

Abstract
The novel selective BCR-ABL Breakpoint cluster region – Abelson murine leukemia viral oncogene homolog 1 (BCR-AML) inhibitor nilotinib (AMN107) is a tyrosine kinase inhibitor that is more potent against leukaemia cells in vitro than imatinib. As nilotinib might be used in the context of allogeneic stem cell transplantation where CD8+ T lymphocytes play a pivotal role in the graft-versus-leukaemia (GVL) effect, we investigated effects of nilotinib on this lymphocyte subpopulation. Nilotinib inhibits phytohemagglutinin (PHA)-induced proliferation of CD8+T lymphocytes in vitro at therapeutically relevant concentrations (0.5–4 μM). The inhibition of CD8+ T lymphocytes specific for leukaemia or viral antigens through nilotinib was associated with a reduced expansion of antigen peptide specific CD8+ T lymphocytes and with a decreased release of interferon—γ and granzyme B by these cells as analysed by flow cytometry and enzyme-linked immunospot (ELISPOT) assays. The inhibitory effect caused by nilotinib was two times stronger than by imatinib. These effects were mediated through the inhibition of the phosphorylation of ZAP-70, Lck and ERK 1/2 and the NF-κβ signalling transduction pathway. Taken together, we observed a strong suppressive impact of nilotinib on the CD8+ T lymphocyte function which should be considered carefully in the framework of allogeneic stem cell transplantation or other T cell based immunotherapies.
Keywords: nilotinib, lymphocyte proliferation, functional activation, healthy donor, chronic myeloid leukaemia
Introduction
Imatinib was the first targeted therapy developed to inhibit the BCR-ABL kinase in Philadelphia chromosome (Ph)-positive chronic myeloid leukaemia (CML). The drug has become the first-line treatment for newly diagnosed CML [1–5]. However, the emergence of resistance to imatinib remains a major problem in the treatment of Ph+ leukaemia [6–8]. The need for alternative or additional treatment options for imatinib-resistant BCR-ABL positive leukaemia has stimulated the design of a second generation of targeted therapies, which has already resulted in the development of the two novel small-molecule inhibitors nilotinib (AMN107), an imatinib mesylate derivative [9–13] and dasatinib (BMS-354825), a combined ABL/SRC kinase inhibitor [14–16].
Nilotinib (Novartis Pharma, Basel, Switzerland) shows a higher potency against CML cells in vitro than does imatinib [11, 17–19]. Nilotinib displays a higher binding affinity and selectivity for the ABL kinase than does imatinib. In a recent dose-escalating Phase-I study, imatinib-resistant CML patients in the chronic phase, accelerated phase and blast crisis were treated with nilotinib resulting in cytogenetic and haematological responses of imatinib-refractory CML patients [13]. The best responses have been seen at a dose of 400 mg once a day and with 400 mg twice a day. Nilotinib is emerging as an important new therapeutic agent in the treatment of imatinib-resistant CML, also after allogeneic stem cell transplantation.
Although nilotinib does not directly inhibit any of the Src family kinases, including Lck, known to be involved in immune cell signalling, several of the hematotoxic molecules phosphorylated by the ABL kinase are also involved in the activation pathways of immune cells. So, one might speculate on potential hematotoxic side effects particularly after daily and long-time exposure to the drug. Several authors have reported the effects of imatinib on T cells [22–26], while the effects of nilotinib on normal CD34+ haematopoietic stem cells [27], but not yet on CD8+ T lymphocytes, have been evaluated. Therefore, we investigated in this study the effect of nilotinib on the proliferation and on the function of CD8+ T lymphocytes in vitro at therapeutically relevant drug concentrations, as well as to investigate the effect on potential signalling pathways affected by nilotinib in BCR-ABL negative cells.
Materials and methods
Samples from healthy donors and patients with CML
All samples, which were human leukocyte antigen A2 (HLA-A2) positive were taken from healthy blood donors and patients with CML in complete molecular remission after their informed consent was obtained. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll – Biocoll Separation Solution (Biochrom, Berlin, Germany) density gradient centrifugation. The viability of PBMCs obtained was always >95%, as determined by trypan blue staining (Trypan Blue Solution 0.4%, Sigma-Aldrich Munich, Germany). For cellular assays, Ficoll separated PBMCs were tested freshly or cryopreserved in RPMI 1640 containing 20% human AB serum (German Red Cross Blood Center, Ulm, Germany) and 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich, Mannheim, Germany), stored in liquid nitrogen.
Nilotinib and imatinib
Nilotinib and imatinib powder were generously provided by Novartis Pharmaceuticals (Basel, Switzerland) and stored at–20°C as 10 mM stock solution in DMSO. Fresh dilutions in X-VIVO 10 medium were prepared prior to the experiments.
T2 cells
T2 cell line used in cellular assays was obtained from the ‘American Type and Culture Collection, http://www.atcc.org’. The T2 cell line was maintained at 37°C in a humidified 5% CO2 atmosphere in a standard medium consisting of RPMI 1640 (Biochrom AG, Berlin, Germany) supplemented with 10% AB serum, 2 mM L-glutamine (Biochrom AG, Berlin, Germany), 100 units/ml penicillin and 100 units/ml streptomycin (Invitrogen Gibco, Grand Island, USA).
Synthetic peptides
Peptides used in our study corresponded to influenza matrix protein (IMP) derived peptide (pos. 58–66: GILGFVFTL), cytomegalovirus (CMV) derived peptide CMV pp65 (pos. 495–503: NLVPMVATV) and RHAMM peptide R3 (pos. 165–173: ILSLELMKL) which are HLA-A*0201 restricted CD8+ T cell epitopes. All peptides were dissolved in DMSO mixed with phosphate buffered saline (PBS) at a concentration of 1 pg/μl for individual experiments. The RHAMM/CD168 derived peptide R3 was chosen because of its high immunogenicity in patients with acute myeloid leukaemia (AML) and CML as well as in healthy donors [28, 29].
