

Background: BCL-3 is an essential negative regulator of inflammation.
Results: A peptide derived from the ankyrin repeat 1 domain of BCL-3 has anti-inflammatory properties.
Conclusion: The interaction of a short region of BCL-3 with p50 has significant functional consequences on inflammatory gene expression.
Significance: Mimicking BCL-3 function has therapeutic potential.
Keywords: inflammation, NF-κB, peptide array, peptides, transcription, BCL-3
Abstract
The NF-κB transcriptional response is tightly regulated by a number of processes including the phosphorylation, ubiquitination, and subsequent proteasomal degradation of NF-κB subunits. The IκB family protein BCL-3 stabilizes a NF-κB p50 homodimer·DNA complex through inhibition of p50 ubiquitination. This complex inhibits the binding of the transcriptionally active NF-κB subunits p65 and c-Rel on the promoters of NF-κB target genes and functions to suppress inflammatory gene expression. We have previously shown that the direct interaction between p50 and BCL-3 is required for BCL-3-mediated inhibition of pro-inflammatory gene expression. In this study we have used immobilized peptide array technology to define regions of BCl-3 that mediate interaction with p50 homodimers. Our data show that BCL-3 makes extensive contacts with p50 homodimers and in particular with ankyrin repeats (ANK) 1, 6, and 7, and the N-terminal region of Bcl-3. Using these data we have designed a BCL-3 mimetic peptide based on a region of the ANK1 of BCL-3 that interacts with p50 and shares low sequence similarity with other IκB proteins. When fused to a cargo carrying peptide sequence this BCL-3-derived peptide, but not a mutated peptide, inhibited Toll-like receptor-induced cytokine expression in vitro. The BCL-3 mimetic peptide was also effective in preventing inflammation in vivo in the carrageenan-induced paw edema mouse model. This study demonstrates that therapeutic strategies aimed at mimicking the functional activity of BCL-3 may be effective in the treatment of inflammatory disease.
Introduction
The NF-κB transcription factor is a critical factor for the normal development and homeostasis of the immune system and is essential for the inflammatory response (1). NF-κB controls the expression of hundreds of genes that encode pro-inflammatory effectors such as cytokines and chemokines, proteins involved in antigen presentation, and regulators of cell death and proliferation. Mice lacking specific NF-κB subunits or components of the NF-κB activation pathway are immunodefective and fail to develop the appropriate immunity to infection and have aberrant inflammatory responses (2). NF-κB is in fact a family of five related factors: p65/RelA, c-Rel, RelB, p50, and p52, all of which possess a Rel homology domain, which mediates subunit dimerization and DNA binding. Each subunit is capable of homodimerization or heterodimerization to yield a theoretical 15 possible dimers, although only 12 specific dimers have so far been demonstrated to exist in cells (3). The p50 and p52 subunits are generated from the limited proteasomal processing of the larger precursor proteins p105 and p100, respectively. Importantly both p50 and p52 lack a transactivation domain and when present as homodimers lack intrinsic transactivation activity (4).
The primary point of control of NF-κB transcriptional activity is the sequestration of NF-κB dimers in the cytoplasm by members of the IκB family of regulatory proteins, of which IκBα is the archetypal member. Upon receipt of a NF-κB activating stimulus, the inhibitor of κB kinase (IKK)3 complex, composed of the kinases IKKα and IKKβ, and the scaffold protein NEMO, phosphorylates IκBα, triggering its ubiquitination and subsequent proteasomal degradation. Free NF-κB dimers then translocate to the nucleus where they bind cognate sites on DNA to regulate the transcription of target genes (4). More recently a number of studies have identified critical regulatory mechanisms in the nucleus that control NF-κB transcriptional activity. Most prominent among these is the regulation of NF-κB stability by the ubiquitin-proteasomal system (5). The ubiquitination of NF-κB is triggered by DNA binding and is also regulated by phosphorylation (6, 7). Ubiquitin-mediated proteasomal degradation of the NF-κB subunit p65/RelA is a major limiting factor in the transcription of target genes (7).
The stability of one NF-κB dimer can also profoundly affect the transcriptional activity of other NF-κB dimers. The IκB protein BCL-3 inhibits the ubiquitination and subsequent proteasomal degradation of p50 homodimers to limit the expression of pro-inflammatory cytokines following activation of Toll-like receptors (8). BCL-3 is an atypical IκB protein that, in contrast to IκBα, is predominantly nuclear in localization and is not degraded following activation of the IKK complex (8). BCL-3-stabilized p50 homodimers form a stable repressor complex at NF-κB binding sites that competes with transcriptionally active NF-κB dimers composed of p65 or c-Rel to inhibit target gene expression. BCL-3 is important for establishing TLR tolerance, a state of altered responsiveness to Toll-like receptor stimulation in macrophage characterized by a block in pro-inflammatory cytokine expression (8). Mice deficient in Bcl3 lack Toll-like receptor tolerance (8), fail to clear infection (9), are more sensitive to the development of type I diabetes (10), and undergo increased granulopoiesis under inflammatory conditions (11). More recently BCL-3 has been identified as an important enforcer of T cell differentiation states (12) and a key factor in promoting dendritic cell priming of T cells (13). Thus BCL-3 is an important regulator of inflammation and immune responses.
We recently employed peptide array techniques to identify critical amino acids of p50, which mediate the interaction with BCL-3 (14). In this study we employ similar techniques to identify residues of BCL-3, which mediate interaction with p50. This approach confirmed previously indicated sites of interaction from in silico modeling studies (15, 16) but also identified additional sites of interaction not predicted by computational methods. Our findings also provide important information on the recognition of NF-κB dimers by IκB proteins. We used the results of our peptide array analysis to generate a short peptide of BCL-3, which mimicked the inhibitory effect of full-length BCL-3 on NF-κB in vitro. Moreover, the BCL-3 mimetic peptide significantly inhibited carrageenan-induced paw inflammation in mice. Our study demonstrates that mimicking BCL-3 function may represent an effective strategy for the inhibition of inflammation.
Experimental Procedures
Cell Culture, Plasmids, and Transfection
HEK293T, RAW 264.7, and HeLa cells were cultured in DMEM containing 10% fetal bovine serum, 2 mm glutamine, and 100 units/ml of penicillin/streptomycin. Transfections were performed using the in vitro transfection reagent Turbofect according to the manufacturer's instructions. Mammalian expression vectors for BCL-3 and p50 were as previously described (8). p50 was subcloned into the pGEX6p1 vector to produce a bacterial GST-p50 expression vector.
