

Abstract
Protein phosphorylation represents one of the key regulatory events in physiological insulin secretion from the islet β-cell. In this context, several classes of protein kinases (e.g. calcium-, cyclic nucleotide- and phospholipid-dependent protein kinases and tyrosine kinases) have been characterized in the β-cell. The majority of phosphorylated amino acids identified include phosphoserine, phosphothreonine and phosphotyrosine. Protein histidine phosphorylation has been implicated in the prokaryotic and eukaryotic cellular signal transduction. Most notably, phoshohistidine accounts for 6% of total protein phosphorylation in eukaryotes, which makes it nearly 100-fold more abundant than phosphotyrosine, but less abundant than phosphoserine and phosphothreonine. However, very little is known about the number of proteins with phosphohistidines, since they are highly labile and are rapidly lost during phosphoamino acid identification under standard experimental conditions. The overall objectives of this review are to: (i) summarize the existing evidence indicating the subcellular distribution and characterization of various histidine kinases in the islet β-cell, (ii) describe evidence for functional regulation of these kinases by agonists of insulin secretion, (iii) present a working model to implicate novel regulatory roles for histidine kinases in the receptor-independent activation, by glucose, of G-proteins endogenous to the β-cell, (iv) summarize evidence supporting the localization of protein histidine phosphatases in the islet β-cell and (v) highlight experimental evidence suggesting potential defects in the histidine kinase signalling cascade in islets derived from the Goto-Kakizaki (GK) rat, a model for type 2 diabetes. Potential avenues for future research to further decipher regulatory roles for protein histidine phosphorylation in physiological insulin secretion are also discussed.
Keywords: islet β-cell, insulin secretion, histidine kinases, histidine phosphatases, nucleoside diphosphate kinase, GTP, GTP-binding proteins
Introduction
Protein histidine phosphorylation in prokaryotes and eukaryotes
-
Identification and characterization of histidine kinases in the pancreatic β-cell
Stability characteristics of phosphoamino acids
Nucleoside diphosphate kinases
Biochemical characterization of the NDPK in β-cell subcellular fractions
Characterization of the mitochondrial isoform of NDPK
A membrane-associated histidine kinase phosphorylates the Gβ-subunit of trimeric G-proteins
A novel histone H4-phosphorylating histidine kinase in islets β-cells
-
Regulation of protein histidine phosphorylation in islet β-cells
Inhibitors of protein histidine phosphorylation
Activators of protein histidine phosphorylation
Activation of histidine kinases by biologically active lipids
Activation of histidine kinases by mastoparan analogues
-
Functional consequences of protein histidine phosphorylation
Compartmentalized generation of GTP
Regulation of islet endogenous G-proteins
Regulation of small G-protein function by histidine phosphorylation
Regulation of trimeric G-protein function by histidine phosphorylation
Regulation of the mitochondrial function via protein histidine phosphorylation
Regulation of ion channels
Regulation of isoprenoid metabolism
Protein histidine phosphatases
Potential defects in histidine phosphorylation in
islets derived from the Goto-Kakizaki rat, a model for type 2 diabetes
Conclusions and future research
Introduction
Insulin secretion from the pancreatic β-cell is regulated principally by the ambient concentration of glucose [1]. However, the molecular and cellular mechanisms underlying the stimulus-secretion coupling of glucose-stimulated insulin secretion (GSIS) remain only partially understood. In this context, it is widely accepted that GSIS is mediated largely via the generation of soluble second messengers, such as cyclic nucleotides, hydrolytic products of phospholipases (PLases) A2, C and D [2–10]. The principal signalling cascade of GSIS is initiated by the glucose transporter protein (i.e. Glut-2)-mediated entry of glucose into the β-cell, followed by an increase in the intracellular adenosine triphosphate (ATP)/adenosine diphosphate (ADP) ratio as a consequence of glucose metabolism. Such an increase in ATP levels culminates in the closure of ATP-sensitive potassium channels localized on the plasma membrane, resulting in membrane depolarization and facilitation of the influx of extracellular calcium through the voltage-sensitive calcium channels, also localized on the plasma membrane. A net increase in intracellular calcium that occurs from the influx of extracellular calcium into the cytosolic compartment, in addition to the mobilization of intracellular calcium from its storage pools, has been shown to be essential for the transport of insulin-laden secretory granules to the plasma membrane for fusion and release of insulin [2–10].
It is well established in most cells that transduction of extracellular signals involves ligand binding to a receptor, often followed by the activation of one or more guanosine triphosphate (GTP)-binding proteins (G-proteins) and their respective effector proteins [11–13]. The pancreatic islet β-cell is unusual in that regard since glucose, the major physiological agonist, lacks an extracellular receptor. Instead, events consequent to glucose metabolism promote insulin secretion (see above). Changes in calcium concentration not only initiate insulin secretion, but also regulate activities of numerous enzymes, including protein kinases, phosphodiesterases, adenylyl cyclases, and PLases, leading to insulin secretion [2–10]. In addition to calcium-dependent protein kinases, several other kinases, including calmodulin-, cyclic nucleotide- and phospholipid-dependent protein kinases, tyrosine kinases and mitogen-activated protein kinases have been identified and characterized in the islet β-cell [14–17 and references therein]. The majority of these protein kinases mediate phosphorylation of endogenous β-cell proteins using ATP as the phosphoryl donor. They catalyse protein phosphorylation at serine (P-Ser), threonine (P-Thr) or tyrosine (P-Tyr) residues. As will be discussed in detail in the following sections, there have been numerous reports [18–39 and references therein] that identified distinct families of protein kinases that mediate the phosphorylation of histidine (phosphohistidine [P-His]) residues. Moreover, earlier studies by Wieland et al.[38] and Kowluru et al.[39] have reported the localization of protein histidine kinases that utilized GTP as the phosphoryl donor.
This review article, therefore, represents an overview of our current knowledge on the regulatory roles for protein histidine phosphorylation in cellular signal transduction, specifically in light of our observations in the area of GSIS in the pancreatic islet β-cell. The overall objectives of this review article are to: (i) summarize the existing evidence indicating subcellular distribution and characterization of various histidine kinases in insulin-secreting cells; (ii) present evidence for functional regulation of such activities by agonists of insulin secretion; (iii) propose a working model to implicate novel regulatory roles for these kinases in the receptor-independent activation, by glucose, of small molecular mass and heterotrimeric G-proteins in the pancreatic β-cell; (iv) describe the available evidence supporting the localization of protein histidine phosphatases in the islet β-cell; (v) summarize experimental data suggesting potential defects in the histidine kinase activation mechanisms in islets derived from the Goto-Kakizaki (GK) rat, a model for type 2 diabetes; and (vi) provide conclusions and identify future directions in this area of research.
Protein histidine phosphorylation in prokaryotes and eukaryotes
A growing body of experimental evidence demonstrates the existence of a number of biological systems that involve P-His. These include, but not limited to, the classical phosphoenolpyru -vate-sugar phosphotransferase system that mediates the transport of glucose (and other sugars) across the bacterial cell as sugar phosphate (i.e. glucose-6-phosphate; reviewed in Reference [18]). In addition to this, the two-component regulatory system is well described in prokaryotes; these signalling steps are involved in coupling the extracellular stimuli (e.g. pH, temperature, chemoattractants and osmolality) to various cellular functions, including transcription, differen tiation and bacterial chemotaxis [19–26]. It is well documented that specific and functionally defined signalling steps involving histidine kinases trigger cellular responses to various environmental stimuli in the two-component regulatory systems [19–26]. Other classes of kinases involved in the N-linked phosphorylation, including arginine kinases, histidine kinases and lysine kinases, have also been identified and characterized (see Reference [18] for a detailed description of these signalling mechanisms).
It is also becoming increasingly evident that protein histidine phosphorylation plays major regulatory roles in mammalian cellular signal transduction [18, 27–33]. Several histidine kinases have been described in the mammalian cells including the nucleoside diphosphate kinase (NDPK), succinyl CoA-synthetase (SCS), his-tone H4 histidine kinases and the mammalian mitochondrial two-component histidine kinases (e.g. branched chain a-ketoacid dehydrogenase kinase and pyruvate dehydrogenase kinase). Some of the phosphoprotein substrates undergoing phosphorylation mediated by NDPK include aldolase, SCS, ATP-citrate lyase and the kinase suppressor of Ras (see References [18], [27–33] and Table 1 for select examples of proteins either containing phos-phohistidines or regulated by histidine kinases). In addition, a membrane-associated kinase that mediates the histidine phospho-rylation of the Gp-subunit of trimeric G-proteins has been reported in HL-60 Human Leukemia-60 [38] and pancreatic islet β-cells [39]. Several other proteins (e.g. P-selectin, annexin-1, 20S proteosome and other metabolic enzymes such as fructose-2,6-bisphosphatase) have also been shown to undergo histidine phos-phorylation (Table 1).
Table 1.
Select examples of proteins known to contain phosphohistidines and/or regulated by histidine kinases
| Protein (reference) |
|---|
| Adenosine monophosphate kinase α-subunit [34] |
| Aldolase C [35] |
| Annexin-1 [36] |
| ATP-citrate lyase [37] |
| β-subunit of trimeric G-proteins [38, 39] |
| Chemotactic factor, CheA [19] |
| Cystic fibrosis conductance regulator [40] |
| Fructose-2,6-bisphosphatase [41, 42] |
| Glucose-6-phosphatase [43] |
| Isocitrate lyase [44] |
| Kinase suppressor of Ras [45] |
| Nitrogen regulation factor II [46] |
| Nucleoside diphosphate kinase [32, 47] |
| p-enolpyruvate-sugar phosphotransferase |
| system [48] |
| 6-phosphofructo-2-kinase [49] |
| Phosphoglycerate mutase [50, 51] |
| Physarium nuclear proteins [52] |
| p38 in rat liver plasma membranes [53] |
| P-selectin [54] |
| Potassium channel KCa3.1 [55] |
| Pyrophosphatase [56] |
| Ras-related protein associated with diabetes [57] |
| Rat liver nuclear proteins [58] |
| Succinic acid thiokinase [59, 60] |
| 20S proteosome [61] |
| Tiam1 guanine nucleotide exchange factor [62, 63] |
Identification and characterization of histidine kinases in the pancreatic β-cell
Stability characteristics of phosphoamino acids
To date, the majority of phosphorylated amino acids identified include P-Ser, P-Thr and P-Tyr. Phosphoamino acids exhibit differential sensitivities to acidic and alkaline pH conditions [18, 27–31, 64–68]. For example, P-Ser and P-Thr, which form O-p(alcoholic O-monoester) linkages, are stable at acidic pH and fairly unstable under alkaline conditions. P-Tyr, which forms O-p(phenolic O-monoester), is stable under acidic and alkaline conditions. Therefore, due to their stability under acidic conditions, P-Ser, P-Thr and P-Tyr are readily identified after acid hydrolysis of phosphorylated proteins. However, acid-labile phosphorami-date linkage has been reported in P-His, arginine (P-Arg) and lysine (P-Lys). Interestingly, the relative abundance of P-His in eukaryotes appears to be significantly higher (nearly 100-fold) than P-Tyr, but certainly less abundant than P-Ser and P-Thr [18, 69]. It is not surprising that very little information is available on the number of proteins with P-His, since its phosphate is acid-labile and is rapidly lost (half-life of 30 min. at pH 3.0) during the identification of phosphoamino acids under standard experimental conditions, including SDS-PAGE fixation conditions, trichloro -acetic acid precipitation, partial acid hydrolysis for the determination of amino acid sequencing, etc. [30]. Other methodological concerns for P-His identification include relatively short half-life of the high-energy bond, which can be lost and/or remain undetected during the routine time course measurements of the enzyme activities. Several alternate experimental approaches have been utilized by investigators in this area [28, 29] including: (i) partial alkali hydrolysis to reduce therelative abundance of base-labile P-Ser and P-Thr, (ii) anion-exchange column chro-matography, (iii) reverse-phase column chromatography, (iv) thin-layer reverse-phase chromatography, (v) thin-layer silica chromatography, (vi) cellulose thin-layer electrophoresis and (viii) mass spectrometry (see References [28] and [29] for reviews and references therein). The reader is referred to several previous reviews [18, 27–31, 64–69] describing the potential methodological hurdles often encountered by researchers in the quantitation of the relative abundance of P-His in cellular lysates and subcellular fractions. Despite these significant methodological issues, a growing body of experimental evidence suggests important roles for protein histidine phosphorylation in cellular signal transduction in multiple cell types [27–31], including the islet β-cell (see below).
Earlier studies from our laboratory have identified at least three distinct classes of histidine kinases [39, 47, 60, 70, 71]. The first group represents the classical NDPK-like enzymes [47, 71] that catalyse the transfer of terminal phosphates from nucleoside triphosphates (NTPs; e.g. ATP) to nucleoside diphosphates (NDPs; e.g. guanosine diphosphate [GDP]) to yield NTPs (e.g. GTP). The transfer of terminal phosphates occurs via a two-step, ping-pong reaction involving the formation of a transient high-energy phosphoprotein intermediate form of NDPK, due to phosphorylation at a histidine residue, followed by transfer of that phosphate to a suitable acceptor [72]. NDPK has been implicated in the direct activation of certain G-proteins as well as phosphorylation and/or regulation of several enzymes of intermediary metabolism, such as ATP-citrate lyase, aldolase, pyruvate kinase and SCS (see below). The second group of histidine kinases, which are yet to be fully characterized in the islet β-cell, catalyses the histidine phosphorylation of the β-subunit of trimeric G-proteins [39]. We have demonstrated that the phosphate from the transiently phosphorylated β-subunit is then transferred to the GDP-bound form of the α-subunit of the trimeric G-proteins (Gα-GDP) to facilitate the formation of GTP-bound, active conformation of the candidate G-protein (i.e. Gα-GTP). The third form of histidine kinase endogenous to the islet β-cell phosphorylates histone H4 at the His-75 residue [70]; this histidine kinase appears to be distinct from the other known histidine kinases in the mammalian cells (see below). The identity, subcellular localization, biochemical properties and functional regulation of these three classes of histidine kinases have been carried out in a variety of insulin-secreting cells, including normal rat islets, human islets and clonal β (HIT-T15, INS-1, INS832/13, βTC3)-cells (see below).
Nucleoside diphosphate kinases (NDPK)
It was originally thought that NDPK is a housekeeping enzyme for the maintenance of intracellular NTP pools [72]. The first NDPK isoform was cloned and its crystal structure was deduced in Dictostelium. Recent studies have identified several NDPK gene products with a wide variety of cellular functions, including differentiation, growth and development, transcriptional regulation, tumor metastasis and programmed cell death [73–80]. For example, nm23-H1, which encodes the human NDPK-A isoform, has been shown to be a suppressor of tumor metastasis [75–80]. nm23-H2, which encodes the human NDPK-B isoform, has been identified as an element in the promoter region of c-myc oncogene and activates its transcription [81]. A series of elegant studies from Lacombe's laboratory have identified two additional homologues of the nm23 gene, termed as nm23-H4 and nm23-H5 [82–85]. The nm23-H4 represents the human mitochondrial NDPK (mNDPK), with a 55–60% identity to other forms of human NDPK. It also consists of an N-terminal extension characteristic of mitochondrial targeting [82, 83]. nm23-H5 is expressed specifically in the testes and has been implicated to play regulatory roles in spermatogenesis [84, 85]. Tsuiki et al. reported a novel isoform mitochondrial NDPK (i.e. nm23-H6) that has been implicated in cell growth and cell cycle progression [86]. In addition, nm23-H7 [87] and nm23-H8 [88] iso-forms have also been identified and appear to control cellular function. Together, these observations from multiple laboratories implicate key functions for NDPK in cellular metabolism and function, including cell survival and demise [see References 73, 74 for reviews].
Biochemical characterization of the NDPK in b-cell subcellular fractions
Original investigations from Metz's laboratory have provided evidence to suggest a permissive role for GTP in nutrient-induced insulin secretion [89, 90]. One of the potential loci at which GTP might exert its regulatory effects include one (or more) of the G-proteins that we and others have identified in various subcellular fractions (e.g. the secretory granules and mitochondria) of pancreatic islets and clonal β-cell preparations [References 91–121 for publications from multiple laboratories and select reviews]. In an attempt to define a role for NDPK in the ‘compartmentalized’ generation of GTP in insulin-secreting cells, we measured NDPK activity in normal rat and human islets and clonal β-cells. We quantitated such an activity by three distinct approaches, namely: (i) quantitation of its catalytic activity (i.e. formation of GTP or GTPγS from GDP and ATP or ATPγS), (ii) determination of its localization via immunodetection methods and (iii) radiometric quantitation of the phosphoenzyme intermediate of NDP kinase, which is involved in a ping-pong phosphotransfer mechanism [47]. The rank order of NDPK catalytic activity in β-cell subcellular fractions was: cytosol > nuclear and plasma membranes > microsomes > mitochondria = secretory granules. Sephadex G-200 size-exclusion chromatographic analysis suggested that the islet holoenzyme is tetrameric in nature, with an apparent molecular mass of 85–90 kD. Such an activity required divalent cations for its maximal activation. Magnesium stimulated the NDPK activity by 17-fold and 8.4-fold in rat islets and human islets, respectively. Uridine diphosphate (UDP), which forms an abortive complex with the enzyme, inhibited its activity in a concentration-dependent manner (Ki= 2 mM). The phosphorylated intermediate of NDPK was differentially sensitive to heat, acidic pH and a histidine-selective reagent, diethyl pyrocarbonate (DEPC), suggesting that (one of) the phosphoamino acid(s) is histidine [47]. Together, our data demonstrated that in β-cells, NDPK undergoes transient phosphorylation at a histidine residue and suggested that this phosphate, in turn, is transferred to GDP to generate GTP. Based on these original observations, we speculated that if GTP, which is formed via NDPK activation, is bound to and/or channelled to relevant G-proteins, it would facilitate the formation of active form of these proteins (see below).