Mixed lymphocyte peptide cultures (MLPCs)
MLPCs were conducted as described earlier [26, 28–31]. Briefly, PBMCs were selected by human CD8 MicroBeads through a magnetic cell sorting (MACS; Miltenyi, Bergisch-Gladbach, Germany). More than 95% purity was reached in the CD8+ fraction confirmed by Fluorescence activated cell sorting (FACS) (Fluorescein isothiocyanate) analysis. CD8 negative antigen-presenting cells (APCs) were irradiated with 30 Gy and pulsed with the IMP, CMV or R3 peptide mentioned above at a concentration of 20 μg/ml for 2 hrs. After coincubation with CD8+ T lymphocytes over night, the MLPC in the presence or absence of nilotinib in different concentrations varying from 0.5 μM to 4 μM was supplemented with 10 U/ml IL-2 and 20 ng/ml IL-7 on day +1. Imatinib was added to MLPCs as previously described [26]. After 8 days of culture, CTLs were evaluated for their interferon-γ (IFN-γ) and granzyme B (Gzm-B) secretion by ELISPOT assays employing T2 cells pulsed with IMP or R3. The frequency of IMP. CMV or R3 specific CD8+ T cells was determined after 8 days of MLPC by staining with HLA-A2/IMP peptide tetramer*PE, HLA-A2/CMV peptide tetramer*PE or HLA-A2/R3 peptide tetramer’PE. CD8*APC-Cy7: CD45RA*APC, CCR7*PE-Cy7 and CD27*FITC Becton Dickinson (BD) Biosciences, California, USA (all from) were added at 4°C for 20 min. in the dark. After washing twice with PBS, stained cells were fixed with 0.5% paraformaldehyde (Sigma, Munich, Germany) for 10 minutes and subsequently analysed by flow cytometry.
Enzyme linked immunospot (ELISPOT) assays for interferon-y and granzyme-B
IFN-7 and Gzm-B ELISPOT assays were performed as previously described to evaluate the inhibitory effects of imatinib on CD8+ T lymphocytes according to the manufacturer's instruction (BD, San Diego, USA) [26, 28–31]. Spots were automatically evaluated by the use of an ELISPOT reader (CTL, Reutlingen, Germany) and expressed as spot-forming cells (SFCs) per 1 × 104 CD8+ T lymphocytes.
Flow cytometry for activation markers and signal transduction cascade
CD8+ T lymphocyte activation following 72 hrs stimulation with PHA (10 μg) and IL-2 (100 U/ml) or anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) antibodies, in the presence or absence of different concentrations of nilotinib, was assessed by quantifying the number of CD8+ T lymphocytes expressing the activation markers CD25 and CD69. Briefly CD8+ T lymphocytes were incubated with the appropriate directly conjugated antibodies for 30 min. on ice. Cells were washed twice with ice-cold PBS and analysed on FACSAria flow cytometer (Becton Dickinson Biosciences, Heidelberg, Germany) using CellQuest® software. For signal transduction, we used NF-κβ p105/p50 antibody phospho-F-κβ p105 (Ser933) (18E6) rabbit mAb, phospho-NF-κβ p65 (Ser536) (93H1) rabbit mAb (Alexa Fluor® 488 Conjugate), RelB antibody, c-Rel antibody, NF-κβ p100/52 (18D10) rabbit mAb (Human Specific), phospho-lIF-KB2 p100 (Ser864/868) antibody, phospho-Zap-70 (Tyr319)/Syk (Tyr352) antibody. All the reagents above were purchased from Cell Signaling Technology®, Beverly, USA. FITC-conjugated donkey anti-rabbit IgG was used as secondary antibody (Jackson ImmunoResearch Laboratories, Hamburg, Germany). Anti-ZAP-70 (1E7.2) FITC, anti-Phospho-Lck (Y505) Alexa Fluor® 488 and anti-Phospho-ERK1/2 (T202/Y204) PE were obtained from BD Biosciences. IntraStain was purchased from DakoCytomation, Glostrup, Denmark. Briefly, the cells were washed twice, first with PBS and then with blocking buffer (PBS 1% foetal calf serum [FCS]). After that, IntraStain Reagent A and IntraStain reagent B were added into the cells according to the manufacturer's instructions. Appropriate antibodies were added to the cells and incubated at 4°C for 30 min. in the dark. When secondary staining was required, cells were counterstained with secondary antibody for 20 min. at 4°C in the dark, following by washing with blocking buffer. Each sample was run with a matched isotype control to exclude non-specific staining. Cells were analysed with FACSAria flow cytometer (Becton Dickinson) after exclusion of dead cells.
Measurement of cell proliferation
An indirect 5-bromo-2-deoxyuridine (BrdU)-FITC flow kit (BD PharmingenTM, Heidelberg, Germany) was used to determine the CD8+ T cell proliferation. CD8+ T cells (5 x105 cells/ml) were stimulated with phytohemagglutinin (PHA) (10 Mg/ml) and IL-2 (60 U/ml) without or with different concentrations of Nilotinib as indicated. After 96 hrs of incubation, cells were harvested and measured according to the manufacturer's protocol. Briefly, cells were labelled with 10 μg/ml BrdU for 1 hr. Cells unlabelled with BrdU served as negative control. Then, the cells were fixed, permeabilized by cytofix/cytoperm buffer and incubated with DNase (30 μg per sample) for 1 hr at 37°C. After that, the cells were stained with FITC-conjugated anti-BrdU, and detected by flow cytometry [32]. Additionally, the division of CD8+ T cells was measured by dilution of carboxyfluorescein diacetate succinimidyl ester (CFSE, Invitrogen Gibco, Grand Island, USA) as described [33] CD8+ T cells were labelled with CFSE at a final concentration of 1 μm for 10 min. at 37°C. Then, the cells were washed with ice-cold culture medium for three times and resuspended in culture medium. 4 μm nilotinib was added at different time points as indicated (0 hr, 24 hrs and 48 hrs). CD8+ T cells untreated with nilotinib served as control. After 96 hrs of stimulation with PHA and IL-2, the proliferation of cells was measured by flow cytometry.
Assessment of apoptosis
For the detection of apoptosis, the annexin V*FITC apoptosis detection kit I (BD Biosciences, California, USA) was used according to the instructions of the manufacturer. Apoptotic cells were defined by flow cytometry as Pl-negative and annexin V*FITC-positive.
