

Abstract
Antimicrobial resistance constitutes one of the major worldwide public health concerns. Bacteria are becoming resistant to the vast majority of antibiotics, and nowadays, a common infection can be fatal. To address this situation, the use of phages for the treatment of bacterial infections has been extensively studied as an alternative therapeutic strategy. Since Pseudomonas aeruginosa is one of the most common causes of health care-associated infections, many studies have reported the in vitro and in vivo antibacterial efficacy of phage therapy against this bacterium. This review collects data of all the P. aeruginosa phages sequenced to date, providing a better understanding about their biodiversity. This review further addresses the in vitro and in vivo results obtained by using phages to treat or prevent P. aeruginosa infections as well as the major hurdles associated with this therapy.
INTRODUCTION
Bacteriophages, or phages, are bacterial viruses that were discovered almost a century ago (1, 2). These viruses are considered to be among the most ubiquitous and diversified organisms on Earth and are believed to exist wherever their hosts thrive (3–6). According to their life cycles, phages can be distributed into two classes: virulent phages (strictly lytic) and temperate phages (7). Lytic phages adsorb to the host cell surface, inject and replicate their DNA, and then induce host cell lysis, resulting in the release of progeny phages that can start another round of infection (7). Temperate phages generally integrate their genome into the host chromosome or sometimes maintain it as a plasmid which is transmitted, by cell division, to the daughter cells (7, 8). For therapeutic purposes, only lytic phages are of interest.
The use of phages as antimicrobial agents was proposed in 1917 by Félix d'Hérelle, and the early results reported were promising (2, 9, 10). However, after the arrival of the golden era of antibiotics, together with the scientific controversy regarding the use of phages due to poorly controlled trials, the interest in phage therapy decreased sharply (11). Antibiotics were cheap and extremely effective against bacterial diseases and thus were considered the perfect solution to fight bacterial diseases. Consequently, following World War II, phage therapy was abandoned in the Western world but kept being used in Poland and the Soviet Union (11–13). The inadequate use of antibiotics has significantly increased the emergence of multidrug-resistant (MDR) bacteria, and new alternative strategies for antibiotherapy are highly desired by the worldwide scientific community (14–16). Consequently, a renewed interest was given to phages and they have been again come to be considered a good option for the treatment of bacterial infections (17–20).
Bacteriophages specific for the Pseudomonas genus were first described in the middle of the 20th century (21, 22), and there are currently (27 January 2015) 137 completely sequenced Pseudomonas phage genomes found in public databases. Since Pseudomonas aeruginosa is one of the most problematic opportunistic pathogens involved in hospital-acquired infections (23–26), large fractions of the phage application studies and genome sequencing projects have been focused on this bacterium (3, 5, 27–33).
This review gathers information about the huge diversity of P. aeruginosa phages, and it further describes in vitro and in vivo phage therapy studies designed to defeat this problematic bacterium that have been reported throughout the years.
OVERVIEW OF P. AERUGINOSA CLINICAL IMPACT: VIRULENCE FACTORS AND ANTIMICROBIAL RESISTANCE
P. aeruginosa is a Gram-negative opportunistic bacterial pathogen frequently present in diverse environments such as water, soil, and plants (34, 35) that is able to infect different organisms, including plants, animals, and humans (34, 36, 37). This opportunistic bacterium is one of the most common causes of health care-associated diseases, including pneumonia, bloodstream infections, urinary tract infections, and surgical site infections, especially in patients with compromised host defenses (34, 35, 37, 38). Most of the infections caused by P. aeruginosa are strikingly difficult to treat using conventional antibiotic therapies, since this microorganism displays high intrinsic resistance to a wide range of antibiotics, including aminoglycosides, fluoroquinolones, and β-lactams (37), which results in significant morbidity and mortality rates (35). According to the U.S. Centers for Disease Control and Prevention, it is estimated that 51,000 health care-associated P. aeruginosa infections occur each year in the United States alone (38). About 13% of these infections are multidrug resistant, with roughly 400 deaths per year attributed to such infections (38). In addition to possessing intrinsic resistance mechanisms (37, 39, 40), P. aeruginosa also has other mechanisms that confer resistance to antibiotics (Fig. 1). For instance, this bacterium quickly develops acquired resistance as a consequence of mutations or through acquisition of antibiotic resistance genes (37, 39). Furthermore, the inadequate use of antibiotics or exposure to different environmental conditions or stresses has induced the development of adaptive resistance (37, 41).
FIG 1.
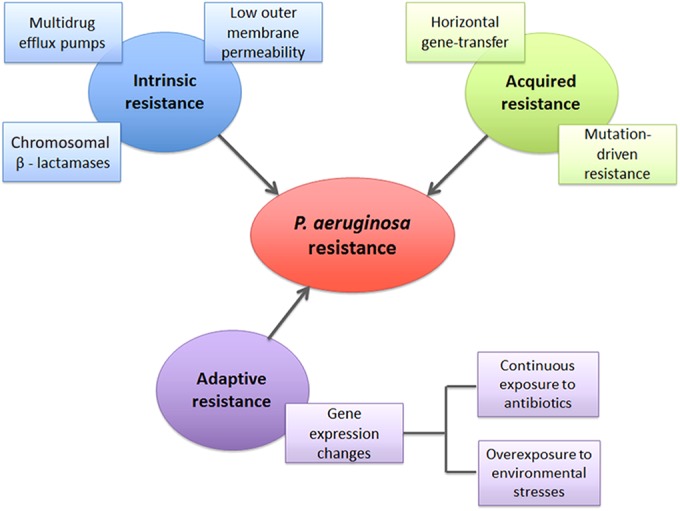
P. aeruginosa resistance mechanisms.
P. aeruginosa possesses an arsenal of virulence factors enabling it to invade host cells and circumvent host defenses (35, 42). These mechanisms of virulence include secreted factors such as proteases, elastase, pyocyanin, exotoxin A, phospholipase C, and exoenzyme S and also cell-associated factors such as lipopolysaccharide (LPS), flagella, and pili (35, 43). Many of these virulence factors are controlled by a cell-cell communication mechanism known as quorum sensing (QS), which allows bacteria to produce these factors in a cell density-dependent manner (35, 43, 44).
