

Background: Articular cartilage vesicles (ACVs) participate in cell communication, protein secretion, and pathologic mineralization.
Results: ACV release from chondrocytes is regulated in concert with autophagy and is caspase-3- and Rho/ROCK-dependent.
Conclusion: Autophagy participates in chondrocyte ACV release.
Significance: This work identifies a potential mechanism of ACV formation and presents opportunities to manipulate quantity and content of these important organelles.
Keywords: articular cartilage, autophagy, chondrocyte, exosome, extracellular vesicles, articular cartilage vesicles, caspase 3
Abstract
Chondrocyte-derived extracellular organelles known as articular cartilage vesicles (ACVs) participate in non-classical protein secretion, intercellular communication, and pathologic calcification. Factors affecting ACV formation and release remain poorly characterized; although in some cell types, the generation of extracellular vesicles is associated with up-regulation of autophagy. We sought to determine the role of autophagy in ACV production by primary articular chondrocytes. Using an innovative dynamic model with a light scatter nanoparticle counting apparatus, we determined the effects of autophagy modulators on ACV number and content in conditioned medium from normal adult porcine and human osteoarthritic chondrocytes. Healthy articular chondrocytes release ACVs into conditioned medium and show significant levels of ongoing autophagy. Rapamycin, which promotes autophagy, increased ACV numbers in a dose- and time-dependent manner associated with increased levels of autophagy markers and autophagosome formation. These effects were suppressed by pharmacologic autophagy inhibitors and short interfering RNA for ATG5. Caspase-3 inhibition and a Rho/ROCK inhibitor prevented rapamycin-induced increases in ACV number. Osteoarthritic chondrocytes, which are deficient in autophagy, did not increase ACV number in response to rapamycin. SMER28, which induces autophagy via an mTOR-independent mechanism, also increased ACV number. ACVs induced under all conditions had similar ecto-enzyme specific activities and types of RNA, and all ACVs contained LC3, an autophagosome-resident protein. These findings identify autophagy as a critical participant in ACV formation, and augment our understanding of ACVs in cartilage disease and repair.
Introduction
Articular cartilage vesicles (ACVs)2 are small, 50–250 nm, extracellular organelles found in normal and osteoarthritic articular cartilage (1) (2). They are similar in size and content to exosomes, and also share many characteristics with the growth plate matrix vesicles that serve as foci of mineral formation in epiphyseal cartilage (3). Initially, ACVs were functionally described in the context of pathologic mineralization in articular cartilage (4), and ACVs isolated from healthy articular cartilage generate calcium-containing crystals identical to those seen in human arthritis (4).
No clear physiologic functions for ACVs were proposed until several years ago, when we demonstrated the presence of RNA in ACVs, and showed that ACV RNA can be transferred to chondrocytes with resultant phenotypic alterations (5). Extracellular vesicles also provide an important mechanism of non-classical protein transport (6), and ACVs contain extracellular proteins, such as type II transglutaminase, that lack typical leader sequences characteristic of secreted proteins (1). Other potential roles for ACVs include providing extracellular matrix repair in pericellular cartilage, and acting to sequester toxic levels of substances such as ATP, calcium, and phosphate, so as to protect nearby chondrocytes from harm.
The potential physiologic importance of ACVs is supported by several findings. ACVs are readily isolated from intact healthy cartilage and are constitutively released into conditioned medium of normal articular chondrocytes (7). There is little evidence that pathologic processes such as cell death or injury contribute to ACV formation (8). We previously showed that the proteomes of ACVs from osteoarthritic and normal human cartilage display only minor differences, which largely reflect alterations in the composition of adherent extracellular matrix in osteoarthritic cartilage (1).
Despite a burgeoning interest in extracellular vesicles, including exosomes and microparticles, surprisingly little is known about the processes through which these extracellular organelles are generated and the factors affecting their formation and release. Recent work supports a potential association of autophagy with exosome formation (9). Autophagy is a constitutive degradative pathway of cellular homeostasis and organelle turnover, and involves formation of autophagosomes containing unwanted proteins and organelles, which fuse with lysosomes to recycle the energy-rich components of these materials (10). Up-regulation of autophagy occurs during infection, starvation, or embryonic development. In endothelial cells, extracellular vesicles are released during cell stress when processes of both autophagy and apoptosis are activated (11, 12). Markers of autophagy in aging retinal cells are found in association with increased evidence of exosome deposition in the retinal extracellular matrix (13).
Differentiated cells have widely varying baseline levels of autophagy (14). We suspected that healthy chondrocytes, which reside in a naturally hypoxic avascular environment, may have high baseline levels of autophagy. There is increasing evidence that defective autophagy contributes to the development of osteoarthritis (15–17). Given the role autophagy plays in release of extracellular vesicles, we hypothesized that autophagy could promote ACV formation and release in primary articular chondrocyte cultures.
Using an innovative dynamic model, we show here that ACV generation is regulated in parallel with autophagy, and that ACVs carry markers of autophagosomes, suggesting they originate from these structures. We also show that ACV formation is dependent on caspase-3 and Rho/Rho-associated protein kinase (ROCK) activities. These findings characterize a critical pathway of ACV formation in cultured articular chondrocytes. Understanding mechanisms involved in ACV formation may shed light on their roles in cartilage health and disease.
Experimental Procedures
Materials
Rapamycin was obtained from Tocris (Bristol, UK) and LC Laboratories (Woburn, MA). SMER28 and autophagy inhibitors were from Tocris. Unless otherwise stated, all other reagents were from Sigma-Aldrich.
Porcine Chondrocytes
Porcine hind legs from 3–5-year-old pigs were generously donated by a local sausage company (Johnsonville Foods Inc, Watertown, WI) and used in accordance with the animal use subcommittee of the Institutional Review Board at the Zablocki VA Medical Center. Normal knee cartilage was removed from the tibial and femoral surfaces of the knee joint. Chondrocytes were enzymatically released from cartilage tissue as previously described (18), and plated at high density (4 × 105 cells/cm2) in Dulbecco's Modified Eagle's medium (DMEM) with 10% fetal calf serum and 1% penicillin-streptomycin-fungizone® (PSF). These conditions maintain the differentiated chondrocyte phenotype (19). Experiments were completed within 3 days of plating.