Measuring total lκβ-α and phospho-lκβ-α (Ser32) by enzyme-linked immunosorbent assay (ELISA)
CD8+ T lymphocytes were resuspended in standard RPM11640 medium at 5 × 105 cells/ml, plated in 24-well plates and rested over night. The CD8+ T lymphocytes were stimulated with PHA (to the final concentration of 10 μg/ml) and IL-2 and incubated at 37°C in the absence or presence of nilotinib in a serial concentration of 0.5–4 μm for 1 hr. The levels of total lκβ-α and phospho-kB-α (Ser32) were assessed according to the instruction manuals of PathScan Total lκβ-α Scanwich ELISA Kit, PathScan® Phospho- lκβ-α (Ser32) Sandwich ELISA Kit (Cell Signaling Technology®, Beverly, USA).
51Cr release cytotoxicity assay
After 8 days of MLPC in the absence or presence of the indicated concentrations of nilotinib, CD8+ T lymphocytes were harvested and evaluated for their specific cytotoxicity in a standard 51Cr release assay against T2 cells pulsed with cognate peptide or irrelevant peptide, or without peptide [26, 28, 29].
Statistical analysis
The results were expressed as the mean ± standard deviation (SD). The significance of the difference between means was determined by the two-tailed t test or analysis of one-way variance (anova) using the Bonferroni multiple comparison correction, and the differences were considered statistically significant at P<0.05.
Results
Nilotinib inhibits the proliferation of CD8+ T lymphocytes
Nilotinib inhibited the CD8+ T lymphocyte proliferation as a function of the concentration (Fig. 1, panels A and C). Significant effects on the cells stimulated with PHA/IL-2 were observed from a concentration ≥1 μM nilotinib. The drug (4 μM) was also added at different time points of CD8+ T cell culture. It could inhibit the proliferation of stimulated CD8+ T lymphocytes even when added 48 hrs after the start of the culture (Fig. 1, panels B, Dand E). Cells divided as demonstrated by CFSE dilution, but then subsequently failed to incorporate BrdU. Nilotinib did not significantly increase apoptosis at concentrations up to 4 μM when compared with untreated cells (data not shown). The fraction of apoptotic cells remained between 1.3 × 104± 4 × 103 to 1.5 × 104± 8 × 103 after the incubation of CD8+T lymphocytes with nilotinib at concentrations of 0.5–4 μM, while the fraction of apoptotic cells was 1.2 × 104± 2 × 103 after incubation of CD8+ T lymphocytes without nilotinib (P> 0.05). Taken into account that nilotinib inhibits proliferation without inducing apoptosis, the cells would resume proliferating following removal of the drug. To test this hypothesis, we incubated PHA/IL-2 stimulated CD8+ T lymphocytes with nilotinib at different concentrations for 72 hrs. Then we washed the cells and after 24 hrs resting of the cells, we restimulated the cells from each sample with PHA/IL-2 and incubated all for additional 72 hrs, before measuring BrdU incorporation. CD8+T lymphocyte proliferation could be completely restored, thus demonstrating that the inhibitory effect was transient (data not shown).
Fig 1.
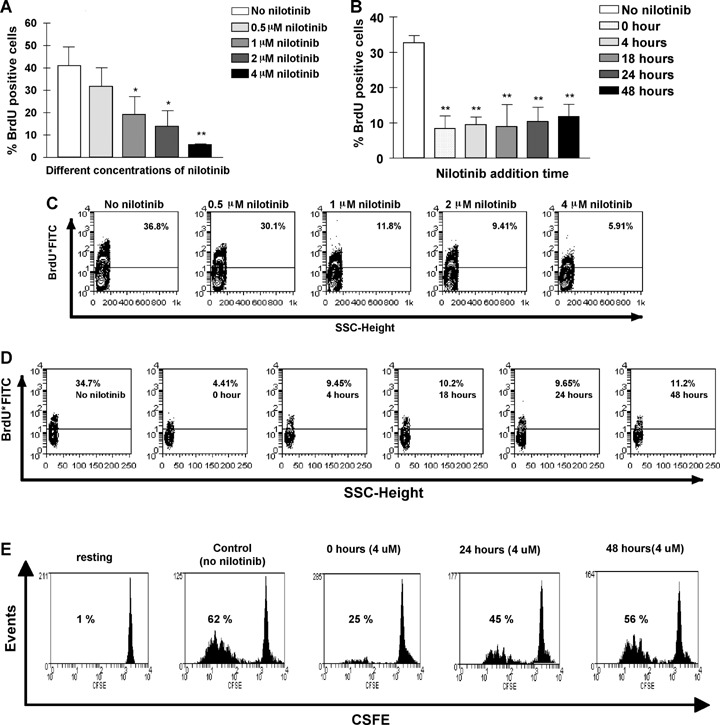
Nilotinib inhibits phytohemagglutinin (PHA) and IL-2-induced proliferation of CD8+ T lymphocytes in a dose-dependent manner and can terminate ongoing proliferation. Panel A+C: CD8+ T lymphocytes from healthy volunteers were purified, and kept in culture for 96 hrs in the presence of PHA/IL-2 without nilotinib or with nilotinib at different concentrations (0.5 μM, 1 μM, 2 μM and 4 μM). BrdU incorporation was analysed by FACS as described in ‘Materials and methods’. Panel A shows the combined results from three independent experiments of three different samples with similar results. The data were expressed as percentage of BrdU positive cells above negative control at each nilotinib concentration. Panel C shows the results of one representative experiment. Panel B+D+E: Nilotinib (4 μM) was also added 0–48 hrs after start of the culture in the presence of PHA/IL-2 for 96 hrs. BrdU positive cells measured by FACS analysis in the presence of nilotinib at all time-points were different from that in nilotinib-free group. Panel B displays the combined results from three independent experiments of three different samples with similar results. Panel D shows the results of one representative experiment. Panel E shows the CFSE uptake as an indicator of cell division at different time points in the absence and presence of 4 μM nilotinib (one representative of three different experiments). Error bars indicate SD. *P<0.05 when compared with no nilotinib group, **P<0.01 when compared with no nilotinib group as the control.