P. aeruginosa also has an ability to adhere to surfaces, such as those of medical devices or epithelial cells, and to form biofilms consisting of microbial communities embedded in a matrix of extracellular polymeric substances (EPS) (45–49). Biofilms are frequently associated with chronic infections, since their structure confers protection of bacterial cells from antimicrobial agents (50).
All of these virulence factors and resistance mechanisms contribute to increased P. aeruginosa pathogenicity, resulting in infections that are consequently very hard to treat (23, 37, 39).
P. AERUGINOSA BACTERIOPHAGES
To date, 94.2% of the 137 known phages targeting the Pseudomonas genus have been found to belong to the Caudovirales order (see Table S1 in the supplemental material), which comprises three families of double-stranded DNA (dsDNA) phages differing in the characteristics of the phage tail: Podoviridae, with a short and noncontractile tail; Myoviridae, with a long and contractile tail, and Siphoviridae, with a long and noncontractile tail (3, 5, 51). So far, only 8 nontailed Pseudomonas phage species have been sequenced: 2 belonging to the Inoviridae family (single-stranded DNA [ssDNA] phages) (52, 53), 2 belonging to the Leviviridae family (ssRNA phages) (54, 55), and 4 belonging to the Cystoviridae family (dsRNA phages) (56–58).
According to the data collected, 85% of the sequenced Pseudomonas genus phages belonging to the Caudovirales order are specific for the P. aeruginosa species (Fig. 2A) and the clear majority (approximately 60%) of these are lytic phages whereas 21.8% are temperate and 18.2% are unclassified (see Table S1 in the supplemental material). Among the P. aeruginosa lytic phages, 41% belong to the Myoviridae family and 38% belong to the Podoviridae family, and the least representative family is the Siphoviridae (20%). Only 1% of the lytic P. aeruginosa phages are nonclassified (Fig. 2B). The genome size distribution of P. aeruginosa lytic phages by family is fairly divergent (Fig. 2C), with Myoviridae phages comprising genome sizes ranging from 64.1 kb to 309.2 kb whereas the Podoviridae and Siphoviridae phage genomes are much smaller and are in the ranges of 41.6 to 74.9 kb and 34.5 to 61.1 kb, respectively. P. aeruginosa lytic phages have been isolated from different sources all over the world (Fig. 2D and E); nevertheless, it seems that sewage, including hospital and wastewater treatment plant sewage, is the best choice for their isolation (56%).
FIG 2.

(A) Distribution of Caudovirales phages targeting the Pseudomonas genus by Pseudomonas species. (B) Distribution of P. aeruginosa lytic phages by family and genus of phages. (C) Genome sizes of P. aeruginosa lytic phages. (D) Isolation sources of P. aeruginosa lytic phages. (E) Distribution of P. aeruginosa lytic phage isolates by country.
IN VITRO PHAGE TRIALS
Many in vitro studies have been conducted in recent years to evaluate the potential of phages against P. aeruginosa clinical isolates, including multidrug-resistant (MDR) strains, in planktonic cultures or even biofilms.
Fu et al. studied the effect of lytic phages in the prevention of P. aeruginosa biofilm formation in hydrogel-coated catheters. For this, they pretreated catheters with Myoviridae phage M4 for 2 h prior to bacterial inoculation and observed a 2.8-log reduction in the number of viable cells compared with control (untreated) catheters after 24 h of biofilm formation. However, a regrowth of biofilms in phage-treated catheters was observed at between 24 and 48 h of biofilm formation. The phage-resistant biofilm isolates were recovered from the pretreated catheters, and, in accordance with their susceptibility profiles with respect to other phages, a cocktail of five phages was developed. The pretreatment of catheters with this cocktail resulted in a 3-log reduction of biofilm cell populations after 48 h, compared with untreated catheters (32).
Pires et al. used a broad-host-range phage for P. aeruginosa biofilm control (59). Although significant reductions in the number of viable cells were observed after 6 h of biofilm treatment, a resurgence in the number of biofilm cells was observed after 24 h of biofilm infection, indicating the emergence of phage-resistant phenotypes (59).
A different approach for the treatment of bacterial infections is the combination of phages with other antimicrobials. Recently, Torres-Barceló et al. reported treatment using a combination of Podoviridae phage LUZ7 and streptomycin against P. aeruginosa PAO1. An exponential-growth culture of P. aeruginosa was challenged with either single or combined treatments, and the bacterial population density was tracked over 70 h. The combined therapy revealed a positive synergism resulting in a bacterial density lower than that seen after the addition of each single treatment (60).
Knezevic et al. also studied the antimicrobial efficacy of therapy consisting of P. aeruginosa-specific phages belonging to the Podoviridae and Siphoviridae families combined with subinhibitory concentrations of gentamicin (an aminoglycoside), ceftriaxone (a cephalosporin), ciprofloxacin (a quinolone), and polymyxin B (a polypeptide) (61). In their work, the effectiveness of the combined treatment was determined by the time-kill curve method and only the combination of the ceftriaxone with one of the phages used in the study revealed a synergistic effect (61). Another work reported a treatment consisting of lytic RNA phages combined with chlorine against P. aeruginosa biofilms that resulted in enhanced efficacy in the reduction of biofilm formation and biofilm eradication using the combined therapy compared with the use of each therapy alone (62).
