

Abstract
Liver cirrhosis is a disease characterized by the loss of functional liver mass. Physiologically based pharmacokinetic (PBPK) modeling was applied to interpret and predict how the interplay among physiological changes in cirrhosis affects pharmacokinetics. However, previous PBPK models under cirrhotic conditions were developed for permeable cytochrome P450 substrates and do not directly apply to substrates of liver transporters. This study characterizes a PBPK model for liver transporter substrates in relation to the severity of liver cirrhosis. A published PBPK model structure for liver transporter substrates under healthy conditions and the physiological changes for cirrhosis are combined to simulate pharmacokinetics of liver transporter substrates in patients with mild and moderate cirrhosis. The simulated pharmacokinetics under liver cirrhosis reasonably approximate observations. This analysis includes meta-analysis to obtain system-dependent parameters in cirrhosis patients and a top-down approach to improve understanding of the effect of cirrhosis on transporter-mediated drug disposition under cirrhotic conditions.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ PBPK models have been developed to simulate pharmacokinetics of liver transporter substrates and more permeable compounds in healthy individuals. Previously developed PBPK models for individuals with liver cirrhosis assume well-stirred conditions in the liver, hence cannot be reasonably expected to describe the pharmacokinetics of transporter substrates. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ This study aims at characterizing a PBPK model capable of simulating the pharmacokinetics of liver transporter substrates under cirrhotic conditions by incorporating changes in physiological and biological parameter values. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ This study presents the first mechanistic model to estimate the impact of liver cirrhosis on human pharmacokinetics of liver transporter substrates. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY THERAPEUTICS ☑ This model can be useful in understanding the changes in liver transporter activity due to cirrhosis, and may aid in predicting systemic and liver exposure for liver transporter substrates under cirrhotic conditions. It can be useful in the design of clinical trials and ultimately for dose adjustments in clinical practice for cirrhotic patients in the future.
Liver cirrhosis is a progressive disease characterized by loss of liver function associated with morphological and physiological changes. The disease progression is usually classified using the Child–Pugh Grades (CP-A (mild), CP-B (moderate), and CP-C (severe)).1 Physiological changes include loss of functional liver size, decreased cytochrome P450 (CYP) expression, reduced glomerular filtration rate (GFR), and altered cardiac output, hepatic blood flow, hematocrit, and plasma albumin and α1-acid glycoprotein concentrations.1,2 The changes may affect systematic and tissue exposure of drugs administered to the patient. Under these pathological conditions, it is necessary to assess drug pharmacokinetics to evaluate potential risk and altered pharmacodynamic effects.
Compared to empirical pharmacokinetic models, physiologically based pharmacokinetic (PBPK) modeling explicitly incorporates physiological information, and can deconvolute multiple mechanisms controlling drug pharmacokinetics.3 As such, PBPK analysis can be invaluable to gain insights into the impact of physiological changes on the pharmacokinetics under disease conditions. PBPK models have been reported in previous publications to predict the pharmacokinetics in patients with liver impairment.4–6 With a “well-stirred” liver model,7 these PBPK models successfully extrapolate pharmacokinetics from healthy individuals to patients, in terms of adequately describing the observed plasma pharmacokinetics under different disease conditions.
In addition to the lipophilic compounds that have been mechanistically modeled,4–6 several groups have investigated the pharmacokinetics of liver transporter substrates under mild and moderate liver cirrhosis conditions, prompted by the fact that these compounds are mainly eliminated hepatically.8–12 In addition, some liver transporter substrates are developed to treat diseases associated with liver cirrhosis. For example, bosentan13 is a dual endothelin receptor antagonist used in the treatment of pulmonary artery hypertension, which has a higher prevalence in more severely cirrhotic patients.14 Repaglinide treats type 2 diabetes.15 In a population-based diabetes study, cirrhosis was the fourth leading cause of death and accounted for 4.4% of diabetes-related deaths; diabetes is also the most common cause of liver disease in the US.16 To date, these compounds have not been modeled under cirrhotic conditions using a PBPK approach, and models for highly permeable compounds cannot be reasonably expected to describe the pharmacokinetics of liver transporter substrates.17
PBPK models for liver transporter substrates have been developed previously, where the distribution of the compounds is modeled as permeability-limited in order to incorporate both passive diffusion and active uptake due to transporter activity.18–21 In this study, we extended published PBPK models18–21 to extrapolate pharmacokinetics from healthy individuals to patients with cirrhosis. The model structure for liver transporter substrate disposition in healthy individuals was combined with adjustments of physiologic parameters. This work updates current understanding of the effect of liver cirrhosis on system parameters, including transporter processes, and has potential usefulness in the individualized dosage adjustment of liver transporter substrates under disease conditions.
METHODS
Model structure
The published PBPK model structure for organic anion transporting polypeptides (OATP) substrates incorporating hepatic uptake clearance (CLact), passive diffusion (CLpass), metabolic (CLmet), and biliary (CLbile) clearances19 is applied for bosentan, olmesartan, repaglinide, and valsartan. The model assumes no active basolateral efflux due to challenges of estimating this activity, although it has been described for at least rosuvastatin.22,23 Enterohepatic recirculation of the parent compound is not modeled for olmesartan and valsartan due to its limited impact on the plasma pharmacokinetic simulations performed with the current model (data not shown). For telmisartan, additional model components for deconjugation of glucuronide metabolite and enterohepatic recirculation of the parent were added to the PBPK model to account for the long half-life and large apparent volume of distribution observed in clinical data as described before.24
First-order absorption kinetics with absorption rate (ka), fraction absorbed (Fa), and assuming no intestinal metabolism or transport (i.e., Fg = 1) are used to describe plasma data after oral dosing of repaglinide and telmisartan.24 Because the first-order absorption cannot adequately describe the plasma concentration–time curves after oral dosing of bosentan and valsartan, a saturable absorption model with maximum absorption rate (Vmax), and Michaelis–Menten saturation constant (Km)25 is used for these two compounds (other empirical absorption models described in ref.25 including a zero-order model, sequential independent zero, and first-order model, sequential linked zero and first-order model, and model with delayed absorption were also tested, but these models did not improve the fitting).
The model is implemented in MATLAB (v. 2010b, MathWorks, Natick, MA). The fitted parameters are estimated by minimizing the squared error between log10 transformed data and simulations, using the pattern search optimizer in the MATLAB Global Optimization Toolbox.
Local sensitivity analysis
Local sensitivity analyses of the area under curve (AUCplasma, 0 – t and AUCliver, 0 – t) were conducted as described previously where each parameter (for healthy individuals) is raised by 1% with respect to its value in the simulations.18 Sensitivity coefficients are normalized to both the parameter value and the model output. To keep the blood flow balanced, when the blood flow to any other organ is increased, the lung blood flow and the cardiac output will be raised accordingly. Similarly, if the lung blood flow or the cardiac output is raised, blood flow in all other organs will be increased. If liver arterial blood, gut blood, or spleen blood flow is raised, both lung blood flow and liver venous blood flow will be increased, and vice versa. To analyze the sensitivity to the liver mass (for healthy individuals the functional liver mass is the same as the physical liver mass), CLpass, CLact, CLmet, and CLbile are raised 1% together with the liver volume. Only parameters with normalized sensitivity coefficients greater than 0.3 or less than −0.3 are reported.