Spot Synthesis of Peptides and Overlay Analysis
BCL-3 peptide arrays were generated by automatic SPOT synthesis as previously described (17). Essentially, a library of overlapping peptides each shifted by 4 amino acids, encompassing the entire murine BCL-3 protein sequence was synthesized on Whatman 50 cellulose supports, using Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry and the AutoSpot-Robot ASS 222 (Intavis Bioanalytical Instruments). BCL-3 peptide arrays were activated by incubation in 100% ethanol for 5 min followed by equilibration in TBS-T for 10 min. Arrays were blocked in a 5% nonfat milk/TBS, 0.05% Tween 20 solution for 1 h at room temperature. To investigate the interaction of GST or GST-p50 with the BCL-3 peptides, arrays were incubated overnight with 10 μg/ml of recombinant protein in a 1% nonfat milk/TBS-T solution. Bound protein was detected by immunoblotting with anti-GST and a secondary anti-rabbit antibody coupled with horseradish peroxidase.
Recombinant Protein Expression and Purification
For purification of GST and GST-p50, Escherichia coli BL21 CodonPlus (Stratagene) were transformed with pGEX-6p1 or pGEX-6p1-p50. Bacteria were incubated in 700 ml of LB at 37 °C with agitation (220–240 rpm) until an A600 of 1.0–2.0 was reached before induction with 1.0 mm isopropyl 3-d-thiogalactopyranoside. Bacteria were grown for a further 16 h at 20 °C with agitation (220–240 rpm) to induce GST protein expression. Bacteria were pelleted at 4 °C and pellets were frozen at −20 °C to aid lysis. Pellets were thawed on ice and resuspended in 70 ml of cold lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, and 1 mm dithiothreitol) supplemented with two complete, mini, EDTA-free protease inhibitor mixture tablets (Roche Applied Science). Pellets were disrupted by sonication on ice for 30 min with 5-min pulses and 5 min rest between each pulse. Lysates were cleared by centrifugation at 15,000 × g for 30 min at 4 °C. Recombinant proteins were affinity purified at 4 °C against GSH-agarose (Sigma), following a 1-h incubation with the cleared lysate, GSH-agarose was washed with 2 liters of wash buffer (pH 7.5) overnight at 4 °C and recombinant proteins were eluted with 10 mm glutathione in 50 mm Tris (pH 8.5) and 150 mm NaCl.
GST Pulldown Assay
HEK293T cells were transiently transfected with FLAG-BCL-3. Whole cell extracts were prepared from cells resuspended in GST lysis and binding buffer (20 mm Tris-Cl, pH 8.0, 200 mm NaCl, 1 mm EDTA, pH 8.0, 0.5% Nonidet P-40) supplemented with 2 μg/ml of aprotinin, 1 μg/ml of pepstatin, and 1 mm PMSF. Equal concentrations of lysates were precleared in 1 ml of binding buffer with 50 μl of GSH-agarose for 2 h rotating at 4 °C. GST or GST-p50 were incubated with precleared lysates and affinity purified with 50 μl of GSH-agarose for 2 h rotating at 4 °C. Following three washes in 1 ml of binding buffer, pulldown complexes were eluted from the GSH-agarose by boiling at 95 °C in 2× SDS gel loading buffer (100 mm Tris-Cl, pH 6.8, 4%(w/v) SDS, 0.2% (w/v) bromphenol blue, 20%(w/v) glycerol, and 200 mm β-mercaptoethanol) and analyzed by Western blot. Anti-FLAG M2, anti-GST, and anti-β-actin were purchased from Sigma.
BCL-3 Peptide Synthesis
The sequences of the Bcl-3 mimetic peptides were as follows: BDP1, YGRKKRRQRRHIAVVQNNIAAVYRILSLFKLGSREVDVHN; BDP2, YGRKKRRQRRAAVYRILSLFKLGSR; mBDP2, YGRKKRRQRRWAWGYILSLDCLGSY. BDP1 with an N-terminal FITC label was synthesized by GL Biochem Ltd. Shangai, BDP2 and mBDP2 were synthesized by Genscript.
Luciferase Assay
RAW 264.7 cells were transiently transfected using Attractene (Qiagen) with an IL23p19 promoter reporter plasmid and the Renilla luciferase expression vector pRL-TK (Promega) for 24 h and cultured with or without 100 ng/ml of LPS (Sigma) for an additional 8 h as previously described (16). Cells were lysed in 1× passive lysis buffer (Promega) and for all samples the firefly luciferase activity (25 mm glycylglycine, 15 mm potassium diphosphate/K2PO4, pH 8, 4 mm EDTA, 15 mm magnesium sulfate, 75 μm luciferin, 1 mm dithiothreitol, 0.1 mm coenzyme A, 2 mm ATP) was divided by that of the Renilla luciferase activity (1.1 m sodium chloride, 2.2 mm EDTA, 220 mm dipotassium phosphate/K2PO4, pH 5.1, 0.44 mg/ml of bovine serum albumin, 1.3 mm sodium azide, 1.43 μm coelentrerazine) to normalize for the transfection efficiency as previously described (18).
ELISA
5 × 104 RAW 264.7 cells were plated per well of a 96-well plate overnight. Cells were pretreated with peptide 2 h before stimulation with 100 ng/ml of LPS and supernatants were harvested after 4 h. The concentration of soluble TNF-α (pg/ml) in the culture medium was determined using the mouse TNF (Mono/Mono) ELISA set (BD Biosciences) and tetramethylbenzidine was purchased from Thermo Fisher.
Gene Expression Analysis
For real-time PCR, total RNA was isolated using RNeasy kits (Qiagen) and reverse transcribed using the NanoScript reverse transcription kit (Primer Design). Real-time PCR was performed with SYBR Green SuperMix with ROX (PerfeCTa) using QuantiTect Primer Assays (Qiagen). Data were normalized to 18S rRNA and gene expression changes calculated using the 2−ΔΔCT method.
WST-1 Cell Viability Assay
5 × 104 RAW 264.7 cells were plated per well in an opaque walled 96-well plate overnight. Cells were treated with increasing concentrations of peptide (0–100 μm) for 6 h and viability was determined using cell proliferation reagent WST-1 (Roche Applied Science) according to the manufacturer's instructions. The absorbance of the sample was measured at 450 nm with a reference wavelength of 630 nm.