The combination of two or more phages with different host ranges in a single suspension—a phage cocktail—has been reported to be more effective than the use of a single phage alone (63–65). Accordingly, Hall et al. studied the effect of using one, two, or four phages, either sequentially or simultaneously, against P. aeruginosa PAO1 planktonic cultures (66). Multiphage therapy was revealed to be more efficient in reducing the bacterial density, and simultaneous application of phages was consistently equal or superior to sequential application with respect to efficacy (66).
CLINICAL TRIALS OF PHAGES IN ANIMAL MODELS
The efficacy and safety of phage therapy have been analyzed using in vivo animal models. Most of the P. aeruginosa phage therapy clinical animal trials that have been reported in the last decade were conducted in mouse models and have shown extremely encouraging results (Table 1).
TABLE 1.
Summary of the most relevant studies with targeted P. aeruginosa phage therapy in animal modelsa
| Phage(s) (taxonomy) | Procedure | Result(s) | Reference |
|---|---|---|---|
| Mouse | |||
| KPP12 (Myoviridae) | Phage at an MOI of 100 was applied in the corneal surface of mice 30 min after infection, and the eyes were examined 1, 3, and 5 days after phage application | The treated mice showed only slight corneal opacities on day 1 which gradually faded by day 5 after treatment; the infected control mice had most of the corneas perforated on day 5 | 67 |
| ØNH-4 (Myoviridae) and ØMR299-2 (Podoviridae) | A phage mix suspension with an MOI of 10 was administered intranasally 2 h after infection | The use of the two phages together was able to reduce P. aeruginosa levels by at least 3 to 4 log units from murine lungs in 6 h | 68 |
| P3_CHA (Myoviridae) | Two doses of phage (MOI of 1 and MOI of 10) were applied intranasally 2 h after the onset of infection in the case of curative treatment or 4 days before infection in the case of preventive treatment | The curative phage treatment allowed over 95% survival of mice, and the preventive treatment resulted in 100% survival in the 16 days of the expt | 31 |
| PA1Ø (Siphoviridae) | Phages were administered via intraperitoneal route in normal mice (MOI of 1, 10, and 100) and neutropenic mice (MOI of 10) | Immunocompetent mice treated with phage achieved 80% to 100% survival rates, and no viable bacteria were found in organ samples after 48 h of the phage treatment; phage treatment was inefficient in neutropenic mice infected with P. aeruginosa PAO1 even though it was able to extend their lives for 12 h | 69 |
| PAK-P1 (Myoviridae) | Phages were applied intranasally 2 h after bacterial infection onset (MOI of 0.1, 1, and 10), with expts performed for up to 12 days, or phages were used as a preventive treatment 24 h before infection (MOI of 10), with expts carried out during 16 days | 100% of the mice treated with an MOI of 10 survived until the end of the expt; in the preventive treatment, 100% of pretreated mice survived until the end of the expt, whereas 100% of untreated mice died within 2 days | 30 |
| MPK1 (Myoviridae) and MPK6 (Podoviridae) | Two different doses of phages (MOI of 1 and 10) were tested by administration through i.m. and i.p. routes after 6 to 12 h postinfection | The survival rate of mice achieved after 48 h of treatment with MK1 phage was up to 80% for i.m. administration or 100% for i.p. administration; the results with phage MK6 were significantly lower (about 20% for i.m. and 70% for i.p.) | 70 |
| Cocktail of 3 phages: ATCC 12175-B1, ATCC 14203-B1, and ATCC 14205-B1 | A phage cocktail containing 1 × 108 PFU of each phage was administered i.m., i.p., or s.c. | A single dose of phage cocktail significantly decreased the mortality of thermally injured P. aeruginosa-infected mice from 6% survival without treatment to 22% to 87% survival with treatment, depending on the route of administration | 29 |
| KPP10 (Myoviridae) | To evaluate the efficacy of phages against gut-derived sepsis, 1 × 1010 PFU was orally administered to each mice 1 day before, 1 day after, or 6 days after P. aeruginosa inoculation | Phage significantly protected mice from mortality, achieving a survival rate of 66.7% for the phage-treated group (1 day after infection) vs 0% for the control group; mice treated with phage also had lower numbers of viable P. aeruginosa cells in their blood, liver, and spleen | 33 |
| Catfish | |||
| MBL (Podoviridae) | A phage suspension (1010 PFU/ml) was applied on the infected skin lesion with a cotton swab | Phage therapy effectively cured the ulcerative lesions in infected fish after 8–10 days of treatment, with a 7-fold reduction of the lesion compared with untreated fish | 28 |
| Wax moth larvae | |||
| 14/1 (Myoviridae); φKZ (Myoviridae); PNM (Podoviridae); PT7 (Myoviridae) | Phages were applied by injection in the abdomen and three treatments were tested: phage φKZ applied alone, all phages applied sequentially, and all phages applied simultaneously in a cocktail | Phage therapy increased the life span of infected wax moth larvae: the avg time to death increased from 12.67 h to 26.67 h, 27.33 h, and 33.33 h, respectively, for larvae treated with φKZ, the multiphage sequence, and the phage cocktail; high phage/bacterium ratios completely cleared otherwise-lethal infections | 66 |
| Dog | |||
| NA | Dogs with chronic otitis were treated with a single dose of a phage cocktail (6 phages), containing 1 × 105 PFU of each phage, by direct application into the external auditory canal | 48 h after treatment, the clinical score and P. aeruginosa counts of all isolates from ears significantly decreased (mean score decrease, 30.1%; mean count decrease, 67%) | 71 |
| NA | A dog with P. aeruginosa chronic bilateral otitis externa was treated with 400 PFU of phage instilled into the right auditory canal | Before phage treatment, the ears were similarly erythematous, with copious discharge; 27 h after treatment, the right ear was dry and did not appear inflamed, while the left ear (not treated) remained unchanged; the left ear was then also treated with phage, which dramatically improved its condition | 72 |
| Drosophila melanogaster | |||
| MPK1 (Myoviridae) and MPK6 (Podoviridae) | The antibacterial efficacy of phages administered by feeding at a concn of 5 × 107 PFU was evaluated in Drosophila melanogaster systemic infection | Both phages significantly delayed the PAO1-induced killing, although MPK1 persisted longer than MPK6 in uninfected D. melanogaster tissue samples | 70 |
MOI, multiplicity of infection; NA, information not available; i.m., intramuscular; i.p., intraperitoneal; s.c., subcutaneous.