Physiological parameters for cirrhosis
Meta-analysis
We used the same source literature reporting changes for physiological parameter values as published before.5 For the functional liver size analysis, only datasets classified using the Child–Pugh system are selected. Studies using functional assays (e.g., galactose elimination capacity or hepatic sorbitol clearance) are not included because the data may be confounded by blood flow, or the expression level of metabolic enzymes. The physiological changes associated with liver cirrhosis are represented as the ratios of mean reported values between the disease group and the healthy group (except for RB/P). To estimate the population mean of the ratios based on the individual studies, one could pool the measured physiological parameter values, and take the ratios between population means of healthy and cirrhosis groups. However, the measurement techniques for these parameters are different from study to study, and as such it may not be proper to pool these measurements directly. To bypass this challenge, instead of pooling the measurements we take the ratios of individually reported values and pool the ratios to estimate the population mean of the ratios.
The ratios are calculated as:
| 1 |
where xcirrohsis and xhealthy represent the reported sample mean values for the cirrhosis group and the healthy group in study i. The standard error (SE) of the ratios in each individual study is calculated through the propagation of uncertainty:
| 2 |
The sample standard deviations of the ratios (SDi) are calculated using the same approach. The population mean value (WX) and population standard deviations (SD) of ratios from N individual studies are estimated as follows using the reciprocal of standard error-weighted mean26 and pooled variance.27
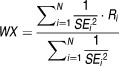 |
3 |
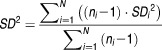 |
4 |
where ni typically indicates the sample size in study i. In the clinical studies, ni was reported for the participants in the healthy and the cirrhotic groups. However, in our analysis, which relies on ratios, we approximated ni as the average number of healthy and cirrhotic participants in each study.
Plasma fraction unbound
Plasma fraction unbound (fu,p) of bosentan, repaglinide, telmisartan, and valsartan have been reported for healthy individuals.18,24 The in-house value of fu,p for olmesartan was used. Assuming the unbound albumin concentration (Calbumin) is approximately equal to the total albumin concentration (Table 3), fu,p values for patients with liver cirrhosis are estimated based on a previously published approach.28
| 5 |
Table 3.
Physiological changes associated with liver cirrhosis (fractions of healthy control values ± standard deviation)a
| Child–Pugh Grade | ||||
|---|---|---|---|---|
| Parameter | A | B | C | |
| Albumin concentration | 0.84±0.15 | 0.69±0.15 | 0.53±0.15 | |
| Hematocrit (%)b | 38±5.0 | 34±5.7 | 34±5.5 | |
| Cardiac output | 1.1±0.39 | 1.2±0.34 | 1.3±0.30 | |
| Portal vein blood flow | 0.72±0.57 | 0.60±0.61 | 0.13±0.57 | |
| Splenic vein blood flow | 1.2±0.29 | 1.5±0.52 | 1.5±0.54 | |
| Liver arterial blood flow | 1.5±1.1 | 1.7±1.5 | 2.1±1.9 | |
| Functional liver size | 0.91±0.26 | 0.81±0.26 | 0.64±0.22 | |
| Liver transporter mRNA levelc | OATP1B1 | 0.65±0.49 | (0.65±0.49) | (0.65±0.49) |
| OATP1B3 | 0.73±0.59 | (0.73±0.59) | (0.73±0.59) | |
| OATP2B1 | 0.77±0.47 | (0.77±0.47) | (0.77±0.47) | |
| MRP2 | 0.54±0.48 | (0.54±0.48) | (0.54±0.48) | |
| BCRP | 0.58±0.45 | (0.58±0.45) | (0.58±0.45) | |
| BSEP | 1.1±0.51 | (1.1±0.51) | (1.1±0.51) | |
| MDR1 | 1.1±0.49 | (1.1±0.49) | (1.1±0.49) | |
| MDR3 | 2.3±0.45 | (2.3±0.45) | (2.3±0.45) | |
| MATE1 | 0.65±0.52 | (0.65±0.52) | (0.65±0.52) | |
| Uptake transporter activityd | 0.78±0.070 | 0.31±0.033 | (0.31±0.033) | |
| Efflux transporter activityd | 0.69±0.12 | 2.6±17 | (2.6±17) | |
The individual values reported in each study and references are given in the supplementary materials.
Hematocrit is given as the volume percentage (%) of red blood cells in blood for CP-A, CP-B, and CP-C groups, rather than the fraction of control value. The hematocrit of healthy individuals is (42±3.3) %.
The reported liver transporter expression level data are not classified into Child–Pugh grade system, as such we assume that CP-A, CP-B, and CP-C groups share the same values. OATP1B1, 1B3, and 2B1 are uptake transporters, and others are biliary efflux transporters.
These values are determined by fitting observed clinical data. Standard deviations are approximated by residual bootstrap. We assume that CP-C group has the same values as CP-B group as mentioned in the text.
Blood to plasma ratio
Blood to plasma ratio (RB/P) is defined as:
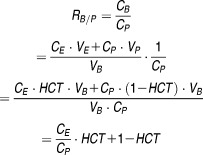 |
6 |
where C and V represent concentration and volume; B, E, and P represent blood, erythrocyte, and plasma; and HCT represents hematocrit (Table 3). The reported RB/P values for bosentan, repaglinide, and valsartan in healthy individuals are 0.48, 0.48, 0.55, and 0.545,18 which would result in CE/CP less than zero. Hence, for these compounds CE/CP is fixed at zero, indicating very limited accumulation of these compounds in the erythrocyte. RB/P of olmesartan is not reported; however, the compound does not penetrate red blood cells,29 hence CE/CP is also fixed at zero. For telmisartan, CE/CP is reported to be 0.5,30 and assumed to be constant and independent of disease conditions.
Tissue to plasma partition coefficient
The tissue to plasma partition coefficients (Kp) for nonliver tissues are calculated using an in silico method reported previously,31under different disease conditions. In the Kp calculation, fu,p and RB/P values are calculated above, pKa and logD7.4 values are in Table 1. For telmisartan, Kp values are derived from human positron emission tomography (PET) data24 due to known misprediction made by in silico methods.
Table 1.