Multiple Sequence Alignment
Multiple sequence alignment and similarity analysis was performed using AlignX software (Vector NTI, Invitrogen) using the BLOSUM62 residue substitution matrix. Similarity index of the ankyrin repeat domain of the murine IκB family was determined following alignment and expressed as a value of 0–1. Residues were scored based on the similarity value where 1 = identical residues, 0.5 = similar residues, and 0.2 = weakly similar residues. The sum value at each residue was then divided by the total number of the sequences in the alignment. The BCL-3 crystal structure, PDB ID 1K1B was annotated using 3D Molecule Viewer (Vector NTI, Invitrogen).
Mice and Carrageenan-induced Paw Edema
Male ICR mice (Harlan), weighing 35–40 g, were used. Animals were provided with food and water ad libitum and maintained on a 12/12-h light/dark cycle. All the procedures were performed in accordance with local ethical and UK Home Office regulations or with Italian and European regulations on protection of animals used for experimental and other scientific purposes. Paw edema was induced by subplantar injection of 50 μl of sterile saline containing 2% λ-carrageenan (Sigma) into the right hind paw (19). Paw volumes or thickness were measured by a plethysmometer (Ugo Basile) or a dial caliper (Kroeplin GmbH), respectively, at varying time intervals. The increase in paw volume/thickness was evaluated as the difference between the paw volume/thickness measured at each time point and the basal paw volume/thickness measured immediately before carrageenan injection. The test agents used in this study, BCL-3-derived peptide 2 (BDP2; 0.1, 1, and 10 mg/kg), mutated BDP2 (mBDP2; 10 mg/kg), and dexamethasone (Dex; 1 mg/kg), were administered intraperitoneally 1 h before the induction of the edema. The control group received an injection of PBS intraperitoneally.
Flow Cytometry and Fluorescence Microscopy
Flow cytometry of FITC-peptide-treated cells was performed using a BD Bioscience FACScan. Data were analyzed using FlowJo software. Fluorescence microscopy was performed using a Nikon Digital Sight DS Fi1C camera and NIS element software (Nikon).
Results
Analysis of BCL-3/p50 Binding by Peptide Array
Previous models of BCL-3 and p50 homodimer interaction have relied on the independently resolved crystal structures of BCL-3 and p50 homodimers (15, 20). These structures do not represent the entire BCL-3 and p50 protein sequences and so these models lack information on the C- and N-terminal regions of both proteins. We recently employed a peptide array based technique to identify amino acids of p50 that mediate interaction with BCL-3 (14). Here, we took a similar approach to identify amino acids of BCL-3 that mediate interaction with p50 homodimers. A library of overlapping peptides 18 amino acids in length, each shifted by 4 amino acids and encompassing the entire sequence of BCL-3, was Spot-synthesized on a cellulose membrane to generate a BCL-3 peptide array. Arrays were probed with either recombinant GST as a control or recombinant GST-p50 and interaction was detected by immunoblotting with anti-GST antibody. GST pulldown experiments demonstrated that GST-p50 but not GST bound to BCL-3 (Fig. 1B). The peptide array analysis revealed that GST-p50 bound specifically and strongly to a number of peptides representing the N terminus and the ANK1, -6, and -7 of BCL-3 (Fig. 1, C and D).
FIGURE 1.
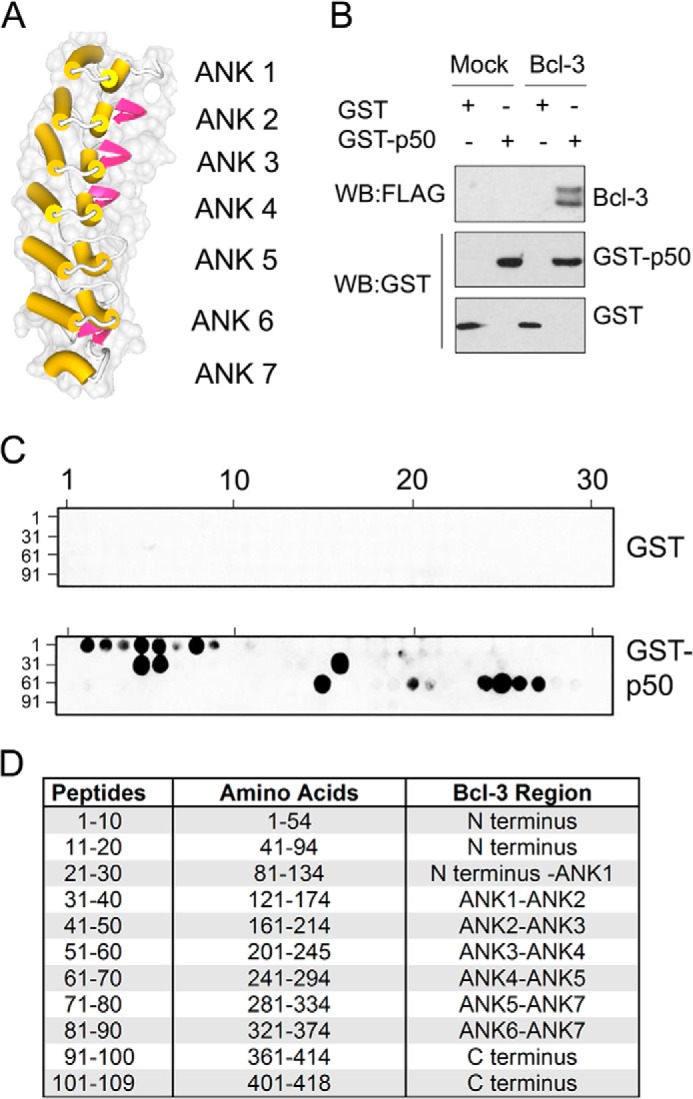
A, crystal structure of human BCL-3 ankyrin repeat domain with β-hairpin (pink) and the inner and outer α helices (yellow) indicated. B, Bcl-3 binds specifically to GST-p50 in a GST pulldown assay. Purified bacterial recombinant GST or GST-p50 was incubated with a whole cell lysate from HEK293T cells transfected with empty vector (Mock) or FLAG-BCL-3 (BCL-3) and affinity purified with GSH-agarose. Pulldown complexes were immunoblotted with anti-FLAG and anti-GST antibodies. C, peptide arrays of immobilized overlapping 18-mer peptides, each shifted to the C terminus by 4 amino acids encompassing the entire BCL-3 sequence were generated. Arrays were probed with GST or GST-p50 and detected by immunoblotting with anti-GST antibody. Positive GST-p50 binding to BCL-3 peptides is shown (black spots) and is representative of duplicate arrays. Peptide identifier numbers are indicated. D, table of BCL-3 peptides 1–109 with corresponding BCL-3 amino acids and the structural region indicated. WB, Western blot.