Proliferation of IMP, CMV or RHAMM specific CD8+T lymphocytes is suppressed by nilotinib
We stimulated purified CD8+T lymphocytes with autologous CD8 negative APCs pulsed with IMP peptide in the absence or presence of different concentrations of nilotinib as described above. IMP-specific CD8+ T lymphocytes proliferated in response to IMP peptide. We found nilotinib to reduce the expansion of IMP-specific CD8+ T lymphocytes in a dose-dependent manner over 8 days of MLPC; representative data are shown in Figure 2, panel A. To further characterize IMP-specific CD8+ T lymphocytes from the gate of the HLA-A2/IMP-tetramer*PE positive CD8+ T lymphocytes were analysed for their expression of CCR7, CD27 and CD45RA Most of the cells (65–93%) revealed to be CD8+ HLA-A2/IMP tetramer CCR7-CD45RA+ effector T cells (data not shown). To confirm that this observation was generally applicable and not limited to the CD8+ T lymphocyte response to IMP, we repeated the experiment with the CMV pp65-derived peptide (Fig. 2, panel B) and the RHAMM-derived R3 peptide (Fig. 2, panel C). We found comparable inhibition of specific CD8+T lymphocyte expansion in response to these three antigens.
Fig 2.
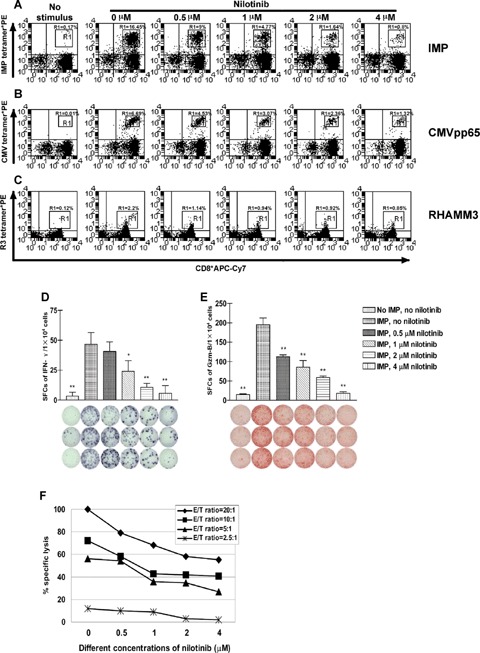
Nilotinib inhibits IMP-, CMV- and RHAMM-specific CD8+ T lymphocytes as a function of the concentration of and the incubation time with the drug. CD8+ T lymphocytes from patients with chronic myeloid leukaemia (CML) were subjected to one round of stimulation with irradiated autolo-gous CD8v− APCs pulsed with influenza matrix protein (IMP), cytomegalovirus (CMV) or RHAMM derived R3 peptide in the absence or presence of 0.5 μM, 1 μM, 2 μM and 4 μM nilotinib as described in ‘Material and methods’. The dot plots show the percentage of HLA-A2/IMP- or HLA-A2/CMV-or HLA-A2/R3- tetramer*PE positive CD8+ T lymphocytes. Effects of nilotinib on the expansion of IMP-specific CD8+ T lymphocytes over 8 days of mixed lymphocyte peptide cultures (MLPC) are displayed in Panel A, effects on the expansion of CMV-specific CD8+ T lymphocytes in Panel B, and effects on the expansion of RHAMM derived R3-specific CD8+T lymphocytes in Panel C. The proliferation of IMP-, CMV- or R3-specific CD8+ T lymphocytes was inhibited in a dose-dependent manner. The figure displays representative results from one of four consecutive experiments with similar results. The T cell reaction was inhibited in a dose-dependent fashion as analysed by ELISPOT assays for spot-forming cells (SFCs) of interferon-γ (IFN-γ) (Panel D) and granzyme B (Gzm-B) (Panel E) after 8 days of MLPC in the absence or presence of different concentrations of nilotinib. As evaluated by 51Cr release assay (Panel F), the lysis of IMP-pulsed T2 cells by cognate peptide specific CD8+ cytotoxic T lymphocytes generated during one round of MLPC was affected by nilotinib in a dose-dependent manner at different E/T ratios. The figures show results from one representative experiment. All assays were performed in triplicate. Error bars indicate the SD. Significant differences from the non-nilotinib-treated group are indicated, *P<0.05, **P<0.01.
Nilotinib inhibits the IFN-γ and Gzm-B production of IMP antigen specific CD8+ T cells
To further evaluate the inhibition of functional activation of CD8+ T lymphocytes by nilotinib, ELISPOT assays were performed to assess the production of IFN-γ and Gzm-B secreted by human IMP specific CD8+ T lymphocytes upon stimulation with autologous CD8 negative APCs which were pulsed with IMP peptide. As shown in representative experiments (Fig. 2, panel D and E), nilotinib reduced the frequency of IFN-γ (Fig. 2, panel D) and Gzm-B (Fig. 2, panel E) SFCs gradually with increasing concentration of nilotinib. When the concentration of nilotinib was as high as 4 μM, the IFN-γ and Gzm-B SFCs were reduced to background level. The secretion of Gzm-B could be inhibited even more drastically. To further confirm the inhibition of function of antigen-specific CD8+ T cells by nilotinib, standard 51Cr release assays were performed. After 8 days of MLPC as indicated in ‘Material and methods’, IMP-specific CD8+T lymphocytes were able to lyse T2 cells pulsed with IMP. Specific lysis could be inhibited by nilotinib in a dose-dependent manner (Fig. 2, panel F). T2 cells pulsed with the unrelated MAGE3 peptide and unpulsed T2 cells were not recognized and lysed by IMP-specific CD8+T lymphocytes (data not shown).
Nilotinib down-regulates CD8+T lymphocyte activation markers
As for the expression of CD8+ T lymphocyte activation markers on CD8+ T lymphocytes isolated from three different healthy volunteers following 72 hrs PHA/IL-2 or anti-CD3/anti-CD28 antibody stimulation, a reduction in both the percentage and mean fluorescence intensity of CD25 and CD69 was observed (Fig. 3). The effect was most pronounced at the highest nilotinib concentration at 4 μM. Significant or highly significant reduction in CD25 (Fig. 3, panel A) and CD69 (Fig. 3, panel B) expression was observed at 4 μM nilotinib. Naive CD8+ T lymphocytes without exposure to nilotinib express only 6.9±4.0% CD25 and 2.6 ± 2.6% CD69 respectively.
Fig 3.