Characterization of the mitochondrial isoform of NDPK (mNDPK)
In subsequent studies, using Western blot analysis, we determined that nm23-H1 (NDPK-A) predominantly localized in the cytosolic fraction isolated from HIT-T15 cells and INS-1 cells. However, nm23-H2 (NDPK-B) appeared to be distributed equally between the cytosolic and membrane compartments [71]. Using specific antisera directed against nm23-H4 (mNDPK), we also reported the localization of mNDPK in a variety of β-cell preparations. Using radiometric assays, we detected a significant NDPK activity in purified mitochondrial fraction isolated from a variety of insulin-secreting cells. Incubation of isolated mitochondrial extracts with either [γ-32P]ATP or GTP resulted in the formation of [32P]NDPK, which could be immunoprecipitated by an anti-NDPK serum. The mNDPK exhibited saturation kinetics with respect to its NDP acceptors and NTP donors and sensitivity to known inhibitors of NDPK (e.g. UDP and cromoglycate). It was found that the mNDPK was uniformly distributed in the β-cell submitochondrial fractions (i.e. inner membrane, outer membrane and soluble compartment) isolated by the swelling and shrinking method [71]. More importantly, a significant amount of NDPK (as determined by the catalytic activity and immunological methods) was recovered in the immunoprecipitates of the mitochondrial fraction derived using an antiserum directed against SCS, suggesting that mNDPK might remain associated with SCS. Together, these studies provided the first evidence for the localization of mNDPK in the islet β-cell, which could constitute an important link between energy metabolism and cellular regulation (see below).
A membrane-associated histidine kinase phosphorylates the Gp-subunit of trimeric G-proteins
Accumulating evidence in multiple cell types, including the islet β-cell, suggests that the β-subunit of trimeric G-proteins (Gβ) undergoes histidine phosphorylation; such a step has been implicated in non-receptor-dependent activation of G-proteins. In 1993, the original investigations by Wieland and coworkers [38] have demonstrated GTP-dependent phosphorylation of the Gβ-subunit in HL-60 cells. The phosphorylation of the Gp, which was specific to either GTP or GTPγS as phosphoryl or thiophos-phoryl donor, also required divalent cations (e.g. magnesium or manganese). Based on the differential sensitivities to heat, acid and hydroxylamine and DEPC (a histidine-modifying agent; see above), these authors identified the phosphorylated amino acid as P-His. Based on the data accrued in the follow-up studies [122], including the reconstitution protocols involving purified α- and βγ-subunits of trimeric G-proteins, these investigators concluded that the Gβ-subunit phosphorylation represents a general phenomenon occurring in various cell types, albeit to different degrees, and that the Gp-subunit phosphorylation requires a membrane-associated cofactor(s).
During this period, studies from our laboratory have demonstrated that a protein with an apparent molecular mass of 37 kD, similar to the size of the Gβ-subunit, underwent phosphorylation in a variety of insulin-secreting β-cells [39]. We noticed that incubation of the β-cell total membrane fraction or the purified secretory granule fraction, but not the cytosolic fraction, with [γ–32P]ATP or GTP resulted in the phosphorylation of the 37 kD protein, which was selectively immunoprecipitated by an anti-serum directed against the common Gβ-subunit. Disruption of the αβγ trimer (by pre-treatment with either fluoroaluminate or GTPγS) prevented the Gβ-subunit phosphorylation. Based on the differential sensitivities to pH, heat and DEPC, the phosphorylated amino acid was identified as P-His [39]. It was also observed that incubation of pure Gβ-subunit alone or in combination with the exogenous purified α-subunit of transducin did not result in the phosphorylation of the Gβ-subunit. However, inclusion of the islet cell membranes into the phosphorylation assay did promote the Gβ-subunit phosphorylation, suggesting that a membrane-associated factor (or kinase) is required for the Gβ-subunit phosphorylation [39]. Incubation of the phosphorylated Gβ-subunit with Gβ-GDP accelerated the dephosphorylation of the Gp-subunit, accompanied by the formation of Ga-GTP. These data, thus, offer a potential alternate mechanism for the activation of G-proteins in β-cells, which contrasts with the classical receptor-agonist mechanism. Such a signalling cascade involves the following: the Gp-subunit undergoes transient phosphorylation at a histidine residue by a GTP-specific protein kinase; this phosphate, in turn, may be transferred via a classical ping-pong mechanism to Ga-GDP (inactive), yielding the active configuration Gα-GTP in secretory granules and/or plasma membrane (i.e. potential strategic locations to modulate exocytosis of insulin).
Together, the findings reviewed in the above section suggest the localization of at least three forms of nm23/NDPK. They also suggest an association of SCS with mNDPK in the purified mitochondrial fraction derived from the insulin-secreting cells. Lastly, a membrane-associated factor/enzyme appears to mediate the histidine phosphorylation of the Gβ-subunit in the β-cell plasma membrane and secretory granule fractions. These findings are summarized in Table 2. Potential significance of these findings in relation to GSIS is discussed in the following sections.
Table 2.
Identification, subcellular distribution and regulation of histidine kinases in the islet β-cell
| Type of NDP kinase | Localization | Mode of histidine phosphorylation |
|---|---|---|
| nm23-H1(NDPK-A) | Predominantly cytosolic | Autophosphorylation |
| nni23-H2 (NDPK-B) | Cytosolic and membranous | Autophosphorylation |
| nm23-H4 | Mitochondrial | Autophosphorylation |
| Gβrsubunit | Membrane and secretory granules | Phosphorylation mediated by NDP kinase or another member of the histidine kinase family (see Table 3) |
| SCS | Mitochondrial | Phosphorylation mediated by NDPK (?) |
A novel histone H4-phosphorylating histidine kinase in islets β-cells
The above data suggested that a membrane-associated factor/protein might facilitate the histidine phosphorylation of the Gp-subunit. This prompted us to quantitate histidine kinase activity in the insulin-secreting cells. A Nitran filter paper assay, originally described by Matthews and coworkers [67], was used to quantitate the ability of islet endogenous histidine kinase(s) to phospho-rylate exogenously added histone H4 as the substrate [70]. The histone H4-phosphorylating activity was detectable in normal rat islets, human islets and clonal β (HIT-T15 and INS-1)-cells that utilized either ATP or GTP as a phosphoryl donor. On a Sephacryl S-100 size-exclusion column, such an activity eluted within the molecular mass range of 60–70 kD. It was stimulated by divalent cations, but was resistant to polyamines. It was inactivated in vitro by known inhibitors of protein histidine phosphorylation (e.g. UDP or cromoglycate). Mastoparan (Mas), a global activator of G-pro-teins [123, 124], but not Mas-17, its inactive analogue, stimulated histone H4-phosphorylating histidine kinase activity in vitro. Under these conditions, Mas, but not Mas-17, markedly stimulated the Gp-subunit phosphorylation and insulin secretion in normal rat islets [70]. These data identified, for the first time, a protein histidine kinase activity in the pancreatic β-cell that does not act on traditional serine, threonine or tyrosine residues. The properties of this novel histidine kinase that we have characterized in the islet β-cell are summarized in Table 3. Additional studies are required to conclusively demonstrate that such a kinase mediates the histidine phosphorylation of the Gβ-subunit.
Table 3.
Properties of the histone H4-phosphorylating histidine kinase in the islet β-cell
| Subcellular distribution | Membrane and cytosol |
|---|---|
| Apparent molecular weight | 60–70 kD |
| Nucleotode specificity | GTP and ATP (affinity for GTP is 3.5 times higher than ATP) |
| pH optimum | 7.0 |
| Metal ion specificity | Manganese > magnesium > calcium |
| Activators | Mas-7 > Mas > Mas-17 = control |
| Inhibitors | UDP, cromoglycate and |
| -SH group modifiers | |
| Potential phosphoprotein substrates | Gβ-subunit and histone H4 |
Accumulating evidence also suggests that several enzymes(proteins of intermediary metabolism undergo phosphorylation at critical histidine residues. Some of these include, but not limited to, ATP-citrate lyase, SCS and glucose-6-phosphatase (Table 1). Therefore, the histidine kinase that we characterized in the islet β-cell could subserve the function of phosphorylating some of these proteins. Clearly, our findings established a biochemical link between the activation of histidine kinase and the activation of the Gβ-subunit phosphorylation through the use of Mas, a global G-protein activator. Additional studies are needed to precisely define the regulation of this enzyme by nutrient insulin secretagogues to establish a link between activation of G-proteins (via activation of this kinase) and insulin secretion from isolated β-cells. Advancement along these lines will, indeed, depend upon the identification of specific inhibitors of this novel protein kinase in intact β-cells (see below).
Are there any known histidine kinases in other cells, and if so, what are their roles in cellular signal transduction? Indeed, the published evidence from multiple laboratories provides support for the localization of histidine kinases in multiple cell types. For example, Huang and coworkers [125] reported purification of a monomeric histidine kinase from Saccharomyces cerevisiae with an apparent molecular mass of 32 kD that specifically used ATP (also GTP, but with minimal affinity) to phosphorylate histone H4. The Km for ATP was 60 μM, and the enzyme required divalent cations, such as magnesium and manganese, for optimal activity. Polyamines (e.g. spermine and spermidine) were ineffective [125]. In another study, Motojima and Goto [126] reported histidine phosphorylation of a 36 kD protein by a histidine kinase in the liver extracts. They also demonstrated the localization of an okadaic acid-resistant phosphatase activity (with an apparent molecular mass of 45 kD). Using an HPLC method, they demonstrated copu-rification of the kinase and P36 substrate at a 70–75 kD size. These data indicate that the liver histidine kinase is different from the yeast enzyme, originally described by Huang and coworkers [125]. Urushidani and Nagao also reported [127] autophosphorylation, at a histidine residue, of a 40 kD protein localized in the membrane fraction derived from the rabbit gastric mucosa. The data from sequence analyses indicated that this protein might represent the a-subunit of extra-mitochondrial SCS or its homologue. Autophosphorylation of this protein was stimulated by GDP, Ras (a small molecular mass G-protein) and myelin basic protein [127]. The phosphoprotein was rapidly dephosphorylated in the presence of ATP, succinate and CoA. Furthermore, Hegde and Das have shown that Ras stimulated the phosphorylation of a 36 kD protein at a histidine residue in the liver membranes [53]. Along these lines, Besant and Attwood purified and characterized a histone H4-phosphorylating histidine kinase activity from the porcine thymus [128]. This enzyme appears to have certain similarities with the yeast enzyme [125], including molecular mass, which was estimated to be approximately 34–41 kD. This kinase [128] and the yeast histidine kinase [125] appear to be distinct from the islet histidine kinase [70; see Table 3] since, based on the elution profile on a size-exclusion column, we estimated its molecular mass to be in the range of 60–70 kD. However, our observations do not rule out the possibility that it might comprise more than one subunit. Additional studies are underway to purify this β-cell kinase and determine its structure and subunit composition.
Regulation of protein histidine phosphorylation in islet β-cells
Inhibitors of protein histidine phosphorylation
A relative lack of specific inhibitors hampered progress in this area considerably. Histidine site-modification reagents such as DEPC have been used to inhibit histidine phosphorylation (see above). The reversal of such an inhibitory step by hydroxylamine was also studied in broken-cell preparations [38, 39]. In addition, -SH-modifying reagents such as p-hydroxy mercuribenzoate and N-ethylmaleimide were studied [47]. UDP and cromoglycate have also been used to inhibit histidine kinase activation and protein histidine phosphorylation in vitro[47, 71]. Genistein, a known inhibitor of protein tyrosine phosphorylation (median inhibition concentration IC50 (2–50 μM), also appears to inhibit protein histidine kinases (IC50= 110 μM) in vitro[129]. Staurosporin, a generic inhibitor of protein kinases, also inhibits protein histidine phosphorylation (IC50= 20 nM) in vitro[18]. In addition to the regulatory effects of these ‘relatively’ non-specific inhibitors, numerous studies from Matthews' laboratory have determined effects of several chemical inhibitors on protein histidine kinases [18]. These include: tyrphostins (i.e. compounds 25 and 48), erb-statin, methyl 2,5-dihydroxycinnamate, ST 271, ST 638 and laven-dustin. The reader is referred to Reference 18 for additional details with regard to the IC50 values for these inhibitors.
In the context of the islet β-cell, we have been able to demonstrate the sensitivity of islet β-cell histidine kinases, including NDPK, the G3-phosphorylating histidine kinase activity and the histone H4-phosphorylating activity by the reagents described above [39, 47, 71]. It should be kept in mind that most of the pharmacological inhibitors used in the studies of histidine kinases (e.g. UDP cromoglycate, DEPC, hydroxylamine, etc.) are fairly non-selective and could exert untoward non-specific effects. Unfortunately, very little data are available with regard to specific pharmacological inhibitors for the mammalian histidine kinases. Interestingly, several recent studies were focussed on the synthesis and characterization of more specific inhibitors for histidine kinases in the bacterial systems and the two-component regulatory systems [130–134]. Additional work, specifically in the area of the development of structure-specific inhibitors of the mammalian histidine kinases, is needed to further evaluate the relative regulatory roles for histidine phosphorylation in islet signal transduction (see below).
Activators of protein histidine phosphorylation
Just as in the case of specific inhibitors of protein histidine phosphorylation, very little is known with regard to specific activators of protein histidine phosphorylation in the mammalian cells. The majority of the mammalian histidine kinases appear to require divalent cations for maximal activity (see above). In the context of the islet β-cell, studies from our laboratory have identified at least two classes of positive modulators (e.g. biologically active lipids and Mas analogues) of protein histidine phosphorylation. The data accrued from the studies involving the use of these modulators are reviewed below.
Activation of histidine phosphorylation by biologically active lipids
Recent studies from our laboratory provided insights into the novel regulation by biologically active lipids of protein histidine phosphorylation in the islet β-cell in vitro[135]. We observed that the phos-phoenzyme formation of NDPK was stimulated by various fatty acids in the following rank order: linoleic acid > arachidonic acid (AA) > oleic acid > palmitic acid = stearic acid = control. Furthermore, the catalytic activity of NDPK was stimulated by these fatty acids in the rank order of: oleic acid > AA > linoleic acid > palmitic acid = stearic acid = control. AA-methyl ester, an inactive analogue of AA, did not significantly affect either the phosphoen-zyme formation or the catalytic activity of NDPK [135]. Interestingly, AA exerted dual effects on the histidine phosphorylation of the Gβ-subunit; it significantly stimulated the phosphorylation at 33 μM, beyond which it was inhibitory. Again, the effects of AA on the Gβ-subunit phosphorylation were specific, since, in a manner akin to NDPK (see above), palmitate (up to 160 μM) had no demonstrable effect on the Gβ-subunit phosphorylation [135].
Our observations of the activation of protein histidine phos-phorylation of NDPK and the Gp-subunit by AA, linoleate and oleate, but not palmitate or stearate, become important since these fatty acids, especially the unsaturated fatty acids, could elicit their effects on insulin secretion by regulating islet endogenous G-protein activation. Based on a convincing body of experimental evidence, we recently proposed that these lipid messengers might exert their positive modulatory effects of G-protein functions at various levels [135]. For example, we previously reported that AA inhibited the GTPase activity in a concentration-dependent manner in both the membrane and cytosolic fractions [98]. A half-maximal inhibition (in both the cytosolic and membrane fractions) was seen at 90–150 μM of AA, the concen trations which fall in the range that has been reported to potently stimulate insulin secretion from isolated rat islets and to be generated by physiological (i.e. glucose) stimulation of the islets [136]. We also observed a 75% inhibition in the GTPase activity by AA in human islet cytosol. Furthermore, of all the four long-chain fatty acids tested, AA inhibited the GTPase activity the most. The rank order of inhibition was AA > linoleic acid > oleic acid > palmitic acid = control, suggesting a role for the degree of unsaturation in the inhibition of GTPase activity, as has been shown for their insulinotropic potency [136]. Our observations of the potential regulation by AA of NDPK and the Gβ-subunit phosphorylation are also physiologically relevant since we observed AA effects at concentrations that are attained in a glucose-stimulated islet [136]. Together, these findings identify additional loci (e.g. NDPK and the Gβ-subunit) at which lipid messengers could exert their intracellular effects and could provide a possible novel regulatory mechanism whereby glucose-induced generation of lipid messengers promotes insulin release. It should also be pointed out that Yaney and Corkey presented experimental evidence indicating that specific members of protein kinase-C family represent the potential effector proteins in the modulation of β-cell function by long-chain fatty CoAs and their esterified derivatives [137]. Our above-described observations provide support for an additional signalling pathway that involves protein histidine phosphorylation, which is regulated, specifically by unsaturated, but not saturated, fatty acids.