To identify amino acids within the identified peptides that are required for binding to p50, a series of alanine scanning arrays were generated. Arrays were derived from a p50 binding 18-mer parent peptide of interest and 18 new peptides containing a single successive alanine substitution were generated. Where an alanine was present on the parental peptide an aspartate was substituted instead. Peptides selected represented the BCL-3 N terminus region (aa 1–30), and ANK-1 (aa 138–160), -6 (aa 297–330), and -7 (aa 330–362). As before, the alanine scanning peptide arrays were incubated with GST-p50 prior to immunoblotting with anti-GST antibody. Detection of GST-p50 binding was then performed using near infrared IR dye-conjugated secondary antibody to facilitate quantification of GST-p50 binding to specific peptides using an infrared scanner. The binding of GST-p50 to the substituted peptide was calculated and expressed as a percentage binding of the control parent peptide contained on the same array (supplemental Table S1). We employed a cut-off of 40% reduction in binding relative to the parental control peptide to identify individual amino acids of BCL-3 that interact with p50 (Fig. 2).
FIGURE 2.

The 18-amino acids of BCL-3-derivied peptides of interest were sequentially substituted with alanine, probed with GST-p50, and detected by immunoblotting with anti-GST antibody. The peptides are numbered in bold as described in the legend to Fig. 1. p50 binding was quantified by densitometry and calculated as a percentage binding of the parent unsubstituted peptide on the same array. Alanine substitutions that resulted in less than 60% binding are underlined. Corresponding BCL-3 amino acids are labeled above each individual substitution peptide series.
Comparison of Peptide Array and in Silico Analysis of BCL-3/p50 Interaction
The current in silico models of the BCL-3·p50 homodimer complex have relied on the partial crystal structures of BCL-3, p50 homodimers, and the IκBα·p65·p50 complex (15, 20). We therefore compared the p50 interacting amino acids of BCL-3 identified in our experiments with those predicted by the studies of Michel et al. (20) and Manavalan et al. (15). To do this, the murine and human BCL-3 protein sequences were aligned to allow comparison of the potential interacting amino acids determined by the peptide array data (murine) and computational modeling analysis (human) (Fig. 3A). Murine and human BCL-3 share 79% sequence identity at the protein level, which readily facilitates the alignment of human and mouse proteins. This comparative analysis of the three models revealed a clear overlap in the amino acids of ANK6 and ANK7 that interact with p50. Specifically, tyrosine 301 (299 human) and glycine 303 (human 301) in ANK6, and arginine 354 (351 human) in ANK7 were identified by three of the 4 models including the peptide array (Fig. 3, A and B). Overall the peptide array data most closely matches a model proposed by Manavalan et al. (15) (Model A) which assumes that the BCL-3·p50 complex is DNA bound (Fig. 3A).
FIGURE 3.
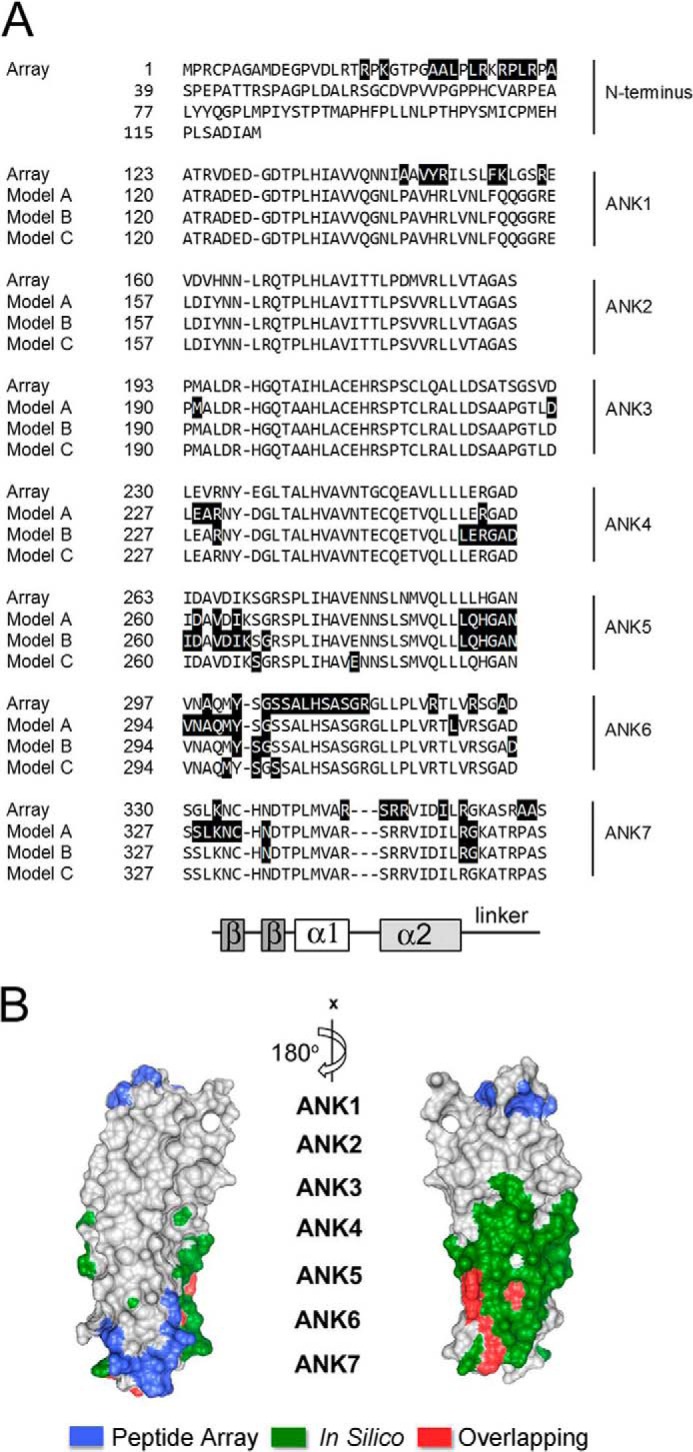
A, alignment of the seven ANK repeats of BCL-3 with a schematic indicating the conserved structural features of the each ANK repeat. Putative p50-binding residues of murine BCL-3 identified by peptide array (Array) in Fig. 2 are shaded in black and compared with p50-binding residues of human Bcl-3 predicted by the three currently available in silico models (Model A–C (15, 20)). The β hairpin (β-β), inner helix (α1), outer helix (α2), and linker regions of each ankyrin repeat are indicated. B, human BCL-3 crystal structure with corresponding unique p50-binding residues determined by peptide array (blue), p50-binding residues predicted by combined in silico Models A–C (green), and overlapping residues identified by both methods (red).