Human Chondrocytes
After obtaining informed consent and with the permission of the local institutional review board at the Zablocki VA Medical Center, cartilage was removed from the femoral and tibial surfaces of de-identified bone fragments obtained at the time of total knee replacement for osteoarthritis. Primary human osteoarthritic (OA) chondrocytes were isolated using an identical protocol to that used for porcine chondrocytes. Cells were counted and plated at similar densities. Normal human chondrocytes from a 50-year-old donor were obtained from Lonza (Rockland, ME), handled according to the manufacturer's directions and plated at similar density in identical medium to OA and porcine chondrocytes.
ACV Quantification
Chondrocytes were incubated in experimental medium consisting of serum-free DMEM with 0.35 mg/ml bovine serum albumin, 1% PSF, and with or without various additives. ACVs were counted in conditioned medium with a NanoSight (UK) detection device which uses laser light scatter to determine size and numbers of particles present in a sample. In selected experiments, ultracentrifugation was used to concentrate ACVs from conditioned medium and collagenase-treated cell layers according to standard published protocols (4).
Western Blotting
Western blotting was used to detect markers of autophagy, including LC3-II, Beclin-1, and ATG5 in chondrocyte cultures treated with various additives. Gels were loaded with 40 μg of protein and transferred to nitrocellulose membranes. Antibody to GAPDH (Cell Signaling, Danvers, MA) was used to ensure equal protein loading in Western blots using cellular protein isolates. Membranes were exposed to antibodies directed against LC3, Beclin-1 (Abcam, Cambridge, MA) and ATG5 (Cell Signaling), at 1:1000 dilution for 1 h (GAPDH) or overnight. After washing, membranes were exposed to peroxidase-labeled goat anti-rabbit (IgG (H+L)) or rabbit anti-mouse for 1 h (1:10,000, Life Technologies, Grand Island, NY) and bands were visualized using chemiluminescence. Western blotting for LC3 was also performed on ACV preparations.
siATG5
Fresh, enzymatically-released porcine chondrocytes were electroporated (AMAXA, Germany) prior to plating with short interfering (si) ATG5 or a scrambled control sequence (Life Technologies). After 48 h, mRNA, and protein levels for ATG5 were measured. Total RNA was extracted using PureLink Mini RNA kit (Life Technologies). cDNA was synthesized from 1 μg of total RNA using QuantiTect Reverse Transcription kit (Qiagen, Valencia, CA). To ensure effective reduction of ATG5 mRNA, relative ATG5 expression was measured by semi-quantitative real-time PCR using a LightCycler 480 (Roche, Indianapolis, IN). Chondrocytes treated with siRNA for ATG5 or scrambled control were exposed to rapamycin for 4 h, and ACVs were enumerated in conditioned medium.
Cell Toxicity
Release of LDH into conditioned medium (Cytoscan™-LDH Cytotoxicity Assay Kit, St. Louis, MO) and the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) formazan conversion assay served as toxicity assays. MTT uptake can also be used to estimate mitogenesis. Both were used according to manufacturer's directions. All toxicity data were collected at the highest concentration and longest exposure time for each reagent.
mTOR Phosphorylation
The effect of rapamycin on mTOR phosphorylation was determined with the PathScan® Phospho-mTOR (Ser-2448) kit (Cell Signaling) according to the manufacturer's directions.
Apoptosis Markers
The Caspase-Glo 3/7 Assay (Promega) was used to detect early apoptosis according to manufacturer's directions.
Electron Microscopy
Chondrocytes treated with or without rapamycin for 4, 6, and 8 h were fixed with 3% glutaraldehyde and sections imaged using a JEOL JEM 1011 transmission electron microscope (JEOL USA, Peabody, MA) at 80 kV fitted with a side mount AMT 2k digital camera (Advanced Microscopy Techniques Woburn, MA).
Ecto-enzyme Activity Levels
Specific activities of the ecto-enzymes that metabolize ATP, and that are often used to characterize ACVs (2), were measured on concentrated ACV fractions from chondrocytes treated with and without rapamycin. Nucleoside triphosphate pyrophosphohydrolase (NTPPPH) activity was measured using 2 mm thymidine 5′-monophosphate p-nitrophenyl ester (TMPNP) as a substrate. Briefly, ACVs were resuspended in TMPNP in HBSS and incubated for 2 h at 37 °C. The reaction was stopped with the addition of 0.1 N NaOH, and absorbance was measured at 410 nm using a Biotek plate reader. Activity of the phosphate generating enzyme, 5′ nucleotidase (5′-NT), was determined with a kit used according to manufacturer's directions (BQ Kits, San Diego, CA) (20). Alkaline phosphatase activity was measured using p-nitrophenol phosphate (PNPP) as a chromogenic substrate. Equal volumes of alkaline buffer solution and PNPP were added and incubated with ACVs for 15 min at 37 °C. The reaction was stopped with 0.05 N NaOH, and absorbance was measured as described above. All results were corrected for protein levels in the samples using the Lowry assay.
RNA Isolation and Characterization
A two-part procedure was used to extract RNA from ACVs. Trizol (Life Technologies) was used to lyse the ACV pellet and total RNA was extracted using PureLink Mini RNA kit (Life Technologies). The FlashGel RNA System (Lonza, Rockland, ME) was used to visualize RNA extracted from ACVs.
Statistical Analysis
All experiments were repeated at least three times with each experiment beginning with different cartilage donors. Results are displayed as representative experiments, while statistical analysis reflects pooled data analyzed with GraphPad Prism® 6.0 software. Statistical differences were assessed using multiple t-tests with Holm-Sidak correction for multiple comparisons for all experiments that included multiple time point analyses. One-way ANOVA was used to analyze the inhibitor and silencing experiments. Student's t test was used for experiments involving ecto-enzyme, LDH, MTT, and caspase activity analyses.
Results
Nanoparticle Counting Accurately Quantifies ACVs
ACVs (Fig. 1) have been traditionally measured by protein quantification of the pellet derived from differential centrifugation of conditioned medium or from enzymatically treated cell layers or cartilage pieces. The NanoSight LM10 was used to count and size particles in conditioned medium that were 50–1200 nm in diameter as illustrated by the curve seen in Fig. 1A. Fresh unconditioned medium demonstrated very low background counts. The mean size of ACVs counted with the NanoSight was 190.7 ± 26 nm. To ensure that we were quantifying the same particles in the nanoparticle counter as in ACV fractions isolated in a traditional manner, we obtained conditioned medium from the one chondrocyte culture, counted the ACVs in one aliquot prior to centrifugation, and then counted ACVs in the pellet after centrifugation. We compared ACV number after differential centrifugation of medium with that of unspun medium (Fig. 1B). As expected, the number of ACVs also correlated with the total number of chondrocytes present (Fig. 1C). As illustrated in Fig. 1D, there are consistent numbers of ACVs in various chondrocyte cultures over time using identical culture conditions. There were no differences in mean particle size under any conditions used in these experiments.