Based on the above discussion, it appears that at least two distinct mechanisms of regulation by fatty acids might underlie the cascade of events leading to insulin secretion. We propose that unsaturated fatty acid-derived, NDPK-mediated pathway represents one such signalling step, presumably involving the regulation of (both inhibitory and stimulatory) the G-proteins within the β-cell. Alternatively, it is possible that fatty acid-potentiated insulin secretion might involve the intermediacy of a G-protein that is activated via a histidine kinase-sensitive mechanism. Indeed, the recent studies by Latour and coworkers [138] provided convincing evidence in support of such a formulation. In these studies, they investigated a role for GPR40, a G-protein-coupled receptor in fatty acid potentiation of insulin secretion. These investigators demonstrated a significant (nearly 50%) inhibition in insulin secretion in response to Intralipid (Fresenius Kabi, Uppsula, Sweden) in the GPR40 knockout mice. Fatty acid-mediated poten-tiation of insulin secretion was also reduced significantly in isolated islets from the GPR40 knockout mice; their response to glucose and other insulin secretagogues, however, remained unaltered. Moreover, YM-254890, a selective inhibitor of Gaq/11, inhibited palmitate potentiation of glucose-stimulated insulin secretion in a dose-dependent fashion. Based on these and other supporting findings, these authors concluded that GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion [138]. The GPR40 knockout mouse should serve as a useful model to undertake a study not only to identify the putative trimeric G-protein coupled to this receptor, but also to verify the potential regulation of the candidate G-protein via phosphorelay mechanisms (see below).
Activation of histidine phosphorylation by Mas analogues
Mas, a tetradecapeptide isolated from wasp venom, is an activator of G-proteins [123, 124]. It has been suggested by Kikkawa et al. that Mas-mediated activation of NDPK provides a mechanism for promoting GTP-for-GDP exchange onto G-proteins [139]. Previously, we demonstrated that Mas stimulated NDPK activity in rat islet homogenates [47]. Furthermore, several earlier studies, including our own, have clearly demonstrated that it promotes insulin secretion from normal islets and clonal β-cell preparations [70, 94, 140–142]. In addition, we have reported Mas-dependent stimulation of a high-affinity guanosine triphosphatase (GTPase) activity in the secretory granules purified from normal rat and human islets [99]. During the same time, Konrad and coworkers [142] also reported a significant stimulation of GTPase activity by Mas-7 and Mas-8 in the secretory granule fraction isolated from the insulin-secreting β-cells. However, Mas-17, an inactive analogue of Mas, failed to exert any effects on this GTPase activity, suggesting a structure-specific activation of this G-protein(s) by Mas [142]. Along these lines, we also observed that Mas analogues stimulated the P-His phosphorylation of the Gβ-subunit in a structure-specific manner [70]. We noticed a significant stimulation in the P-His phosphorylation of the Gβ-subunit with the following rank order: Mas-7 > Mas > Mas-17 = control. Compatible with these data are our observations that demonstrated a similar stimulatory effect of Mas, but not Mas-17, on the histone H4-phosphorylating histidine kinase activity and insulin secretion from normal rat islets [70]. Thus, these findings establish a potential relationship between histidine kinase activation and Gp-sub-unit phosphorylation lysates derived from rat islets. Furthermore, they identify additional regulatory loci for Mas, which has been shown to activate G-proteins, specifically via the activation of GTP/GDP exchange processes [123, 124, 139]. While several previous studies, including our own have demonstrated insulinotropic effects of Mas, the observations reviewed in this section suggest, for the first time, that Mas-mediated signalling events could include activation of protein histidine phosphorylation in the pancreatic β-cell. Additional studies are needed to identify those G-proteins and their subcellular localization (e.g. secretory granules) that might require Mas-(or glucose ?) sensitive-histidine phosphorylation for optimal activation and propagation of signals necessary for insulin secretion.
In support of our hypothesis that cellular activation leads to rapid protein histidine phosphorylation, Crovello et al. provided the first direct evidence for the induction of rapid and reversible histidine phosphorylation in the mammalian cells upon activation [54]. They demonstrated that exposure of platelets to thrombin or collagen results in transient phosphorylation of histidine residues on the cytoplasmic tail of P-selectin. These investigators provided convincing evidence indicating that the kinetics of histidine phosphorylation and dephosphorylation on P-selectin is rapid. Thus, these studies provide further support to the hypothesis that protein histidine (de)phosphorylation could represent one of the key signalling steps in cellular activation in specific cell types, including the islet β-cell.
Functional consequences of protein histidine phosphorylation
Based on the existing evidence, we propose that protein histidine phosphorylation could play significant regulatory roles in islet function at several levels, leading to insulin secretion under physiological conditions. These include, but not limited to: (i) compartmentalized generation of GTP (via transphosphorylation of GDP in the presence of ATP), (ii) activation of islet endogenous G-proteins that exert essential roles in GSIS, (iii) regulation of the mitochondrial function and key enzymes of intermediary metabolism, (iv) regulation of ion channels and (v) regulation of other aspects of cellular metabolism, including isoprenoid metabolism. These aspects are briefly discussed below.
Compartmentalized generation of GTP
Using specific inhibitors of inosine monophosphate dehydroge-nase, Metz and colleagues [89, 90] have suggested a permissive role for GTP for physiological insulin secretion; these findings were confirmed by studies from Sharp's laboratory [143]. However, there are remarkably little data available regarding the mechanism of conversion of NDPs to NTPs, the active form of purine nucleotides in the islets. While the mitochondria convert a substantial degree of ADP to ATP in most cells, there are virtually no extant studies conducted with regard to the conversion of GDP to GTP in the islet β-cell. Potential sites for the latter include substrate-level phosphorylation via SCS in the mitochondria [60; see below for additional discussion] and NDPK in the islet β-cell sub-cellular compartments (e.g. the mitochondria or cytosol). Although electron-transport chain could, in principle, use GDP as substrate, it is not a substrate for the mitochondrial nucleotide translocase [144], which is coupled to oxidative phosphorylation. On the basis of the data that we accrued on islet NDPK (see above), one might ask the question whether there is a link between NDPK and intracellular GTP levels. Since the intra-islet GTP concentrations are rather high (about 0.75 mM), we feel that the conversion of GDP to GTP (from GDP and ATP) by NDPK may not be required for all GTP-mediated events in the islet β-cell. However, it seems likely that NDPK might play a critical regulatory role in maintaining the GTP/GDP ratio in the ‘vicinity’ of putative G-proteins; such ‘newly formed’ GTP may then be channelled to the G-proteins [145]. Moreover, as implicated by Srere [146], it is also possible that NDPK may play a role in coupling the mitochondrial events to the cytosolic events necessary for insulin exocytosis. This may be achieved by transferring the terminal phosphate from the mitochondrially derived ATP to cytosolic GDP in the proximity of key intracellular organelles (i.e. the plasma membrane), thus ‘buffering’ a high local GTP/GDP ratio that may be required for insulin exocytosis. In support of this formulation, previous data suggest that the GTP/GDP ratio might be more important than absolute GTP content in modulating secretion [89, 90]. Furthermore, these studies have shown that maneuvers that reduce mitochondrial ATP formation in islets are accompanied by parallel changes in GTP, UTP and GTP/GDP ratio, presumably consumed to regenerate ATP viathe actions of NDPK [89, 90].
Regulation of islet endogenous G-proteins
Although the precise molecular and cellular mechanisms underlying the role of GTP in GSIS remain to be defined, available evidence indicates that it might involve activation of one or more G-proteins endogenous to the islet β-cell (see the above descriptions, and References [91–121]). At least two major groups of G-proteins have been identified and characterized in the islet β-cell (see Reference [39] for a review). The first group consists of trimeric G-proteins comprising a- (39–43 kD), β- (35–37 kD) and γ- (5–10 kD) subunits. These are involved in the coupling of various G-protein-coupled receptors to their intracellular effector proteins, including adenylate cyclase, phosphodiesterase and several forms of PLases. The second group of G-proteins comprises small molecular mass-G-proteins (20–25 kD) that are involved in sorting of proteins as well as trafficking of secretory vesicles. In support of this postulation that G-proteins, specifically the small G-proteins, are involved in GSIS is the well-established experimental support which suggests that the signalling steps involved in GSIS from the β-cell involve well-regulated trafficking of insulin-laden secretory granules for their docking and fusion with the plasma membrane [33].
Original observations from multiple laboratories, including our own, demonstrated critical involvement of small G-proteins, such as Rac1, Cdc42, Rap1 and ADP-ribosylation factor 6 (ARF 6), in GSIS from normal rat islets, human islets and clonal β-cell preparations [91–121]. Such conclusions were drawn primarily based on the data from three mutually complementary experimental approaches. The first approach involved the use of clostridial toxins (e.g. toxin A or B) that monoglucosylate and inactivate specific G-proteins [105]. The second experimental manipulation involved molecular biological approaches including expression of dominant-negative mutants and(or selective knockdown (i.e. siRNA methodology) of candidate G-proteins [115, 118, 120, 121]. The third approach involved the use of pharmacological inhibitors of G-protein activation to further decipher their regulatory roles in GSIS (summarized in References [33] and [119]). Potential regulation of G-protein function via histidine phosphory-lation is discussed below.
Regulation of small G-protein function by histidine phosphorylation
A growing body of evidence from multiple laboratories implicates a significant cross-talk between various isoforms of nm23/NDPK and small molecular mass-G-proteins and/or their regulatory proteins/factors. For example, Otsuki et al. [62] have reported interaction between Tiam1, a guanine nucleotide exchange factor for Rac1, and nm23-H1 in 293T human embryonal kidney cells. They reported a significant inhibition of Tiam1-induced formation of GTP-bound Rac1 and c-Jun kinase activation in cells overexpress-ing nm23-H1. Furthermore, forced expression of the wild-type nm23-H1, but not its kinase- deficient mutant, converted the GDP-bound forms of Rac1, Cdc42 and Rho-A to their active GTP-bound forms in vitro. Based on these findings, these authors concluded that nm23-H1 plays a negative modulatory role in the Tiam1-induced activation of Rac1 [62].
Palacios et al.[63] demonstrated a potential cross-talk between nm23-H1 and ARF 6. They demonstrated that constitu-tively active ARF 6 binds and recruits nm23-H1 to cell junctions. Further studies revealed that the localization of nm23-H1 at these sites facilitates dynamin-dependent endocytosis and down-regulation of Rac1 activity. It has also been reported that nm23-H2 associates with β-integrins through integrin cytoplasmic domain-associated protein 1-α, resulting in inhibition of Rac1 and Cdc42 activation during integrin-mediated cell adhesion [63].
Using NIH-3T3 cells, Gallagher and coworkers have recently reported novel roles for NDPK in Rac-related cortical events as well as GTP-dependent intracellular vesicular trafficking [147]. Using immunofluorescence approach, they reported two distinct pools of cytosolic NDPK that appear to be differentially regulated. The first pool of NDPK associates with membrane ruffles and lamellipodia following cellular activation, which is elicited by serum supplementation, growth factors or bombesin. Interestingly, overexpression of activated Rac1, but not its inactive mutant, in these cells markedly promoted NDPK translocation to the cell cortex, thus mimicking the effects of cellular activation. The second pool of cytosolic NDPK appears to sequester around intracellular vesicles independent of cellular activation, but seem to be governed by microtubule integrity, and intracellular GTP levels [147]. These findings clearly suggest distinct regulatory roles of NDPK in cellular functions, including intracellular vesicular trafficking.
Small G-proteins such as Ras have been implicated in human cancers. In specific cancerous cells, Ras proteins remain activated in their GTP-bound conformation since the bound GTP is resistant to hydrolysis due to mutations in the GTPase functional domain. Fischbach and Settleman [148] have recently reported the inactiva-tion of oncogenic Ras proteins by NDPK. They also demonstrated the localization of an enzymatic activity in Escherichia coli (E. coli) extracts that can efficiently hydrolyse the mutationally activated GTP-bound Ras to its inactive GDP-bound conformation. Purification of this GTPase activity from the E. coli extracts led to its identity as NDPK. The purified NDPK effectively inactivated several Ras forms associated with human cancers including the RasD12, but not the wild-type Ras, or the other GTP-bound G-proteins belonging to the Rho subfamily [148]. Together, these observations suggest novel GTPase-like properties of NDPK.
Finally, studies from Kahn's laboratory yielded additional regulatory roles for nm23 in cellular function. They first reported localization of a 35 kD protein, named Rad (Ras associated with diabetes) in skeletal muscle of type 2 diabetics [149]. Subsequently they reported GTPase-activating protein (GAP)-like properties for nm23 towards Rad [57]. Immunodepletion of nm23 in human skeletal muscle cytosol completely prevented Rad-GAP activity which was recovered following incubation with bacterial nm23. Furthermore, incubation of Rad-GDP with ATP yielded the active GTP-bound form via a transphosphorylation reaction catalysed by nm23. Together, these data suggest dual roles (i.e. modulation of GTP binding and hydrolysis) for nm23 in the regulation of Rad function. Interestingly, Rad also elicited direct effects on NDPK activity of nm23. Based on these observations, these investigators concluded that the interaction of nm23 and Rad provides a novel mechanism for bidirectional, bimolecular regulation in which nm23 stimulates both GTP binding and hydrolysis of Rad, whereas Rad regulates the activity of nm23 [57].
From the above discussion, it is apparent that nm23/NDPK signalling cascade plays central roles in cellular function, specifically at the level of functional regulation of small G-proteins. It is likely that such interactions between nm23/NDPK and small G-proteins (e.g. Rac1, Cdc42 and ARF 6, etc.) might be taking place in the pancreatic β-cell, as well. As stated above, numerous studies from different laboratories have documented evidence to suggest regulatory roles for these G-proteins in GSIS. Specifically, we have been able to demonstrate essential roles for Rac1 in signalling events leading to insulin secretion [33, 108, 115, 118, 119]. We also reported the localization of regulatory factors for glucose-induced activation of Rac1 in insulin-secreting cells, including the GDP-dis-sociation inhibitor (GDI; 121], and obtained evidence of regulatory roles for Tiam1 in glucose-induced Rac1 activation in islet β-cells (Presented at the 67th Annual Scientific Session of the American Diabetes Association – held in Chicago in June, 2007.).
Recent evidence from our laboratory further supports the formulation that nm23/NDPK exerts direct regulatory roles in GSIS. For example, we found that forced expression of the wild-type nm23-H1, but not its kinase-deficient mutant (H118F), markedly potentiated GSIS from INS 832/13 cells [150]. No effects of nm23-H1 expression were seen in cells incubated in the presence of basal glucose, suggesting that signals derived from glucose metabolism are needed for the potentiating effects of nm23-H1 [150]. We also obtained preliminary evidence to demonstrate glucose-mediated translocation of nm23-H1 to the membrane fraction in INS 832/13 cells under conditions in which it promoted translocation and membrane association of small G-proteins (e.g. Rac1 and Cdc42; 115, 118, 120] (Presented at the 7th International Congress of the NDP Kinase/NM23/awd Family: Emerging links between cancer, metassasis, metabolic syndrome, cystic fibrosis and fetal development – held at the University of Dundee, Scotland in September, 2007.). Based on these observations, I propose a model (Fig. 1) for nm23/NDPK-mediated, G-protein-sensitive signalling steps leading to GSIS in the islet β-cell. Potential roles for these histidine kinases as GEFs, GAP and other G-protein regulatory mechanisms (see the above discussion) remain to be determined in the islet. Follow-up studies are underway to further decipher the potential glucose-induced crosstalk between various islet G-proteins (e.g. Rac1, Cdc42 and ARF 6) and NDPK (e.g. nm23-H1, H2 and H4) to determine the roles for these signalling steps in insulin secretion.
Fig 1.
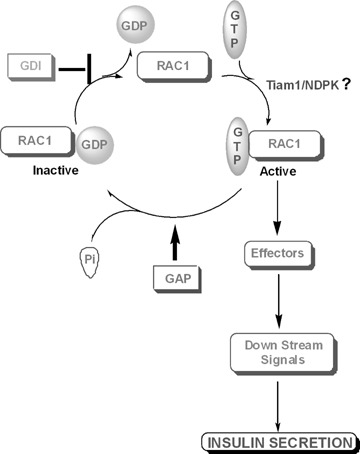
Functional regulation of small molecular weight-G-proteins (e.g. Rac1) in the islet β-cell: potential involvement of a histidine kinase. Recent evidence suggests key regulatory roles for small molecular weight-G-proteins (e.g. Cdc42, Rac1, Rap1 and ARF 6) in GSIS (see text for additional details). Recently, we have also identified additional regulatory factors in the islet β-cell that are involved in the activation of some of these G-proteins (e.g. Rac1). These include the GDP dissociation inhibitor (GDI), which remains associated with the GDP-bound Rac1 (inactive) in the cytosolic fraction [119, 121]. Upon receipt of appropriate signals consequent to glucose metabolism (e.g. biologically active lipids), Rac1 translocates to the membrane fraction where it is dissociated from the GDI to gain the GTP-bound active conformation. Our data also suggest that the GTP/GDP exchange is catalysed by guanine nucleotide exchange factors (e.g. Tiam1). The GTP-bound Rac1 then activates various effector proteins to generate downstream signals necessary for GSIS. Following this, the GTP bound to Rac1 is hydrolysed to GDP by its intrinsic GTPase activity. Although not identified in the β-cell, evidence in other cell types suggests that the GTPase activity is subjected to further activation by the GTPase-activating protein (GAP). Evidence in other cell types also suggests a potential interaction between the G-protein regulatory factors and NDPK, which might be necessary for the functional regulation of specific G-proteins. Our own observations in the islet β-cell indicate a potential association of small G-proteins with NDPK, which could mediate direct activation of these G-proteins via transphorylation of GDP to GTP.
Regulation of trimeric G-protein function by histidine phosphorylation
The published evidence from several laboratories suggests functional activation of trimeric G-proteins via histidine phosphorylation [151, 152]. As recently reviewed by Hippe and Wieland [153], at least two potential mechanisms might exist for the activation of this class of G-proteins by NDPK/nm23-like enzymes. The first one involves the generation of GTP from ATP and GDP by NDPK in the close vicinity of the G-protein, which in turn, could be transferred onto the Ga-subunit via GTP/GDP exchange. The second mechanism might underlie the transphosphorylation of GDP bound to the Ga-subunit by NDPK/nm23 to GTP in the presence of intracellular ATP. While the first mechanism of activation still appears to be plausible, the second one was met with a considerable skepticism (see Reference [153] for a detailed discussion of this topic). Furthermore, a three-dimensional structural data analysis indicates that the P-His in the NDPK and the GDP bound to the Gα-subunit are deeply buried within their cognate structures, thus making the phosphotransfer from phospho-NDPK to GDP bound to the Ga-subunit rather unlikely [153].