Our peptide array experiments also identified two potential p50 interacting binding regions not identified by the previous computational models. These sites are comprised of residues in ANK1 and the N terminus of BCL-3 (Fig. 3, A and B). Of note, the ANK1 region of BCL-3 has previously been demonstrated to be required for interaction with p50 (21). Neither of these two regions of BCL-3 are represented in the available BCL-3 crystal structure, however, the in silico models of BCL-3:p50 homodimers would suggest that they may interact with the C-terminal region of p50 (15, 20). In addition, we have recently demonstrated the C-terminal region of p50 and in particular amino acids 359–361 and 363, to be required for interaction with BCL-3 (14). This, combined with the peptide data presented here suggests that the N terminus and ANK1 of BCL-3 may mediate the interaction with the C-terminal region of p50 to promote the formation of a BCL-3·p50 homodimer complex.
The ANK1 Region of BCL-3 Is Not Conserved across Other IκB Proteins
In mammals the IκB family of proteins includes 9 members characterized by the presence of 6 or 7 central ANK motifs. The ANK motif is a 33-amino acid motif with a high degree of conservation and that adopts a canonical helix-turn-helix conformation in which the two α-helices are arranged in an antiparallel fashion with an almost perpendicular outward projecting β-hairpin loop (22). We analyzed the ANK1 region identified in our peptide array experiments for sequence similarity with the corresponding regions in IκBα, IκBβ, IκBϵ, IκBNS, IκBζ, p100, and p105. Multiple sequence alignment of IκB family proteins was performed and this analysis revealed that this region is of low similarity across all IκB proteins (Fig. 4). These data indicate that this short region of IκB proteins is largely protein specific and may contribute to dimer selective properties of individual IκB proteins.
FIGURE 4.
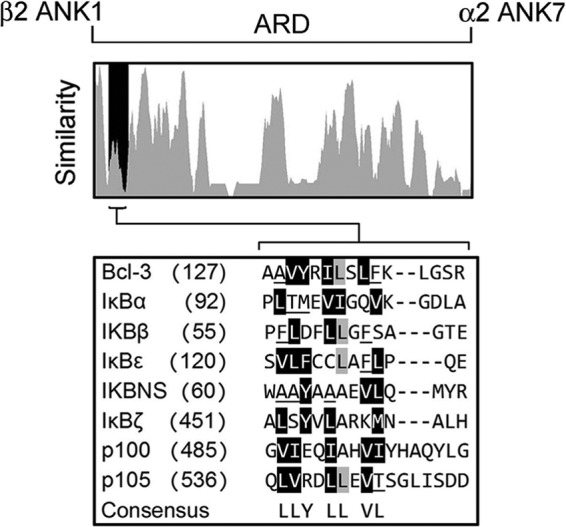
Similarity index of ANK1-ANK7 of the ARD of murine IκB family members, BCL-3, IκBα, IκBβ, IκBϵ, IκBNS, p100, and p105. Bcl-3 residues 144–158 are shaded in black and a multiple sequence alignment of this section expanded below. Non-homologous/non-similar residues are indicated by black text on white background, conservative residues derived from a block of similar residues at a given position are black text on gray background, blocks of similar residues derived from a single conservative residue at a given position are white text on black background and weakly similar residues are underlined. The consensus sequence was determined using identical and conservative amino acids.
Generation of a Bcl-3 Mimetic Peptide
We next assessed whether the short region of the ANK1 motif of BCL-3 identified by peptide array possessed any of the inhibitory activity on the transcription of NF-κB target genes. To do this we first generated a BCL-3-derived peptide (BDP1) spanning both α-helices of ANK1 and the first β sheet of ANK2 of BCL-3. This region incorporates the p50 interacting residues identified by our peptide array analysis and spans residues 135 to 164 of murine Bcl-3 (Fig. 5, A–C). This peptide sequence was fused to the HIV-tat cargo carrying the sequence YGRKKRRQRR, which mediates the cellular uptake and subsequent nuclear localization of fused peptides (23). Initially we assessed the cellular uptake and subcellular localization of a FITC-labeled BDP1 peptide using flow cytometry and fluorescence microscopy. RAW 264.7 macrophage cells were incubated with 2 μm FITC-BDP1 for 2 h prior to analysis by flow cytometry. 98% of cells were FITC positive indicating strong uptake of the peptide as expected (Fig. 5D). Fluorescence microscopy of HeLa cells treated with FITC-BDP1 further demonstrated nuclear localization of the peptide (Fig. 5E).
FIGURE 5.

A, sequences of Bcl-3 peptides 33–40 from the BCL-3 peptide array described in the legend to Fig. 1 with corresponding BCL-3 amino acid positions indicated. Amino acids 144–158 are underlined and amino acids 135–164 are indicated by bold type. B, BCL-3 crystal structure showing residues corresponding to BDP1 shaded in blue. C, schematic representation of murine BCL-3 with amino acids 135–164 of BDP1 indicated. D, flow cytometry analysis of RAW 264.7 following treatment with 2 μm-FITC-labeled BDP1 or vehicle control for 2 h. E, immunofluorescence analysis of HeLa cells treated with 20 μm FITC-labeled BDP for 2.5 h. F, RAW 264.7 cells were transiently transfected with the pLucp19 plasmid and BCL-3 expression vector (BCL-3) or with empty vector (Mock, BDP) for 24 h. Cells were left untreated (Mock and BCL-3) or pre-treated with 20 μm peptide as indicated for 1.5 h before stimulation with 100 ng/ml of LPS. Cells were cultured for an additional 8 h before luciferase activity was measured. The Renilla luciferase expression vector pRL-TK was used as an internal control to normalize the transfection efficiency across all samples. Il23p19 reporter activity is represented as fold-increase over untreated cells transfected with pLucp19 plasmid and empty vector expression (Mock). Transfections were performed in triplicate per experiment and data shown are mean ± S.D. and are representative of independent experiments. Statistical significance was determined two-way analysis of variance with Bonferroni post-hoc analysis, p < 0.001 (***).