FIGURE 1.

The NanoSight detection system accurately quantifies ACVs from chondrocyte-conditioned medium. A, medium aliquots were tested in the NanoSight before (“unconditioned”) and after (“conditioned”) exposure to chondrocytes. Representative curves show typical results. B, conditioned medium were removed from chondrocyte cultures and counted in the NanoSight without further preparation (“media-derived”) or after centrifugation at 2000 × g for 15 min and then at 130,000 g for 60 min. The resultant pellet was resuspended in DMEM and counted in the NanoSight (“pellet-derived”). Values represent means ± S.D. There were no differences in the numbers of ACVs counted in the resuspended pellet and un-centrifuged medium (n = 3, p > 0.05). C, ACVs were counted in conditioned medium from chondrocytes plated at 4 × 105/cm2 or 2 × 105/cm 2. The numbers of ACVs correlated with the number of plated cells (n = 3, ***, p < 0.001). D, consistent increase in numbers of ACVs are identified under similar culture conditions, as shown by the scatter plot of ACV number at various time points.
Rapamycin Increases ACV Number
Rapamycin is a potent and commonly used agent which induces autophagy by inhibiting signaling by the mechanistic Target of Rapamycin (mTOR), a serine/threonine kinase that coordinates control of multiple cellular pathways (21). As shown in Fig. 2A, chondrocytes treated with 50 μm rapamycin for 4 h produce an average of 2.4 ± 0.03-fold (p < 0.01) more ACVs than untreated chondrocytes. No change in particle size was observed. Rapamycin had a strong stimulatory effect on ACV number at all time points tested. Increasing doses of rapamycin from 25 to 50 μm resulted in a stepwise increase in ACV number (n = 3, p < 0.05) (Fig. 2B). ACV numbers in the cell layer with 50 μm rapamycin also increased 4.03 ± 0.7-fold over control values at 4 h (n = 7), indicating that rapamycin increases the total number of ACVs released by chondrocytes and does not simply alter their location.
FIGURE 2.
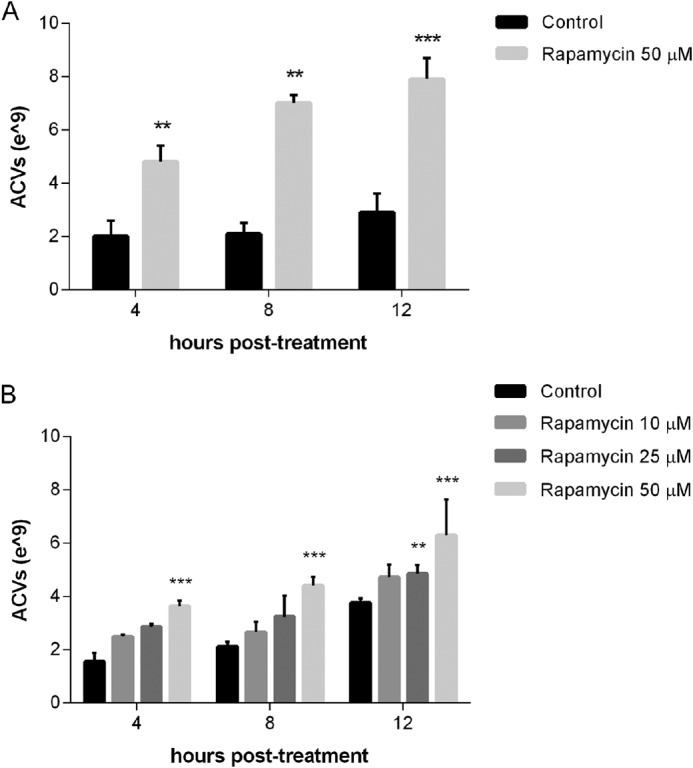
Rapamycin increases ACV number in a time and dose-dependent manner. A, porcine chondrocytes were incubated with no additives or 50 μm rapamycin for 4–12 h. Aliquots of medium were removed and ACVs were counted. Values represent means ± S.D. Rapamycin increased ACV numbers relative to control at all time points tested (n = 3, **, p < 0.01; ***, p < 0.001). B, porcine chondrocytes were incubated with no additives or 10–50 μm rapamycin for 4–12 h. Aliquots of medium were removed and ACVs were counted. Values represent means ± S.D. Rapamycin increased ACV numbers relative to control at 12 h at 25 μm (n = 3, **, p < 0.01) and all time points at 50 μm (n = 3, ***, p < 0.001).
Rapamycin Induces Autophagy without Causing Cell Toxicity
Rapamycin induces autophagy by suppressing mTOR phosphorylation (22, 23). Rapamycin concentrations which increase ACV number significantly reduce mTOR phosphoryation in chondrocytes. Levels of phosphorylated mTOR were 30.8 ± 0.55% below controls with 50 μm rapamycin (p < 0.0001) (data not shown). This concentration of rapamycin is consistent with those commonly employed in primary chondrocytes (15, 24). As expected, no signs of cell toxicity as measured by LDH release or MTT conversion were noted at these concentrations (Fig. 3A). LC3 is a commonly used marker of autophagy signaling. Induction of autophagy produces increased levels of a lipidated form of LC3, known as LC3-II, which runs as a higher mobility band on SDS-PAGE. As we predicted, untreated chondrocytes demonstrate a relatively high level of baseline autophagy signal, as illustrated by a visible LC3-II band. Rapamycin treatment decreased levels of LC3-I and increased levels of LC3-II, as shown in the Western blot in Fig. 3B. More modest increases were observed at 12 h in protein levels of Beclin, another commonly used autophagy marker (Fig. 3B). Small increases in LC3 mRNA were noted at 8 h, while no changes in Beclin-1 mRNA were detectable (Fig. 3C). Electron micrographs of resting chondrocytes reveal a variety of cytoplasmic membrane structures, including electron-dense and electron-light vesicles and many elongated, curved structures, which are reminiscent of the “pre-autophagosomal structures” which eventually self-fuse to encompass cytosol in a double-membraned structure (Fig. 4). Upon rapamycin treatment for 8 h, cells demonstrate clear electron-dense autophagolysosomes, morphologically confirming our quantitative data and suggesting overall progression of the autophagic pathway.