What then are the potential mechanisms whereby histidine phosphorylation leads to activation of trimeric G-proteins? It has been proposed that the Gβ-subunit undergoes histidine phosphorylation; this phosphate, in turn, is transferred to GDP bound to the Ga-subunit to yield the GTP-bound active conformation (see the above sections for additional background information). Experimental evidence accrued in multiple cells, including the islet β-cell, in support of such a formulation is briefly described below.
Original studies by Wieland and colleagues [38] have demonstrated that the Gβ-subunit undergoes phosphorylation in HL-60 cell membranes in a GTP-dependent manner. They also suggested a requirement for an additional protein/factor for this signalling event to take place [122]. In 1996, we demonstrated that the Gβ-subunit undergoes histidine phosphorylation in the membrane and secretory granule fractions purified from normal rat islets, human islets and clonal β-cell preparations [39]. In the reconsti-tution protocols involving the purified αβγ-subunits and islet sub-cellular fractions, we also demonstrated that a membrane-associated factor (kinase ?) is required for Gβ phosphorylation to occur [39]. We were also able to demonstrate that incubation of the phosphorylated Gβ-subunit with the GDP-bound Ga-subunit accelerated not only the dephosphorylation of the Gβ-subunit, but also was accompanied by the formation of Gα-GTP [39]. These data, however, do not provide adequate information with regard to the identity or properties of the putative histidine kinase that mediates the phosphorylation of the Gβ-subunit. In an attempt to further understand these signalling steps, we were able to identify and characterize a histone H4-phosphorylating histidine kinase in insulin-secreting cells (Table 3). It remains to be seen if this kinase subserves the function of the Gβ-subunit phosphory-lating kinase.
Klinker and Seifert [154] investigated additional roles for NDPK in the activation of transducin, a retinal G-protein via histidine phosphorylation of the Gβ-subunit. They were able to measure a significant NDPK activity in soluble transducin preparations; this activity was referred to as transducin-NDPK. Remarkably, transducin-NDPK exhibited preference to GDP as the phosphoryl acceptor and ATPγS as the most effective thiophosphoryl donor. Using [γ–32P]ATP or GTP or [7–35S]ATPγS, they further demonstrated (thio)phosphoryla-tion of a 36 kD protein, similar to the molecular size of the Gβ-sub-32 unit. 32P-labelling of this protein demonstrated characteristics of histidine phosphorylation. Based on these findings, these authors concluded that soluble preparations of transducin contain a guanine nucleotide-sensitive NDPK, and that the transducin β-subunit serves as a phosphorylated NDPK intermediate in promoting the activation of this retinal trimeric G-protein [154].
More recent studies by Cuello et al. demonstrated [155] activation of trimeric G-proteins by a high-energy phosphate transfer from the histidine-phosphorylated NDPK to the Gβ-subunit of trimeric G-proteins. Using the bovine retinal and brain preparations, these investigators observed that NDPK-B forms complexes with the β7-subunits of trimeric G-proteins and contributes to the activation of the respective G-protein by increasing the high-energy phosphate transfer from a transiently phosphorylated Gβ-subunit (at His-266 residue) to Gα-GDP, to yield an active, GTP-bound conformation (i.e. Gα-GTP). Additional studies along these lines by Hippe et al. provided further evidence in support of the above formulation that NDPK contributes to G-protein activation in intact H10 cells [156]. They found that overexpression of either NDPK-A or NDPK-B had no effects on the intracellular ATP, GTP, adenylyl cyclase or cyclic adenosine monophosphate (cAMP) levels. Overexpression of the α-subunit of the stimulatory G-protein (i.e. Gsa) resulted in a significant increase in the intracellular cAMP levels, which was further potentiated in the cells overex-pressing NDPK-B, but not NDPK-A. Overexpression of the kinase-dead mutant of NDPK-B (H118N) inhibited Gsa-mediated increase in cAMP levels. These studies also demonstrated association between NDPK and the βγ complex. Finally, overexpression of NDPK-B, but not NDPK-A, in these cells resulted in a significant increase in the phosphorylation of the Gβ-subunit. Together, observations of Cuello et al. [155] and Hippe et al. [156] clearly indicate that NDPK-B promotes activation of trimeric G-proteins. Based on these findings, these investigators proposed a mechanism for the receptor-independent activation of trimeric G-proteins [153].
Lastly, studies by Marciniak and Edwardson in zymogen granule, indeed, shed further insights into the potential roles for histidine phosphorylation in the control of exocytosis [157]. They observed that NDPK is associated with the cytoplasmic face of pancreatic zymogen granules. It also behaved as a 21 kD phos-phoprotein and was able to generate GTP from ATP and GDP. In a manner akin to the islet NDPK (see above), the zymogen granule NDPK is stimulated by Mas. Further studies revealed that the GTP produced (from ATP and GDP) by endogenous NDPK not only promoted the histidine phosphorylation of a 37 kD zymogen granule protein, but also stimulated the fusion of zymogen granules with pancreatic plasma membranes in vitro. The identity of the 37 kD protein was not determined, but could represent the Gβ-subunit. Based on these observations, the authors concluded that the granule-associated NDPK might be involved in the control of exocytosis in the pancreatic acinar cell [157].
Together, it is plausible that nm23/NDPK-like enzymes that we characterized in the islet β-cell (Table 1) could subserve the function of histidine phosphorylation of key proteins, leading to the generation of appropriate signals necessary for physiological insulin secretion, including fusion of insulin-laden secretory granules with the plasma membrane. In addition, our previously published evidence [39] indicating that the incubation of phosphory-lated Gβ-subunit with Gα-GDP accelerated the dephosphorylation of the Gβ-subunit, accompanied by the formation of Gα-GTP, further substantiates such a formulation. Taken together, based on our own observations in normal rat islets, human islets and clonal β-cells and their subcellular fractions (e.g. plasma membrane and secretory granules), we propose a model for glucose-induced, non-receptor-mediated activation of trimeric G-proteins in the islet β-cell (Fig. 2). A detailed description of our proposed model is provided in the caption to Fig. 2.
Fig 2.
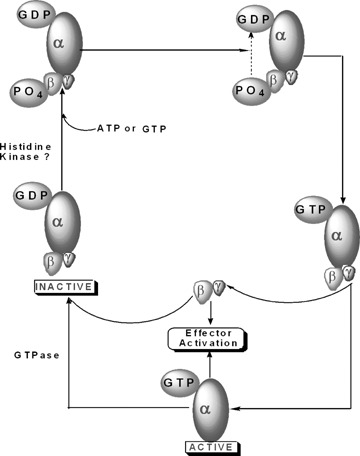
Proposed mechanism for an alternate, receptor-independent activation of trimeric G proteins in the pancreatic β-cell by glucose. Trimeric G-proteins remain inactive when their respective α-subunit is bound to GDP. In the context of physiological (i.e. glucose-induced) insulin secretion, we propose that either a member of the histi-dine kinase superfamily (Table 3) or NDPK phosphorylates the Gβ-subunit at a histidine residue via a phosphorami-date linkage. This phosphate, in turn, is relayed to the GDP bound to the Gα-subunit to yield the active, GTP-bound conformation of the G-protein [33]. It is proposed that such a mechanism is similar to the classical ping-pong mechanism of activation of NDPK. Following this, the Gα-gtp dissociates from the Gβγ complex for regulation of its respective effector proteins. It should be also noted that the βγ complex, by itself, is able to regulate various effector proteins. Following hydrolysis of the GTP bound to the Gα-subunit by its intrinsic GTPase activity, the Gα-GDP re-associates with the βγ-complex to complete the activation cycle. Not shown in the figure is the possibility of a membrane-associated NDPK (nm23-H2; Table 2) mediating the phosphorylation of the Gβ-subunits (see text for additional details and relevant citations in this area).
Regulation of the mitochondrial function via protein histidine phosphorylation
As reviewed above, available evidence suggests that P-His regulates functional activation of several enzymes on intermediary metabolism, including ATP-citrate lyase, aldolase, SCS and fruc-tose-2,6-bisphosphatase (see Table 1 for a list of some of these enzymes that are relevant to glucose metabolism in the islet β-cell). Recent data from our laboratory have identified potential association between SCS and mNDPK in the insulin-secreting cells [71]. These findings are important, especially in the light of the previous observations from our laboratory that identified and characterized both ATP- and GTP-sensitive SCS activities in the mitochondrial fraction purified from isolated β-cells [45]. More recent observations by Kibbey et al. [158] indicating central roles for mitochondrial GTP in GSIS, indeed, warrant further studies to examine the relative significance of mitochondrial protein histidine phosphorylation, potentially mediated by mitochondrial histidine kinases, in signalling events leading to GSIS.
What are the functional roles of the mitochondrial isoform of NDPK (mNDPK) in β-cell stimulus-secretion coupling? First, mNDPK could contribute to the compartmentalized synthesis of GTP in the mitochondria for various GTP-requiring functions, including activation of the mitochondrial G-proteins. Second, it could subserve the function of a protein histidine kinase, thereby regulating the functional properties of the mitochondrial proteins such as SCS. These aspects are discussed below.
A role for mNDPK in the generation of intramitochondrial GTP is an important function since unlike ATP, GTP is not transported across the mitochondrial inner membrane via the classical nucleotide translocase [71]. Therefore, it is likely that GTP is formed within the mitochondria via the transphosphorylation of GDP in the presence of ATP, a reaction catalysed by mNDPK. Along these lines, multiple functions for GTP in the mitochondria have been described previously. For example, GTP is required for the activation of specific mitochondrial G-proteins that have been implicated in several mitochondrial functions, including protein synthesis, steroidogenesis, protein transport, membrane fusion, contact point alignment and permeability transition [159–163]. Three previous studies from our laboratory provided evidence in support of the localization of G-proteins in the mitochondrial fractions isolated from normal rat islets and clonal β-cells. In the first study, we have detected significant amounts of high-affinity GTPase activity in the normal rat islet mitochondrial fraction [96]. In the second study, we observed enrichment of prenyl-cysteine methyltransferase activity in the mitochondrial fraction derived from INS-1 cells; this enzyme carboxylmethylates and activates specific G-proteins in a GTP-sensitive manner [164]. In the third study, we reported immunological evidence to indicate the localization of specific Rho subfamily of small G-proteins (e.g. Rac) in isolated mitochondrial fractions from βTC3 cells [112]. Based on these findings, we propose that mNDPK could play contributory roles in maintaining the GTP/GDP ratio in the ‘vicinity’ of candidate GTP-binding and/or other GTP-requiring proteins.
The above formulation is further supported by our findings suggesting inhibition, by G-protein inhibitors, of insulin secretion elicited by mitochondrial fuels such as the succinic acid methyl ester (SAME). Pre-incubation of insulin-secreting βTC3 cells with Clostridium difficile toxin-B, which monoglucosylates and inactivates Cdc42 and Rac1 [112], markedly decreased (> 70%) SAME-induced insulin secretion. Furthermore, exposure of these cells to GGTI-2147, a selective inhibitor of the requisite prenylation of Rac1 and Cdc42, significantly reduced (80%) SAME-induced insulin release, suggesting that post-translational prenylation of these proteins is necessary for SAME-induced insulin release [112]. Western blot analysis indicated the localization of Cdc42, Rac1 and Ras in the β-cell mitochondrial fraction. Confocal microscopy revealed a modest, but inconsistent, increase in the association of either Rac1 or Cdc42 with Mitotracker (Invitrogen, Carlsbad, CA (USA)), a mitochondrial marker, following exposure to SAME. These data suggest that the activation of pre-existing intra-mitochondrial Rac1 and Cdc42 may be sufficient to regulate SAME-induced insulin secretion. It is reasonable then to speculate that intra-mitochondrial GTP, derived potentially from mNDPK, is utilized for the activation of mitochondrial G-proteins. Together, these findings support a role for G-proteins in insulin secretion at a step dependent on the mitochondrial metabolism [112]. They also identify mevalonate-derived, isoprenoid-modified (i.e. ger-anyl-geranylated) Rho G-proteins as specific signalling molecules in insulin secretion; these findings further substantiate the succi-nate mechanism of insulin release originally proposed by Fahien and MacDonald [165].
In addition to the activation of mitochondrial G-proteins, GTP is also required for the optimal functioning of other mitochondrial proteins such as the GTP-specific SCS activity that we have characterized in the β-cell previously [60]. Studies by Alarcon et al. [166] and Kibbey et al.[158] further characterized SCS in the islet β-cell and provided additional insights into the regulatory roles for this enzyme in the islet function. For example, Kibbey et al. provided conclusive evidence to suggest that mitochondrial GTP regulates GSIS [158]. They demonstrated that siRNA-mediated suppression of the GTP-producing pathway markedly reduced (by 50%) GSIS in INS-1 832/13 cells. Based on these and other supporting evidence, these investigators concluded that mitochondrial GTP plays an important role in insulin secretion, possibly involving intra-mitochondrial calcium [158]. On the basis of these findings, one might ask the question if SCS represents the primary source of GTP in the islet β-cell, which is essential for GSIS to occur [89, 90, 143]. As demonstrated clearly by Kibbey et al. [158], it would definitively serve as the main source for the mitochondrial GTP (also see above for a detailed discussion). Along these lines, earlier studies by Alarcon and coworkers [166] identified SCS activity in the cytosolic fraction derived from normal rat islets. Such an activity represented nearly 25% of the total SCS activity in islets, thus raising a possibility that it might serve as a source for GTP in the cytosolic fraction as well. In this context, studies by Metz and colleagues [89, 90] reported greater than 80% inhibition in intra-islet GTP content in the presence of mycophenolic acid, an inhibitor of de novo GTP biosynthesis. Together, these data suggest that the de novo as well as the salvage GTP biosynthetic pathways and the SCS could represent potential sources for GTP within the β-cell. It is emphasized that while NDPK-derived GTP may not contribute significantly to the increase in the levels of GTP in a stimulated islet, it likely plays a regulatory role in the generation of GTP in the ‘vicinity’ of candidate G-proteins [145], whose functional activation is necessary for GSIS (see the above discussion).
Previous studies have demonstrated that mNDPK might subserve the function of a histidine kinase as well. For example, it has also been shown that mNDPK associates with (and/or copurifies with) the putative phosphoprotein substrates. Kavanaugh-Black et al. [167] have demonstrated copurification of NDPK with SCS activity and provided additional evidence for the phosphorylation of SCS by NDPK. Using rat liver mitochondrial preparations, Kadrmas and coworkers [168] also described the copurification of SCS with NDPK on sucrose gradients. Therefore, it is likely that mNDPK might be regulating SCS activity in the β-cell via phosphorylation of its β-subunits at a histidine residue [60]. Our findings of the submitochondrial colocalization of these two proteins (i.e. mNDPK and GTP-specific SCS) in the mitochondrial fraction further support the possibility that they might remain associated with each other. It remains to be seen whether it undergoes autophosphorylation or an NDPK-mediated phosphorylation. A close association of NDPK and SCS, as demonstrated in our earlier studies, argues in favour of the latter [60]. Furthermore, the studies by Kavanaugh-Black suggested that even though SCS undergoes ATP- or GTP-dependent autophosphorylation, the presence of small amounts of NDPK influences the level of SCS autophosphorylation [167]. Together, these data support our formulation that histidine phosphorylation is critical to β-cell stimulus-secretion coupling not only in the generation of GTP, but also in the functional regulation of several key proteins of intermediary metabolism, such as SCS. Lastly, it is likely that mNDPK might interact with other mitochondrial proteins, as well. This is true, especially based on the studies of Srere and coworkers [169, 170] that clearly indicated the existence of complexes (appropriately termed as ‘metabolons’) of sequential metabolic enzymes involved in the tricarboxylic acid cycle. Such possibilities are being explored currently in our laboratory in relation to the glucose-stimulated insulin secretion. Alternatively, like ATP the intra-mitochondrial GTP could provide signal functions independent from interaction with and activation of β-cell G-proteins to facilitate insulin secretion. Potential intracellular functions and roles of GTP, in the context of insulin secretion, have been reviewed [171].
Regulation of ion channels
Recently, Srivastava et al. [55] provided the first evidence for NDPK-mediated histidine phosphorylation and activation of the calcium-activated potassium channel KCa3.1, which has been shown to play a regulatory role in promoting calcium influx in multiple cell types. These investigators reported direct binding of NDPK-B, which in turn, activates the channel by phosphorylation at the His-358 residue. Such an activation step appears to be fairly specific for NDPK-B since neither NDPK-A nor the kinase-dead NDPK-B mutant [H-118N] elicited the stimulatory effects. Together, these findings provide basis for future research in the area of ion channel regulation by histidine phosphorylation.