The impact of the BDP1 peptide on the expression of NF-κB target genes was initially assessed using a luciferase-based reporter assay incorporating the Il23p19 promoter. Il23p19 is a NF-κB target gene and is negatively regulated by BCL-3 (24). RAW 264.7 macrophage were transfected with the Il23p19 reporter plasmid prior to treatment with the BDP1 (20 μm) for 1.5 h prior to stimulation with LPS (100 ng/ml) for an additional 8 h. Measurement of luciferase activity showed that the BDP1 peptide effectively inhibited LPS-inducible reporter activity and that this level of inhibition was similar to that observed following the overexpression of full-length BCL-3 (Fig. 5F). Together these data identify the BDP1 peptide as a mimic of the inhibitory action of BCL-3 on NF-κB target gene expression following LPS treatment.
BCL-3 Mimetic Peptide Inhibits Inflammation in Vivo
Peptide cargo size can effect cellular uptake and cytotoxicity of cell permeable peptides (25) and so we next sought to further optimize the BDP1 peptide by reducing it in size. A 25-amino acid BCL-3-derived peptide (BDP2) was generated that consisted of BCL-3 amino acids 144–158 fused with the HIV tat cargo carrying sequence (Fig. 6A). The BDP2 peptide was tested as before using the Il23p19 luciferase reporter assay, which demonstrated that the shorter BDP2 peptide retained inhibitory activity (Fig. 6B). Peptide treatment did not significantly affect cell viability at the concentrations used in our studies, although cytotoxcity was observed using higher concentrations of peptide (LC50 = 80 μm) as measured by the WST-1 cell viability assay (Fig. 6C). To further assess the BDP2 peptide we next evaluated the effects of the BDP2 peptide using an in vivo model of acute inflammation in the mouse. We employed the carrageenan-induced paw edema mouse model that invokes an NF-κB-dependent acute inflammatory response, which peaks ∼6 h post administration of carrageenan (19, 26). Mice were administered 0.1, 1, or 10 mg/kg of BDP2 via intraperitoneal injection 1 h prior to injection of carrageenan in the foot pad. An additional group of mice received intraperitoneal injection of the anti-inflammatory steroid dexamethasone (1 mg/ml). The control group received an injection of PBS intraperitoneally. Inflammation was assessed by measuring paw size (edema) over a range of time points up to 6 h post-carrageenan administration. The results of this analysis demonstrated a significant and dose-dependent inhibition of inflammation in mice receiving the BDP2 when compared with control mice (Fig. 6D). At the highest dose of BDP2 (10 mg/kg) the inhibitory effects on carageenan-induced edema were comparable with dexamethasone (1 mg/ml) treatment.
FIGURE 6.

A, schematic representation of the full-length (BDP1) and shortened (BDP2) BCL-3-derived peptide sequences with corresponding murine Bcl-3 amino acid positions indicated. B, RAW 264.7 cells were transiently transfected with the pLucp19 plasmid and Bcl-3 expression vector (Bcl-3) or with empty vector (Mock and BDP2) for 24 h. Cells were left untreated (Mock and BCL-3) or pre-treated with 20 μm BDP2 peptide for 2 h before stimulation with 100 ng/ml of LPS overnight. Transfections were performed in triplicate per experiment and data shown are mean ± S.D. and representative of independent experiments. Il23p19 reporter activity was determined as described in the legend to Fig. 5. Statistical significance was determined two-way analysis of variance with Bonferroni post hoc analysis, p < 0.001 (***). C, RAW 264.7 cells were treated with increasing concentrations of BDP2 peptide (0–100 μm) for 6 h and viability was assessed by WST-1 assay. Viability is expressed as a percentage of the control (no peptide) absorbance A450 nm to A630 nm and data shown are mean ± S.D. of 3 independent experiments. Peptide concentration is presented on a Log2 scale. D, PBS (Control), BDP2 (0.1, 1, and 10 mg/kg), and 1 mg/kg of Dex were administered intraperitoneally 1 h before edema induction. 2% λ-carrageenan was administered by subplantar injection into the hind paw and edema was evaluated as the difference between basal paw volume and paw volume measured at 2, 4, and 6 h following carrageenan injection. Data shown are mean ± S.E. of individual mice. Statistical significance between control and treatment groups was determined by two-way analysis of variance with Bonferroni post hoc analysis. For clarity only statistical significance at 6 h was graphed. There was no statistical significant difference (p > 0.05) between control (n = 20) and the 0.1 mg/kg of BDP2 group (n = 8) at any time point. 1 mg/kg of BDP2 (n = 8) was significant at 4 (*) and 6 h (**), 10 mg/kg of BDP2 (n = 20) was significant at 2 (***), 4 (***), and 6 (***) h. 1 mg/kg of Dex (n = 20) was significant at 2 (**), 4 (***), and 6 (***) h. Not significant (ns), p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). E, RNA was extracted from paw tissue at 6 h post-carrageenan injection. Tnf, Il6, Ccl2, and Il1b mRNA levels were determined by real-time PCR. Data are represented as the mean ± S.E. of individual mice; control, BDP2, and Dex (n = 5) and untreated (n = 4), BDP2 (10 mg/kg) and Dex (1 mg/kg). Statistical significance was determined by one-way analysis of variance with Tukey's post hoc analysis, p < 0.05 (*), p < 0.01 (**).
To further explore the effects of BDP2 in this model of inflammation we analyzed the expression of the NF-κB target genes Tnf in whole soft tissue from the paws of mice 6 h following carageenan treatment. For this analysis we employed a 10 mg/kg dose of BDP2 peptide. Quantitative PCR analysis revealed a large increase in mRNA for Tnf in the paws of mice receiving carageenan treatment compared with untreated mice. This carageenan-induced Tnf expression was significantly inhibited by pre-treatment of mice with BDP2 peptide to a level comparable with pre-treatment with dexamethasone (Fig. 6E). Similar data were obtained for Il6, whereas a trend toward significant inhibition of Ccl2 and Il1b expression by BDP2 was also apparent (Fig. 6E). These data reveal that the BDP2 peptide has potent anti-inflammatory activity in vivo that results in the significant inhibition of the key pro-inflammatory cytokines Tnf and Il6.
