously unrecognized mechanism whereby HSV profoundly interferes with T cell activation.
HSV infects CD4+ as well as CD8+ T cells in human diseases (24). Previous studies have suggest that T cells infection by HSV-1 occurs efficiently through cell-to-cell spread that relies on the virological synapse (24). Coincubation of T cells with HSV-infected fibroblast cells impairs cytokine production and cytotoxic T cell function (25–27, 45). A proposed model suggests that Us3 in HSV-infected cells transmits an inhibitory signal to uninfected T cells, leading to T cell dysfunction (27). Nevertheless, Us3 has also been reported to prevent apoptosis induced by cytotoxic T cells via granzyme B (46). The fact that Us3 inhibits T cell activation suggests an additional layer of HSV mechanisms. We speculate that perturbation of TCR signal transduction by Us3 may lead to aberrant or defective adaptive immunity.
As a membrane-associated adaptor, LAT plays a central role in T cell activation (20). When phosphorylated on its cytoplasmic domain, LAT forms a complex with multiple proteins, which activates downstream effectors (47). Intriguingly, infection with wild-type virus, but not the Us3-null mutant, precluded the phosphorylation of LAT upon TCR stimulation (Fig. 5A). Consistently, this paralleled with the phosphorylation status of PLCγ1 and ERK1/2. However, such an effect was not detectable for ZAP-70. These results support the notion that, when expressed in T cells, Us3 functions at a site proximal to LAT, which may result in the hypophosphorylation of LAT.
It has been reported recently that, besides phosphorylation, LAT is subjected to ubiquitination by TRAF6, an essential ubiquitin E3 ligase, upon TCR stimulation (21). This process relies on the association of TRAF6 with LAT via its TRAF domain. Interpreted within this framework, it is noteworthy that the wild-type virus abolished the ubiquitination of TRAF6 and LAT, whereas the Us3-null mutant reversed the phenotypes (Fig. 6). Overexpression of TRAF6 restored LAT phosphorylation upon HSV infection. On the other hand, when expressed ectopically, Us3 abolished the LAT-TRAF6 interaction by binding to LAT. A simple explanation to reconcile these observations is that Us3 may compete with TRAF6 for LAT binding. Accordingly, the Us3-LAT interaction generates a structural hindrance to TRAF6. Alternatively, by docking to LAT, Us3 may phosphorylate TRAF6, which inactivates TRAF6 and its interaction with LAT. Notably, the interaction between Us3 and TRAF6 was detected negatively by Knipe and co-workers (32) and our group. These models are not mutually exclusive. Additional work is needed to test these possibilities.
In summary, we uncover that the Us3 protein of HSV-1 negatively modulates T cell signal transduction through the linker for activation of T cells. As a result of this interaction, HSV-1 infection suppresses cytokine production and calcium mobilization and subsequently impairs T cell function. Therefore, our results reveal a novel HSV mechanism that may contribute to viral evasion of antiviral immunity-mediated T cell activation.
Acknowledgments
We thank Prof. Chunfu Zheng (Soochow University, China) for the Us3-null virus and valuable discussions. We also thank Dr. Yingchi Zhang (Institute of Hematology and Blood Diseases Hospital, Peking Union College, China) for help with separating human CD3+ T cells.
This work was supported by National Natural Science Foundation of China Grants 81171556 and 31370862 (to Y. C.) and Ministry of Science and Technology of China Grant 2012CB917204 (to Y. C.). This work was also supported by Program of Introducing Talents of Discipline to Universities B08011.
- TCR
- T cell receptor
- LAT
- linker for activation of T cells
- h.p.i.
- hours post-infection.