Compound-specific parameters for healthy individuals
| Bosentan | Repaglinide | Telmisartane | Valsartan | Olmesartanf | |
|---|---|---|---|---|---|
| pKa a | 5.2 | 4.4 (acidic) | 3.8 | 4.6 (acidic) | 4.14 |
| 6.1 (basic) | 3.8 (acidic) | ||||
| logD7.4 a | 1.3 | 2.1 | 2.5 | −0.88 | 0.513 |
| fu,p a | 0.0053 | 0.0074 | 0.0050 | 0.0015 | 0.0045 |
| RB/P | 0.58 | 0.58 | 0.79 | 0.58 | 0.58 |
| fu,liver b | 0.018 | 0.015 | 0.013 | 0.048 | 0.0090 |
| CLR (L/h) c | 0 | 0 | 0 | 0.62 | 0.60 |
| CLact (L/h) d | 4,400 | 12,000 | 31,000 | 1,300 | 290 (250, 330) |
| CLpass (L/h) d | 11 | 120 | 70 | 0.46 | 1.3 (0.53, 2.1) |
| CLmet (L/h) d | 11 | 120 | 530 | 0 | 0 |
| CLbile (L/h) d | 1.5 | 0 | 0.62 | 0.77 | 8.2 (6.0, 9.1) |
| ka (h−1) d | — | 1.0 | 0.38 | — | — |
| Vmax (μg· h−1) d | 41,000 | — | — | 70,000 | — |
| Km (μg) d | 16,000 | — | — | 27,000 | — |
| Fa d | 0.95 | 1.0 | 0.86 | 0.32 | — |
Except for olmesartan and telmisartan, values are reported in ref.18.
All values are reported in ref.19 except for olmesartan, for which fu,liver is predicted using the method described in ref.18.
CLR of valsartan and olmesartan are reported in refs. 29, 32.
These values are estimated as described in the method section.
The values for telmisartan hepatic clearances are reported previously,24 while absorption parameters are reestimated using healthy individuals reported in the liver cirrhosis pharmacokinetic study. For glucuronide, CLbile is 62 L/h, and fu,liver is 0.0073.
CLpass, CLact, and CLbile of olmesartan in healthy individuals are estimated by fitting observed plasma PK data without boundaries based on SCHH study. The values given in parentheses for healthy individuals are the 95% confidence intervals approximated by a bootstrap method. logD7.4 and fu,p value of olmesartan are in house data, while pKa has been published before.29
All values are rounded to two significant figures.
Blood flow
Tissue blood flows are modeled as fractions of cardiac output. The cardiac output, portal vein, splenic vein, and liver arterial blood flow under liver cirrhosis conditions are adjusted using values given in Table 3. Blood flows in other tissues are also adjusted accordingly such that the sum of blood flow fractions contributed by the tissues is 1.
Renal clearance
Renal clearance (CLR) of prodrug of olmesartan (olmesartan medoxomil) under healthy and cirrhosis conditions has been reported.29 Since CLR has not been reported for olmesartan, we assume that CLR values of olmesartan and its prodrug are the same considering the fast conversion from prodrug to olmesartan in the human body.29 There is no reported CLR of valsartan under conditions of cirrhosis but only for healthy individuals,32 hence the ratios of valsartan CLR between healthy and cirrhotic conditions are estimated as the weighted mean values of rosuvastatin and olmesartan (0.85 and 0.79 for CP-A and B groups, see Supplemental Materials).11,29 Alternatively, assuming changes in CLR in cirrhosis are similar to those for glomerular filtration rate (GFR), valsartan CLR can be predicted using ratios for GFR changes in the previous meta-analysis (0.70 and 0.58 for CP-A and B in ref.5). However, such an approach leads to greater changes in CLR than observed for rosuvastatin and olmesartan.11,29 The pharmacokinetics of rosuvastatin in healthy11 and liver cirrhosis individuals11 have been published; however, under cirrhotic conditions derived pharmacokinetic parameters but not concentration–time course are reported. As such, rosuvastatin is not included in this study, because of the additional challenge in estimating absorption and hepatic clearance without time-course data. We assume there is no renal excretion for the other three compounds.18,24 The unbound renal clearance (CLR,u) is calculated using reported or estimated clinical renal clearance assuming well-stirred conditions as described before:18
| 7 |
where Qrenal,blood is renal blood flow.
Hepatic clearance
CLpass and CLact of bosentan, repaglinide, and valsartan, CLmet of bosentan and repaglinide, as well as CLbile of olmesartan and valsartan for healthy individuals are estimated by fitting the model to the observed plasma concentration time profiles after intravenous dosing.11,13,15,32 CLmet of olmesartan and valsartan, and CLbile of repaglinide are fixed at zero.19,29 CLbile of bosentan is fixed at a previously predicted value using sandwich-cultured human hepatocyte (SCHH).19 If we simultaneously estimate CLpass, CLact, CLmet (or CLbile) by fitting the clinical data for each compound individually, the model may be over-parameterized as described before,17 leading to unreliable estimates. On the other hand, if we fix CLpass, CLact, CLmet, and CLbile at SCHH predicted values given in a previous study,19 there are substantial discrepancies between simulations and data. We want to minimize such discrepancies, since the following simulations and predictions for cirrhotic patients rely on the accuracy of simulation for healthy individuals. To address these issues, CLpass, CLact, CLmet, and CLbile are estimated by fitting plasma concentration time profiles of each compound individually; however, they are bounded by their confidence intervals obtained in the previous study (i.e., in vitro SCHH (or human liver microsome, HLM) clearance × physiological scaling factor × confidence intervals of empirical scaling factors). The details of the estimation of the confidence intervals are given in the original publication.19 CLpass, CLact, CLmet, and CLbile used for telmisartan prediction under healthy conditions are fixed at values given in the previous study due to the good agreement between data30 and simulations.24 There is no published SCHH data of olmesartan, hence its CLpass, CLact, and CLbile are estimated by fitting observed plasma concentration time course29 without boundaries based on SCHH. To assess the variability of the estimates, confidence intervals for CLpass, CLact, and CLbile are approximated by using a residual bootstrap method as described before.19 Bootstrap is not performed for other compounds because their fitting has been bounded by confidence intervals generated previously.
Passive permeability of the compound is assumed to be independent of the disease status, hence CLpass under cirrhosis conditions is assumed to be only affected by the functional liver mass.
| 8 |
where Rliver mass is the fraction of the functional liver size in healthy control.