Overall, phage therapy has significantly contributed to control and even prevention of P. aeruginosa infections in vivo (Table 1). Regarding the clinical trials in mouse models, most of the reported cases showed survival rates that ranged between 80% and 100% after phage treatment, and intraperitoneal or intranasal administration seemed to be the most efficient route of administration.
The great potential of phage therapy was also observed in catfish and dogs, since phages were able to successfully treat ulcerative lesions and chronic otitis, respectively. Phages were also able to increase the life span of both P. aeruginosa-infected wax moth larvae and P. aeruginosa-infected Drosophila melanogaster. Overall, the in vivo studies (Table 1) have demonstrated that broad-host-range phage cocktails potentiate the efficacy of phage therapy to control P. aeruginosa infections.
CLINICAL TRIALS OF PHAGES IN HUMAN PATIENTS
The therapeutic use of phages started in Paris in 1919 when d'Herelle used oral phage preparations to treat bacterial dysentery (73). Patients treated with a single dose of phage preparation started to recover within 24 h of treatment (11, 20). Later, d'Herelle also introduced treatment with intravenous phage for invasive infections (74). However, the first report about the use of phages to treat bacterial infections in humans was published in 1921 by Bruynoghe and Maisin (9). In that study, phages were administered by injection to treat staphylococcal skin disease and clinical improvements were clearly visible within 24 to 48 h (9). Following those early reports, many other studies have shown that phages can be used successfully for therapeutic purposes (75, 76).
Even after the arrival of antibiotics, phage therapy continued to be extensively used to treat bacterial infections in eastern Europe and the former Soviet Union, and the Eliava Institute in Tbilisi (Georgia) was one of the crucial centers (11, 77). The increasing problem of bacterial resistance to antibiotics revitalized interest in phage therapy, and many clinical trials in humans have been conducted (72, 78–82).
The use of phages against P. aeruginosa has also been examined in clinical trials. For instance, the efficacy and safety of a therapeutic phage preparation (Biophage-PA) were reported by Wright et al. (82). According to those authors, 12 patients with antibiotic-resistant P. aeruginosa chronic otitis who were treated with a single dose of phage preparation and followed at 7, 21, and 42 days after treatment (82) showed significant clinical improvements compared to 12 placebo group individuals. Furthermore, no related side effects or local systemic toxicities were observed, which highlights the safety of phage therapy (82).
Previously, Sivera Marza et al. reported the successful topical use of phage to treat a burn patient who had been colonized by P. aeruginosa months after skin grafts had been applied (72). In this case, the purified phage preparation, adsorbed to filter paper discs, was placed in the patient's colonized burned areas, and 3 days later, P. aeruginosa was no further isolated, resulting in a subsequent successful grafting (72).
Merabishvili et al. described a quality-controlled small-scale production of a phage cocktail (BFC-1) for use in human clinical practice (83). The cocktail was designed to treat P. aeruginosa and Staphylococcus aureus burn wound infections, and the quality control parameters of the phage cocktail, such as stability, pyrogenicity, sterility, and cytotoxicity, were evaluated (83). The BFC-1 phage cocktail was then topically applied in the wounds of nine acute burn patients with MDR P. aeruginosa and/or S. aureus burn wound colonization (84). Although that study did not enable any conclusion regarding the efficacy of the phage cocktail, no adverse effects or clinical abnormalities were observed after phage application (84).
CURRENT CHALLENGES FACED BY PHAGE THERAPY
Despite the remarkable positive results obtained by phage therapy, there are still some major concerns regarding the use of phages in clinical practice. The fast development of bacterial resistance to phage, the immune response that induces the production of antibodies neutralizing phage action, and the safety concerns regarding phage preparations that might contain bacterial endo- and exotoxins, as well as the limited spectrum of activity, are some of the major drawbacks of phage therapy that limit its broad application. Some of these bottlenecks are already being addressed by the introduction of molecular biology techniques that enable the genetic engineering of phage genomes. For example, phages have been genetically engineered to enhance their performance against biofilms (85), to improve antibiotic activity (86, 87), and to expand their host ranges (88–91). The use of well-defined and purified phage preparations is also crucial for therapeutic applications and for regulatory approval (83, 92). Furthermore, a deep understanding of the phage-host interaction, phage diversity, phage dynamics, and genome function is crucial for a better understanding of the limitations of phage therapy and for designing new strategies to overcome it.
CONCLUDING REMARKS
The continuous development of antibiotic-resistant bacteria, together with the current low rate of antibiotic discovery, urgently demands novel and effective strategies to combat bacterial infections. The huge diversity of phages, the easy discovery of new ones, their low-cost production, and their effectiveness against the target bacteria make them a very attractive alternative option to antibiotics.
The in vitro and animal model studies reported to date are good indicators that phages effectively constitute a powerful tool to fight P. aeruginosa infections. Early work in human patients also showed promising results. Furthermore, phages can be used in combination with antibiotics or other antimicrobials for improved performance.