FIGURE 3.

Rapamycin increases markers of autophagy without inducing cell toxicity. A, porcine chondrocytes treated with no additives or 50 μm rapamycin for 12 h were assessed for injury by measuring LDH levels in medium and uptake and conversion of MTT into a colored formazan. Results are expressed as a percent of control (no rapamycin). There were no differences in cell injury markers or caspase-3 levels with rapamycin (n = 3, p > 0.05). B, porcine chondrocytes treated with no additives or 50 μm rapamycin for 4–12 h were lysed and lysates were used in Western blotting with antibodies to LC3 and Beclin. Increased levels of LC3-II occur with rapamycin treatment at 4, 6, and 8 h. Increased levels of Beclin were noted at 12 h. C, RNA was isolated from porcine chondrocytes incubated with and without rapamycin for 4–12 h. mRNA for LC3 and Beclin were measured with end-point PCR and showed no changes in Beclin mRNA levels and increases in LC3 at 8 h. D, chondrocytes were isolated and plated in iso-osmolar medium containing normal (5 mm) or high (25 mm) glucose. After 18 h, medium were removed, identical fresh medium was added, and ACVs were enumerated in conditioned medium for an additional 4–12 h.
FIGURE 4.

Rapamcyin induces morphologic changes of increased autophagy by electron microscopy (EM). Porcine chondrocytes were incubated with or without rapamycin for 4–8 h. After fixation with glutaraldehyde, they were examined with TEM. In these representative images, typical autophagosomes (solid arrows) can be identified by the presence of double membranes and cytoplasmic components such as ribosomes and mitochondria and are visible at 4 and 8 h after rapamycin treatment. Pre-autophagosomal structures (open arrows), which are identified by their typical crescent shape, are present 4 h after rapamycin treatment.
Glucose Levels Do Not Significantly Affect ACV Number or Induce Autophagy
In some cell types, exposure to high glucose medium promotes cell division and autophagy (25). Noting the high basal levels of LC3-II and ACV number and the high glucose content of our usual culture medium, we investigated the effect of ambient glucose levels on ACV number. Porcine chondrocytes were isolated and plated in normal glucose medium (5 mm) or in the iso-osmolar high glucose medium (25 mm) used for the majority of these experiments. As shown in Fig. 3D, ACV numbers were not different in normal and high glucose medium, and LC3-II levels were unchanged (data not shown).
Pharmacologic Inhibitors of Autophagy Suppress the Rapamycin-induced Increase in ACV Number
Chondrocytes were initially treated for 2 h with various autophagy inhibitors including 5 mm 3-methyladenine (3-MA), 100 nm bafilomycin, 20 μm chloroquine, 10 μm SP600125, and 10 μm spautin or no additives. Rapamycin was then added for an additional 2 to 10 h. Medium was collected at 3 time points, and ACVs were enumerated. As shown in the representative 2-h experiment in Fig. 5A, all inhibitors tested except 3-MA significantly suppressed the increase in ACV number seen with rapamycin (n = 3, p < 0.05). Similar changes were noted at other time points (data not shown).
FIGURE 5.

Autophagy inhibition suppresses rapamycin-induced ACV production. A, porcine chondrocytes were incubated with no additives or autophagy inhibitors for 2 h, and then with rapamycin for an additional 2 h. ACVs were enumerated in conditioned medium. Values represent means ± standard deviations. All inhibitors tested except 3-MA significantly suppressed rapamycin-induced ACV numbers (n = 3, *, p < 0.05; **, p < 0.01). B, porcine chondrocytes were transfected with siATG5 or a scrambled control for 24 h. Medium was removed, and chondrocytes were exposed to no additives or to rapamycin for 8 h. ACVs in the conditioned medium were enumerated. Values represent means ± S.D. Cell treated with siATG5 produced less ACVs in the presence of rapamycin than those treated with scrambled control (n = 3, **, p < 0.01). C, RNA was isolated from chondrocytes treated with siATG5 or scrambled control. mRNA for ATG5 was quantified by real-time PCR. Values represent means ± S.D. siATG5 significantly reduced mRNA for ATG5 (n = 3, **, p < 0.01) and protein (inset). D, OA chondrocytes and E, normal human chondrocytes were treated with 50 μm rapamycin for 4–8 h. ACVs were enumerated. Values represent means ± S.D. Rapamycin did not increase ACV number from OA chondrocytes (n = 4, p > 0.05), but significantly increased ACV number in normal human chondrocytes (n = 3, ***, p < 0.001).
Reducing ATG5 Levels Suppresses Rapamycin-induced ACV Generation
Because pharmacologic inhibition may have off-target effects, we treated chondrocytes with an RNAi silencer for ATG5 (siATG5), a gene required for autophagy. As shown in Fig. 5B, siATG5 significantly reduced the effect of rapamycin on ACV number at 8 h compared with a scrambled control (n = 3, p < 0.05). Effective silencing was documented by a dramatic reduction in mRNA and protein for ATG5 (Fig. 5C).
Osteoarthritic (OA) Chondrocytes Do Not Produce Increased Numbers of ACVs upon Rapamycin Treatment
If ACV formation is related to autophagy, then cells with defective autophagy should show no increase in ACV numbers after treatment with rapamycin. OA chondrocytes have been demonstrated to be deficient in autophagic degradation, although markers for autophagic signaling are activated (12, 17, 18). As shown in the representative experiment in Fig. 5D, OA chondrocytes did not increase ACV number in response to rapamycin, in contrast to the vigorous response seen in normal human chondrocytes (Fig. 5E).
Induction of Autophagy through mTOR-independent Pathways Also Promotes ACV Release
To determine if ACV generation and release required mTOR, we investigated the effect of SMER28, which induces autophagy through mTOR-independent mechanisms (26, 27). As shown in Fig. 6, SMER28 increases ACV formation (Fig. 6A) as well as LC3-II levels (Fig. 6B) without causing cell toxicity (Fig. 6C).
FIGURE 6.