Regulation of isoprenoid metabolism
It is well established that the majority of small molecular weight-G-proteins and the G-γ-subunits of trimeric G-proteins undergo a series of post-translational modifications at their C-terminal cys-teines that appear to be essential for the optimal interaction of these proteins with their effector proteins and/or membranous sites [33, 110, 118, 119]. Using generic as well as more site-specific inhibitors of protein fanesylation and geranyl geranylation, we have been able to demonstrate that isoprenoid-derived modification of these proteins is critical for GSIS; these findings were confirmed via molecular biological approaches, as well [33, 110, 118, 119]. Moreover, the stimulatory concentrations of glucose markedly augment C-terminal modification (e.g. carboxylmethyla-tion) of small G-proteins as well as the G-y-subunits, suggesting a central role for this signalling step in GSIS [104]. Previous studies by Wagner and Wu [172] have demonstrated that nm23-H1 mediates the phosphorylation of farnesyl- and geranyl pyrophos-phates to their respective triphosphates. These investigators also reported a significant increase in the farnesyl triphosphate and geranyl triphosphate in cells with elevated levels of nm23-H1. Further, these cells also contained relatively higher abundance of farnesylated proteins (24 kD and 46 kD) than the control cells. Together, these data appear to indicate novel regulatory roles of NDPK at the level of isoprenoid metabolism; this remains to be verified in the islet β-cell.
Taken together, it is becoming increasingly evident that protein histidine phosphorylation plays key roles in cellular functions in the islet β-cell, including compartmentalized generation of GTP (via transphosphorylation of GDP in the presence of ATP) and activation of islet endogenous G-proteins, which exert essential roles in GSIS. In this context, our findings implicate roles for histidine kinases in the β-cell subcellular fractions (e.g. the mitochondria and cytosol) in the regulation of the islet function. As stated above, a significant amount of NDPK appears to reside in the secretory granule fraction [47]. We are currently examining if such an activity could contribute towards the fusion of secretory granules with the plasma membrane, as demonstrated by Marciniak and Edwardson in the zymogen granule model system [157]. It also appears that it is involved in the regulation of the mitochondrial function (e.g. G-protein activation) and key enzymes of intermediary metabolism (e.g. SCS). Regulatory roles of this signalling step in other cellular functions, including ion channel activation, remain to be explored (see below).
Protein histidine phosphatases
In a manner akin to protein histidine kinases, a growing body of evidence appears to suggest the localization of protein histidine phosphatases in prokaryotes as well eukaryotes (see Reference [24] for a review). The SixA protein, consisting of 161 amino acids chain length with an arginine-histidine-glycine signature at the N-terminus, from E. coli was the first bacterial histidine phosphatase that has been implicated in the histidine-aspartate phosphorelay mechanisms [173]. In 1993, Ohmori et al. reported the localization of a 150 kD protein in rat brain with a dual 6-P-Lys and 3-P-His phosphatase properties [174]. Protein phosphatases with similar catalytic functional properties were also reported in rat tissues by Wong et al. [175]. A series of investigations from Matthews' laboratory have identified protein phosphatases 1, 2A and C as protein P-His phosphatases [176]. These studies also determined inhibitory constants for various known inhibitors of phosphatases (e.g. inhibitors 1, 2, okadaic acid and mictocystin-LR) against P-His dephosphorylation using phosphorylated histone IV as the substrate [18, 176]. As appropriately pointed out by these investigators, potential caveats in these studies include the non-specific nature of the chemical inhibitors used to characterize the putative histidine phosphatases.
The observations from Klumpp's laboratory have provided additional insights into P-His phosphatases in the mammalian cells [27, 177, 178]. In 2002, they reported the identification and functional characterization of vertebrate P-His phosphatase from rabbit liver [27]. Based on extensive studies, including amino acid analysis and inhibitor sensitivity, these investigators concluded that this phosphatase is distinct from other known phosphatases acting on P-Ser, P-Thr or P-Tyr. This phosphatase appears to be highly expressed throughout different tissues. More recent investigations by these researchers suggested that this 16 kD phosphatase mediates the P-His dephosphorylation of ATP-citrate lyase [177].
In this context, Maurer et al. have provided evidence to implicate that the aforementioned P-His phosphatase catalyses the dephosphorylation of the Gβ-subunit, as well [179]. Using an antiserum directed against this phosphatase, these investigators demonstrated the colocalization of the Gβ-subunit and the phosphatase in various rat tissues, including the brain, heart, kidney, spleen and lung. In subsequent experiments, they were able to demonstrate marked dephosphorylation of the Gβ-subunit in H10 and C3 cells overexpressing the protein histidine phosphatase [179].
The above findings are of immediate significance to our published evidence on P-His phosphorylation of the Gβ-subunit in the islet β-cell plasma membrane and secretory granule fractions [39]. Therein, we reported a time-dependent dephosphorylation of the Gβ-subunit (>50% within 20 min.) in the rat islet membranes that was accelerated in the presence of either free GDP or Gα-GDP (40–50% within 10 min.). We also noticed similar dephosphorylation kinetics for the Gβ-subunit in the secretory granule fraction [39]. NDP specificity studies revealed that only GDP, and not ADP, IDP, CDP and UDP subserved the roles of ‘receivers' of the phosphate [39]. Similar NDP specificity has been demonstrated by Wieland et al. for the dephosphorylation of the Gβ-subunit from HL-60 membranes [38]. Together, our findings provide the first evidence for the localization of putative P-His phosphatase in the islet β-cell. As reviewed by the author recently [180], protein phosphatases (e.g. protein phosphatase-2A) play significant roles in islet signal transduction. Indeed, studies from Matthews' laboratory have identified protein phosphatase-2A as one of the P-His phosphatases [176]. Additional studies are needed, however, to identify, characterize and further understand the contributory roles for histidine phosphatase in GSIS.
Potential defects in histidine phosphorylation in islets derived from the Goto-Kakizaki rat, a model for type 2 diabetes
The lesion intrinsic to the pancreatic β-cell in human type 2 diabetes still remains only partially understood. In the search to identify such a defect, animal models of diabetes have frequently been studied. The most widely accepted of these models is the GK rat that is developed by repeated inbreeding of glucose-intolerant Wistar rats [181, 182]. We previously investigated the potential abnormalities in protein histidine phosphorylation in islets derived from GK rats [183, 184]. We noticed a substantial reduction (> 50%) in P-His phosphorylation of NDPK in GK rat islets in the presence of either [γ32P]ATP or GTP. Compatible with these data are our observations of a significant attenuation (−40%) in the catalytic activity of NDPK in the diabetic GK islets when compared to islets from the control Wistar islets. In addition to NDPK, we also observed a marked reduction (>90%) in the degree of endogenous protein histidine phosphorylation in GK rat islets when [y-3 P]ATP was used as the phosphoryl donor [183]. Further, under similar experimental conditions, we observed a substantial degree of reduction (-64%) in histidine phosphorylation of exogenously added histone H4 in the homogenates of diabetic GK rat islets in comparison to those from the control Wistar rats when [γ–32P]ATP or GTP was used as a phosphoryl donor [183]. Together, these data suggest significant alterations in the protein histidine phosphorylation in the diabetic GK rat islets.
What then is the significance of these findings, specifically at the level of impaired insulin secretion seen in these animals? We offer two potential possibilities. First, a reduction in the histidine phosphorylation of specific endogenous proteins (e.g. NDPK, SCS, ATP-citrate lyase, etc) in the GK islet could translate into defects in their functional activation, leading to impaired insulin secretion. Second, the observed defects in protein histidine phosphorylation could lead to abnormal activation of candidate G-pro-teins (both monomeric as well as trimeric forms), which appear to be mediated via the intermediacy of protein histidine phosphorylation (see above).
Along these lines, studies by Metz and coworkers [184] have demonstrated that Mas, but not Mas-17, an inactive analogue, completely restored insulin secretory defects in GK islets. Based on these observations and our findings of a significant stimulatory effect of Mas, but not Mas 17, on islet endogenous NDPK, histone H4-phosphorylating histidine kinase, and the Gβ-phos-phorylating kinase [70], we conclude that the insulin secretory defects in the GK islet might underlie at the site of activation by protein histidine kinase of a Mas-sensitive G-protein-dependent step in exocytosis. Additional studies are needed to further substantiate this formulation.
In this context, previous studies by Ostenson and coworkers provided additional insights into the modes of action-specific chimeric peptides (e.g. galparan) in reversing the insulin secretory defects in GK rat islets. Galparan is a 21-amino acid-containing peptide that is derived by linking galanin (1–13 residues) to Mas via a peptide bond to provide the Mas and galanin effector parts of the molecule [185]. The studies involving this chimeric peptide revealed that it stimulates insulin secretion from normal rat islets by nearly 26-fold in the presence of basal glucose (3.3 mM) and by 6-fold in the presence of stimulatory glucose (16.7 mM) concentrations [185]. Such effects of galparan were independent of extracellular calcium and were also insensitive to pertussis toxin. Interestingly, galparan also stimulated insulin secretion in islets derived from the GK rats to a comparable degree seen in normal rat islets. The reversible nature of defects in insulin secretion seen in GK rat by Mas [184] and galparan [185] could form the basis for the development of novel therapeutics for the treatment of insulin secretory defects in diabetes.
Finally, it remains to be determined whether mNDPK activity is also altered in the diabetic β-cell, thereby contributing to alterations in the high-energy phosphate metabolism and the associated signal transduction steps culminating in impaired insulin secretion in these cells. Putative genetic, biochemical and clinical implications of the signalling events involved in the high-energy metabolism in the context of mitochondrial diabetes have been reviewed recently by Gerbitz et al.[186] and Maechler and Wollheim [187].
Conclusions and future research
The experimental evidence reviewed in this article indicates the localization of at least three forms of nm23/NDPK (e.g. nm23-H1, nm23-H2 and nm23-H4) in the islet β-cell. I also described evidence for the localization of other histidine kinases in the islet and proposed mechanisms for the regulation of endogenous G-protein activation by these kinases. It appears that biologically active lipids modulate the functional activation of these enzymes and other G-protein functions in a structure-specific manner.
On the basis of our current understanding of the biochemical properties and physiological regulation of nm23/NDPK in the islet β-cell, I propose a model for potential contributory roles of histidine kinases in GSIS, specifically at the level of activation of G-proteins (Fig. 3). I propose that glucose-induced increases in the GTP/GDP ratio may, in part, be due to the activation of histidine kinases endogenous to the islet β-cell, which generate GTP via transphosphorylation of GDP from ATP. Such an increase in GTP levels favours either the facilitation of GTP/GDP exchange onto a relevant G-protein(s) or the channelling of GTP to candidate G-protein (s), culminating in their activation. It is also likely that glucose-derived second messengers (e.g. biologically active lipids that are generated via activation of specific PLases) could directly activate histidine kinases, which in turn, may regulate G-protein functional activation. In addition, previous studies from our laboratory have documented evidence to suggest regulation by biologically active lipids, including activation of GTP/GDP exchange (see above) and translocation of the cytosolic G-proteins (e.g. Rac1) to the membrane fraction [188] for activation and interaction with various effector proteins. Lastly, it is possible that glucose metabolism-derived biologically active lipids promote a unique cross-talk among various guanine nucleotide regulatory factors (GRFs; e.g. Tiam1) and NDPK-like enzymes in the modulation of specific G-proteins (e.g. Rac1, Cdc42 and ARF-6) that are essential for GSIS (see Fig. 1 for additional details).
Fig 3.
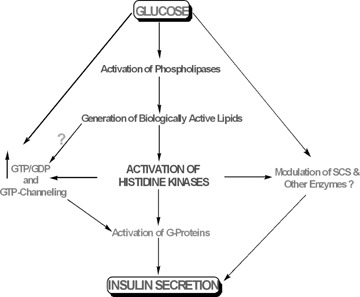
Our proposed model for novel roles for protein histidine phos-phorylation in GSIS. We propose a model for the glucose-mediated activation of histidine kinases leading to GSIS. Glucose metabolism leads to the activation of specific PLases in the islet β-cell, leading to the generation of biologically active lipids, which in turn, activate protein histidine kinases. These kinases regulate specific signalling steps including generation of GTP in the ‘vicinity’ of candidate G-protein for their functional activation. An increase in protein histidine kinase activity also can mediate regulation of several enzymes (e.g. SCS) that are essential for glucose metabolism. Such histidine phosphorylation-mediated activation steps (i.e. activation of specific G-proteins and enzymes involved in glucose metabolic steps) promote insulin secretion. It is also likely that biologically active lipids could exert direct effects on G-protein function independent of histidine kinases; these include stimulation of GTP/GDP exchange and translocation of specific G-proteins (e.g. Rac1) to the membrane fraction for interaction with their respective effector proteins [150].
Also, it is likely that protein histidine phosphorylation plays a critical regulatory role in the activation of trimeric G-proteins in the pancreatic β-cell (Fig. 2). I described evidence [39] to suggest that the Gβ-subunit undergoes histidine phosphorylation mediated by a membrane-associated kinase, such as a high-energy phosphate, which in turn, is transferred to the Gα-GDP to yield its active GTP-bound conformation. The identity of the histidine kinase that catalyses this phosphorylation step remains unknown at the present time. The histone H4-phosphorylating kinase that I described above might represent such a kinase. Alternatively, the nm23/NDPK isoforms, specifically the nm23-H2, might subserve that function; but this remains to be investigated. Existing evidence in other cell types clearly support our working model, but a methodical investigation of glucose- (and other nutrient) mediated regulatory effects on G-protein (small molecular as well as trimeric forms) activation via protein histidine phosphorylation are still needed to definitively understand the in vivo relevance of the in vitro observations.
A considerable amount of work still lies ahead to conclusively demonstrate the potential regulatory roles for protein histidine phosphorylation in cellular signal transduction in general. There are at least two issues that need immediate attention, namely, a refinement in the methodology for the identification and quantita-tion of P-His and the development of novel, cell-permeable inhibitors of protein histidine kinases and phosphatases. As stated above, a significant hurdle still exists with regard to the quantita-tion of histidine kinase activity and P-His phosphorylation due to the labile nature of the high-energy phosphate bond and the acid-labile nature of P-His. The current techniques of SDS-PAGE and/or phosphoamino acid analysis are reasonable but not adequate, and thus, require further refinements. Recently, Kleinnijenhuis and coworkers reported more sensitive methods for the analysis of histidine phosphorylation using synthetic peptides and in more complex samples [69]. In this study, they utilized four different mass spectrometric fragmentation techniques for the detection and quantitation of histidine phosphorylated samples. These methods include, collision-induced dissociation, electron-capture dissociation, electron-transfer dissociation and electron-detachment dissociation. As a logical extension to these studies involving synthetic peptides, they also utilized these methodologies for the detection and quantitation of P-His phosphorylation in tryptic digests of the cytoplasmic domain of the histidine kinase EnvZ, which undergoes autophosphorylation at a histidine residue. In the same study, they introduced a new method for non-retentive solid-phase extraction of histidine phosphorylated peptides using polymeric Strata-X microcolumns [69]. While these novel methodologies provide fresh insights into the detection of P-His residues, their utility in the quantification of P-His in cell lysates and subcellular fractions remains to be verified.
The synthesis and characterization (e.g. structure and activity relationships) of specific cell-permeable inhibitors for the mammalian protein histidine kinases and phosphatases remain an open area of investigation. Indeed, studies from the Matthews laboratory [18] provided insights into these aspects, but a considerable amount of work is still needed in the development of novel pharmacological probes with minimal cytotoxicity for studies involving intact cells. The emergence of recent information [130–134] on the development of specific inhibitors for the two-component phosphorelay system might potentially be useful in addressing this question.
In the context of the islet β-cell and insulin secretion, I believe that the identification and characterization of protein histidine kinases and phosphatases remain an important area of investigation, and we are currently working on some of these aspects. Our goal is to further understand the nature and properties of the histone H4 histidine kinase (Table 3) that we identified in the islet and verify its role in the phosphorylation of the Gβ-subunit to promote trimeric G-protein activation. Experiments are also underway to determine potential sequence homology between the islet histidine kinase (Table 3) and known histidine kinases, including NDPK. Finally, I am optimistic that recent studies from Klumpp's laboratory on the mammalian protein histidine phosphatases [27, 177–179] will aid us in further characterization of such a phos-phatase in the islet β-cell.
In conclusion, the available evidence from multiple laboratories lend evidence to support the overall hypothesis that signals derived from glucose metabolism (e.g. changes in intracellular calcium, cyclic nucleotides, generation of biologically active lipids and increase in intracellular adenine and guanine nucleotides) promote a cascade of events, leading to the transport of insulin-laden secretory granules to the plasma membrane for fusion and release of insulin via exocytosis. Protein (de)phosphorylation, specifically at tyrosine, threonine and serine residues, mediated by specific kinases, has also been recognized as one of the major regulatory steps involved in GSIS. The current review summarizes the potential novel roles for histidine kinases in these signalling events, specifically at the level of compartmentalized generation of GTP from GDP and ATP via the transphosphorylation reactions. A net increase in GTP levels, specifically in the ‘vicinity’ of GTP-requiring proteins, would favour their functional regulation, leading to cellular activation (see above for a detailed discussion).
It may be germane to point out that most of the work reviewed above on the histidine phosphorylation in islet stimulus-secretion coupling comes from the author's laboratory. We have been able to identify and characterize various forms of nm23 and a novel histone H4-phosphorylating histidine kinase in the islet β-cell. We also identified potential endogenous substrates (e.g. SCS, Gβ-subunit) for such kinases in the pancreatic islet β-cell. I would like to emphasize, however, that while our findings support the localization and regulation of several protein histidine kinases and their phosphoproteins in vitro, considerable work is still needed to conclusively demonstrate histidine kinase-mediated regulation of insulin secretion in intact β-cells. I hope that this review will form the basis for additional investigations in this area, specifically in furthering not only our current understanding of this signalling cascade in normal islets, but also in determining the nature and consequences of nm23/NDPK defects that we observed in the diabetic GK rat islet. The data accrued from these investigations may ultimately prove to be important towards the development of novel therapeutics for the prevention and onset of diabetes. Such probes might include, but not limited to, the development of potent and specific stimulators of nm23/NDPK (e.g. Mas-like or galparan-like compounds), with a potential for the reversal of insulin secretory defects in diabetics.