CLmet under liver cirrhosis conditions is assumed to be decreased due to both reduced functional liver mass and the reduced metabolic enzyme activity as described previously.4,5
| 9 |
where Fenzyme is the fraction of the total metabolism contributed by each metabolic enzyme involved in the hepatic metabolism; Rmetabolism is the ratio of metabolic enzyme activity between the diseased group and the healthy control, i represents each metabolic enzyme, and n is the number of metabolic enzymes. According to previous HLM studies, 60% of dosed bosentan is metabolized through CYP3A4, and the remaining 40% metabolized through CYP2C9,33 while 63% of repaglinide is metabolized by CYP2C8, 18% by CYP3A4, and 19% by UDP-glucuronosyltransferase (UGT).34 Telmisartan is metabolized exclusively by UGT.35 We used reported Rmetabolism of CYP3A4, CYP2C8, and CYP2C9 activity in CP-A, CP-B, and CP-C patients.5 The UGT activity is assumed to be not affected by liver cirrhosis as suggested by a previous study.36
Similar to CLmet, in this study CLact and CLbile are assumed to depend on both functional liver mass and the transporter activity.
 |
10 |
where Ftransporter is the fraction of each transporter made to CLact (or CLbile), and Rtransporter is the ratio of transporter activity between the diseased group and the healthy control. Rtransporter has been reported using mRNA measurements but the diseased individuals were not assessed based on the Child–Pugh classification system (Table 3),37,38 hence in the prospective predictions (Method 1), we assume the three disease groups have the same Rtransporter and standard deviation. Ftransporter has been reported for olmesartan (OATP1B1, 0.53; OATP1B3, 0.47; no OATP2B1 and NTCP uptake39,40) but not for the other four compounds, hence for other compounds Ftransporter are generated randomly from a uniform distribution.
Assuming liver cirrhosis has a similar impact on all uptake transporters, Rtransporter values of CP-A and CP-B are estimated by fitting observed concentration–time courses of patients while fixing other parameters at their mean values in Table 3 (Method 2). Because the current simulations are not sensitive to CLbile as they are to CLact (Table 2), a similar assumption is also made on biliary transporters, although this assumption is not entirely consistent with mRNA data (Table 3). The uncertainty of fitted parameters are approximated by residual bootstrap.19 The pharmacokinetics of repaglinide were reported as the average values of nine CP-B and three CP-C patients.9 Studies for the other four compounds did not include CP-C individuals. We assumed Rtransporter of CP-C patients has the same value as that of CP-B patients, because adding separated parameters for CP-C patients does not improve the fitting (data not shown).
Table 2.
The local sensitivity coefficient of AUCplasma, 0 – 12 hours and AUCliver, 0 – 12 hours to parameters
| Bosentan | Repaglinidea | Valsartan | Telmisartanb | Olmesartanc | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AUCplasma | AUCliver | AUCplasma | AUCliver | AUCplasma | AUCliver | AUCplasma | AUCliver | AUCplasma | AUCliver | |
| fu, p | −1.1 | — | −1.3 | — | −0.66 | 0.36 | −1.72 | — | −0.93 | — |
| RB/P | — | — | 0.33 | — | — | −0.30 | 0.80 | — | — | — |
| fu, liver | — | −0.56 | — | −1.0 | −0.98 | — | −0.96 | — | −0.97 | |
| CLact | −1.1 | — | −1.3 | — | −0.43 | 0.57 | −1.7 | — | −0.57 | 0.38 |
| CLpass | 0.30 | — | 0.63 | — | — | — | — | — | — | |
| CLmet/CLbile | — | −0.51 | −0.64 | −1.0 | — | −0.97 | — | −0.95 | — | −0.91 |
| Vlt | −0.86 | −0.81 | −1.0 | −0.78 | −0.34 | −0.33 | −1.4 | −0.79 | −0.45 | −0.48 |
| Qhv | — | — | 0.34 | — | — | — | 0.82 | — | — | — |
| Qlung | — | — | 0.33 | — | — | — | 0.81 | — | — | — |
| Fa | 1.0 | 0.94 | 1.0 | 1.0 | 0.93 | 0.93 | 1.4 | 1.4 | — | — |
Only sensitivity coefficients less than −0.3 or greater than 0.3 are reported here.
The sensitive analysis for all compounds except for olmesartan is performed following oral dosing. The analysis for olmesartan is performed following intravenous bolus dosing.
Repaglinide AUCplasma has sensitivity coefficient to liver blood volume (Vlb) of 0.31.
Telmisartan AUCplasma has sensitivity coefficient to gut blood flow (Qgut) and ka of 0.56 and 0.40; and telmisartan AUCliver has sensitivity coefficient to ka of 0.40.
The sensitivity analysis for olmesartan AUCplasma and AUCliver is performed from 0 to 96 hours. AUCplasma and AUCliver have sensitivity coefficient of CLR,u of −0.37 and −0.38.
The previously reported fractions unbound in liver tissue (fu,liver)19,24 are used for healthy individuals and the two cirrhosis groups, assuming fu,liver is not affected by the liver cirrhosis. Since there is no published value of fu,liver for olmesartan, it is predicted from fu,p as described previously.18
Drug absorption
The absorption parameters ka (or Vmax, Km) and Fa are estimated by fitting the observed plasma concentration–time profiles following oral dosing from healthy individuals in the liver cirrhosis studies,8–12 simultaneously with CLpass, CLact, CLmet, and CLbile estimation by fitting intravenous data from healthy individuals as described above (with the exception of telmisartan fitting, where all parameters except for ka and Fa are fixed as described above). Due to the lack of intestinal transporter data under cirrhotic conditions at this time, assuming liver is the major disposition organ, and no change of the intestinal transporter activity, the absorption parameters are kept the same for the healthy and liver cirrhosis groups. Absorption parameters are not estimated for olmesartan, because the human pharmacokinetic study including individuals with cirrhosis was performed following intravenous bolus dosing.29
Simulations of pharmacokinetics under liver cirrhotic conditions
Assuming all parameters are independent of each other and follow normal distributions, the values of albumin concentrations as the fraction of healthy control are randomly generated from a normal distribution with mean and standard deviation in Table 3, as are values of other parameters with relatively high sensitivity coefficients including HCT values, cardiac output, portal vein blood flow, splenic vein blood flow, liver arterial blood flow, functional liver size, and hepatic transporter activity. For a few parameters with standard deviations of a similar magnitude as the mean values, negative values may be generated occasionally but were discarded. With generated parameter values, concentration–time profiles are simulated using the PBPK model.
RESULTS
Published plasma time course data in healthy individuals and cirrhotic patients for liver transporter substrates bosentan,12,13 olmesartan,29 repaglinide,9,15 and telmisartan,10,30 and valsartan8,32 were analyzed. The values of compound-specific parameters for healthy individuals are given in Table 1 (except for tissue–plasma partition coefficients (Kp) for nonliver tissues, shown in the Supplemental Materials). With fitted hepatic clearances and oral absorption parameters, the simulated bosentan, olmesartan, repaglinide, telmisartan, and valsartan plasma concentrations can reasonably match the observations from healthy individuals (Figure 1).
Figure 1.