Supplementary Material
ACKNOWLEDGMENTS
D.P.P. acknowledges financial support from the Portuguese Foundation for Science and Technology (FCT) (grant SFRH/BD/76440/2011). S.S. is an FCT investigator (IF/01413/2013). The authors also thank FCT for the Strategic Project of the UID/BIO/04469/2013 unit, FCT and the European Union (FEDER/COMPETE) for funds for the RECI/BBB-EBI/0179/2012 (FCOMP-01-0124-FEDER-027462) project, and the “BioHealth-Biotechnology and Bioengineering approaches to improve health quality” (NORTE-07-0124-FEDER-000027) project, cofunded by the Programa Operacional Regional do Norte (ON.2–O Novo Norte), QREN, and FEDER.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00385-15.
REFERENCES
- 1.Twort FW. 1915. An investigation on the nature of ultra-microscopic viruses. Lancet 186:1241–1243. doi: 10.1016/S0140-6736(01)20383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.d'Hérelle F. 1917. Sur un microbe invisible antagoniste des bacilles dysentériques. C R Hebd Seances Acad Sci Ser D 165:373–375. [Google Scholar]
- 3.Ceyssens P-J, Lavigne R. 2010. Bacteriophages of Pseudomonas. Future Microbiol 5:1041–1055. doi: 10.2217/fmb.10.66. [DOI] [PubMed] [Google Scholar]
- 4.Clokie MR, Millard AD, Letarov AV, Heaphy S. 2011. Phages in nature. Bacteriophage 1:31–45. doi: 10.4161/bact.1.1.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azeredo J, Sillankorva S, Pires DP. 2014. Pseudomonas bacteriophage isolation and production, p 23–32. In Filloux A, Ramos J-L (ed), Pseudomonas methods and protocols. Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 6.Holmfeldt K, Solonenko N, Shah M, Corrier K, Riemann L, Verberkmoes NC, Sullivan MB. 2013. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc Natl Acad Sci U S A 110:12798–12803. doi: 10.1073/pnas.1305956110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guttman B, Raya R, Kutter E. 2004. Basic phage biology, p 29–66. In Kutter E, Sulakvelidze A (ed), Bacteriophages: biology and applications. CRC Press LLC, Boca Raton, FL. [Google Scholar]
- 8.Little J. 2005. Lysogeny, prophage induction, and lysogenic conversion, p 37–54. In Waldor M, Friedman D, Adhya S (ed), Phages: their role in bacterial pathogenesis and biotechnology. ASM Press, Washington, DC. [Google Scholar]
- 9.Bruynoghe R, Maisin J. 1921. Essais de thérapeutique au moyen du bacteriophage. C R Soc Biol 85:1120–1121. [Google Scholar]
- 10.d'Hérelle F. 1921. Le bactériophage: son rôle dans l'immunité. Masson et Cie, Paris, France. [Google Scholar]
- 11.Sulakvelidze A, Alavidze Z, Morris JG. 2001. Bacteriophage therapy. Antimicrob Agents Chemother 45:649–659. doi: 10.1128/AAC.45.3.649-659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dublanchet A, Fruciano E. 2008. A short history of phage therapy. Med Mal Infect 38:415–420. doi: 10.1016/j.medmal.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 13.Chanishvili N. 2012. Phage therapy—history from Twort and d'Herelle through Soviet experience to current approaches. Adv Virus Res 83:3–40. doi: 10.1016/B978-0-12-394438-2.00001-3. [DOI] [PubMed] [Google Scholar]
- 14.Levy SB, Marshall B. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med 10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 15.Davies J, Davies D. 2010. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wright GD, Sutherland AD. 2007. New strategies for combating multidrug-resistant bacteria. Trends Mol Med 13:260–267. doi: 10.1016/j.molmed.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Kutateladze M, Adamia R. 2010. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol 28:591–595. doi: 10.1016/j.tibtech.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Potera C. 2013. Phage renaissance: new hope against antibiotic resistance. Environ Health Perspect 121:a48–a53. doi: 10.1289/ehp.121-a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuzaki S, Rashel M, Uchiyama J, Sakurai S, Ujihara T, Kuroda M, Ikeuchi M, Tani T, Fujieda M, Wakiguchi H, Imai S. 2005. Bacteriophage therapy: a revitalized therapy against bacterial infectious diseases. J Infect Chemother 11:211–219. doi: 10.1007/s10156-005-0408-9. [DOI] [PubMed] [Google Scholar]
- 20.Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM. 2011. Phage treatment of human infections. Bacteriophage 1:66–85. doi: 10.4161/bact.1.2.15845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kellenberger G, Kellenberger E. 1957. Electron microscopical studies of phage multiplication. Virology 3:275–285. doi: 10.1016/0042-6822(57)90093-4. [DOI] [PubMed] [Google Scholar]
- 22.Holloway B, Egan J, Monk M. 1960. Lysogeny in Pseudomonas aeruginosa. Aust J Exp Biol Med Sci 38:321–330. doi: 10.1038/icb.1960.34. [DOI] [PubMed] [Google Scholar]
- 23.Driscoll JA, Brody SL, Kollef MH. 2007. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs 67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- 24.Mah T-F, Pitts B, Pellock B, Walker GC, Stewart PS, O'Toole GA. 2003. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 426:306–310. doi: 10.1038/nature02122. [DOI] [PubMed] [Google Scholar]
- 25.Obritsch MD, Fish DN, MacLaren R, Jung R. 2005. Nosocomial infections due to multidrug-resistant Pseudomonas aeruginosa: epidemiology and treatment options. Pharmacotherapy 25:1353–1364. doi: 10.1592/phco.2005.25.10.1353. [DOI] [PubMed] [Google Scholar]