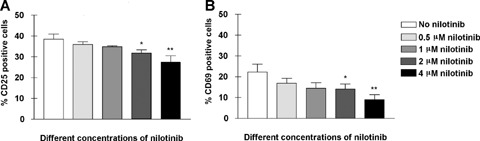
Nilotinib inhibits the expansion of the activation markers CD69 and CD25 in a dose-dependent manner. CD8+ T lymphocytes from patients with CML were stimulated with anti-CD3/CD28 antibodies for 72 hrs. Cell surface expression of activation markers was assessed by immunofluorescence staining. Cells were incubated with CD25*PE and CD69*PerCP and isotype-matched mAbs. Cell surface expression was analysed using a BD FACS can flow cytometer. The percentages of CD8+ T lymphocytes expressing the relevant marker from three independent experiments are shown. The percentage of cells expressing CD25 (Panel A) and percentage of cells expressing CD69 (Panel B) are shown for several concentrations (0–4 μM) of nilotinib. Error bars indicate the SD.
Nilotinib reduces levels of phosphorylated Lck, ERK1/2, ZAP-70 and blocks the NF-κβ pathway
In an attempt to identify a nilotinib-sensitive intracellular signalling pathway, we measured the level of phosphorylation of the Lck molecule which is the first tyrosine kinase activated by T cell receptor (TCR) signalling, as well as ERK 1/2 involved in further TCR-mediated signalling [34]. We exposed PHA-stimulated CD8+ T lymphocytes to 4 μM nilotinib and determined the relative levels of phosphorylated Lck and ERK 1/2 by FACS analysis. Nilotinib reduced the phosphorylation of both ERK 1/2 (Fig. 4, panel A) and Lck (Fig. 4, panel B). Moreover, we observed a down-regulation of phospho-NF-κβ p105 (Ser933) (18E6) (Fig. 4, panel C), phospho-NF-kB2 p100 (Ser864/868) (Fig. 6, panel D), phospho-ZAP-70 (Fig. 4, panel E) by nilotinib. Even so, ELISA demonstrated a reduction in phosphorylation of lκβ-α after incubation with nilotinib (Fig. 4, panel G). In contrast, we did not detect down-regulation of other molecules involved in the further T cell signalling cascade ZAP-70 (Fig. 4, panel F), and total Iκβ-α (Fig. 4, panel G), Rel-B, c-Rel, NF-κβ p105/p50, NF-kB2 p100/52, phospho-NF-κβ p65, (data not shown). For the posphorylated forms of NF-kB p105, ERK1/2, ZAP70and Lck the results were confirmed by western blotting (Fig. 4, panel H).
Fig 4.
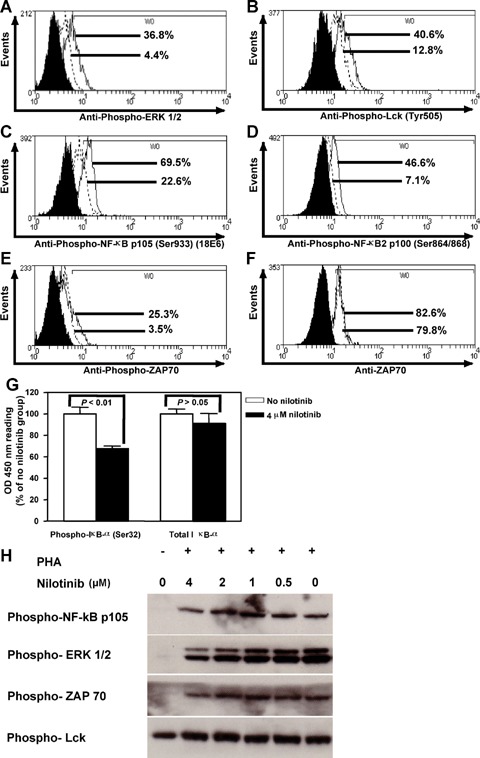
Nilotinib inhibits the phosphorylation of molecules participating in T cell receptor down-stream signalling. To identify nilo-tinib-sensitive intracellular signalling pathways, we measured the levels of ZAP70 expression and phosphorylation of ERK 1/2, Lck, ZAP-70, NF-κβ p105 (Ser933) (18E6), NF-kB2 p100 (Ser864/868) by FACS analysis and phospho-lκβ-α (Ser32) and total lκβ-α by ELISA. We exposed PHA-stimulated CD8+ T lymphocytes to 4 μM nilotinib for 1 hr and found that the drug did not affect the level of ZAP70 (Panel F) and total lκβ-α (Panel G), but inhibited the phosphorylation of ERK 1/2 (Panel A), Lck (Tyr505) (Panel B), NF-kB p105 (Ser933) (18E6) (Panel C), NF-kB2 p100 (Ser864/868) (Panel D), ZAP70 (Panel E) and lκβ-α (Ser32) (Panel G). As for the FACS analysis, solid histograms represent the background staining with the isotype control, dotted lines the 4 μM nilotinib group, and solid lines the group without nilotinib. For NF-κβ p105, ZAP70, ERK1/2 and Lck western blotting was performed to confirm the results (Panel H).
Comparison of the inhibitory effects on CD8+ T lymphocytes through nilotinib versus imatinib
To compare the inhibitory effects on CD8+T lymphocytes between nilotinib and imatinib, we used the Stata 7.0 statistical package (Stata Corp, College Station, TX, USA) to get the exponential curve for the definition of the median effective concentration (EC50) (Fig. 5). The expansion of IMP-specific CD8+ lymphocytes was measured by tetramer FACS analysis and by ELISPOT assays for IFN-γ and Gzm-B after MLPC. Comparing the proliferation of IMP-specific CD8+ T lymphocytes with respect to EC50s for nilotinib and imatinib, nilotinib revealed an EC50nilotinib of 0.75 μM (R2= 0.99) (Fig. 5, panel A), and imatinib an EC50imatinib of 1.40 μM (R2= 0.98) (Fig. 5, panel B). Nilotinib and imatinib inhibited the expansion of SFCs of IFN-7 with an EC50 of 0.84 μM (tf = 0.99) (Fig. 5, panel C) and 2.05 μM (R2= 0.98) (Fig. 5, panel D), and of Gzm-B with an EC50 of 0.85 μM (R2= 0.99) (Fig. 5, panel E), and 1.51 μM (R2= 0.99) (Fig. 5, panel F) respectively. All the EC50 values for nilotinib were lower than those observed for imatinib, suggesting that nilotinib is a more potent inhibitor of CD8+ T lymphocytes than imatinib at the same drug concentration.
Fig 5.