Anti-inflammatory Action of BCL-3 Mimetic Peptide Requires Amino Acids That Interact with p50
Our initial peptide array experiments identified individual amino acids in the BDP2 peptide that are critical for interaction with p50 (Fig. 2). To assess the importance of these same amino acids to the anti-inflammatory action of the BDP2 peptide we next generated a peptide in which these 7 amino acids of BDP2 were mutated (mBDP2) (Fig. 7A). We pre-treated RAW 264.7 cells with BDP2 and mBDP2 for 2 h prior to stimulation with LPS before measuring TNFα production by ELISA. This analysis revealed that the mBDP2 peptide had no effect on TNFα production following LPS stimulation, whereas BDP2 pre-treatment resulted in a significant inhibition of LPS-induced TNFα production (Fig. 7B). Of note, the cytotoxicity of high BDP2 concentrations was not observed for the mBDP2 peptide suggesting that BDP2 cytotoxicity may be associated with its inhibitory effect on NF-κB, which is also an important pro-survival factor (Fig. 7C). To further extend this analysis we repeated the carageenan-induced edema experiments described above and pre-treating mice with BDP2 or mBDP2 (10 mg/kg) as before. This experiment demonstrated that in contrast to the BDP2 peptide the mBDP2 peptide fails to inhibit inflammation in vivo (Fig. 7D). Together these data show that the BDP2 peptide has potent anti-inflammatory properties that depends on the amino acids that interact with p50 and that mimicking BCL-3 function may be a viable therapeutic strategy in the treatment of inflammatory disease.
FIGURE 7.

A, schematic representation of the BDP1, BDP2, and mutated BDP2 (mBDP2) BCL-3-derived peptide sequences with corresponding murine BCL-3 amino acid positions indicated and mutations underlined. B, RAW 264.7 cells were left untreated or pre-treated for 2 h with increasing concentrations (10 μm-40 μm) of either BDP2 or mBDP2 peptide before stimulation with 100 ng/ml of LPS. Supernatants were harvested 4 h after stimulation and the concentration of TNFα was determined by ELISA. ND, not detected. C, RAW 264.7 cells were treated with increasing concentrations of BDP2 or mBDP2 peptides (0–100 μm) for 6 h and viability was assessed by WST-1 assay. Viability is expressed as a percentage of the control (no peptide) absorbance A450 nm to A630 nm and data shown are representative of 3 independent experiments. Peptide concentration is presented on a Log2 scale. D, PBS (Control), BDP2 (10 mg/kg), and mBDP2 (10 mg/kg) were administered intraperitoneally 1 h before edema induction. 2% λ-carrageenan was administered by subplantar injection into the hind paw, and edema was evaluated as the difference between basal paw thickness and paw thickness measured at 2, 4, and 6 h following carrageenan injection. Data shown are mean ± S.E. of individual mice, n = 15/group. Statistical significance between control and treatment groups was determined by two-way analysis of variance with Bonferroni post hoc analysis. NS, not significant and p < 0.01 (**).
Discussion
BCL-3 mediates inhibition of p50 homodimer ubiquitination, leading to the formation of a stable p50·BCL-3 repressor complex bound to the promoters of NF-κB target genes (8). Interaction with p50 is necessary and sufficient for this anti-inflammatory function of BCL-3 (14). To further investigate the role of this complex in the regulation of NF-κB-mediated gene transcription we employed a BCL-3 peptomimetic strategy. A peptide array approach identified short peptides of BCL-3 with p50 binding activity representing the N terminus, ANK1, ANK6, and ANK7. Following alanine substitution analysis, critical residues within these peptides were identified. There was considerable overlap between the individual residues identified by peptide array and those predicted from computation modeling particularly in ANK6 and ANK7, further supporting the peptide array as a method to identify critical p50 binding regions.
Current computational models of BCL-3/p50 are constrained by the available crystal structures, which are limited to the independent crystal structures of DNA bound p50 homodimer and the central ankyrin repeat domain of BCL-3 (15, 20). In both cases the extreme N- and C-terminal regions are unstructured and so are not represented in either the resolved structures or in silico models. The data from the experiments performed here along with previous studies on the interaction of BCL-3 and p50 (14, 15, 20) suggests that the N-terminal domain of BCL-3 most likely interacts with the extreme C terminus of p50.
Our peptide array data have identified amino acids of the N-terminal and ANK1 domain of BCL-3, which interact with p50. These data are supported by earlier studies indicating that the first ANK domain is required for interaction with p50 (21). Multiple sequence alignment of this region of BCL-3-(144–158) with the homologous regions of the other IκB proteins identifies it as an area of low sequence similarity between IκB proteins. This region has also been suggested to contribute to the functional divergence of IκB factors and has also been implicated in BCL-3-mediated survival of activated T and B cells (27, 28). These previous studies and the data presented here suggested that this region of BCL-3 may contain important functional activity.
To explore the functional activity of peptides comprising the 144–148 sequence of BCL-3 we utilized the HIV tat cargo carrying peptide sequence. Because BCL-3 is a predominantly nuclear protein that regulates NF-κB activity at gene promoters we reasoned that the tat peptide sequence was most suited to delivering the BCL-3-derived peptide to the site of BCL-3 activity as it rapidly translocates through the plasma membrane and accumulates in the nucleus (23). Our initial analyses demonstrated potent inhibitory activity of the BDP peptide in both luciferase-based reporter and ELISA-based assays of Toll-like receptor-induced expression of the Il23 and TNFα cytokines, respectively. Of note, both of these genes have previously been established as targets of BCL-3 inhibitory activity (8, 16). The BDP peptide was also a potent inhibitor of cytokine gene expression and inflammation in vivo. Treatment of mice with the BDP2 peptide was effective in preventing carageenan-induced cytokine expression and paw edema. By mutating the amino acids identified as important for peptide interaction with p50 we were able to demonstrate that the functional effects of the BDP peptide, both in vitro and in vivo, were dependent on interaction with p50.
NF-κB regulates the transcription of a number of genes critical for the inflammatory response and is considered a potential therapeutic target in a range of human diseases where inflammation plays a role. Hundreds of inhibitors of NF-κB activation have been described but are limited by broad specificity (29). The BCL-3-derived peptide described here, through mimicking BCL-3 function, represents a novel class of anti-inflammatory agents that target a nuclear regulatory event in the NF-κB pathway. This BCL-3-derived peptide provides an important proof of principle that targeting transcriptional regulators of inflammation is a valid strategy for developing novel anti-inflammatory compounds of therapeutic value. The BDP peptide is also a valuable tool for further research on BCL-3 function and for the development of BCL-3-based therapeutic agents for inflammatory disease.
Supplementary Material
Acknowledgment
We thank Karen Keeshan for critical evaluation of the manuscript.
This work was supported in part by Science Foundation Ireland Grant 08/IN.1/B1843, Biotechnology and Biological Sciences Research Council Grant BB/M003671/1 and the Institute of Infection, Immunity and Inflammation at the University of Glasgow. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
- IKK
- IκB kinase
- ANK
- ankyrin repeat
- Dex
- dexamethasone
- BDP
- Bcl-3-derived peptide.