- 26.Aloush V, Navon-Venezia S, Seigman-Igra Y, Cabili S, Carmeli Y. 2006. Multidrug-resistant Pseudomonas aeruginosa: risk factors and clinical impact. Antimicrob Agents Chemother 50:43–48. doi: 10.1128/AAC.50.1.43-48.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Essoh C, Blouin Y, Loukou G, Cablanmian A, Lathro S, Kutter E, Thien HV, Vergnaud G, Pourcel C. 2013. The susceptibility of Pseudomonas aeruginosa strains from cystic fibrosis patients to bacteriophages. PLoS One 8:e60575. doi: 10.1371/journal.pone.0060575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khairnar K, Raut MP, Chandekar RH, Sanmukh SG, Paunikar WN. 2013. Novel bacteriophage therapy for controlling metallo-beta-lactamase producing Pseudomonas aeruginosa infection in catfish. BMC Vet Res 9:264. doi: 10.1186/1746-6148-9-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McVay CS, Velásquez M, Fralick JA. 2007. Phage therapy of Pseudomonas aeruginosa infection in a mouse burn wound model. Antimicrob Agents Chemother 51:1934–1938. doi: 10.1128/AAC.01028-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Debarbieux L, Leduc D, Maura D, Morello E, Criscuolo A, Grossi O, Balloy V, Touqui L. 2010. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J Infect Dis 201:1096–1104. doi: 10.1086/651135. [DOI] [PubMed] [Google Scholar]
- 31.Morello E, Saussereau E, Maura D, Huerre M, Touqui L, Debarbieux L. 2011. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PLoS One 6:e16963. doi: 10.1371/journal.pone.0016963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu W, Forster T, Mayer O, Curtin JJ, Lehman SM, Donlan RM. 2010. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob Agents Chemother 54:397–404. doi: 10.1128/AAC.00669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe R, Matsumoto T, Sano G, Ishii Y, Tateda K, Sumiyama Y, Uchiyama J, Sakurai S, Matsuzaki S, Imai S, Yamaguchi K. 2007. Efficacy of bacteriophage therapy against gut-derived sepsis caused by Pseudomonas aeruginosa in mice. Antimicrob Agents Chemother 51:446–452. doi: 10.1128/AAC.00635-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pereira SG, Rosa AC, Ferreira AS, Moreira LM, Proença DN, Morais PV, Cardoso O. 2014. Virulence factors and infection ability of Pseudomonas aeruginosa isolates from a hydropathic facility and respiratory infections. J Appl Microbiol 116:1359–1368. doi: 10.1111/jam.12463. [DOI] [PubMed] [Google Scholar]
- 35.Wagner VE, Filiatrault MJ, Picardo KF, Iglewski BH. 2008. Pseudomonas aeruginosa virulence and pathogenesis issues, p 129–158. In Cornelis P. (ed), Pseudomonas genomics and molecular biology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 36.Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breidenstein EBM, de la Fuente-Núñez C, Hancock REW. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 38.CDC. 2013. Antibiotic resistance threats in the United States. CDC, Atlanta, GA. [Google Scholar]
- 39.Fajardo A, Martínez JL. 2008. Antibiotic resistance in Pseudomonas, p 177–211. In Cornelis P. (ed), Pseudomonas genomics and molecular biology. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 40.Fajardo A, Martínez-Martín N, Mercadillo M, Galán JC, Ghysels B, Matthijs S, Cornelis P, Wiehlmann L, Tümmler B, Baquero F, Martínez JL. 2008. The neglected intrinsic resistome of bacterial pathogens. PLoS One 3:e1619. doi: 10.1371/journal.pone.0001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernández L, Breidenstein EBM, Hancock REW. 2011. Creeping baselines and adaptive resistance to antibiotics. Drug Resist Updat 14:1–21. doi: 10.1016/j.drup.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Ballok AE, O'Toole GA. 2013. Pouring salt on a wound: Pseudomonas aeruginosa virulence factors alter Na+ and Cl− flux in the lung. J Bacteriol 195:4013–4019. doi: 10.1128/JB.00339-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strateva T, Mitov I. 2011. Contribution of an arsenal of virulence factors to pathogenesis of Pseudomonas aeruginosa infections. Ann Microbiol 61:717–732. doi: 10.1007/s13213-011-0273-y. [DOI] [Google Scholar]
- 44.Bjarnsholt T, Jensen PØ, Jakobsen TH, Phipps R, Nielsen AK, Rybtke MT, Tolker-Nielsen T, Givskov M, Høiby N, Ciofu O. 2010. Quorum sensing and virulence of Pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PLoS One 5:e10115. doi: 10.1371/journal.pone.0010115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Medina R, Dunne WM, Singh PK, Brody SL. 2005. Pseudomonas aeruginosa acquires biofilm-like properties within airway epithelial cells. Infect Immun 73:8298–8305. doi: 10.1128/IAI.73.12.8298-8305.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreau-Marquis S, Stanton BA, O'Toole GA. 2008. Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm Pharmacol Ther 21:595–599. doi: 10.1016/j.pupt.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson GG, Moreau-Marquis S, Stanton BA, O'Toole GA. 2008. In vitro analysis of tobramycin-treated Pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect Immun 76:1423–1433. doi: 10.1128/IAI.01373-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drenkard E, Ausubel FM. 2002. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416:740–743. doi: 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- 49.Watnick P, Kolter R. 2000. Biofilm, city of microbes. J Bacteriol 182:2675–2679. doi: 10.1128/JB.182.10.2675-2679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 51.Ackermann H-W. 2009. Phage classification and characterization. Methods Mol Biol 501:127–140. doi: 10.1007/978-1-60327-164-6_13. [DOI] [PubMed] [Google Scholar]