Acknowledgments
The research work described in this review was supported by grants to the author from the Department of VA (Merit Review and REAP Awards), National Institutes of Health (RO1 DK 56005 and RO1 DK 74921), the American Diabetes Association, the Juvenile Diabetes Research Foundation and the Grodman Cure Foundation. AK is a recipient of the Senior Research Career Scientist Award from the Department of VA. The author would like to offer sincere thanks to all the colleagues and collaborators at the William S. Middleton VA Medical Center and the University of Wisconsin in Madison and the John D. Dingell VA Medical Center and Wayne State University in Detroit for their continued support and the necessary assistance in generating the experimental data reviewed in this article.
References
- 1.Malaisse WJ. Hormonal and environmental modification of islet activity. In: Steiner DF, Frankel N, editors. Handbook of physiology. American Physiological Society; 1972. pp. 237–60. In:, editors.. New York:. [Google Scholar]
- 2.Prentki M, Matschinsky FM. Calcium, cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev. 1987;67:1223–6. doi: 10.1152/physrev.1987.67.4.1185. [DOI] [PubMed] [Google Scholar]
- 3.Metz SA. Membrane phospholipid turnover as an intermediary step in insulin secretion. Putative roles of phospholipases in cell signaling. Am J Med. 1988;85:9–21. doi: 10.1016/0002-9343(88)90393-2. [DOI] [PubMed] [Google Scholar]
- 4.MacDonald MJ. Elusive proximal signals of beta-cells for insulin secretion. Diabetes. 1990;39:1461–6. doi: 10.2337/diab.39.12.1461. [DOI] [PubMed] [Google Scholar]
- 5.Laychock SG. Glucose metabolism, second messengers and insulin secretion. Life Sci. 1990;47:2307–16. doi: 10.1016/0024-3205(90)90269-w. [DOI] [PubMed] [Google Scholar]
- 6.Metz SA. The pancreatic islet as a Rubik's cube. Is phospholipid hydrolysis a piece of the puzzle. Diabetes. 1991;40:1565–73. doi: 10.2337/diab.40.12.1565. [DOI] [PubMed] [Google Scholar]
- 7.Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 8.Deeney JT, Prentki M, Corkey BE. Metabolic control of beta-cell function. Semin Cell Dev Biol. 2000;11:267–75. doi: 10.1006/scdb.2000.0175. [DOI] [PubMed] [Google Scholar]
- 9.Newgard CB, Lu D, Jensen MV, Schissler J, Boucher A, Burgess S, Sherry AD. Stimulus(secretion coupling factors in glucose-stimulated insulin secretion: insights gained from a multidisciplinary approach. Diabetes. 2002;51:S389–93. doi: 10.2337/diabetes.51.2007.s389. [DOI] [PubMed] [Google Scholar]
- 10.Berggren PO, Leibiger IB. Novel aspects on signal transduction in the pancreatic beta cell. Nutr Metab Cardiovasc Dis. 2006;16:S7–10. doi: 10.1016/j.numecd.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–49. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 12.Birnbaumer L. Receptor-to-effector signaling through G proteins: roles for beta gamma dimers as well as alpha subunits. Cell. 1992;71:1069–72. doi: 10.1016/s0092-8674(05)80056-x. [DOI] [PubMed] [Google Scholar]
- 13.Clapham DE, Neer EJ. New roles for G-protein beta gamma-dimers in transmem-brane signaling. Nature. 1993;365:403–6. doi: 10.1038/365403a0. [DOI] [PubMed] [Google Scholar]
- 14.Jones PM, Persaud SJ. Protein kinases, protein phosphorylation, and the regulation of insulin secretion from pancreatic beta-cells. Endocr Rev. 1998;19:429–61. doi: 10.1210/edrv.19.4.0339. [DOI] [PubMed] [Google Scholar]
- 15.Easom RA. CaM kinase II: a protein kinase with extraordinary talents germane to insulin exocytosis. Diabetes. 1999;48:674–84. doi: 10.2337/diabetes.48.4.675. [DOI] [PubMed] [Google Scholar]
- 16.Nesher R, Anteby E, Yedozvizky M, Warwar N, Cerasi E. Beta-cell protein kinases and the dynamics of the insulin response to glucose. Diabetes. 2002;51:S63–73. doi: 10.2337/diabetes.51.2007.s68. [DOI] [PubMed] [Google Scholar]
- 17.Khoo S, Gibson TB, Arnette D, Lawrence M, January B, McGlynn K, Vanderbilt CA, Griffen SC, German MS, Cobb MH. MAP kinases and their roles in pancreatic beta-cells. Cell Biochem Biophys. 2004;40:191–200. doi: 10.1385/cbb:40:3:191. [DOI] [PubMed] [Google Scholar]
- 18.Matthews HR. Protein kinases and phos-phatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: a possible regulator of the mitogen activated protein kinase cascade. Pharmacol Ther. 1995;67:323–50. doi: 10.1016/0163-7258(95)00020-8. [DOI] [PubMed] [Google Scholar]
- 19.Hess JF, Bourret RB, Simon MI. Histidine phosphorylation and phosphoryl group transfer in bacterial chemotaxis. Nature. 1988;336:139–43. doi: 10.1038/336139a0. [DOI] [PubMed] [Google Scholar]
- 20.Stock JB, Ninfa AJ, Stock AM. Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol Rev. 1989;53:450–90. doi: 10.1128/mr.53.4.450-490.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker MD, Wolanin PM, Stock JB. Signal transduction in bacterial chemotaxis. Bioessays. 2006;28:9–22. doi: 10.1002/bies.20343. [DOI] [PubMed] [Google Scholar]
- 22.Wolanin PM, Thomason PA, Stock JB. Histidine protein kinases: key signal transducers outside the animal kingdom. Genome Biol. 2002;3:1–8. doi: 10.1186/gb-2002-3-10-reviews3013. : 3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grebe TW, Stock JB. The histidine protein kinase superfamily. Adv Microb Physiol. 1999;41:139–227. doi: 10.1016/s0065-2911(08)60167-8. [DOI] [PubMed] [Google Scholar]
- 24.Alex LA, Simon MI. Protein histidine kinases and signal transduction in prokaryotes and eukaryotes. Trends Genet. 1994;10:133–8. doi: 10.1016/0168-9525(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 25.Khorchid A, Ikura M. Bacterial histidine kinase as signal sensor and transducer. Int J Biochem Cell Biol. 2006;38:307–12. doi: 10.1016/j.biocel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 26.Swanson RV, Alex LA, Simon MI. Histidine and aspartate phosphorylation: two component systems and the limits of homology. Trends Biochem Sci. 1994;19:485–90. doi: 10.1016/0968-0004(94)90135-x. [DOI] [PubMed] [Google Scholar]
- 27.Klumpp S, Krieglstein J. Phosphorylation and dephosphorylation of histidine residues in proteins. Eur J Biochem. 2002;269:1067–71. doi: 10.1046/j.1432-1033.2002.02755.x. [DOI] [PubMed] [Google Scholar]
- 28.Besant PG, Attwood PV. Mammalian histi-dine kinases. Biochim Biophys Acta. 2005;1754:281–90. doi: 10.1016/j.bbapap.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 29.Besant PG, Tan E, Attwood PV. Mammalian protein histidine kinases. Int J Biochem and Cell Biol. 2003;35:297–309. doi: 10.1016/s1357-2725(02)00257-1. [DOI] [PubMed] [Google Scholar]
- 30.Steeg PS, Palmieri D, Ouatas T, Salerno M. Histidine kinases and histidine phos-phorylated proteins in mammalian cell biology, signal transduction and cancer. Can Lett. 2003;190:1–12. doi: 10.1016/s0304-3835(02)00499-8. [DOI] [PubMed] [Google Scholar]
- 31.Tan E, Besant PG, Attwood PV. Mammalian histidine kinases: do they really exist? Biochemistry. 2002;41:3843–51. doi: 10.1021/bi012021r. [DOI] [PubMed] [Google Scholar]
- 32.Kimura N, Shimada N, Fukuda M, Ishijima Y, Miyazaki H, Ishii A, Takagi Y, Ishikawa N. Regulation of cellular functions by nucleoside diphosphate kinases in mammals. J Bioenerg Biomembr. 2000;32:309–15. doi: 10.1023/a:1005549315846. [DOI] [PubMed] [Google Scholar]
- 33.Kowluru A. Regulatory roles for small G-proteins in the pancreatic beta cell: lessons from models of impaired insulin secretion. Am J Physiol Endocrinol Metab. 2003;285:E669–84. doi: 10.1152/ajpendo.00196.2003. [DOI] [PubMed] [Google Scholar]
- 34.Crawford RM, Treharne KJ, Best OG, Muimo R, Riemen CE, Mehta A. A novel physical and functional association between nucleoside diphosphate kinase A and AMP-activated protein kinase alpha1 in liver and lung. Biochem J. 2005;392:201–9. doi: 10.1042/BJ20050269. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Wagner P, Vu ND. Histidine to aspartate phosphotransferase activity of nm23: phos-phorylation of aldolase C on Asp 319. Biochem J. 2000;346:623–30. [PMC free article] [PubMed] [Google Scholar]
- 36.Muimo R, Hornickova Z, Riement C, Gerke V, Matthews H, Mehta A. Histidine phosphorylation of annexin 1 in airway epithelia. J Biol Chem. 2000;275:36632–6. doi: 10.1074/jbc.M000829200. [DOI] [PubMed] [Google Scholar]
- 37.Wagner PD, Vu ND. Phosphorylation of ATP-citrate lyase by nucleoside diphos-phate kinase. J Biol Chem. 1995;270:21758–64. doi: 10.1074/jbc.270.37.21758. [DOI] [PubMed] [Google Scholar]
- 38.Wieland T, Nunberg B, Ulibarri I, Kaldenberg-Stasch S, Schultz G, Jakobs KH. Guanine nucleotide specific phosphate transfer by guanine-binding regulatory protein beta-subunits. Characterization of the phosphorylated amino acid. J Biol Chem. 1993;268:18111–8. [PubMed] [Google Scholar]
- 39.Kowluru A, Seavey SE, Rhodes CJ, Metz SA. A novel mechanism for trimeric GTP-binding proteins in the membrane and secretory granule fractions of human and rodent beta cells. J Biochem. 1996;313:97–107. doi: 10.1042/bj3130097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crawford RM, Treharne KJ, Best OG, Riemen CE, Muimo R, Gruenert DC, Arnaud-Dabernat S, Daniel JY, Mehta A. NDPK-A (but not NDPK-B) and AMPK alpha1 (but not AMPK alpha2) bind the cystic fibrosis transmembrane conductance regulator in epithelial cell membranes. Cell Signal. 2006;18:1595–603. doi: 10.1016/j.cellsig.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Yuen MH, Mizuguchi H, Lee YH, Cook PF, Uyeda K, Hasemann CA. Crystal structure of the H256A mutant of rat testis fructose-6-phosphate, 2-kinase/fructose-2,6-diphosphatase. Fructose 6-phosphate in the active site leads to mechanisms for both mutant and wild type bisphosphatase activities. J Biol Chem. 1999;274:2176–84. doi: 10.1074/jbc.274.4.2176. [DOI] [PubMed] [Google Scholar]
- 42.Mizuguchi H, Cook PF, Tai CH, Haseman CA, Uyeda K. Reaction mechanism of fructose-2,6-bisphosphatase. A mutation of nucleophilic catalyst, histidine 256, induces an alteration in the reaction pathway. J Biol Chem. 1999;274:2166–75. doi: 10.1074/jbc.274.4.2166. [DOI] [PubMed] [Google Scholar]
- 43.Feldman F, Butler LG. Detection and characterization of the phosphorylated form of microsomal glucose-6-phosphatase. Biochem Biophys Res Commun. 1969;36:119–25. doi: 10.1016/0006-291x(69)90657-3. [DOI] [PubMed] [Google Scholar]
- 44.Robertson EF, Hoyt JC, Reeves HC. Evidence of histidine phosphorylation in isocitrate lyase from Escherichia coli. J Biol Chem. 1988;263:2477–82. [PubMed] [Google Scholar]
- 45.Hartsough M, Morrison D, Salerno M, Palmeri D, Quatas T, Mair M, Patrick J, Steeg P. Nm23-H1 metastassis suppressor phosphorylation of kinase suppressor ras (KSR), via a histidine kinase pathway. J Biol Chem. 2002;277:32389–99. doi: 10.1074/jbc.M203115200. [DOI] [PubMed] [Google Scholar]
- 46.Weiss V, Magasanik B. Phosphorylation of nitrogen regulator I (NRI) of Escherichia coli. Proc Natl Acad Sci USA. 1988;85:8919–23. doi: 10.1073/pnas.85.23.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kowluru A, Metz SA. Characterization of nucleoside diphosphokinase activity in human and rodent pancreatic beta cells: evidence for its role in the formation of guanosine triphosphate, a permissive factor for nutrient-induced insulin secretion. Biochemistry. 1994;33:12495–503. doi: 10.1021/bi00207a017. [DOI] [PubMed] [Google Scholar]
- 48.Anderson B, Weigel N, Kundig W, Roseman S. Sugar transport. 3. Purification and properties of a phospho carrier protein (HPr) of the phospho-enolpyruvate-dependent phosphotrans-ferase system in Escherichia coli. J Biol Chem. 1971;246:7023–33. [PubMed] [Google Scholar]
- 49.Pilkis SJ, Regan DM, Stewart HB, Pilkis J, Pate TM, El-Maghrabi MR. Evidence for two catalytic sites on 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase. Dynamics of substrate exchange and phosphoryl enzyme formation. J Biol Chem. 1984;259:949–58. [PubMed] [Google Scholar]
- 50.Rose ZB. Evidence for a phosphohistidine protein intermediate in the phosphoglycer-ate mutase reaction. Arch Biochem Biophys. 1970;140:508–13. doi: 10.1016/0003-9861(70)90095-0. [DOI] [PubMed] [Google Scholar]
- 51.Han CH, Rose ZB. Active site phosphohis-tidine peptides from red cell bisphospho-glycerate synthase and yeast phospho-glycerate mutase. J Biol Chem. 1979;254:8836–40. [PubMed] [Google Scholar]
- 52.Huebner VD, Matthews HR. Phosphory-lation of histidine in proteins by a nuclear extract of Physarium polycephalum plas-modia. J Biol Chem. 1985;260:16106–13. [PubMed] [Google Scholar]
- 53.Hegde AN, Das MR. Ras proteins enhance the phosphorylation of a 38 kDa protein (p38) in rat liver plasma membrane. FEBS Lett. 1987;217:74–80. doi: 10.1016/0014-5793(87)81246-2. [DOI] [PubMed] [Google Scholar]
- 54.Crovello CS, Furie BC, Furie B. Histidine phosphorylation of P-selectin upon stimulation of human platelets: a novel pathway for activation-dependent signal transduc-tion. Cell. 1995;82:279–86. doi: 10.1016/0092-8674(95)90315-1. [DOI] [PubMed] [Google Scholar]
- 55.Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AL, Yan Y, Backer JM, Unutmaz D, Coetzee WA, Skolnik EY. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol Cell. 2006;24:665–75. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 56.Verner AV, Ichetovkina LE, Komissarov AA, Nazarova TI, Avaeva SM. Phosphorylation as a method of regulating inorganic pyrophosphatase activity in E. coli. II. Identification of the types of chemical bonds between phosphate and the enzyme. Bioorg Khim. 1996;12:200–5. [PubMed] [Google Scholar]
- 57.Zhu J, Tseng YH, Kantor JD, Rhodes CJ, Zetter BR, Moyers JS, Kahn CR. Interaction of the Ras-related protein associated with diabetes rad and the putative tumor metastasis suppressor NM23 provides a novel mechanism of GTPase regulation. Proc Natl Acad Sci USA. 1999;96:14911–8. doi: 10.1073/pnas.96.26.14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zetterqvist O, Engstrom L. Isolation of N-e[32P]phosphoryl-lysine from rat-liver cell sap after incubation with [32P]adenosine triphosphate. Biochim Biophys Acta. 1967;141:523–32. doi: 10.1016/0304-4165(67)90181-x. [DOI] [PubMed] [Google Scholar]
- 59.Boyer PD, Deluca M, Ebner KE, Hultquist DE, Peter JB. Identification of phospho-histidine in digests from a probable intermediate of oxidative phosphorylation. J Biol Chem. 1962;237:3306–8. [PubMed] [Google Scholar]
- 60.Kowluru A. Adenine and guanine nucleotide-specific succinyl-CoA syn-thetases in the clonal beta-cell mitochondria: implications in the beta-cell high-energy phosphate metabolism in relation to physiological insulin secretion. Diabetologia. 2001;44:89–94. doi: 10.1007/s001250051584. [DOI] [PubMed] [Google Scholar]
- 61.Yano M, Mori S, Kido H. Intrinsic nucleo-side diphosphate kinase-like activity is a novel function of the 20S proteosome. J Biol Chem. 1999;274:34375–82. doi: 10.1074/jbc.274.48.34375. [DOI] [PubMed] [Google Scholar]