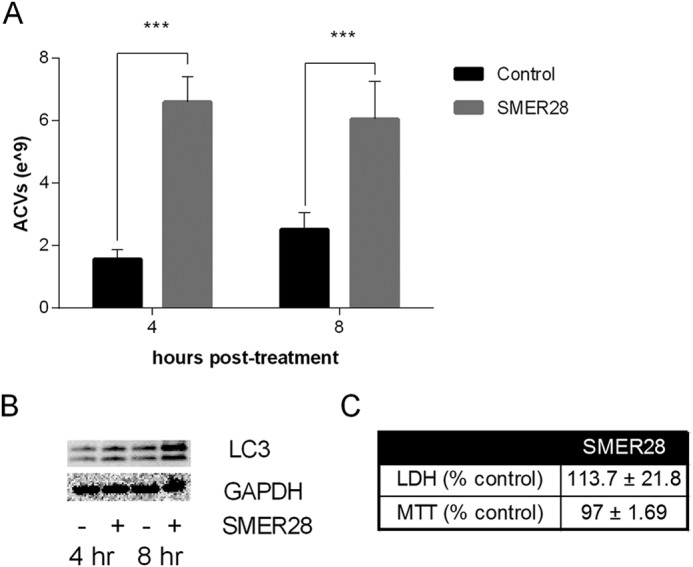
SMER28, which induces autophagy independent of mTOR, also increases ACV number without cell toxicity. A, porcine chondrocytes were incubated with no additives or 250 μm SMER28 for 4–8 h. ACVs were counted in conditioned medium. SMER28 increased ACV number (n = 9, ***, p < 0.001). B, porcine chondrocytes were incubated with no additives or 250 μm SMER28 for 4–8 h. Cells were lysed, and the lysate used in Western blotting to measure levels of LC3. SMER28 increased levels of LC3-II. C, toxicity was assessed using LDH levels in conditioned medium and the MTT assay. SMER28 did not increase cell toxicity (n = 3, p > 0.05).
Rapamycin-induced Increases in ACV Number Are Dependent upon Caspase-3, but Independent of Apoptosis
In endothelial cells, release of autophagosomes to the extracellular space is dependent on active caspase 3. Caspase 3 may be necessary for autophagosome maturation (12), but is also an early marker of apoptosis. Increased levels of caspase 3 coincide with the membrane blebbing that occurs in the early phases of apoptosis. We explored the effects of Z-VAD-FMK, a potent caspase-3 inhibitor, on ACV production with and without rapamycin. As shown in Fig. 7A, exposure of chondrocytes to 50 μm Z-VAD-FMK for 2 h suppressed the increase of ACV numbers normally induced by rapamycin without affecting the rapamycin-induced increase in LC3-II levels (Fig. 7B). We also examined the effects of the apoptosis agonists, doxorubicin, and staurosporine, on ACV formation in the absence and presence of rapamycin (Fig. 7, C and D). These agents did not augment ACV numbers after 4–12 h of exposure with and without rapamycin at doses, which induce classic markers of early apoptosis (Fig. 7E).
FIGURE 7.

The effect of rapamycin on ACV number is dependent on caspase-3 but not mimicked by apoptosis agonists. A, porcine chondrocytes were exposed to rapamycin for 2 h and then either no additional additives or 50 μm of the caspase inhibitor Z-VAD-FMK for 2 h. ACVs were enumerated. Values represent means ± standard deviations. Z-VAD-FMK reduced the effect of rapamycin on ACV number (n = 3, *, p < 0.05) (B) without altering levels of LC3-II. C, porcine chondrocytes were incubated with doxorubicin or staurosporine for 4–12 h. ACVs were counted in conditioned medium. Values represent means ± S.D. Neither doxorubicin nor staurosporine affected ACV number (n = 9, p > 0.05). D, porcine chondrocytes were incubated with doxorubicin or staurosporine for 4–12 h with and without rapamycin. ACVs were counted in conditioned medium. Neither doxorubicin nor staurosporine increased ACV number (n = 9). E, porcine chondrocytes were incubated with doxorubicin or staurosporine for 6 h. Caspase-3 levels were measured to assess apoptosis (according to manufacturer's instructions). Both agents increased caspase levels in comparison to control (n = 3, p < 0.05).
Rho/ROCK Inhibition Suppresses Rapamcyin Induced ACV Number
We observed cytoplasmic extensions and extracellular vesicles in chondrocytes treated with rapamycin (Fig. 8A). These images are reminiscent of the phenomenon of membrane blebbing, which may be involved in exosome formation. As shown in Fig. 8B, Y27632, Rho/ROCK inhibitor, suppressed the effect of rapamycin on ACV number, suggesting that these mediators participate in this process. To determine if other factors known to produce membrane blebbing also increased ACV number, we investigated the effect of purinergic stimulants on ACV generation. Activation of purinergic receptor P2X7 by exposure to high ATP concentrations produces membrane blebs (28). As seen in Fig. 8C, 1 mm ATP produced no changes in ACV numbers. Taken together, these data suggest that Rho/ROCK activation is necessary but not sufficient for ACV formation
FIGURE 8.

Rho/ROCK-dependent cell-blebbing is necessary but not sufficient for ACV release. A, cytoplasmic extensions are visible by EM in porcine chondrocytes at 12 h after rapapmycin treatment. B, porcine chondrocytes were incubated with or without the Rho/ROCK inhibitor, 10 μm Y27632 for 4–12 h in the presence and absence of rapamycin. ACVs were counted in conditioned medium. Values represent means ± S.D. Y27632 suppressed the effect of rapamycin on ACV number (n = 9, *, p < 0.05; **, p < 0.01). C, porcine chondrocytes were incubated with or without 100 μm or 1 mm ATP. ACVs were counted in conditioned medium after 4–12 h. Values represent means ± S.D. ATP had no effect on ACV number (n = 9, p > 0.05).
Rapamycin-induced ACVs Contain LC3 and Display Characteristic Ecto-enzymes and RNA
LC3 is present on isolated ACVs from control as well as rapamycin-treated chondrocyte cultures (Fig. 9A). This suggests that ACVs originate as autophagosomes and are externalized. ACVs have historically been characterized by levels of ATP-metabolizing enzymes involved in mineralization. These include the nucleoside triphosphate pyrophosphohydrolase enzymes that cleave ATP into AMP and inorganic pyrophophosphate and 5′ nucleotidase, which generates adenosine from AMP. Alkaline phosphatase is another common marker of ACVs, and hydrolyzes pyrophosphate into inorganic phosphate and cleaves phosphates off ATP. As shown in Fig. 9B, there were no differences in any of these enzymes' specific activities in rapamycin-induced ACVs compared with controls. We previously demonstrated heterogenous sizes of RNA in ACVs. We confirmed the presence of similar RNA types in ACVs from rapamycin-treated cells (Fig. 9C).