- 52.Luiten RG, Putterman DG, Schoenmakers JG, Konings RN, Day LA. 1985. Nucleotide sequence of the genome of Pf3, an IncP-1 plasmid-specific filamentous bacteriophage of Pseudomonas aeruginosa. J Virol 56:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holland SJ, Sanz C, Perham RN. 2006. Identification and specificity of pilus adsorption proteins of filamentous bacteriophages infecting Pseudomonas aeruginosa. Virology 345:540–548. doi: 10.1016/j.virol.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 54.Olsthoorn RC, Garde G, Dayhuff T, Atkins JF, Van Duin J. 1995. Nucleotide sequence of a single-stranded RNA phage from Pseudomonas aeruginosa: kinship to coliphages and conservation of regulatory RNA structures. Virology 206:611–625. doi: 10.1016/S0042-6822(95)80078-6. [DOI] [PubMed] [Google Scholar]
- 55.Ruokoranta TM, Grahn AM, Ravantti JJ, Poranen MM, Bamford DH. 2006. Complete genome sequence of the broad host range single-stranded RNA phage PRR1 places it in the Levivirus genus with characteristics shared with alloleviviruses. J Virol 80:9326–9330. doi: 10.1128/JVI.01005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoogstraten D, Qiao X, Sun Y, Hu A, Onodera S, Mindich L. 2000. Characterization of phi8, a bacteriophage containing three double-stranded RNA genomic segments and distantly related to Phi6. Virology 272:218–224. doi: 10.1006/viro.2000.0374. [DOI] [PubMed] [Google Scholar]
- 57.Qiao X, Qiao J, Onodera S, Mindich L. 2000. Characterization of phi 13, a bacteriophage related to phi 6 and containing three dsRNA genomic segments. Virology 275:218–224. doi: 10.1006/viro.2000.0501. [DOI] [PubMed] [Google Scholar]
- 58.Gottlieb P, Wei H, Potgieter C, Toporovsky I. 2002. Characterization of phi 12, a bacteriophage related to phi 6: nucleotide sequence of the small and middle double-stranded RNA. Virology 293:118–124. doi: 10.1006/viro.2001.1288. [DOI] [PubMed] [Google Scholar]
- 59.Pires D, Sillankorva S, Faustino A, Azeredo J. 2011. Use of newly isolated phages for control of Pseudomonas aeruginosa PAO1 and ATCC 10145 biofilms. Res Microbiol 162:798–806. doi: 10.1016/j.resmic.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 60.Torres-Barceló C, Arias-Sánchez FI, Vasse M, Ramsayer J, Kaltz O, Hochberg ME. 2014. A window of opportunity to control the bacterial pathogen Pseudomonas aeruginosa combining antibiotics and phages. PLoS One 9:e106628. doi: 10.1371/journal.pone.0106628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knezevic P, Curcin S, Aleksic V, Petrusic M, Vlaski L. 2013. Phage-antibiotic synergism: a possible approach to combatting Pseudomonas aeruginosa. Res Microbiol 164:55–60. doi: 10.1016/j.resmic.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Hu Z. 2013. Combined treatment of Pseudomonas aeruginosa biofilms with bacteriophages and chlorine. Biotechnol Bioeng 110:286–295. doi: 10.1002/bit.24630. [DOI] [PubMed] [Google Scholar]
- 63.Gu J, Liu X, Li Y, Han W, Lei L, Yang Y, Zhao H, Gao Y, Song J, Lu R, Sun C, Feng X. 2012. A method for generation phage cocktail with great therapeutic potential. PLoS One 7:e31698. doi: 10.1371/journal.pone.0031698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan BK, Abedon ST, Loc-Carrillo C. 2013. Phage cocktails and the future of phage therapy. Future Microbiol 8:769–783. doi: 10.2217/fmb.13.47. [DOI] [PubMed] [Google Scholar]
- 65.Jaiswal A, Koley H, Ghosh A, Palit A, Sarkar B. 2013. Efficacy of cocktail phage therapy in treating Vibrio cholerae infection in rabbit model. Microbes Infect 15:152–156. doi: 10.1016/j.micinf.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 66.Hall AR, De Vos D, Friman V-P, Pirnay J-P, Buckling A. 2012. Effects of sequential and simultaneous applications of bacteriophages on populations of Pseudomonas aeruginosa in vitro and in wax moth larvae. Appl Environ Microbiol 78:5646–5652. doi: 10.1128/AEM.00757-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fukuda K, Ishida W, Uchiyama J, Rashel M, Kato S, Morita T, Muraoka A, Sumi T, Matsuzaki S, Daibata M, Fukushima A. 2012. Pseudomonas aeruginosa keratitis in mice: effects of topical bacteriophage KPP12 administration. PLoS One 7:e47742. doi: 10.1371/journal.pone.0047742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alemayehu D, Casey PG, McAuliffe O, Guinane CM, Martin JG, Shanahan F, Coffey A, Ross RP, Hill C. 2012. Bacteriophages φMR299-2 and φNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. mBio 3:e00029-12. doi: 10.1128/mBio.00029-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tiwari BR, Kim S, Rahman M, Kim J. 2011. Antibacterial efficacy of lytic Pseudomonas bacteriophage in normal and neutropenic mice models. J Microbiol 49:994–999. doi: 10.1007/s12275-011-1512-4. [DOI] [PubMed] [Google Scholar]