References
- 1. Whitley R. J., Roizman B. (2001) Herpes simplex virus infections. Lancet 357, 1513–1518 [DOI] [PubMed] [Google Scholar]
- 2. Melroe G. T., DeLuca N. A., Knipe D. M. (2004) Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 78, 8411–8420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin R., Noyce R. S., Collins S. E., Everett R. D., Mossman K. L. (2004) The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78, 1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taylor K. E., Chew M. V., Ashkar A. A., Mossman K. L. (2014) Novel roles of cytoplasmic ICP0: proteasome-independent functions of the RING finger are required to block interferon-stimulated gene production but not to promote viral replication. J. Virol. 88, 8091–8101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ma Y., Jin H., Valyi-Nagy T., Cao Y., Yan Z., He B. (2012) Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J. Virol. 86, 2188–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verpooten D., Ma Y., Hou S., Yan Z., He B. (2009) Control of TANK-binding kinase 1-mediated signaling by the γ(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 284, 1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin H., Yan Z., Ma Y., Cao Y., He B. (2011) A herpesvirus virulence factor inhibits dendritic cell maturation through protein phosphatase 1 and IκB kinase. J. Virol. 85, 3397–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poppers J., Mulvey M., Khoo D., Mohr I. (2000) Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 74, 11215–11221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang S., Wang K., Lin R., Zheng C. (2013) Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits β interferon production. J. Virol. 87, 12814–12827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Y., Zhang C., Chen X., Yu J., Wang Y., Yang Y., Du M., Jin H., Ma Y., He B., Cao Y. (2011) ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2α (eIF2α) and protein phosphatase 1. J. Biol. Chem. 286, 24785–24792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He B., Gross M., Roizman B. (1998) The γ134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 273, 20737–20743 [DOI] [PubMed] [Google Scholar]
- 12. York I. A., Roop C., Andrews D. W., Riddell S. R., Graham F. L., Johnson D. C. (1994) A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 77, 525–535 [DOI] [PubMed] [Google Scholar]
- 13. Hill A., Jugovic P., York I., Russ G., Bennink J., Yewdell J., Ploegh H., Johnson D. (1995) Herpes simplex virus turns off the TAP to evade host immunity. Nature 375, 411–415 [DOI] [PubMed] [Google Scholar]
- 14. Barcy S., Corey L. (2001) Herpes simplex inhibits the capacity of lymphoblastoid B cell lines to stimulate CD4+ T cells. J. Immunol. 166, 6242–6249 [DOI] [PubMed] [Google Scholar]
- 15. Neumann J., Eis-Hübinger A. M., Koch N. (2003) Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J. Immunol. 171, 3075–3083 [DOI] [PubMed] [Google Scholar]
- 16. Jin H., Ma Y., Prabhakar B. S., Feng Z., Valyi-Nagy T., Yan Z., Verpooten D., Zhang C., Cao Y., He B. (2009) The γ 1 34.5 protein of herpes simplex virus 1 is required to interfere with dendritic cell maturation during productive infection. J. Virol. 83, 4984–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nel A. E. (2002) T-cell activation through the antigen receptor: Part 1: signaling components, signaling pathways, and signal integration at the T-cell antigen receptor synapse. J. Allergy Clin. Immunol. 109, 758–770 [DOI] [PubMed] [Google Scholar]
- 18. Samelson L. E. (2002) Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu. Rev. Immunol. 20, 371–394 [DOI] [PubMed] [Google Scholar]
- 19. Chan A. C., Dalton M., Johnson R., Kong G. H., Wang T., Thoma R., Kurosaki T. (1995) Activation of ZAP-70 kinase activity by phosphorylation of tyrosine 493 is required for lymphocyte antigen receptor function. EMBO J. 14, 2499–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang W., Sloan-Lancaster J., Kitchen J., Trible R. P., Samelson L. E. (1998) LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92, 83–92 [DOI] [PubMed] [Google Scholar]
- 21. Xie J. J., Liang J. Q., Diao L. H., Altman A., Li Y. (2013) TNFR-associated factor 6 regulates TCR signaling via interaction with and modification of LAT adapter. J. Immunol. 190, 4027–4036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hammer S. M., Carney W. P., Iacoviello V. R., Lowe B. R., Hirsch M. S. (1982) Herpes simplex virus infection of human T-cell subpopulations. Infect. Immun. 38, 795–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Braun R. W., Teute H. K., Kirchner H., Munk K. (1984) Replication of herpes simplex virus in human T lymphocytes: characterization of the viral target cell. J. Immunol. 132, 914–919 [PubMed] [Google Scholar]