FIGURE 9.
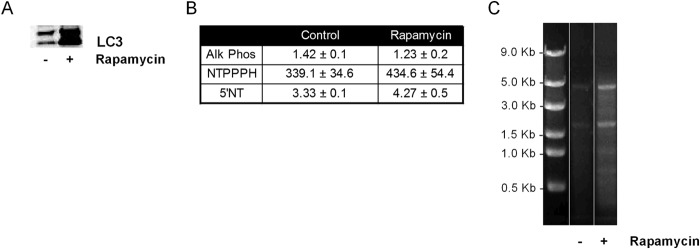
ACVs from rapamycin-treated and control chondrocytes contain autophagosome markers, characteristic ecto-enzymes, and RNA. A, protein isolates from ACVs formed in the absence (lane 1) and presence (lane 2) of rapamycin were run in Western blots with anti-LC3 antibody. B, enzyme levels on ACVs from chondrocytes treated with or without rapamycin. NTPPPH, alkaline phosphatase, and 5′ nucleotidase specific activities were measured on ACVs isolated from untreated and rapamycin-treated chondrocytes (n = 3). There were no differences in enzyme specific activity levels between groups. C, RNA was isolated from ACVs derived from chondrocytes treated with or without rapamycin and run on an agarose gel.
Discussion
We show here that rapamycin induces autophagy, and increases the release of extracellular vesicles similar to the normal ACVs released from cultured chondrocytes. This effect is caspase-3- and Rho/ROCK- dependent, and ACVs display markers consistent with having origins in the autophagic pathway (Fig. 10). SMER28, which induces autophagy independent of mTOR, produces similar effects. Autophagy signaling is primed at significant constitutive levels in cultured chondrocytes, whose normal environs are nutrient-poor and hypoxic. The identification of autophagy as a critical factor in ACV generation, serves as a necessary first step in exploiting the function of these organelles to modulate cartilage health and repair.
FIGURE 10.

The proposed pathway of ACV formation in articular chondrocytes. Articular chondrocytes display ongoing autophagy and low levels of ACV formation and release. Factors which increase autophagy produce increased numbers of autophagosomes, which are released as extracellular ACVs in a caspase- and Rho/ROCK- dependent process.
Previous studies on the formation and release of ACVs and other small vesicles from primary cells have been hampered by cumbersome methodology that required ultracentrifugation of large amounts of conditioned medium to yield very small numbers of ACVs. Unlike microparticles, ACVs, and exosomes at diameters of 50–250 nm are too small to be detected by conventional flow cytometry. Thus, the nanoparticle counting technology allowed us to assess ACV generation from chondrocytes in real-time, and for the first time, to identify quantitative differences in ACV release in response to pharmacological agents.
A connection between autophagy and ACV formation is suggested by the dramatic effect of rapamycin and SMER28 on chondrocyte ACV production. Using both pharmacologic and genetic approaches, we demonstrate that inhibitors of autophagy abrogate the effects of rapamycin on ACV number. Interestingly, none of the inhibitors significantly affected basal ACV numbers. It is possible that these ACVs are formed and ready to be elaborated before autophagy inhibitors are added. Alternatively, another process is responsible for basal ACV formation. Taken together with the presence of LC3-II, a marker of autophagosomes, on ACVs, these findings strongly support the hypothesis that autophagic membranes contribute to ACV formation in articular chondrocytes.
While markers of autophagic signaling may be increased in osteoarthritic chondrocytes (24) (29), functional autophagic degradation is deficient (15). The factors responsible for defective autophagy in OA chondrocytes have not been clearly delineated. Taking advantage of the natural occurrence of deficient autophagy in this setting, we demonstrated the absence of an ACV response to rapamycin in chondrocytes from patients with osteoarthritis. Thus dys-regulation of ACV generation and release may occur in OA cartilage.
Our studies support prior findings of Sirois et al. and Pallet et al. who noted a positive association of autophagy and exosome formation in human umbilical vascular endothelial cells (11, 12). Their work was based on reports of non-classical protein secretion in yeast, which were the first studies to suggest that autophagosomes that fail to fuse with lysosomes may have an extracellular fate (30, 31). This is however, not a universal phenomenon. For example, in an erythroleukemic cell line, Fader et al. showed that the release of exosomes derived from multivesicular bodies is suppressed by the induction of autophagy (32). Although chondrocytes contain multivesicular bodies (33), they behave in a manner similar to endothelial cells in regards to autophagy and ACV formation. It is likely that the origin of exosomes varies between different cell types and tissues. Because exosome formation and function may be radically altered by malignant cellular transformation (34), we have used primary cells in this study, increasing the likelihood of physiologic relevance of our findings to articular cartilage.
Despite their important role in mineralization, very little is known about extracellular vesicle formation in cartilage and chondrocytes. In growth plate chondrocytes and osteoblast-like cells, matrix vesicles were postulated to be formed by zeoitic blebbing of apical microvilli (35) (36). These studies showed similarities in biochemical composition between matrix vesicles and microvilli, and that retraction of actin filaments was necessary for the generation and release of matrix vesicles. Autophagy was not specifically addressed in this work, and many of the pharmacologic inhibitors used in these investigations have broad metabolic sequellae.
Our work supports a possible role for membrane blebbing in ACV formation. The Rho/ROCK pathway is a key component of many membrane blebbing processes (37). We show that Rho/ROCK-dependent membrane blebbing is necessary for rapamycin-induced ACV release in articular chondrocytes. However, membrane blebbing does not appear to be sufficient for ACV production in our system. This is demonstrated by the lack of increased ACVs in response to the P2X7 agonist, ATP (38). Membrane blebbing is also seen in the early phases of apoptosis (39), but apoptosis agonists did not increase ACV number.