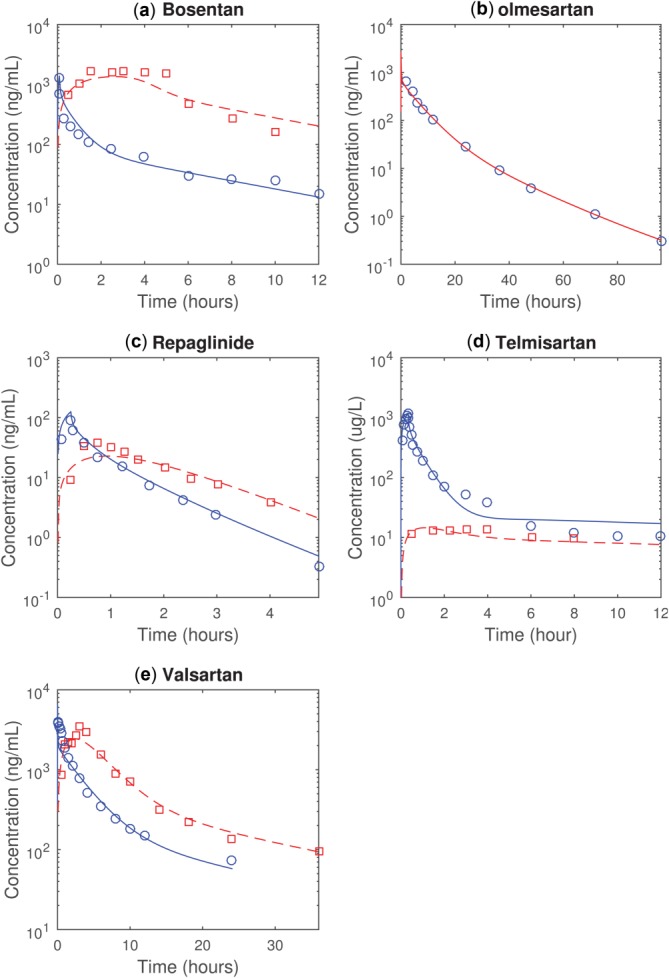
The observed and simulated mean plasma concentration time profiles in healthy individuals of (a) bosentan (10 mg 5-minute intravenous infusion and 125 mg oral dosing), (b) olmesartan (8 mg intravenous bolus dosing), (c) repaglinide (2 mg 15-minute intravenous infusion and 4 mg oral dosing), (d) telmisartan (40 mg 20-minute intravenous infusion and 40 mg oral dosing), and (e) valsartan (20 mg intravenous bolus and 160 mg oral dosing). The blue circles and solid lines represent the observations and simulations following intravenous dosing. The red squares and dashed lines represent the observations and simulations following oral dosing. The simulations are performed with parameter values for healthy individuals given in Table 1.
The local sensitivity analysis (Table 2) indicates that AUCplasma is in general sensitive to parameters including plasma fraction unbound (fu,p), blood to plasma ratio (RB/P), hepatic active uptake (CLact), passive diffusion (CLpass), metabolic (CLmet) (or biliary (CLbile)) clearances, the functional liver tissue volume (Vlt), liver venous blood flow (Qhv), cardiac output, and fraction absorbed (Fa). These results are consistent with published findings.18,24 The AUCliver of most compounds is sensitive to liver tissue fraction unbound (fu,liver), CLmet (or CLbile), Vlt, and Fa (Table 2).
To prospectively predict pharmacokinetics under liver cirrhosis conditions, the parameters with high sensitivity coefficients are adjusted. The ratios (and the standard deviations of ratios) between healthy individuals and liver cirrhosis patients (CP-A, CP-B, and CP-C) are estimated from published studies (Table 3). We used published mRNA data37,38 to estimate the ratios of transporter activities (Rtransporter) between healthy individuals and the diseased groups (Method 1). Unfortunately, these data are not classified based on the Child–Pugh system so the same ratio is applied to all cirrhotic groups (Table 3). The parameter values under cirrhosis are predicted as the products of healthy values (Table 1) and the ratios (Table 3). The fraction each transporter contributes to hepatic uptake or biliary efflux is unknown except for olmesartan uptake,39,40 hence is randomly generated in the simulations. Assuming the parameters are independent of each other and follow normal distributions with means and standard deviations given in Table 3, 1,000 simulated plasma concentration–time profiles are generated for each compound. The averaged simulations for liver cirrhosis patients, 2.5 and 97.5 percentiles are superimposed on the observations (Figure 2). The observed and predicted AUCplasma with 95% prediction intervals are reported in Table 4. The predictions more reasonably match observations from the CP-A groups than CP-B groups. This is probably due to misprediction of liver transporter activity under CP-B conditions using mRNA data.
Figure 2.
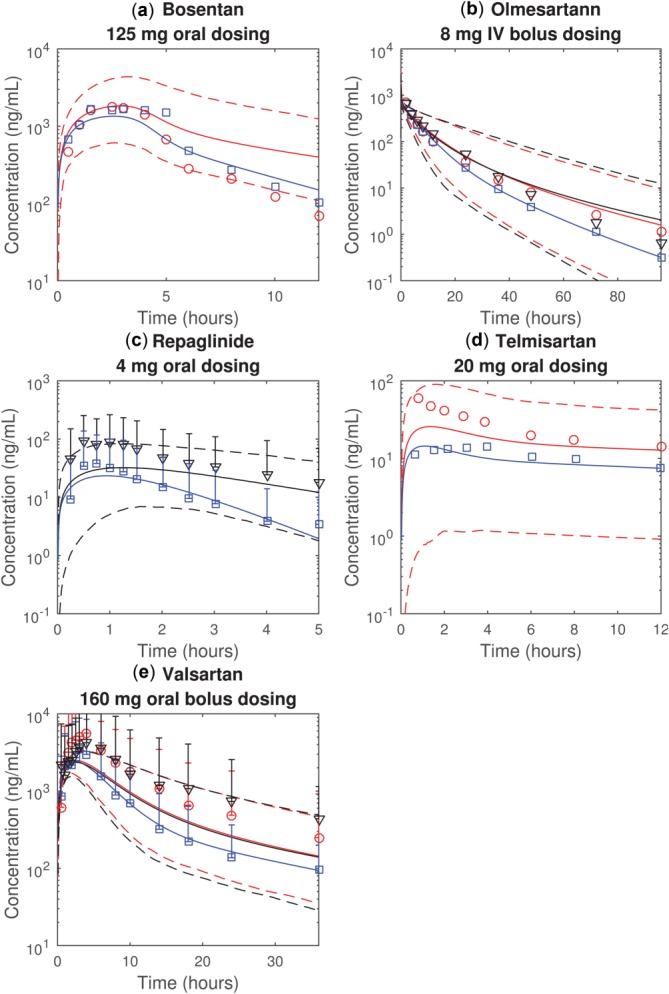
The observed and predicted plasma concentration time profiles of (a) bosentan, (b) olmesartan, (c) repaglinide, (d) telmisartan, and (e) valsartan. The blue, red, and black represent the healthy individuals, patients with CP-A liver cirrhosis, and the patients with CP-B liver cirrhosis, respectively. The markers (i.e., squares, circles, and triangles) represent the observations. The solid and dotted lines represent the average and 95% prediction intervals (approximated by 2.5 and 97.5 percentile) of 1,000 simulations where means and standard deviations of mRNA-derived Rtransporter (Method 1) and other parameters (Table 3) are used to generate random values. Error bars indicate 95% prediction intervals estimated using observed standard deviations.