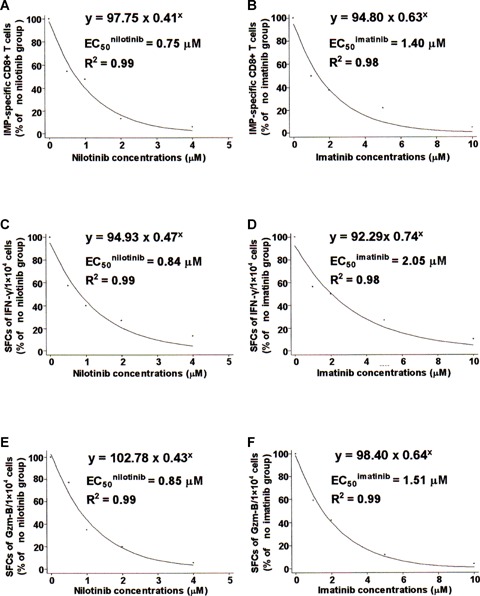
Comparison of inhibitory effects on CD8+ T lymphocytes between nilotinib and imatinib as evaluated by EC50. The expansion of IMP-specific CD8+ T lymphocytes from CML patients (Panels A and B) was measured by tetramer FACS analysis, and the expansion of SFCs of IFN-γ (Panels C and D) or Gzm-B (Panels E and F) was assessed by ELISPOT assays. The values decreased by augmenting the dose of nilotinib (Panels A, C and E) or imatinib (Panels B, D and F). The inhibitory effect was evaluated by comparison of the EC50 after MLPCs as described in ‘Materials and methods’. The expansion of IMP-specific CD8+ T lymphocytes and SFCs of IFN-γ or Gzm-B are shown on the Y-axis with ‘% of no nilotinib group’ or’% of no imatinib group’; concentrations of nilotinib or imatinib are indicated on the X-axis. Values represent the combined results of at least four independent experiments.
Discussion
Imatinib inhibits the tyrosine kinase activity of the BCR-ABL protein and is an effective frontline therapy for chronic-phase CML. However, CML patients and Ph+ ALL patients in an accelerated phase or in a blast-crisis often relapse due to drug resistance resulting from the emergence of imatinib-resistant point mutations within the BCR-ABL tyrosine kinase domain [8, 9, 14]. This has stimulated the development of new kinase inhibitors overriding the resistance to imatinib. The novel, selective BCR-ABL inhibitor nilotinib has produced haematological and cytogenetic responses in CML patients, who either did not initially respond to imatinib or developed imatinib resistance [11, 13]. As nilotinib might be used in the framework of allo-transplantation and other T cell-based immunotherapies, we wondered whether inhibition of ABL kinase by nilotinib might have a detrimental effect on the immune response. In several studies, T lymphocytes from CML patients have been found to be Ph chromosome negative and/or BCR-ABL negative [35–37]. On the other hand, several of intracellular signalling molecules triggered by the ABL kinase are also involved in the activation pathways of immune cells. Effects of imatinib on T cells have been extensively reported [22–26]. Here, we report that nilotinib inhibits CD8+ T lymphocyte proliferation and functional activation in response to non-specific PHA stimulation and TCR engagement by anti-CD3/anti-CD28. These effects were dose-dependent with some inhibition of proliferation and function at 1 μM, which corresponds to the mean steady-state plasma level achieved by daily administration of 400 mg nilotinib; and no dose-limiting toxic effects were seen at dosages of up to 600 mg daily [13]. The mean plasma level at steady-state were 1.0 μM with 400 mg nilotinib administration daily and 2.3 μM with 600 mg daily, while administration of nilotinib at 400 mg twice daily provided maximal and minimal plasma drug concentrations of 3.6 and 1.7 μM respectively. All levels exceeded the 50 percent inhibitory concentration (IC50) of cellular phosphorylation of BCR-ABL (20–57 nM, depending on cell type) and of 32 out of 33 BCR-ABL kinase mutants (19–709 nM) [13, 36, 38].
As demonstrated in the present study, the CD8+ T lymphocyte inhibitory effect in the presence of nilotinib was not mediated through the induction of apoptosis in CD8+ T lymphocytes. Under these conditions, nilotinib was cytostatic, but not cytotoxic. After the removal of nilotinib, the proliferation of CD8+ T lymphocytes could be resumed. In this study we found that the up-regulation of the activation markers CD25 and CD69 on CD8+ T lymphocytes was significantly inhibited by nilotinib after stimulation [39, 40]. The dose-dependent inhibition of cognate peptide-specific CD8+ T lymphocytes suggests that nilotinib could interfere with the clinically important T lymphocyte effector function, as is supported by the release of IFN-γ and Gzm-B, as well as by 51Cr cytotoxicity assays.
While dissecting the TCR signalling pathway inhibited by nilotinib, we observed a dose-dependent reduction in phosphorylation of ZAP-70, Lck and ERK1/2 showing a direct inhibition of the TCR signalling pathway. Phospho-NF-κβ p105 (Ser933) (18E6), phos-pho-NF-KB2 p100 (Ser864/868) and phospho-kB (Ser 32) were down-regulated in a dose-dependent fashion. In comparison with imatinib [22, 24], nilotinib showed a similar pattern of inhibition of the TCR signal transduction.
When comparing the IC50 for nilotinib and imatinib, the inhibition of the proliferation of IMP-specific CD8+ T lymphocytes as assessed by SFCs for IFN-γ and of SFCs for Gzm-B was twice as strong by nilotinib. The mean plasma level achieved at steady-state was about 1 μM after daily dosing with 400 mg nilotinib and imatinib respectively. Given, that nilotinib is in vitro it least 20-fold more potent than imatinib against unmutated Abl [11] and that nilotinib is active against many imatinib-resistant BCR-ABL mutants [10, 11, 15], nilotinib would be relatively less detrimental to CD8+ T lymphocytes than imatinib.
Effects of nilotinib on the immune reconstitution and CD8+ T lymphocyte function may be especially relevant for the administration in the treatment of patients where the regulation of immune functions is critical and has a direct impact on morbidity and mortality, for example in the setting of allogeneic transplantation. We have shown here that nilotinib directly inhibits the proliferation and TCR signalling after non-specific stimulation, and this effect might even potentiate the effects of immunosuppressive drugs. Although an increase in opportunistic infections has not been reported in patients treated with nilotinib, close monitoring for CMV re-activation or other infections in patients under nilotinib treatment, especially in stem cell transplant recipients, might be of value.