Autophagy is a complex multistep process that shares some components with other cellular stress responses including apoptosis. In chondrocytes, these two processes are particularly closely aligned. Indeed, the term chondroptosis was coined to describe unusual aspects of apoptotic cell death in chondrocytes, including some related to active autophagy (40) (41). Caspase-3 typically regulates apoptosis. Our observation that inhibition of caspase-3 suppressed the effects of rapamycin on ACV production without altering LC3-II levels suggests that either some induction of apoptosis is necessary for the autophagy-ACV process in chondrocytes, or that this enzyme is involved in another aspect of ACV formation or release. Work by Sirois et al. (12) demonstrated a potential role for caspase in autophagosome maturation in their system, but further work to explore the role of caspase in ACV generation is necessary.
Several lines of evidence make it unlikely that ACVs are apoptotic bodies. First, apoptotic bodies are considerably larger than ACVs. Furthermore, typical apoptosis agonists did not increase ACV numbers. Finally, published work demonstrated functional differences between cartilage extracellular vesicles and apoptotic bodies (42), and Jaovisidha et al. demonstrated no increase in calcification in ACVs derived from apoptotic cells (8). Our studies show no evidence of cell death during induction of autophagy or significant cell toxicity resulting from any culture conditions during the time periods in which we see rapamycin-induced ACV production.
This work is not without limitations. Chondrocytes in monolayer cultures have been stressed by exposure to mechanical and enzymatic digestion procedures, and thus, their behavior may not fully mimic that of healthy chondrocytes in vivo. Although we used conditions that maintain the highly differentiated chondrocyte phenotype in short term culture, chondrocytes typically reside in a three-dimensional matrix under low oxygen conditions. We did not fully replicate these conditions, and further work to determine the role of oxygen tension in ACV production is warranted. A three-dimensional matrix renders the dynamic assessment of ACV release challenging and will require additional model development, but will be clearly important in the future. The absence of a response to rapamycin in OA chondrocytes may be due to age alone, as we did not know the ages of the OA donors, and the mechanism through which autophagy is blocked in OA chondrocytes requires further study. The complex relationship between autophagy and apoptosis in chondrocytes also warrants further analysis.
In summary, we show here that ACV generation in chondrocytes occurs in parallel with autophagy, that ACVs carry markers of autophagosomes, and are released in a caspase-3- and Rho/ROCK-dependent manner. As ACVs likely participate in secretion of extracellular proteins important in matrix repair and contain highly protected RNA, identifying the mechanisms involved in their generation and release may result in improved understanding of the roles of ACVs in cartilage health and disease.
Acknowledgments
We thank Aidan McCarty for expert technical assistance and Dr. Vincent C. Hascall, PhD, for the thoughtful and detailed review of the manuscript.
This work was supported by the Veteran's Affairs Research Service with a Merit Review Grant from the Dept. of Veteran's Affairs (to A. K. R., 101BX000812) and National Institutes of Health Grant R01-AI104928 (to W. T. J.).
- ACVs
- articular cartilage vesicles
- PSF
- Penicillin-Streptomycin-Fungizone®
- OA
- osteoarthritis
- TMPNP
- thymidine 5′-monophosphate p-nitrophenyl ester
- 5′-NT
- 5 ′-nucleotidase
- PNPP
- p-nitrophenol phosphate
- NTPPPH
- nucleoside triphosphate pyrophosphohydrolase
- ROCK
- Rho-associated kinase
- GAPDH
- glyceraldehyde phosphate dehydrogenase
- ATG5
- autophagy-related 5
- LC3
- microtubule-associated protein 1 light chain 3
- 3-MA
- 3-methyladenine.
References
- 1. Rosenthal A. K., Gohr C. M., Ninomiya J., Wakim B. T. (2011) Proteomic analysis of articular cartilage vesicles from normal and osteoarthritic cartilage. Arthritis Rheum. 63, 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Derfus B. A., Kurtin S. M., Camacho N. P., Kurup I., Ryan L. M. (1996) Comparison of matrix vesicles derived from normal and osteoarthritic human articular cartilage. Connect Tissue Res. 35, 337–342 [DOI] [PubMed] [Google Scholar]
- 3. Derfus B., Kranendonk S., Camacho N., Mandel N., Kushnaryov V., Lynch K., Ryan L. (1998) Human osteoarthritic cartilage matrix vesicles generate both calcium pyrophosphate dihydrate and apatite in vitro. Calcified Tissue International 63, 258–262 [DOI] [PubMed] [Google Scholar]
- 4. Derfus B. A., Rachow J. W., Mandel N. S., Boskey A. L., Buday M., Kushnaryov V. M., Ryan L. M. (1992) Articular cartilage vesicles generate calcium pyrophosphate dihydrate-like crystals in vitro. Arthritis Rheum. 35, 231–240 [DOI] [PubMed] [Google Scholar]
- 5. Mitton E., Gohr C. M., McNally M. T., Rosenthal A. K. (2009) Articular cartilage vesicles contain RNA. Biochem. Biophys. Res. Commun. 388, 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang M., Schekman R. (2013) Cell biology. Unconventional secretion, unconventional solutions. Science 340, 559–561 [DOI] [PubMed] [Google Scholar]
- 7. Derfus B. A., Camacho N. P., Olmez U., Kushnaryov V. M., Westfall P. R., Ryan L. M., Rosenthal A. K. (2001) Transforming growth factor beta-1 stimulates articular chondrocyte elaboration of matrix vesicles capable of greater calcium pyrophosphate precipitation. Osteoarthritis Cartilage 9, 189–194 [DOI] [PubMed] [Google Scholar]
- 8. Jaovisidha K., Hung J., Ning G., Ryan L. M., Derfus B. A. (2002) Comparative calcification of native articular cartilage matrix vesicles and nitroprusside-generated vesicles. Osteoarthritis Cartilage 10, 646–652 [DOI] [PubMed] [Google Scholar]
- 9. Kulich I., Žársky V. (2014) Autophagy-related direct membrane import from ER/cytoplasm into the vacuole or apoplast: a hidden gateway also for secondary metabolites and phytohormones? Int. J. Mol. Sci. 15, 7462–7474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gottlieb R. (2013) Overview: Selective Removal of Aggregates and Organelles in Autophagy in Health and Disease (Gottlieb R., ed), pp. 3–9, Acadmic Press; London, UK [Google Scholar]