Table 4.
Observed and simulated AUCplasma (mean and 95% prediction interval)
| Simulations | ||||
|---|---|---|---|---|
| Observations | Method 1 | Method 2 | ||
| Bosentan | Healthy | 11957 (1803, 23035) | 8313 | 8313 |
| CP-A | 10781 (1577, 20931) | 13389 (4870, 45698) | 12825 (6811, 32887) | |
| Olmesartan | Healthy | 5964 (3657, 8271) | 4811 | 4811 |
| CP-A | 6780 (0, 15640) | 5795 (2698, 12648) | 5294 (3457, 10907) | |
| CP-B | 6972 (4093, 9851) | 4965 (2256, 13245) | 7394 (3686, 17451) | |
| Repaglinide | Healthy | 92 (0, 245) | 62 | 62 |
| CP-B | 369 (0, 903) | 173 (32, 694) | 303 (129, 910) | |
| Telmisartan | Healthy | 471 (0, 1282) | 392 | 392 |
| CP-A | 1290 (0, 4060) | 785 (49, 3079) | 766 (83, 3762) | |
| Valsartan | Healthy | 21200 (6309, 36091) | 21640 | 21640 |
| CP-A | 46800 (25698, 67902) | 26817 (11829, 52989) | 25568 (15995, 47824) | |
| CP-B | 45400 (37827, 52973) | 25436 (10618, 53145) | 39796 (19739, 72470) | |
AUCs of healthy individuals are generated using values given in Table 1.
95% prediction intervals of observations are estimated using reported standard deviations and t statistic, and prediction intervals of simulations are approximated using 2.5 and 97.5 percentile of 1000 simulations.
Method 1 represents 1000 simulations with mRNA-derived Rtransporter, contributions of individual transporters made to total uptake are randomly generated from a uniform distribution, and other parameters listed in Table 3 are randomly generated using reported mean and standard deviations.
Method 2 represents the single simulations with fitted Rtransporter, and other parameters listed in Table 3 fixed at their mean values. The prediction intervals are approximated using the same method as Method 1 except that Rtransporter were prepared with residual bootstrap results.
For bosentan, the observed AUCs are reported as geometric mean values, as are the simulated AUCs. For the other four compounds, the observed AUC are reported as arithmetic mean values, as are the simulated AUCs.
For bosentan and telmisartan, the observed AUCs are reported as AUC0 – ∞, as are the simulations. For olmesartan, repaglinide, and valsartan, the observed AUCs are reported up to 96, 48, and 36 hours, respectively, as are the simulations.
Assuming in liver cirrhosis that all uptake (or efflux) transporters are similarly affected within a grade but differences exist between grades, we estimated Rtransporter by simultaneously fitting observed pharmacokinetics of the five compounds (Method 2) (Table 3). Fitted values of Rtransporter for uptake transporters in the CP-A groups are similar to the values estimated using published mRNA data; however, fitted values for CP-B groups are different from mRNA-estimated values. With the fitted Rtransporter, the overall agreement between simulations and observations is improved (Figure 3) as are predictions for AUCplasma (Table 4).
Figure 3.
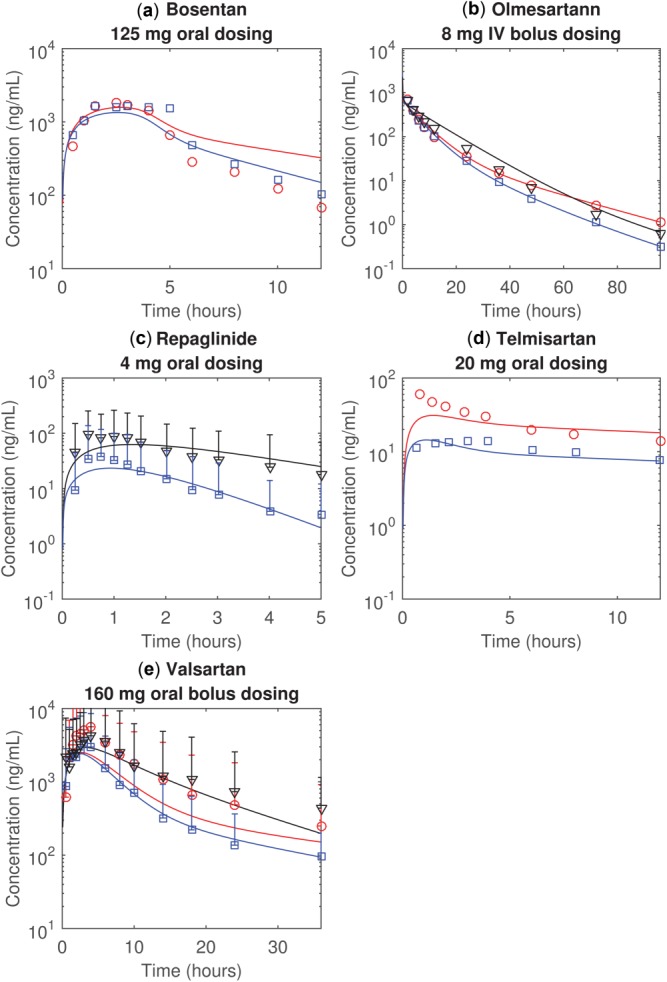
The observed and simulated plasma concentration time profiles of (a) bosentan, (b) olmesartan, (c) repaglinide, (d) telmisartan, and (e) valsartan. The blue, red, and black represent the healthy individuals, patients with CP-A liver cirrhosis, and the patients with CP-B liver cirrhosis, respectively. The markers (i.e., squares, circles, and triangles) represent the observations. The solid lines represent the simulations assuming all uptake transporters are equally affected by the liver cirrhosis conditions and Rtransporter is fitted (Method 2). All simulations assume the biliary efflux transporters are equally affected by the liver cirrhosis, and the impact of cirrhosis on all transporters depends on Child–Pugh scores. Error bars indicate 95% prediction intervals estimated using observed standard deviations.
DISCUSSION
We established a PBPK model to simulate how liver cirrhosis affects the pharmacokinetics of liver transporter substrates. A major challenge is that the effect of liver cirrhosis on the in vivo transporter activity is unknown. Although previous studies have reported that liver cirrhosis may change mRNA and protein expression levels,37,38 it is unclear whether changes in transporter expression levels due to liver disease can be directly translated into pharmacokinetics in patients.