References
- 1. Carmody R. J., Chen Y. H. (2007) Nuclear factor-κB: activation and regulation during Toll-like receptor signaling. Cell Mol. Immunol. 4, 31–41 [PubMed] [Google Scholar]
- 2. Gerondakis S., Grossmann M., Nakamura Y., Pohl T., Grumont R. (1999) Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene 18, 6888–6895 [DOI] [PubMed] [Google Scholar]
- 3. Huxford T., Ghosh G. (2009) A structural guide to proteins of the NF-κB signaling module. Cold Spring Harb. Perspect. Biol. 1, a000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hayden M. S., Ghosh S. (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Natoli G., Chiocca S. (2008) Nuclear ubiquitin ligases, NF-κB degradation, and the control of inflammation. Sci. Signal 1, pe1. [DOI] [PubMed] [Google Scholar]
- 6. Bosisio D., Marazzi I., Agresti A., Shimizu N., Bianchi M. E., Natoli G. (2006) A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-κB-dependent gene activity. EMBO J. 25, 798–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colleran A., Collins P. E., O'Carroll C., Ahmed A., Mao X., McManus B., Kiely P. A., Burstein E., Carmody R. J. (2013) Deubiquitination of NF-κB by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. U.S.A. 110, 618–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carmody R. J., Ruan Q., Palmer S., Hilliard B., Chen Y. H. (2007) Negative regulation of Toll-like receptor signaling by NF-κB p50 ubiquitination blockade. Science 317, 675–678 [DOI] [PubMed] [Google Scholar]
- 9. Pene F., Paun A., Sønder S. U., Rikhi N., Wang H., Claudio E., Siebenlist U. (2011) The IκB family member Bcl-3 coordinates the pulmonary defense against Klebsiella pneumoniae infection. J. Immunol. 186, 2412–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruan Q., Zheng S. J., Palmer S., Carmody R. J., Chen Y. H. (2010) Roles of Bcl-3 in the pathogenesis of murine type 1 diabetes. Diabetes 59, 2549–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kreisel D., Sugimoto S., Tietjens J., Zhu J., Yamamoto S., Krupnick A. S., Carmody R. J., Gelman A. E. (2011) Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J. Clin. Invest. 121, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang W., Wang H., Claudio E., Tassi I., Ha H. L., Saret S., Siebenlist U. (2014) The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells. Immunity 41, 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tassi I., Claudio E., Wang H., Tang W., Ha H. L., Saret S., Ramaswamy M., Siegel R., Siebenlist U. (2014) The NF-κB regulator Bcl-3 governs dendritic cell antigen presentation functions in adaptive immunity. J. Immunol. 193, 4303–4311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Collins P. E., Kiely P. A., Carmody R. J. (2014) Inhibition of transcription by B cell leukemia 3 (Bcl-3) protein requires interaction with nuclear factor κB (NF-κB) p50. J. Biol. Chem. 289, 7059–7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Manavalan B., Basith S., Choi Y. M., Lee G., Choi S. (2010) Structure-function relationship of cytoplasmic and nuclear IκB proteins: an in silico analysis. PLoS One 5, e15782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carmody R. J., Ruan Q., Liou H. C., Chen Y. H. (2007) Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J. Immunol. 178, 186–191 [DOI] [PubMed] [Google Scholar]
- 17. Kramer A., Schneider-Mergener J. (1998) Synthesis and screening of peptide libraries on continuous cellulose membrane supports. Methods Mol. Biol. 87, 25–39 [DOI] [PubMed] [Google Scholar]
- 18. Dyer B. W., Ferrer F. A., Klinedinst D. K., Rodriguez R. (2000) A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal. Biochem. 282, 158–161 [DOI] [PubMed] [Google Scholar]
- 19. Ialenti A., Grassia G., Di Meglio P., Maffia P., Di Rosa M., Ianaro A. (2005) Mechanism of the anti-inflammatory effect of thiazolidinediones: relationship with the glucocorticoid pathway. Mol. Pharmacol. 67, 1620–1628 [DOI] [PubMed] [Google Scholar]
- 20. Michel F., Soler-Lopez M., Petosa C., Cramer P., Siebenlist U., Müller C. W. (2001) Crystal structure of the ankyrin repeat domain of Bcl-3: a unique member of the IκB protein family. EMBO J. 20, 6180–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Q., Didonato J. A., Karin M., McKeithan T. W. (1994) BCL3 encodes a nuclear protein which can alter the subcellular location of NF-κB proteins. Mol. Cell Biol. 14, 3915–3926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J., Mahajan A., Tsai M. D. (2006) Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45, 15168–15178 [DOI] [PubMed] [Google Scholar]
- 23. Vivès E., Brodin P., Lebleu B. (1997) A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 272, 16010–16017 [DOI] [PubMed] [Google Scholar]
- 24. Mühlbauer M., Chilton P. M., Mitchell T. C., Jobin C. (2008) Impaired Bcl3 up-regulation leads to enhanced lipopolysaccharide-induced interleukin (IL)-23P19 gene expression in IL-10−/− mice. J. Biol. Chem. 283, 14182–14189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tünnemann G., Martin R. M., Haupt S., Patsch C., Edenhofer F., Cardoso M. C. (2006) Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 20, 1775–1784 [DOI] [PubMed] [Google Scholar]
- 26. Posadas I., Bucci M., Roviezzo F., Rossi A., Parente L., Sautebin L., Cirino G. (2004) Carrageenan-induced mouse paw oedema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase-2 expression. Br. J. Pharmacol. 142, 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitchell T. C., Teague T. K., Hildeman D. A., Bender J., Rees W. A., Kedl R. M., Swanson B., Kappler J. W., Marrack P. (2002) Stronger correlation of bcl-3 than bcl-2, bcl-xL, costimulation, or antioxidants with adjuvant-induced T cell survival. Ann. N.Y. Acad. Sci. 975, 114–131 [DOI] [PubMed] [Google Scholar]
- 28. Mitchell T. C., Thompson B. S., Trent J. O., Casella C. R. (2002) A short domain within Bcl-3 is responsible for its lymphocyte survival activity. Ann. N.Y. Acad. Sci. 975, 132–147 [DOI] [PubMed] [Google Scholar]
- 29. Gilmore T. D., Herscovitch M. (2006) Inhibitors of NF-κB signaling: 785 and counting. Oncogene 25, 6887–6899 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.