- 11. Pallet N., Sirois I., Bell C., Hanafi L. A., Hamelin K., Dieudé M., Rondeau C., Thibault P., Desjardins M., Hebert M. J. (2013) A comprehensive characterization of membrane vesicles released by autophagic human endothelial cells. Proteomics 13, 1108–1120 [DOI] [PubMed] [Google Scholar]
- 12. Sirois I., Groleau J., Pallet N., Brassard N., Hamelin K., Londono I., Pshezhetsky A. V., Bendayan M., Hébert M. J. (2012) Caspase activation regulates the extracellular export of autophagic vacuoles. Autophagy 8, 927–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang A. L., Lukas T. J., Yuan M., Du N., Tso M. O., Neufeld A. H. (2009) Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One 4, e4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanida I., Minematsu-Ikeguchi N., Ueno T., Kominami E. (2005) Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1, 84–91 [DOI] [PubMed] [Google Scholar]
- 15. Caramés B., Taniguchi N., Otsuki S., Blanco F. J., Lotz M. (2010) Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 62, 791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Caramés B., Hasegawa A., Taniguchi N., Miyaki S., Blanco F. J., Lotz M. (2012) Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 71, 575–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lopez de Figueroa P., Lotz M., Blanco F. J., Carames B. (2015) Autophagy activation protects from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 67, 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenthal A. K., Cheung H. S., Ryan L. M. (1991) Transforming growth factor beta 1 stimulates inorganic pyrophosphate elaboration by porcine cartilage. Arthritis Rheum. 34, 904–911 [DOI] [PubMed] [Google Scholar]
- 19. Mitchell P. G., Struve J. A., McCarthy G. M., Cheung H. S. (1992) Basic calcium phosphate crystals stimulate cell proliferation and collagenase message accumulation in cultured adult articular chondrocytes. Arthritis Rheum. 35, 343–350 [DOI] [PubMed] [Google Scholar]
- 20. Arkesteijn C. (1976) A kinetic method for serum 5′ -nucleotidase using stabilized glutamate dehydrogenase. J. Clin. Chem. Clin. Biochem. 14, 155–159 [DOI] [PubMed] [Google Scholar]
- 21. Huang K., Fingar D. C. (2014) Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 36, 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen J., Zheng X. F., Brown E. J., Schreiber S. L. (1995) Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl. Acad. Sci. U.S.A. 92, 4947–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choi J., Chen J., Schreiber S. L., Clardy J. (1996) Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242 [DOI] [PubMed] [Google Scholar]
- 24. Sasaki H., Takayama K., Matsushita T., Ishida K., Kubo S., Matsumoto T., Fujita N., Oka S., Kurosaka M., Kuroda R. (2012) Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 64, 1920–1928 [DOI] [PubMed] [Google Scholar]
- 25. Wang A., Hascall V. C. (2009) Hyperglycemia, intracellular hyaluronan synthesis, cyclin D3 and autophagy. Autophagy 5, 864–865 [DOI] [PubMed] [Google Scholar]
- 26. Kalamida D., Karagounis I. V., Giatromanolaki A., Koukourakis M. I. (2014) Important role of autophagy in endothelial cell response to ionizing radiation. PLoS. One 9, e102408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shen D., Coleman J., Chan E., Nicholson T. P., Dai L., Sheppard P. W., Patton W. F. (2011) Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem. Biophys. 60, 173–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pfeiffer Z. A., Aga M., Prabhu U., Watters J. J., Hall D. J., Bertics P. J. (2004) The nucleotide receptor P2X7 mediates actin reorganization and membrane blebbing in RAW 264.7 macrophages via p38 MAP kinase and Rho. J. Leukoc. Biol. 75, 1173–1182 [DOI] [PubMed] [Google Scholar]
- 29. Lee S. W., Song Y. S., Lee S. Y., Yoon Y. G., Lee S. H., Park B. S., Yun I., Choi H., Kim K., Chung W. T., Yoo Y. H. (2011) Downregulation of protein kinase CK2 activity facilitates tumor necrosis factor-α-mediated chondrocyte death through apoptosis and autophagy. PLoS One 6, e19163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duran J. M., Anjard C., Stefan C., Loomis W. F., Malhotra V. (2010) Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 188, 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manjithaya R., Subramani S. (2010) Role of autophagy in unconventional protein secretion. Autophagy 6, 650–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fader C. M., Sánchez D., Furlán M., Colombo M. I. (2008) Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 9, 230–250 [DOI] [PubMed] [Google Scholar]
- 33. Roy S., Meachim G. (1968) Chondrocyte ultrastructure in adult human articular cartilage. Ann. Rheum. Dis. 27, 544–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Antonyak M. A., Li B., Boroughs L. K., Johnson J. L., Druso J. E., Bryant K. L., Holowka D. A., Cerione R. A. (2011) Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hale J. E., Wuthier R. E. (1987) The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J. Biol. Chem. 262, 1916–1925 [PubMed] [Google Scholar]
- 36. Thouverey C., Strzelecka-Kiliszek A., Balcerzak M., Buchet R., Pikula S. (2009) Matrix vesicles originate from apical membrane microvilli of mineralizing osteoblast-like Saos-2 cells. J. Cell Biochem. 106, 127–138 [DOI] [PubMed] [Google Scholar]
- 37. Godin C. M., Ferguson S. S. (2010) The angiotensin II type 1 receptor induces membrane blebbing by coupling to Rho A, Rho kinase, and myosin light chain kinase. Mol. Pharmacol. 77, 903–911 [DOI] [PubMed] [Google Scholar]
- 38. Wiley J. S., Sluyter R., Gu B. J., Stokes L., Fuller S. J. (2011) The human P2X7 receptor and its role in innate immunity. Tissue Antigens 78, 321–332 [DOI] [PubMed] [Google Scholar]
- 39. Bovellan M., Fritzsche M., Stevens C., Charras G. (2010) Death-associated protein kinase (DAPK) and signal transduction: blebbing in programmed cell death. FEBS J 277, 58–65 [DOI] [PubMed] [Google Scholar]
- 40. Roach H. I., Aigner T., Kouri J. B. (2004) Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis 9, 265–277 [DOI] [PubMed] [Google Scholar]
- 41. Almonte-Becerril M., Navarro-Garcia F., Gonzalez-Robles A., Vega-Lopez M. A., Lavalle C., Kouri J. B. (2010) Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis 15, 631–638 [DOI] [PubMed] [Google Scholar]
- 42. Kirsch T., Wang W., Pfander D. (2003) Functional differences between growth plate apoptotic bodies and matrix vesicles. J. Bone Miner. Res. 18, 1872–1881 [DOI] [PubMed] [Google Scholar]