We initially prospectively predicted the change of in vivo liver transporter activity (Rtransporter) using mRNA data. The mean predictions can more reasonably match mean observations from the CP-A than CP-B groups. This might arise if more mRNA samples in these studies were taken from CP-A patients.37,38 To improve prediction accuracy, transporter mRNA or protein expression data classified using the Child–Pugh system would be highly desirable in the future. In addition, because the fractional contributions that transporters made to the total uptake are unknown, they are randomly generated in this analysis. This leads to additional uncertainty in predictions. Several different in vitro assays have been developed to deconvolute the contribution made by individual transporters to the overall uptake.17 It could be useful to incorporate these data into the model once they are available. The Rtransporter value may also be predicted using experimentally determined protein expression levels, but would be only slightly less than the value estimated from mRNA data.37 In this model, because we assumed that all parameters in Table 1 follow normal distributions and are independent of each other, the variability of the simulations may be overpredicted. However, the approximated variability is in general comparable to the observed variability (Table 4 and Figure 2).
To better describe the data, we reestimated Rtransporter for the CP-A and CP-B groups by fitting the observed pharmacokinetics and assuming all uptake transporters are similarly affected in the diseased conditions. As expected, the fitted value for CP-A groups (0.78) is similar to the mRNA-predicted values for uptake transporters (0.65 for OATP1B1, 0.73 for OATP1B3, and 0.77 for OATP2B1); however, the fitted value for the CP-B groups (0.31) is lower than these mRNA-predicted values. This top-down fitting approach provides an alternative method to the mRNA-predicted Rtransporter. The advantage of the top-down approach is to characterize the impact of cirrhosis on the transporter activity based on the CP system, which is not available from current mRNA data. The two approaches should be further tested when data for more compounds are available.
We assumed that the differences were due to liver transporter parameters, but given the very similar results with IV olmesartan and a lack of data on intestinal changes, we cannot rule out as an alternative hypothesis that absorption is affected to varying degrees in the diseased patients.
Additionally, liver cirrhosis has a pronounced impact on the observed pharmacokinetics of some compounds (e.g., valsartan) but not others (e.g., olmesartan), which makes it challenging to use a unique Rtransporter value to describe the change in the pharmacokinetics of all the compounds. It may be that the Child–Pugh score is not an accurate enough indicator of hepatic function, considering the score is not directly assessed on liver function. For example, a large body of conflicting literature on changes in hepatic blood flows in cirrhosis have been generated based on the Child–Pugh classification,5 indicating that under the current paradigms, the Child–Pugh score cannot properly reflect the physiological changes due to cirrhosis. On the other hand, although not reflected in the mRNA data, it is also possible that the disease can more significantly affect the activity of some transporters than others. As such, because the contribution each transporter makes to the total hepatic uptake varies for different compounds, liver cirrhosis could also have different impacts on these compounds. To test this hypothesis, we assumed that there are type-1 and type-2 uptake transporters responsible for the hepatic uptake of these compounds that have different Rtransporter values, and estimated parameters by fitting the clinical observations. This alternative analysis resulted in improved agreement between the mean simulations and observations (data not shown) for valsartan. However, the improvement is not significant for other drugs. Furthermore, such an approach is not practical to apply as a prospective prediction because of the lack of information about the contributions each transporter made to the total uptake.
In conclusion, a PBPK model for liver transporter substrates to simulate pharmacokinetics under liver cirrhosis conditions was established, and a meta-analysis was performed to obtain system-dependent parameters. This model can be useful in understanding the changes in the liver transporter activity due to cirrhosis and may aid in predicting systemic and potentially liver exposure of the liver transporter substrates under cirrhotic conditions in the future.
Acknowledgments
The authors thank Dr Veena Somayaji (Pfizer) and Dr Vinicius Bonato (Pfizer) for helpful discussions about statistical meta-analysis.
Author Contributions
R.L., H.B., and T.M. wrote the article; R.L., H.B., and T.M. designed the research; R.L. performed the research; R.L. analyzed the data.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- 1.Schuppan D. Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodighiero V. Effects of liver disease on pharmacokinetics. An update. Clin. Pharmacokinet. 1999;37:399–431. doi: 10.2165/00003088-199937050-00004. [DOI] [PubMed] [Google Scholar]
- 3.Jones H. Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT: Pharmacometrics Syst. Pharmacol. 2013;2:e63. doi: 10.1038/psp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edginton AN. Willmann S. Physiology-based simulations of a pathological condition: prediction of pharmacokinetics in patients with liver cirrhosis. Clin. Pharmacokinet. 2008;47:743–752. doi: 10.2165/00003088-200847110-00005. [DOI] [PubMed] [Google Scholar]
- 5.Johnson TN, Boussery K, Rowland-Yeo K, Tucker GT. Rostami-Hodjegan A. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharmacokinet. 2010;49:189–206. doi: 10.2165/11318160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 6.Strougo A, Yassen A, Krauwinkel W, Danhof M. Freijer J. A semiphysiological population model for prediction of the pharmacokinetics of drugs under liver and renal disease conditions. Drug Metab. Dispos. 2011;39:1278–1287. doi: 10.1124/dmd.110.037838. [DOI] [PubMed] [Google Scholar]
- 7.Wilkinson GR. Shand DG. Commentary: a physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 1975;18:377–390. doi: 10.1002/cpt1975184377. [DOI] [PubMed] [Google Scholar]
- 8.Brookman LJ, et al. Pharmacokinetics of valsartan in patients with liver disease. Clin. Pharmacol. Ther. 1997;62:272–278. doi: 10.1016/S0009-9236(97)90029-1. [DOI] [PubMed] [Google Scholar]
- 9.Hatorp V, Walther KH, Christensen MS. Haug-Pihale G. Single-dose pharmacokinetics of repaglinide in subjects with chronic liver disease. J. Clin. Pharmacol. 2000;40:142–152. doi: 10.1177/00912700022008793. [DOI] [PubMed] [Google Scholar]