- 24. Aubert M., Yoon M., Sloan D. D., Spear P. G., Jerome K. R. (2009) The virological synapse facilitates herpes simplex virus entry into T cells. J. Virol. 83, 6171–6183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Posavad C. M., Newton J. J., Rosenthal K. L. (1993) Inhibition of human CTL-mediated lysis by fibroblasts infected with herpes simplex virus. J. Immunol. 151, 4865–4873 [PubMed] [Google Scholar]
- 26. Posavad C. M., Newton J. J., Rosenthal K. L. (1994) Infection and inhibition of human cytotoxic T lymphocytes by herpes simplex virus. J. Virol. 68, 4072–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sloan D. D., Zahariadis G., Posavad C. M., Pate N. T., Kussick S. J., Jerome K. R. (2003) CTL are inactivated by herpes simplex virus-infected cells expressing a viral protein kinase. J. Immunol. 171, 6733–6741 [DOI] [PubMed] [Google Scholar]
- 28. Sloan D. D., Han J. Y., Sandifer T. K., Stewart M., Hinz A. J., Yoon M., Johnson D. C., Spear P. G., Jerome K. R. (2006) Inhibition of TCR signaling by herpes simplex virus. J. Immunol. 176, 1825–1833 [DOI] [PubMed] [Google Scholar]
- 29. Giulietti A., Overbergh L., Valckx D., Decallonne B., Bouillon R., Mathieu C. (2001) An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods 25, 386–401 [DOI] [PubMed] [Google Scholar]
- 30. Wang Y., Yang Y., Wu S., Pan S., Zhou C., Ma Y., Ru Y., Dong S., He B., Zhang C., Cao Y. (2014) p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J. Biol. Chem. 289, 35795–35805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He B., Gross M., Roizman B. (1997) The γ(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A. 94, 843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sen J., Liu X., Roller R., Knipe D. M. (2013) Herpes simplex virus US3 tegument protein inhibits Toll-like receptor 2 signaling at or before TRAF6 ubiquitination. Virology 439, 65–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang K., Ni L., Wang S., Zheng C. (2014) Herpes simplex virus 1 protein kinase US3 hyperphosphorylates p65/RelA and dampens NF-κB activation. J. Virol. 88, 7941–7951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang Y., Wange R. L. (2004) T cell receptor signaling: beyond complex complexes. J. Biol. Chem. 279, 28827–28830 [DOI] [PubMed] [Google Scholar]
- 35. Lamothe B., Besse A., Campos A. D., Webster W. K., Wu H., Darnay B. G. (2007) Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of IκB kinase activation. J. Biol. Chem. 282, 4102–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reynolds A. E., Wills E. G., Roller R. J., Ryckman B. J., Baines J. D. (2002) Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76, 8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Imai T., Arii J., Minowa A., Kakimoto A., Koyanagi N., Kato A., Kawaguchi Y. (2011) Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B. J. Virol. 85, 5003–5015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chuluunbaatar U., Roller R., Feldman M. E., Brown S., Shokat K. M., Mohr I. (2010) Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 24, 2627–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leopardi R., Van Sant C., Roizman B. (1997) The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. U.S.A. 94, 7891–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hata S., Koyama A. H., Shiota H., Adachi A., Goshima F., Nishiyama Y. (1999) Antiapoptotic activity of herpes simplex virus type 2: the role of US3 protein kinase gene. Microbes Infect. 1, 601–607 [DOI] [PubMed] [Google Scholar]
- 41. Ogg P. D., McDonell P. J., Ryckman B. J., Knudson C. M., Roller R. J. (2004) The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology 319, 212–224 [DOI] [PubMed] [Google Scholar]
- 42. Poon A. P., Gu H., Roizman B. (2006) ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 9993–9998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walters M. S., Kinchington P. R., Banfield B. W., Silverstein S. (2010) Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J. Virol. 84, 9666–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Piroozmand A., Koyama A. H., Shimada Y., Fujita M., Arakawa T., Adachi A. (2004) Role of Us3 gene of herpes simplex virus type 1 for resistance to interferon. Int. J. Mol. Med. 14, 641–645 [PubMed] [Google Scholar]
- 45. Posavad C. M., Rosenthal K. L. (1992) Herpes simplex virus-infected human fibroblasts are resistant to and inhibit cytotoxic T-lymphocyte activity. J. Virol. 66, 6264–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jerome K. R., Fox R., Chen Z., Sears A. E., Lee H., Corey L. (1999) Herpes simplex virus inhibits apoptosis through the action of two genes, Us5 and Us3. J. Virol. 73, 8950–8957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Finco T. S., Kadlecek T., Zhang W., Samelson L. E., Weiss A. (1998) LAT is required for TCR-mediated activation of PLCγ1 and the Ras pathway. Immunity 9, 617–626 [DOI] [PubMed] [Google Scholar]