- 62.Otsuki Y, Tanaka M, Yoshii S, Kawazoe N, Nakaya K, Sugimura H. Tumor metastas-sis suppressor nm23H1 regulates Rac1 GTPase by interaction with Tiam1. Proc Natl Acad Sci USA. 2001;98:4385–90. doi: 10.1073/pnas.071411598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Palacios F, Schweitzer JK, Boshans RL, D’Souza-Schorey C. ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly. Nat Cell Biol. 2002;4:929–36. doi: 10.1038/ncb881. [DOI] [PubMed] [Google Scholar]
- 64.Attwood PV, Piggott MJ, Zu XL, Besant PG. Focus on phosphohistidines. Amino Acids. 2007;32:145–56. doi: 10.1007/s00726-006-0443-6. [DOI] [PubMed] [Google Scholar]
- 65.Besant PG, Attwood PV. Problems with phosphoamino acid analysis using alkaline hydrolysis. Anal Biochem. 1998;265:187–90. doi: 10.1006/abio.1998.2880. [DOI] [PubMed] [Google Scholar]
- 66.Munoz-Dorado Y, Almaula N, Inouye S, Inouye M. Autophosphorylation of nucleo-side diphosphate kinase from Myxococcus xanthus. J Bacteriol. 1993;175:1176–81. doi: 10.1128/jb.175.4.1176-1181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei YF, Matthews HR. A filter-based protein kinase assay selective for alkali-stable protein phosphorylation and suitable for acid-labile protein phosphorylation. Anal Biochem. 1990;190:188–92. doi: 10.1016/0003-2697(90)90179-d. [DOI] [PubMed] [Google Scholar]
- 68.Matthews HR, Chan K. Protein histidine kinase. Meth Mol Biol. 1998;124:171–82. doi: 10.1385/1-59259-059-4:171. [DOI] [PubMed] [Google Scholar]
- 69.Kleinnijenhuis AJ, Kjeldsen F, Kallipoltis B, Haselmann KF, Jensen ON. Analysis of histidine phosphorylation using tandem MS and ion-electron reactions. Anal Chem. 2007;79:7450–6. doi: 10.1021/ac0707838. [DOI] [PubMed] [Google Scholar]
- 70.Kowluru A. Identification and characterization of a novel protein histidine kinase in the islet beta-cell: evidence for its regulation by mastoparan, an activator of G-pro-teins and insulin secretion. Biochem Pharmacol. 2002;63:2091–100. doi: 10.1016/s0006-2952(02)01025-0. [DOI] [PubMed] [Google Scholar]
- 71.Kowluru A, Tannous M, Chen HQ. Localization and characterization of the mitochondrial isoform of the nucleoside diphosphate kinase in the pancreatic beta-cell: evidence for its complexation with mitochondrial succinyl-CoA synthetase. Arch Biochem Biophys. 2002;398:160–9. doi: 10.1006/abbi.2001.2710. [DOI] [PubMed] [Google Scholar]
- 72.Agarwal RP, Parks RE., Jr Erythrocytic nucleoside diphosphokinase. V. Some properties and behavior of the pI 7.3 isozyme. J Biol Chem. 1971;246:2258–64. [PubMed] [Google Scholar]
- 73.Kimura N, Shimada N, Fukuda M, Ishijima Y, Miyazaki H, Ishii A, Takagi Y, Ishikawa N. Regulation of cellular functions by nucleoside diphosphate kinases in mammals. J Bioenerg Biomemb. 2000;32:307–15. doi: 10.1023/a:1005549315846. [DOI] [PubMed] [Google Scholar]
- 74.Kimura N, Shimada N, Ishijima Y, Fukuda M, Takagi Y, Ishikawa N. Nucleoside diphosphate kinases in mammalian signal transduction systems: recent development and perspective. J Bioenerg Biomemb. 2003;35:41–7. doi: 10.1023/a:1023489722460. [DOI] [PubMed] [Google Scholar]
- 75.Steeg PS, Cohn KH, Leone A. Tumor metas-tassis and nm23: current concepts. Cancer Cells. 1991;3:257–62. [PubMed] [Google Scholar]
- 76.MacDonald NJ, de la Rosa A, Steeg PS. The potential roles of nm23 in cancer metastassis and cellular differentiation. Eur J Cancer. 1995;31:1096–100. doi: 10.1016/0959-8049(95)00152-9. [DOI] [PubMed] [Google Scholar]
- 77.de la Rosa A, Mikhak B, Steeg PS. Identification and characterization of the promoter for the human metastassis suppressor gene nm23-H1. Arch Med Res. 1996;27:395–401. [PubMed] [Google Scholar]
- 78.Martin KK, Pilkington GJ. Nm23: an invasion suppressor gene in CNS tumors? Anticancer Res. 1998;18:919–26. [PubMed] [Google Scholar]
- 79.Wagner PD, Steeg PS, Vu ND. Two component kinase-like activity of nm23 correlates with its motility-suppressing activity. Proc Natl Acad Sci USA. 1997;94:9000–5. doi: 10.1073/pnas.94.17.9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hartsough MT, Steeg PS. Nm23/nucleoside diphosphate kinase in human cancers. J Bioenerg Biomembr. 2000;32:301–8. doi: 10.1023/a:1005597231776. [DOI] [PubMed] [Google Scholar]
- 81.Postel EH, Berberich SJ, Flint SJ, Ferrone CA. Human c-myc transcription factor PuF identified as nm23-H2 nucleo-side diphosphate kinase, a candidate suppressor of tumor metastassis. Science. 1993;261:478–80. doi: 10.1126/science.8392752. [DOI] [PubMed] [Google Scholar]
- 82.Milon L, Rousseau-Merck MF, Munier A, Erent M, Lascu I, Capeau J, Lacombe ML. nm23-H4, a new member of the family of human nm23/nucleoside diphosphate kinase genes localised on chromosome 16p13. Human Genet. 1997;99:550–7. doi: 10.1007/s004390050405. [DOI] [PubMed] [Google Scholar]
- 83.Milon L, Meyer P, Chiadmi M, Munier A, Johansson M, Karlsson A, Lascu I, Capeau J, Janin J, Lacombe ML. The human nm23-H4 gene product is a mito-chondrial nucleoside diphosphate kinase. J Biol Chem. 2000;275:14264–72. doi: 10.1074/jbc.275.19.14264. [DOI] [PubMed] [Google Scholar]
- 84.Munier A, Feral C, Milon L, Pinon VP, Gyapay G, Capeau J, Guellaen G, Lacombe ML. A new human nm23 homo-logue (nm23-H5) specifically expressed in testis germinal cells. FEBS Lett. 1998;434:289–94. doi: 10.1016/s0014-5793(98)00996-x. [DOI] [PubMed] [Google Scholar]
- 85.Munier A, Serres C, Kann ML, Boissan M, Lesaffre C, Capeau J, Fouquet JP, Lacombe ML. Nm23/NDP kinases in human male germ cells: role in spermato-genesis and sperm motility. Exp Cell Res. 2003;289:295–306. doi: 10.1016/s0014-4827(03)00268-4. [DOI] [PubMed] [Google Scholar]
- 86.Tsuiki H, Nitta M, Furuya A, Hanai N, Fujiwara T, Inagaki M, Kochi M, Ushio Y, Saya H, Nakamura H. A novel human nucleoside diphosphate (NDP) kinase, nm23-H6, localizes in mitochondria and affects cytokinesis. J Cell Biochem. 1999;76:254–69. doi: 10.1002/(sici)1097-4644(20000201)76:2<254::aid-jcb9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 87.Seifert M, Welter C, Mehraein Y, Seitz G. Expression of the nm23 homologues nm23-H4, nm23-H6 and nm23-H7 in human gastric and colon cancer. J Pathol. 2005;205:623–32. doi: 10.1002/path.1724. [DOI] [PubMed] [Google Scholar]
- 88.Padma P, Hozumi A, Ogawa K, Inaba K. Molecular cloning and characterization of a thioredoxin(nucleoside diphosphate kinase related dynein intermediate chain from the ascidian, Ciona intestinalis. Gene. 2001;275:177–83. doi: 10.1016/s0378-1119(01)00661-8. [DOI] [PubMed] [Google Scholar]
- 89.Metz SA, Rabaglia ME, Pintar T. Selective inhibitors of GTP synthesis impede exocy-totic insulin release from intact rat islets. J Biol Chem. 1992;267:12517–27. [PubMed] [Google Scholar]
- 90.Metz SA, Meredith M, Rabaglia ME, Kowluru A. Small elevations of glucose concentration redirect and amplify the synthesis of guanosine 5’-triphosphate in rat islets. J Clin Invest. 1993;92:872–82. doi: 10.1172/JCI116662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robertson RP, Seaquist ER, Walseth TF. G proteins and modulation of insulin secretion. Diabetes. 1991;40:1–6. doi: 10.2337/diab.40.1.1. [DOI] [PubMed] [Google Scholar]
- 92.Regazzi R, Kikuchi A, Takai Y, Wollheim CB. The small GTP-binding proteins in the cytosol of insulin-secreting cells are com-plexed to GDP dissociation inhibitor proteins. J Biol Chem. 1992;267:17512–9. [PubMed] [Google Scholar]
- 93.Li G, Regazzi R, Roche E, Wollheim CB. Blockade of mevalonate production by lovastatin attenuates bombesin and vaso-pressin potentiation of nutrient-induced insulin secretion in HIT-T15 cells. Probable involvement of small GTP-binding proteins. J Biochem. 1993;289:379–85. doi: 10.1042/bj2890379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Metz SA, Rabaglia ME, Stock JB, Kowluru A. Modulation of insulin secretion from normal rat islets by inhibitors of the post-translational modifications of GBPs. J Biochem. 1993;295:31–40. doi: 10.1042/bj2950031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Seaquist ER, Walseth TF, Redmon JB, Robertson RP. G-protein regulation of insulin secretion. J Lab Clin Med. 1994;123:338–45. [PubMed] [Google Scholar]
- 96.Kowluru A, Rabaglia ME, Muse KE, Metz SA. Subcellular localization and kinetic characterization of guanine nucleotide binding proteins in normal rat and human pancreatic islets and transformed beta cells. Biochim Biophys Acta. 1994;1222:348–59. doi: 10.1016/0167-4889(94)90040-x. [DOI] [PubMed] [Google Scholar]
- 97.Kowluru A, Metz SA. GTP and its binding proteins in the regulation of insulin exocy-tosis. In: Draznin B, LeRoith D, editors. Molecular biology of diabetes. Humana; 1994. pp. 249–83. In:, editors.. Totowa, NJ:. [Google Scholar]
- 98.Kowluru A, Metz SA. Regulation of gua-nine-nucleotide binding proteins in islet subcellular fractions by phospholipase-derived lipid mediators of insulin secretion. Biochim Biophys Acta. 1994;1222:360–8. doi: 10.1016/0167-4889(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 99.Kowluru A, Metz SA. Stimulation of prostaglandin E2 of a high-affinity GTPase in the secretory granules of normal rat and human pancreatic islets. Biochem J. 1994;297:399–406. doi: 10.1042/bj2970399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kowluru A, Seavey SE, Li G, Sorenson RL, Weinhaus AJ, Nesher R, Rabaglia ME, Vadakekalam J, Metz SA. Glucose-and GTP-dependent stimulation of the car-boxyl methylation of Cdc42 in rodent and human pancreatic islets and pure beta-cells. J Clin Invest. 1996;98:540–55. doi: 10.1172/JCI118822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leiser M, Efrat S, Fleischer N. Evidence that Rap1 carboxylmethylation is involved in regulated insulin secretion. Endocrinology. 1995;136:2521–30. doi: 10.1210/endo.136.6.7750474. [DOI] [PubMed] [Google Scholar]
- 102.Regazzi R, Ravazzola M, Lezzi M, Lang J, Zahraoui A, Andereggen E, Morel P, Takai Y, Wollheim CB. Expression, localization and functional role of small GTPases of the Rab3 family in insulin-secreting cells. J Cell Sci. 1996;109:2265–73. doi: 10.1242/jcs.109.9.2265. [DOI] [PubMed] [Google Scholar]
- 103.Sharp GW. Mechanisms of inhibition of insulin release. Am J Physiol. 1996;271:C1781–99. doi: 10.1152/ajpcell.1996.271.6.C1781. [DOI] [PubMed] [Google Scholar]
- 104.Kowluru A, Li G, Metz SA. Glucose activates the carboxyl methylation of gamma-subunits of trimeric GTP-binding proteins in pancreatic beta-cells. J Clin Invest. 1997;100:596–610. doi: 10.1172/JCI119684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kowluru A, Li G, Rabaglia ME, Hofmann F, Aktories K, Metz SA. Evidence for differential roles of the Rho subfamily of GTP-binding proteins in glucose- and calcium-induced insulin secretion from pancreatic beta cells. Biochem Pharmacol. 1997;54:1097–108. doi: 10.1016/s0006-2952(97)00314-6. [DOI] [PubMed] [Google Scholar]
- 106.Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem. 1999;259:3–17. doi: 10.1046/j.1432-1327.1999.00043.x. [DOI] [PubMed] [Google Scholar]
- 107.Kowluru A, Robertson RP, Metz SA. GTP-binding proteins in the regulation of pancreatic beta cell function. In: LeRoith D, Olefsky JM, Taylor S, editors. Diabetes mellitus—fundamental and clinical text. J.B. Lippincott Co; 2000. pp. 78–94. In:, editors.. Philadelphia, PA:. [Google Scholar]
- 108.Amin R, Chen HQ, Tannous M, Gibbs R, Kowluru A. Inhibition of glucose- and calcium-induced insulin secretion from betaTC3 cells by novel inhibitors of protein isopreny-lation. J Pharmacol Exp Ther. 2002;303:82–8. doi: 10.1124/jpet.102.036160. [DOI] [PubMed] [Google Scholar]
- 109.Kowluru A, Morgan NG. GTP-binding proteins in cell survival and demise: the emerging picture in the pancreatic beta-cell. Biochem Pharmacol. 2002;63:1027–35. doi: 10.1016/s0006-2952(02)00849-3. [DOI] [PubMed] [Google Scholar]
- 110.Kowluru A, Amin R. Inhibitors of post-translational modifications of G-proteins as probes to study the pancreatic beta cell function: potential therapeutic implications. Curr Drug Targets Immune Endocr Metabol Disord. 2002;2:129–39. [PubMed] [Google Scholar]
- 111.Daniel S, Noda M, Cerione RA, Sharp GW. A link between Cdc42 and syntaxin is involved in mastoparan-stimulated insulin release. Biochemistry. 2002;41:9663–71. doi: 10.1021/bi025604p. [DOI] [PubMed] [Google Scholar]
- 112.Kowluru A, Chen H-Q, Tannous M. Novel roles for the Rho subfamily of GTP-binding proteins in succinate-induced insulin secretion from betaTC3 cells: further evidence in support of succinate mechanism of insulin release. Endocr Res. 2003;29:363–76. doi: 10.1081/erc-120025043. [DOI] [PubMed] [Google Scholar]
- 113.Nevins AK, Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol. 2003;285:C698–710. doi: 10.1152/ajpcell.00093.2003. [DOI] [PubMed] [Google Scholar]
- 114.Lawrence JT, Birnbaum MJ. ADP-ribosy-lation factor 6 regulates insulin secretion through plasma membrane phosphatidyli-nositol 4,5-bisphosphate. Proc Natl Acad Sci USA. 2003;100:13320–5. doi: 10.1073/pnas.2232129100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li J, Luo R, Kowluru A, Li G. Novel regulation by Rac1 of glucose-and forskolin-induced insulin secretion in INS-1 beta cell. Am J Physiol Endocrinol Metab. 2004;286:E818–27. doi: 10.1152/ajpendo.00307.2003. [DOI] [PubMed] [Google Scholar]
- 116.Aizawa T, Komatsu M. Rab27a: a new face in beta cell metabolism and secretion coupling. J Clin Invest. 2005;115:227–30. doi: 10.1172/JCI24269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nevins AK, Thurmond DC. A direct interaction between Cdc42 and vesicle-associated membrane protein 2 regulates SNARE-dependent insulin exocytosis. J Biol Chem. 2005;280:1944–52. doi: 10.1074/jbc.M409528200. [DOI] [PubMed] [Google Scholar]
- 118.Veluthakal R, Kaur H, Goalstone M, Kowluru A. Dominant negative alpha-sub-unit of farnesyl-and geranyl geranyltrans-ferase inhibits glucose-stimulated, but not KCl-stimulated, insulin secretion in INS 832/13 cells. Diabetes. 2007;56:204–10. doi: 10.2337/db06-0668. [DOI] [PubMed] [Google Scholar]
- 119.Kowluru A. Protein prenylation in glucose-induced insulin secretion from pancreatic islet beta cell: a perspective. J Cell Mol Med. 2008;12:164–73. doi: 10.1111/j.1582-4934.2007.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang Z, Oh E, Thurmond D. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J Biol Chem. 2007;282:9536–46. doi: 10.1074/jbc.M610553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kowluru A, Veluthakal R. Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes. 2005;54:3523–9. doi: 10.2337/diabetes.54.12.3523. [DOI] [PubMed] [Google Scholar]
- 122.Nurnberg B, Harhammer R, Exner T, Schulze RA, Wieland T. Species-and tissue-dependent diversity of G-protein beta subunit phosphorylation: evidence for a cofactor. Biochem J. 1996;318:717–22. doi: 10.1042/bj3180717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Higashijima T, Burnier J, Ross EM. Regulation of Gi and Go by mastoparan, related amphiphilic peptides, and hydrophobic amines. J Biol Chem. 1990;265:14176–81. [PubMed] [Google Scholar]
- 124.Higashijima T, Uzu S, Nakajima T, Ross EM. Mastoparan, a peptide toxin from wasp venom mimics receptors by activating GTP-binding regulatory proteins (G-proteins) J Biol Chem. 1988;263:6491–4. [PubMed] [Google Scholar]