Nilotinib might potentially interfere with the expansion of effector T cells responsible for the graft-versus-leukaemia (GVL), thus hampering or at least reducing the therapeutic efficacy of the transplantation and/or donor lymphocyte infusions (DLIs), but might also down-regulate the graft-versus-host disease (GVHD).
Further research will increase the understanding of how nilotinib might affect normal haematopoietic stem cells as well as CML progenitor cells in concert with dendritic cells and T cells. It remains to be elucidated how the immunological control of CML in nilotinib-treated patients can be achieved and how nilotinib should be integrated in the treatment of residual disease in the post-transplant setting. As a result of the reported nilotinib-induced impairment of CD8+ T lymphocyte proliferation and function, nilotinib-free therapeutic windows after stem cell transplantation and other CD8+ T lymphocyte based immunotherapies might be required for the expansion of leukaemia-specific CD8+ T lymphocytes.
Acknowledgments
This research was kindly supported by Novartis, Nuremberg, Germany.
References
- 1.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. NEngl JMed. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Savage DG, Antman KH. Imatinib mesylate-a new oral targeted therapy. N Engl J Med. 2002;346:683–93. doi: 10.1056/NEJMra013339. [DOI] [PubMed] [Google Scholar]
- 3.Magnusson MK, Meade KE, Nakamura R, Barrett J, Dunbar CE. Activity of STI571 in chronic myelomonocytic leukemia with a platelet-derived growth factor beta receptor fusion oncogene. Blood. 2002;100:1088–91. doi: 10.1182/blood-2002-01-0165. [DOI] [PubMed] [Google Scholar]
- 4.Millot F, Guilhot J, Nelken B, Leblanc T, De Bont ES, Békassy AN, Gadner H, Sufliarska S, Stary J, Gschaidmeier H, Guilhot F, Suttorp M. Imatinib mesylate is effective in children with chronic myelogenous leukemia in late chronic and advanced phase and in relapse after stem cell transplantation. Leukemia. 2006;20:187–92. doi: 10.1038/sj.leu.2404051. [DOI] [PubMed] [Google Scholar]
- 5.Ramanarayanan J, Dunford LM, Baer MR, Sait SN, Lawrence W, McCarthy PL. Chronic myeloid leukemia after treatment of lymphoid malignancies: Response to imatinib mesylate and favorable outcomes in three patients. Leuk Res. 2006;30:701–5. doi: 10.1016/j.leukres.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 6.Chung YJ, Kim TM, Kim DW, Namkoong H, Kim HK, Ha SA, Kim S, Shin SM, Kim JH, Lee YJ, Kang HM, Kim JW. Gene expression signatures associated with the resistance to imatinib. Leukemia. 2006;20:1542–50. doi: 10.1038/sj.leu.2404310. [DOI] [PubMed] [Google Scholar]
- 7.Druker BJ. Circumventing resistance to kinase-inhibitor therapy. N Engl J Med. 2006;354:2594–6. doi: 10.1056/NEJMe068073. [DOI] [PubMed] [Google Scholar]
- 8.Mishra S, Zhang B, Cunnick JM, Heisterkamp N, Groffen J. Resistance to imatinib of bcr/abl p190 lymphoblastic leukemia cells. Cancer Res. 2006;66:5387–93. doi: 10.1158/0008-5472.CAN-05-3058. [DOI] [PubMed] [Google Scholar]
- 9.O’Hare T, Walters DK, Deininger MW, Druker BJ. AMN107: Tightening the grip of imatinib. Cancer Cell. 2005;7:117–9. doi: 10.1016/j.ccr.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Weisberg E, Manley PW, Breitenstein W, Briiggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG, Griffin JD. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–41. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 11.O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC, Deininger MW, Druker BJ. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–5. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 12.Stover EH, Chen J, Lee BH, Cools J, McDowell E, Adelsperger J, Cullen D, Coburn A, Moore SA, Okabe R, Fabbro D, Manley PW, Griffin JD, Gilliland DG. The small molecule tyrosine kinase inhibitor AMN107 inhibits TEL-PDGFRbeta and FIP1 L1-PDGFRalpha in vitro and in vivo. Blood. 2005;106:3206–13. doi: 10.1182/blood-2005-05-1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, Cortes J, Alland L, Ottmann OG. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–51. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 14.Travis J. Gleevec, chapter two: new leukemia drug aims to overcome resistance. Science. 2004;305:319–21. doi: 10.1126/science.305.5682.319a. [DOI] [PubMed] [Google Scholar]
- 15.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding Imatinib Resistance with a Novel ABL Kinase Inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 16.Gambacorti-Passerini C, Gasser M, Ahmed S, Assouline S, Scapozza L. Abl inhibitor BMS354825 binding mode in Abelson kinase revealed by molecular docking studies. Leukemia. 2005;19:1267–9. doi: 10.1038/sj.leu.2403775. [DOI] [PubMed] [Google Scholar]
- 17.Verstovsek S, Golemovic M, Kantarjian H, Manshouri T, Estrov Z, Manley P, Sun T, Arlinghaus RB, Alland L, Dugan M, Cortes J, Giles F, Beran M. AMN107, a novel aminopyrimidine inhibitor of p190 BCR-ABL activation and of in vitro proliferation of Philadelphia-positive acute lymphoblastic leukemia cells. Cancer. 2005;104:1230–6. doi: 10.1002/cncr.21299. [DOI] [PubMed] [Google Scholar]
- 18.Golemovic M, Verstovsek S, Giles F, Cortes J, Manshouri T, Manley PW, Mestan J, Dugan M, Alland L, Griffin JD. Arlinghaus RB, Sun T, Kantarjian H, Beran M. AMN107, a novel aminopyrimidine inhibitor of Bcr-Abl, has in vitro activity against imatinib-resistant chronic myeloid leukemia. Clin Cancer Res. 2005;11:4941–7. doi: 10.1158/1078-0432.CCR-04-2601. [DOI] [PubMed] [Google Scholar]
- 19.Von Bubnoff N, Manley PW, Mestan J, Sanger J, Peschel C, Duyster J. Bcr-Abl resistance screening predicts a limited spectrum of point mutations to be associated with clinical resistance to the Abl kinase inhibitor nilotinib (AMN107) Blood. 2006;108:1328–33. doi: 10.1182/blood-2005-12-010132. [DOI] [PubMed] [Google Scholar]