- 10.Stangier J, Su CA, Schondorfer G. Roth W. Pharmacokinetics and safety of intravenous and oral telmisartan 20 mg and 120 mg in subjects with hepatic impairment compared with healthy volunteers. J. Clin. Pharmacol. 2000;40:1355–1364. [PubMed] [Google Scholar]
- 11.Simonson SG, Martin PD, Mitchell P, Schneck DW, Lasseter KC. Warwick MJ. Pharmacokinetics and pharmacodynamics of rosuvastatin in subjects with hepatic impairment. Eur. J. Clin. Pharmacol. 2003;58:669–675. doi: 10.1007/s00228-002-0541-7. [DOI] [PubMed] [Google Scholar]
- 12.van Giersbergen PL, Popescu G, Bodin F. Dingemanse J. Influence of mild liver impairment on the pharmacokinetics and metabolism of bosentan, a dual endothelin receptor antagonist. J. Clin. Pharmacol. 2003;43:15–22. doi: 10.1177/0091270002239701. [DOI] [PubMed] [Google Scholar]
- 13.Weber C, et al. Pharmacokinetics and pharmacodynamics of the endothelin-receptor antagonist bosentan in healthy human subjects. Clin. Pharmacol. Ther. 1996;60:124–137. doi: 10.1016/S0009-9236(96)90127-7. [DOI] [PubMed] [Google Scholar]
- 14.Auletta M, Oliviero U, Iasiuolo L, Scherillo G. Antoniello S. Pulmonary hypertension associated with liver cirrhosis: an echocardiographic study. Angiology. 2000;51:1013–1020. doi: 10.1177/000331970005101206. [DOI] [PubMed] [Google Scholar]
- 15.Hatorp V, Oliver S. Su CA. Bioavailability of repaglinide, a novel antidiabetic agent, administered orally in tablet or solution form or intravenously in healthy male volunteers. Int. J. Clin. Pharmacol. Ther. 1998;36:636–641. [PubMed] [Google Scholar]
- 16.Tolman KG, Fonseca V, Dalpiaz A. Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care. 2007;30:734–743. doi: 10.2337/dc06-1539. [DOI] [PubMed] [Google Scholar]
- 17.Li R, Barton H. Varma M. Prediction of pharmacokinetics and drug–drug interactions when hepatic transporters are involved. Clin. Pharmacokinet. 2014:1–20. doi: 10.1007/s40262-014-0156-z. [DOI] [PubMed] [Google Scholar]
- 18.Jones HM, et al. Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 2012;40:1007–1017. doi: 10.1124/dmd.111.042994. [DOI] [PubMed] [Google Scholar]
- 19.Li R, et al. A "middle-out" approach to human pharmacokinetic predictions for OATP substrates using physiologically-based pharmacokinetic modeling. J. Pharmacokinet. Pharmacodyn. 2014;41:197–209. doi: 10.1007/s10928-014-9357-1. [DOI] [PubMed] [Google Scholar]
- 20.Poirier A, Cascais AC, Funk C. Lave T. Prediction of pharmacokinetic profile of valsartan in human based on in vitro uptake transport data. J Pharmacokinet. Pharmacodyn. 2009;36:585–611. doi: 10.1007/s10928-009-9139-3. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe T, Kusuhara H, Maeda K, Shitara Y. Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol. Exp. Ther. 2009;328:652–662. doi: 10.1124/jpet.108.146647. [DOI] [PubMed] [Google Scholar]
- 22.Pfeifer ND, Bridges AS, Ferslew BC, Hardwick RN. Brouwer KL. Hepatic basolateral efflux contributes significantly to rosuvastatin disposition II: characterization of hepatic elimination by basolateral, biliary, and metabolic clearance pathways in rat isolated perfused liver. J. Pharmacol. Exp. Ther. 2013;347:737–745. doi: 10.1124/jpet.113.208314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfeifer ND, Yang K. Brouwer KL. Hepatic basolateral efflux contributes significantly to rosuvastatin disposition I: characterization of basolateral versus biliary clearance using a novel protocol in sandwich-cultured hepatocytes. J. Pharmacol. Exp. Ther. 2013;347:727–736. doi: 10.1124/jpet.113.207472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li R, Ghosh A, Maurer TS, Kimoto E. Barton HA. Physiologically Based Pharmacokinetic Prediction of Telmisartan in Human. Drug Metab. Dis. doi: 10.1124/dmd.114.058461. e-pub ahead of print 2014. [DOI] [PubMed] [Google Scholar]
- 25.Holford NH, Ambros RJ. Stoeckel K. Models for describing absorption rate and estimating extent of bioavailability: application to cefetamet pivoxil. J. Pharmacokinet. Biopharm. 1992;20:421–442. doi: 10.1007/BF01061464. [DOI] [PubMed] [Google Scholar]
- 26.Hedges LV. Meta-analysis. J. Educ. Behav. Stat. 1992;17:279–296. [Google Scholar]
- 27.Killeen PR. An alternative to null-hypothesis significance tests. Psychol. Sci. 2005;16:345–353. doi: 10.1111/j.0956-7976.2005.01538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalvass JC. Maurer TS. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm. Drug Dis. 2002;23:327–338. doi: 10.1002/bdd.325. [DOI] [PubMed] [Google Scholar]
- 29.U.S. Food and Drug Administration & Center for Drug Evaluation and Research. 2002. Benicar NDA 21–286 Clinical Pharmacology Biopharmaceutics Review. ( )
- 30.Stangier J, et al. Absorption, metabolism, and excretion of intravenously and orally administered [14C]telmisartan in healthy volunteers. J. Clin. Pharmacol. 2000;40:1312–1322. [PubMed] [Google Scholar]
- 31.Rodgers T. Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006;95:1238–1257. doi: 10.1002/jps.20502. [DOI] [PubMed] [Google Scholar]
- 32.Flesch G, Muller P. Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur. J. Clin. Pharmacol. 1997;52:115–120. doi: 10.1007/s002280050259. [DOI] [PubMed] [Google Scholar]
- 33.Dingemanse J. van Giersbergen PL. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin. Pharmacokinet. 2004;43:1089–10115. doi: 10.2165/00003088-200443150-00003. [DOI] [PubMed] [Google Scholar]
- 34.Sall C, Houston JB. Galetin A. A comprehensive assessment of repaglinide metabolic pathways: impact of choice of in vitro system and relative enzyme contribution to in vitro clearance. Drug Metab. Dispos. 2012;40:1279–1289. doi: 10.1124/dmd.112.045286. [DOI] [PubMed] [Google Scholar]
- 35.Wienen W, et al. A review on telmisartan: a novel, long-acting angiotensin ii-receptor antagonist. Cardiovasc. Drug Rev. 2000;18:127–154. [Google Scholar]
- 36.Strassburg CP, et al. Developmental aspects of human hepatic drug glucuronidation in young children and adults. Gut. 2002;50:259–265. doi: 10.1136/gut.50.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zollner G, et al. Adaptive changes in hepatobiliary transporter expression in primary biliary cirrhosis. J. Hepatol. 2003;38:717–727. doi: 10.1016/s0168-8278(03)00096-5. [DOI] [PubMed] [Google Scholar]
- 38.Ogasawara K, et al. Hepatitis C virus-related cirrhosis is a major determinant of the expression levels of hepatic drug transporters. Drug Metab. Pharmacokinet. 2010;25:190–199. doi: 10.2133/dmpk.25.190. [DOI] [PubMed] [Google Scholar]
- 39.Yamada A, et al. Multiple human isoforms of drug transporters contribute to the hepatic and renal transport of olmesartan, a selective antagonist of the angiotensin II AT1-receptor. Drug Metab. Dispos. 2007;35:2166–2176. doi: 10.1124/dmd.107.017459. [DOI] [PubMed] [Google Scholar]
- 40.Nakagomi-Hagihara R, et al. OATP1B1, OATP1B3, and mrp2 are involved in hepatobiliary transport of olmesartan, a novel angiotensin II blocker. Drug Metab. Dispos. 2006;34:862–869. doi: 10.1124/dmd.105.008888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information