- 125.Huang J, Wei Y, Kim Y, Osterberg L, Matthews HR. Purification of a protein histidine kinase from the yeast Saccharomyces cerevisiae. The first member of this class of protein kinases. J Biol Chem. 1991;266:9023–31. [PubMed] [Google Scholar]
- 126.Motojima K, Goto S. Histidyl phosphory-lation and dephosphorylation of P36 in liver extracts. J Biol Chem. 1994;269:9030–7. [PubMed] [Google Scholar]
- 127.Urushidani T, Nagao T. Calcium-dependent membrane bound protein fraction from rabbit gastric mucosa contains a protein whose histidyl residue is phosphorylated. Biochim Biophys Acta. 1997;1356:71–83. doi: 10.1016/s0167-4889(96)00161-9. [DOI] [PubMed] [Google Scholar]
- 128.Besant PG, Attwood PV. Detection of a mammalian histone H4 kinase that has yeast histidine kinase-like enzymic activity. Int J Biochem Cell Biol. 2000;32:243–53. doi: 10.1016/s1357-2725(99)00119-3. [DOI] [PubMed] [Google Scholar]
- 129.Huang J, Nasir M, Kaimnd Y, Matthew HR. Genistein inhibits protein histidine kinase. J Biol Chem. 1992;267:15511–5. [PubMed] [Google Scholar]
- 130.Gilmour R, Foster JE, Sheng Q, McClain JR, Riley A, Sun PM, Ng WL, Yan D, Nicas TI, Henry K, Winkler ME. New class of competitive inhibitor of bacterial histi-dine kinases. J Bacteriol. 2005;187:8196–200. doi: 10.1128/JB.187.23.8196-8200.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Okada A, Gotoh Y, Watanabe T, Furuta E, Yamamoto K, Utsumi R. Targeting two-component signal transduction: a novel drug discovery system. Methods Enzymol. 2007;422:386–95. doi: 10.1016/S0076-6879(06)22019-6. [DOI] [PubMed] [Google Scholar]
- 132.Ramstrom H, Bourotte M, Philippe C, Schmitt M, Haiech J, Bourguignon JJ. Heterocyclic bis-cations as starting hits for design of inhibitors of the bifunctional enzyme histidine-containing protein kinase/ phosphatase from Bacillus subtilis. J Med Chem. 2004;47:2264–75. doi: 10.1021/jm021043o. [DOI] [PubMed] [Google Scholar]
- 133.Stephenson K, Hoch JA. Developing inhibitors to selectively target two-component and phosphorelay signal transduction systems of pathogenic microorganisms. Curr Med Chem. 2004;11:765–73. doi: 10.2174/0929867043455765. [DOI] [PubMed] [Google Scholar]
- 134.Macielag MJ, Goldschmidt R. Inhibitors of bacterial two-component signalling systems. Expert Opin Investig Drugs. 2000;9:2351–69. doi: 10.1517/13543784.9.10.2351. [DOI] [PubMed] [Google Scholar]
- 135.Kowluru A. Differential regulation by fatty acids of protein histidine phosphorylation in rat pancreatic islets. Mol Cell Biochem. 2004;266:175–82. doi: 10.1023/b:mcbi.0000049157.03855.74. [DOI] [PubMed] [Google Scholar]
- 136.Metz SA. Exogenous arachidonic acid promotes insulin release from intact or per-meabilized rat islets by dual mechanisms. Putative activation of calcium mobilization and protein kinase C. Diabetes. 1988;37:1453–69. doi: 10.2337/diab.37.11.1453. [DOI] [PubMed] [Google Scholar]
- 137.Yaney GC, Corkey BE. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia. 2003;46:1297–312. doi: 10.1007/s00125-003-1207-4. [DOI] [PubMed] [Google Scholar]
- 138.Latour MG, Alquier T, Oseid E, Tremblay C, Jetton TL, Luo J, Lin DC-H, Poitout V. GPR40 is necessary but not suffiecient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56:1087–94. doi: 10.2337/db06-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kikkawa S, Takahashi K, Takahashi K, Shimada N, Ui M, Kimura N, Katada T. Activation of nucleoside diphosphate kinase by mastoparan, a peptide isolated from wasp venom. FEBS Lett. 1992;305:237–40. doi: 10.1016/0014-5793(92)80676-8. [DOI] [PubMed] [Google Scholar]
- 140.Komatsu M, McDermott AM, Gillison SL, Sharp GWG. Mastoparan stimulates exo-cytosis at a Ca2+-independent late site in stimulus—secretion coupling. Studies with RINm5F beta-cell line. J Biol Chem. 1993;268:23297–306. [PubMed] [Google Scholar]
- 141.Jones PM, Mann FM, Persaud SJ, Wheeler-Jones CPD. Mastoparan stimulates insulin secretion from pancreatic beta-cells by effects at a late stage in the secretory pathway. Mol Cell Endocrinol. 1993;94:97–103. doi: 10.1016/0303-7207(93)90056-p. [DOI] [PubMed] [Google Scholar]
- 142.Konrad RJ, Young RA, Record RD, Smith RM, Butkerait P, Manning D, Jarett L, Wolf BA. The heterotrimeric G-protein Gi is localized to the insulin secretory granules of beta-cells and is involved in insulin exocyto-sis. J Biol Chem. 1995;270:12869–76. doi: 10.1074/jbc.270.21.12869. [DOI] [PubMed] [Google Scholar]
- 143.Straub SG, James RF, Dunne MJ, Sharp GW. Glucose augmentation of mastoparan-stimulated insulin secretion in rat and human pancreatic islets. Diabetes. 1998;47:1053–7. doi: 10.2337/diabetes.47.7.1053. [DOI] [PubMed] [Google Scholar]
- 144.Heldt HW. Tager JM, Papa S, Qualiariello E, Slater EC, editors. Elsvier. 1966. pp. 51–66. In:, editors. Amsterdam: Regulation of metabolic processes in mitochondria.
- 145.Heidbuchel H, Callewaert G, Vereecke J, Carmeliet E. Acetylcholine-mediated K+ channel activity in guinea-pig atrial cells is supported by nucleoside diphosphate kinase. Pflugers Arch. 1993;422:316–24. doi: 10.1007/BF00374286. [DOI] [PubMed] [Google Scholar]
- 146.Srere PA. Complexes of sequential metabolic enzymes. Annu Rev Biochem. 1987;56:89–124. doi: 10.1146/annurev.bi.56.070187.000513. [DOI] [PubMed] [Google Scholar]
- 147.Gallagher BC, Parrott KA, Szabo G, Otero ADS. Receptor activation regulates cortical, but not vesicular localization of NDP kinase. J Cell Sci. 2003;116:3239–50. doi: 10.1242/jcs.00630. [DOI] [PubMed] [Google Scholar]
- 148.Fischbach MA, Settleman J. Specific biochemical inactivation of oncogenic Ra proteins by nucleoside diphosphate kinase. Cancer Res. 2003;63:4089–94. [PubMed] [Google Scholar]
- 149.Reynet C, Kahn CR. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science. 1993;262:1441–4. doi: 10.1126/science.8248782. [DOI] [PubMed] [Google Scholar]
- 150.Kowluru A, Veluthakal R, Kaetzel DM. Regulatory roles for nm23/nucleoside diphosphate kinase-like enzymes in insulin secretion from the pancreatic islet beta cell. J Bioenerg Biomembr. 2006;38:227–32. doi: 10.1007/s10863-006-9038-x. [DOI] [PubMed] [Google Scholar]
- 151.Piacentini L, Niroomand F. Phospho-transfer reactions as a means of G protein activation. Mol Cell Biochem. 1996;157:59–63. doi: 10.1007/BF00227881. [DOI] [PubMed] [Google Scholar]
- 152.Nederkoorn PHJ, Timmerman H, Timms D, Wilkinson AJ, Kelly DR, Broadley KJ, Davies RH. Stepwise phosphorylation mechanisms and signal transmission within a ligand receptor Gbeta-gamma-protein complex. J Mol Struct. 1998;452:25–47. [Google Scholar]
- 153.Hippe H-J, Wieland T. High energy phosphate transfer by NDPK B/Gbeta-gamma complexes-an alternative signaling pathway involved in the regulation of basal cAMP production. J Bioenerg Biomembr. 2006;38:197–203. doi: 10.1007/s10863-006-9035-0. [DOI] [PubMed] [Google Scholar]
- 154.Klinker JF, Seifert R. Nucleoside diphos-phate kinase activity in soluble transducin preparations. Biochemical properties and possible role of transducin-beta as phos-phorylated enzyme intermediate. Eur J Biochem. 1999;261:72–80. doi: 10.1046/j.1432-1327.1999.00209.x. [DOI] [PubMed] [Google Scholar]
- 155.Cuello F, Schulze RA, Heemeyer F, Meyer HE, Lutz S, Jakobs KH, Niroomand F, Wieland T. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Complex formation of NDPK B with Gbeta-gamma dimmers and phosphorylation of His-266 in Gbeta. J Biol Chem. 2003;278:7220–6. doi: 10.1074/jbc.M210304200. [DOI] [PubMed] [Google Scholar]
- 156.Hippe H-J, Lutz S, Cuello F, Knorr K, Vogt A, Jakobs KH, Wieland T, Niroomand F. Activation of heterotrimeric G proteins by a high energy phosphate transfer vianucleo-side diphosphate kinase (NDPK) B and Gbeta subunits. Specific activation of Gsalpha by an NDPK B Gbeta-gamma complex in H10 cells. J Biol Chem. 2003;278:7227–33. doi: 10.1074/jbc.M210305200. [DOI] [PubMed] [Google Scholar]
- 157.Marciniak SJ, Edwardson JM. Association of nucleoside diphosphate kinase with pancreatic zymogen granules: effects of local GTP generation on granule membrane characteristics. Biochem J. 1996;316:99–106. doi: 10.1042/bj3160099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, Shulman GI. Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab. 2007;5:253–64. doi: 10.1016/j.cmet.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Thomson M. What are guanosine triphos-phate-binding proteins doing in mitochondria? Biochim Biophys Acta. 1998;1403:211–8. doi: 10.1016/s0167-4889(98)00069-x. [DOI] [PubMed] [Google Scholar]
- 160.Thomson M. The regulation of mitochon-drial physiology by organelle-associated GTP-binding proteins. Cell Biochem Funct. 2002;20:273–8. doi: 10.1002/cbf.974. [DOI] [PubMed] [Google Scholar]
- 161.Thomson M. Evidence of undiscovered cell regulatory mechanisms: phosphoproteins and protein kinases in mitochondria. Cell Mol Life Sci. 2002;59:213–9. doi: 10.1007/s00018-002-8417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–60. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 163.Lyssand JS, Bajjalieh SM. The het-erotrimeric G protein subunit Galphai is present on mitochondria. FEBS Lett. 2007;581:5765–8. doi: 10.1016/j.febslet.2007.11.044. [DOI] [PubMed] [Google Scholar]
- 164.Li G, Kowluru A, Metz SA. Characterization of prenylcysteine methyltransferase in insulin-secreting cells. Biochem J. 1996;316:345–51. doi: 10.1042/bj3160345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fahien LA, MacDonald MJ. The succinate mechanism of insulin release. Diabetes. 2002;51:2669–76. doi: 10.2337/diabetes.51.9.2669. [DOI] [PubMed] [Google Scholar]
- 166.Alarcon C, Wicksteed B, Prentki M, Corkey BE, Rhodes CJ. Succinate is a preferential metabolic stimulus-coupling signal for glucose-induced proinsulin biosynthesis translation. Diabetes. 2002;51:2496–504. doi: 10.2337/diabetes.51.8.2496. [DOI] [PubMed] [Google Scholar]
- 167.Kavanaugh-Black A, Connolly DM, Chugani SA, Chakrabarty AM. Charac-ter-ization of nucleoside-diphosphate kinase from Pseudomonas aeruginosa: complex formation with succinyl-CoA synthetase. Proc Natl Acad Sci USA. 1994;91:5883–7. doi: 10.1073/pnas.91.13.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kadrmas EF, Ray PD, Lambeth DO. Apparent ATP-linked succinate thiokinase activity and its relation to nucleoside diphosphate kinase in mitochondrial matrix preparations from rabbit. Biochim Biophys Acta. 1991;1074:339–46. doi: 10.1016/0304-4165(91)90083-s. [DOI] [PubMed] [Google Scholar]
- 169.Srere PA. Complexities of metabolic regulation. Trends Biochem Sci. 1994;19:519–20. doi: 10.1016/0968-0004(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 170.Srere PA. 17th Fritz Lipman Lecture. Wanderings (wonderings) in metabolism. Biol Chem Hoppe Seyler. 1993;374:833–42. [PubMed] [Google Scholar]
- 171.Metz SA, Kowluru A. Inosine monophos-phate dehydrogenase: a molecular switch integrating pleotropic GTP-dependent beta-cell functions. Proc Assoc Am Physicians. 1999;111:335–46. doi: 10.1046/j.1525-1381.1999.99245.x. [DOI] [PubMed] [Google Scholar]
- 172.Wagner PD, Vu ND. Phosphorylation of geranyl and farnesyl pyrophosphates by Nm23 proteins/nucleoside diphos -phate kinases. J Biol Chem. 2000;275:35570–6. doi: 10.1074/jbc.M006106200. [DOI] [PubMed] [Google Scholar]
- 173.Matsubara M, Mizuno T. The SixA phos-pho-histidine phosphatase modulates the ArcB phosphorelay signal transduction in Escherichia coli. FEBS Lett. 2000;470:118–24. doi: 10.1016/s0014-5793(00)01303-x. [DOI] [PubMed] [Google Scholar]
- 174.Ohmori H, Kuba M, Kumon A. Two phos-phatases for 6-phospholysine and 3-phos-phohistidine from rat brain. J Biol Chem. 1993;268:7625–7. [PubMed] [Google Scholar]
- 175.Wong C, Faiola B, Wu W, Kennelly PJ. Phosphohistidine and phospholysine phosphatase activities in the rat: potential protein-lysine and protein-histidine phos-phatases? Biochem J. 1993;296:293–6. doi: 10.1042/bj2960293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kim Y, Huang J, Cohen P, Matthews HR. Protein phosphatases 1, 2A and 2C are protein histidine kinases. J Biol Chem. 1993;268:18513–8. [PubMed] [Google Scholar]
- 177.Klumpp S, Bechmann G, Maurer A, Selke D, Krieglstein J. ATP-citrate lyase as a substrate of protein histidine phosphatase in vertebrates. Biochem Biophy Res Comm. 2003;306:110–5. doi: 10.1016/s0006-291x(03)00920-3. [DOI] [PubMed] [Google Scholar]
- 178.Bäumer N, Mäurer A, Krieglstein J, Klumpp S. Expression of protein histidine phosphatase in Escherichia coli, purification, and determination of enzyme activity. Methods Mol Biol. 2007;365:247–60. doi: 10.1385/1-59745-267-X:247. [DOI] [PubMed] [Google Scholar]
- 179.Maurer A, Wieland T, Meissl F, Niroomand F, Mehringer R, Krieglstein J, Klumpp S. The beta-subunit of G proteins is a substrate of protein histidine phos-phatase. Biochem Biophy Res Comm. 2005;334:1115–20. doi: 10.1016/j.bbrc.2005.06.200. [DOI] [PubMed] [Google Scholar]
- 180.Kowluru A. Novel regulatory roles for protein phosphatase-2A in the islet beta cell. Biochem Pharmacol. 2005;69:1681–91. doi: 10.1016/j.bcp.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 181.Movassat J, Saulnier C, Portha B. Beta-cell mass depletion precedes the onset of hyperglycemia in the GK rat, a genetic model of non-insulin-dependent diabetes mellitus. Diabetes Metab. 1995;21:365–70. [PubMed] [Google Scholar]
- 182.Movassat J, Saulnier C, Serradas P, Portha B. Impaired development of pancreatic beta-cell mass is a primary event during the progression to diabetes in GK rat. Diabetologia. 1997;40:916–25. doi: 10.1007/s001250050768. [DOI] [PubMed] [Google Scholar]
- 183.Kowluru A. Defective protein histidine phosphorylation in islets from the Goto-Kakizaki diabetic rat. Am J Physiol Endocrinol Metab. 2003;285:E498–503. doi: 10.1152/ajpendo.00121.2003. [DOI] [PubMed] [Google Scholar]
- 184.Metz SA, Meredith ME, Vadakekalam J, Rabaglia ME, Kowluru A. A defect late in stimulus-secretion coupling impairs insulin secretion in Goto Kakizaki diabetic rats. Diabetes. 1999;48:1754–62. doi: 10.2337/diabetes.48.9.1754. [DOI] [PubMed] [Google Scholar]
- 185.Ostenson CG, Zaitsev S, Berggren PO, Efendic S, Langel U, Bartfai T. Galparan: a powerful insulin-releasing chimeric pep-tide acting at a novel site. Endocrinology. 1997;138:3308–13. doi: 10.1210/endo.138.8.5307. [DOI] [PubMed] [Google Scholar]
- 186.Gerbitz KD, Gempel K, Brdiczka D. Mitochondria betes. Genetic, biochemical, and clinical implications of the cellular energy circuit. Diabetes. 1996;45:113–26. doi: 10.2337/diab.45.2.113. and dia. [DOI] [PubMed] [Google Scholar]
- 187.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic beta-cells. Nature. 2001;414:807–12. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 188.McDonald P, Veluthakal R, Kaur H, Kowluru A. Biologically active lipids promote trafficking and membrane association of Rac1 in insulin-secreting INS832/13 cells. Am J Physiol Cell Physiol. 2007;292:C1216–20. doi: 10.1152/ajpcell.00467.2006. [DOI] [PubMed] [Google Scholar]