- 20.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl protooncogene. Cell. 1991;65:1153–63. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 21.Schwartzberg PL, Stall AM, Hardin JD, Bowdish KS, Humaran T, Boast S, Harbison ML, Robertson EJ, Goff SP. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell. 1991;65:1165–75. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 22.Dietz AB, Souan L, Knutson GJ, Bulur PA, Litzow MR, Vuk-Pavlovic S. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood. 2004;104:1094–9. doi: 10.1182/blood-2003-12-4266. [DOI] [PubMed] [Google Scholar]
- 23.Cwynarski K, Laylor R, Macchiarulo E, Goldman J, Lombardi G, Melo JV, Dazzi F. Imatinib inhibits the activation and proliferation of normal T lymphocytes in vitro. Leukemia. 2004;18:1332–9. doi: 10.1038/sj.leu.2403401. [DOI] [PubMed] [Google Scholar]
- 24.Seggewiss R, Loré K, Greiner E, Magnusson MK, Price DA, Douek DC, Dunbar CE, Wiestner A. Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood. 2005;105:2473–9. doi: 10.1182/blood-2004-07-2527. [DOI] [PubMed] [Google Scholar]
- 25.Boissel N, Rousselot P, Raffoux E, Cayuela JM, Soulier J, Mooney N, Charron D, Dombret H, Toubert A, Rea D. Imatinib mesylate minimally affects bcrabl+ and normal monocyte-derived dendritic cells but strongly inhibits T cell expansion despite reciprocal dendritic cell-T cell activation. J Leukoc Biol. 2006;79:747–56. doi: 10.1189/jlb.0705419. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Schmitt A, Chen B, Rojewski M, Ringhoffer M, von Harsdorf S, Greiner J, Guillaume P, Döhner H, Bunjes D, Schmitt M. Imatinib impairs CD8+ T lymphocytes specifically directed against the leukemia-associated antigen RHAMM/CD168 in vitro. Cancer Immunol Immunother. 2007;56:849–61. doi: 10.1007/s00262-006-0232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent anti-proliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109:4016–9. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- 28.Greiner J, Li L, Ringhoffer M, Barth TF, Giannopoulos K, Guillaume P, Ritter G, Wiesneth M, Döhner H, Schmitt M. Identification and characterization of epitopes of the receptor for hyaluronic acid-mediated motility (RHAMM/CD168) recognized by CD8+ T cells of HLA-A2-positive patients with acute myeloid leukemia. Blood. 2005;106:938–45. doi: 10.1182/blood-2004-12-4787. [DOI] [PubMed] [Google Scholar]
- 29.Greiner J, Schmitt M, Li L, Giannopoulos K, Bosch K, Schmitt A, Döhner K, Schlenk RF, Pollack JR, Dohner H, Bullinger L. Expression of tumor-associated antigens in acute myeloid leukemia: Implications for specific immunotherapeutic approaches. Blood. 2006;108:4109–17. doi: 10.1182/blood-2006-01-023127. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Reinhardt P, Schmitt A, Barth TF, Greiner J, Ringhoffer M, Dohner H, Wiesneth M, Schmitt M. Dendritic cells generated from acute myeloid leukemia (AML) maintain the expression of immunogenic leukemia associated antigens. Cancer Immunol Immunother. 2005;54:685–93. doi: 10.1007/s00262-004-0631-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt M, Schmitt A, Reinhardt P, Thess B, Manfras B, Lindhofer H, Riechelmann H, Wiesneth M, Gronau S. Opsonization with a trifunctional bispecific (alphaCD3 x alphaEpCAM) antibody results in efficient lysis in vitro and in vivo of EpCAM positive tumor cells by cytotoxic T lymphocytes. IntJ Oncol. 2004;25:841–8. [PubMed] [Google Scholar]
- 32.Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, Donehower LA. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–42. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lyons AB, Hasbold J, Hodgkin PD. Flow cytometric analysis of cell division history using dilution of carboxyfluoresceindiacetate succinimidyl ester, a stably integrated fluorescent probe. Methods in Cell Biol. 2001;63:375–98. doi: 10.1016/s0091-679x(01)63021-8. [DOI] [PubMed] [Google Scholar]
- 34.Singer AL, Koretzky GA. Control of T cell function by positive and negative regulators. Science. 2002;296:1639–40. doi: 10.1126/science.1071551. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi N, Miura I, Saitoh K, Miura AB. Lineage involvement of stem cells bearing the Philadelphia chromosome in chronic myeloid leukemia in the chronic phase as shown by a combination of fluorescence-activated cell sorting and fluorescence in situ hybridization. Blood. 1998;92:4758–63. [PubMed] [Google Scholar]
- 36.Garicochea B, Chase A, Lazaridou A, Goldman JM. T lymphocytes in chronic myelogenous leukaemia (CML): no evidence of the BCR/ABL fusion gene detected by fluorescence in situ hybridization in 14 patients. Leukemia. 1994;8:1197–201. [PubMed] [Google Scholar]
- 37.Cho EK, Heo DS, Seol JG, Seo EJ, Chi HS, Kim ES, Lee YY, Kim BK, Kim NK. Ontogeny of natural killer cells and T cells by analysis of BCR-ABL rearrangement from patients with chronic myelogenous leukaemia. Br J Haeinatol. 2000;111:216–22. doi: 10.1046/j.1365-2141.2000.02341.x. [DOI] [PubMed] [Google Scholar]
- 38.Kantarjian HM, Ottmann O, Cortes J, Wassmann B, Jones D, Hochhaus A, Alland L, Dugan M, Albitar M, Giles F. AMN107, a novel aminopyrimidine inhibitor of Bcr-Abl, has significant activity in imatinib-resistant bcr-abl positive chronic myeloid leukemia (CML) American Society of Clinical Oncology. 2005;23:3014. [Google Scholar]
- 39.Brenchley JM, Douek DC, Ambrozak DR, Chatterji M, Betts MR, Davis LS, Koup RA. Expansion of activated human naive T-cells precedes effector function. Clin Exp Immunol. 2002;130:432–40. doi: 10.1046/j.1365-2249.2002.02015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayward AR, Kurnick JT, Clarke DR. T cell growth factor-enhanced PHA response of human thymus cells: requirement for T3+ cells. J Immunol. 1981;127:2079–82. [PubMed] [Google Scholar]