- 70.Heo Y-J, Lee Y-R, Jung H-H, Lee J, Ko G, Cho Y-H. 2009. Antibacterial efficacy of phages against Pseudomonas aeruginosa infections in mice and Drosophila melanogaster. Antimicrob Agents Chemother 53:2469–2474. doi: 10.1128/AAC.01646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hawkins C, Harper D, Burch D, Anggård E, Soothill J. 2010. Topical treatment of Pseudomonas aeruginosa otitis of dogs with a bacteriophage mixture: a before/after clinical trial. Vet Microbiol 146:309–313. doi: 10.1016/j.vetmic.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 72.Sivera Marza JA, Soothill JS, Boydell P, Collyns TA. 2006. Multiplication of therapeutically administered bacteriophages in Pseudomonas aeruginosa infected patients. Burns 32:644–646. doi: 10.1016/j.burns.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 73.Sulakvelidze A, Kutter E. 2004. Bacteriophage therapy in humans, p 381–436. In Kutter E, Sulakvelidze A (ed), Bacteriophages: Biology and Applications. CRC Press LLC, Boca Raton, FL. [Google Scholar]
- 74.d'Herelle F. 1931. Bacteriophage as a treatment in acute medical and surgical infections. Bull N Y Acad Med 7:329–348. [PMC free article] [PubMed] [Google Scholar]
- 75.Rice TB. 1930. Use of bacteriophage filtrates in treatment of suppurative conditions: report of 300 cases. Am J Med Sci 179:345–360. [Google Scholar]
- 76.Schless RA. 1932. Staphylococcus aureus meningitis: treatment with specific bacteriophage. Am J Dis Child 44:813–822. doi: 10.1001/archpedi.1932.01950110115011. [DOI] [Google Scholar]
- 77.Pirnay J-P, De Vos D, Verbeken G, Merabishvili M, Chanishvili N, Vaneechoutte M, Zizi M, Laire G, Lavigne R, Huys I, Van den Mooter G, Buckling A, Debarbieux L, Pouillot F, Azeredo J, Kutter E, Dublanchet A, Górski A, Adamia R. 2011. The phage therapy paradigm: prêt-à-porter or sur-mesure? Pharm Res 28:934–937. doi: 10.1007/s11095-010-0313-5. [DOI] [PubMed] [Google Scholar]
- 78.Bruttin A, Brüssow H. 2005. Human volunteers receiving Escherichia coli phage T4 orally: a safety test of phage therapy. Antimicrob Agents Chemother 49:2874–2878. doi: 10.1128/AAC.49.7.2874-2878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rhoads DD, Wolcott RD, Kuskowski MA, Wolcott BM, Ward LS, Sulakvelidze A. 2009. Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J Wound Care 18:237–238, 240–243. doi: 10.12968/jowc.2009.18.6.42801. [DOI] [PubMed] [Google Scholar]
- 80.Weber-Dabrowska B, Mulczyk M, Górski A. 2001. Bacteriophage therapy for infections in cancer patients. Clin Appl Immunol Rev 1:131–134. doi: 10.1016/S1529-1049(01)00015-0. [DOI] [Google Scholar]
- 81.Sarker SA, McCallin S, Barretto C, Berger B, Pittet A-C, Sultana S, Krause L, Huq S, Bibiloni R, Bruttin A, Reuteler G, Brüssow H. 2012. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 434:222–232. doi: 10.1016/j.virol.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 82.Wright A, Hawkins CH, Anggård EE, Harper DR. 2009. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin Otolaryngol 34:349–357. doi: 10.1111/j.1749-4486.2009.01973.x. [DOI] [PubMed] [Google Scholar]
- 83.Merabishvili M, Pirnay J-P, Verbeken G, Chanishvili N, Tediashvili M, Lashkhi N, Glonti T, Krylov V, Mast J, Van Parys L, Lavigne R, Volckaert G, Mattheus W, Verween G, De Corte P, Rose T, Jennes S, Zizi M, De Vos D, Vaneechoutte M. 2009. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS One 4:e4944. doi: 10.1371/journal.pone.0004944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rose T, Verbeken G, Vos DD, Merabishvili M, Vaneechoutte M, Lavigne R, Jennes S, Zizi M, Pirnay J-P. 2014. Experimental phage therapy of burn wound infection: difficult first steps. Int J Burns Trauma 4:66–73. [PMC free article] [PubMed] [Google Scholar]
- 85.Lu TK, Collins JJ. 2007. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci U S A 104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu TK, Collins JJ. 2009. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci U S A 106:4629–4634. doi: 10.1073/pnas.0800442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Edgar R, Friedman N, Molshanski-Mor S, Qimron U. 2012. Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl Environ Microbiol 78:744–751. doi: 10.1128/AEM.05741-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoichi M, Abe M, Miyanaga K, Unno H, Tanji Y. 2005. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157:H7. J Biotechnol 115:101–107. doi: 10.1016/j.jbiotec.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 89.Mahichi F, Synnott AJ, Yamamichi K, Osada T, Tanji Y. 2009. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol Lett 295:211–217. doi: 10.1111/j.1574-6968.2009.01588.x. [DOI] [PubMed] [Google Scholar]
- 90.Lin T-Y, Lo Y-H, Tseng P-W, Chang S-F, Lin Y-T, Chen T-S. 2012. A T3 and T7 recombinant phage acquires efficient adsorption and a broader host range. PLoS One 7:e30954. doi: 10.1371/journal.pone.0030954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Heilpern AJ, Waldor MK. 2003. pIIICTX, a predicted CTXphi minor coat protein, can expand the host range of coliphage fd to include Vibrio cholerae. J Bacteriol 185:1037–1044. doi: 10.1128/JB.185.3.1037-1044.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Merril CR, Scholl D, Adhya SL. 2003. The prospect for bacteriophage therapy in Western medicine. Nat Rev Drug Discov 2:489–497. doi: 10.1038/nrd1111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.