

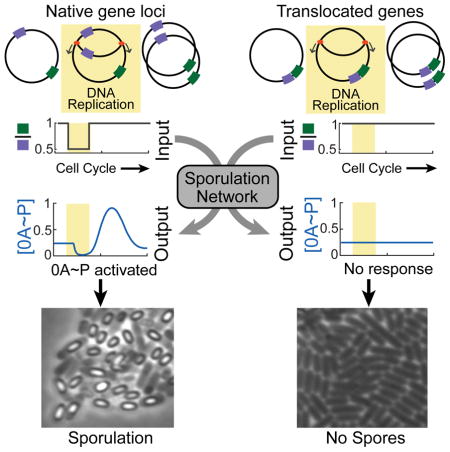
Summary
Genes encoding proteins in a common regulatory network are frequently located close to one another on the chromosome to facilitate co-regulation or couple gene expression to growth rate. Contrasting with these observations, here we demonstrate a functional role for the arrangement of Bacillus subtilis sporulation network genes on opposite sides of the chromosome. We show that the arrangement of two sporulation network genes, one located close to the origin, the other close to the terminus leads to a transient gene dosage imbalance during chromosome replication. This imbalance is detected by the sporulation network to produce cell-cycle coordinated pulses of the sporulation master regulator Spo0A~P. This pulsed response allows cells to decide between sporulation and continued vegetative growth during each cell-cycle spent in starvation. The simplicity of this coordination mechanism suggests that it may be widely applicable in a variety of gene regulatory and stress-response settings.
Graphical Abstract
Introduction
A recurring theme in recent studies of cellular differentiation networks is that these networks respond to stimuli in a highly dynamic, time-dependent manner (Kuchina et al., 2011a; Kuchina et al., 2011b). One of the main roles of this dynamical response is to coordinate the expression of genes in the differentiation program with cell-cycle events such as DNA replication and cell division (Doncic et al., 2011; Toettcher et al., 2009; Veening et al., 2009). Lack of coordination between the differentiation program and the cell-cycle can often result in incomplete/abortive differentiation or even cell death. As a result, uncovering the system-level mechanisms of cell-cycle coordinated differentiation is essential to understand this process. Keeping this in mind, we investigated sporulation in soil bacterium Bacillus subtilis to uncover how this differentiation program is coordinated with the cell-cycle.
Sporulation is the last resort of starving B. subtilis cells. In response to starvation, B. subtilis cells cease vegetative growth and asymmetrically divide to initiate a multistage program that produces stress-resistant and metabolically inert spores (Figure 1A) (Higgins and Dworkin, 2012). At the molecular level, the sporulation program is controlled by the master regulator Spo0A (0A) which is active in its phosphorylated (0A~P) form (Errington, 2003). Expression of many downstream sporulation program genes is controlled by 0A~P, and it has been shown that a threshold level of 0A~P commits cells to sporulation (Eswaramoorthy et al., 2010; Fujita and Losick, 2005; Narula et al., 2012). The activation of 0A itself is regulated by a complex network known as the sporulation phosphorelay (Burbulys et al., 1991). Since cells need two complete chromosomes to produce a viable spore (Hauser and Errington, 1995), the dynamics of 0A activation must be temporally coordinated with the completion of DNA replication. Previous studies have suggested that Sda, an inhibitor of the phosphorelay kinase that activates 0A may be responsible for coordinating 0A activation with DNA replication (Veening et al., 2009). However, it has been recently shown that deletion of sda does not completely abolish cell-cycle coupling of 0A activation (Levine et al., 2012). As a result, the central question of how the phosphorelay coordinates 0A activation with the cell-cycle remains unaddressed.
Figure 1.

Coordination of the sporulation response with the cell-cycle in B. subtilis.
A. In starvation, B. subtilis make cell-fate decision either to divide medially continuing vegetative growth or to divide asymmetrically committing to sporulation. The decision is based on the level of phosphorylated master regulator Spo0A (0A~P). 0A~P exceeding a threshold commit cells to sporulation whereas lower levels allows cells to continue growth. The decision must be made after the completion of DNA replication phase (yellow bar) since two complete chromosomes are needed to produce a viable spore. B–C Single cell time-lapse microscopy using a P0A-cfp reporter for 0A~P with vertical dashed lines indicate cell divisions and yellow shaded regions indicate periods of DNA replication (detected by the presence of DnaN-YFP foci). (B) Expression level of P0A-cfp increases in a pulsatile fashion over multiple cell-cycles in starvation media. (C) Its promoter activity (defined as production rate, an indicator of 0A~P level) also shows pulses of increasing amplitude over multiple generations. Note that DNA replication is sometimes initiated just before cell division. For each cell cycle, we can determine the time from birth to end of DNA replication (Tr) and time from birth to peak P0A-cfp Promoter activity (Tp) represent respectively. D. Measurements of time from birth to end of DNA replication (Tr) and time from birth to peak P0A-cfp Promoter activity (Tp) show that Tp>Tr for the vast majority of the cell-cycles implying that 0A activity peaks occurs after DNA replication is complete.
Here we show that the chromosomal arrangement of two phosphorelay genes plays a critical role in coupling 0A activation to DNA replication. The location of these genes on opposite sides of the chromosome – one located close to the origin of replication, the other close to the terminus, leads to a transient imbalance in their gene dosage during chromosome replication. Combined with a delayed negative feedback loop in the phosphorelay this transient imbalance results in the pulsatile activation of 0A that is responsible for coordinating the commitment to sporulation with the cell-cycle.
Results
0A activity pulses follow the completion of DNA replication
To understand the dynamics of sporulation network response, we employed time-lapse microscopy and simultaneously tracked 0A activity and DNA replication in single B. subtilis cells (see Methods). We used fluorescent reporters to measure gene expression from 0A~P-regulated promoters for spo0A and spo0F (P0A and P0F). In addition, we fluorescently tagged a replisome component, DnaN, so that periods of DNA replication could be detected by the presence of fluorescent DnaN foci (Su'etsugu and Errington, 2011). In agreement with previous studies (Levine et al., 2012; Veening et al., 2008), we found that in these conditions cells do not sporulate immediately upon exposure to starvation, but rather complete multiple divisions before finally producing spores. Measurements of the expression level of P0A-cfp (Figure 1B) and the cell growth rate (inferred from cell elongation rate) enabled us to compute 0A activity defined as production rate of the reporter protein (Figure 1C). The results revealed that during the multi-cycle progression to spore formation a single pulse of 0A activity is produced every cell-cycle in starvation conditions (Figure 1C). Similar pulsing was observed in the production of fluorescent reporters of other 0A~P-regulated promoters such as P0F (Figure S1). We measured the pulse timing during the cell-cycle (Figure 1C) and found that 0A activity pulses always follow the completion of DNA replication (Figure 1D).
This type of 0A activity pulsing has also been reported by other recent studies (Kuchina et al., 2011b; Levine et al., 2012; Veening et al., 2009). Veening and coworkers reported that this pulsatile response is the result of pulsing of Sda, an inhibitor of the kinase that activates 0A (Veening et al., 2009). However a more recent study (Levine et al., 2012), has shown that Sda might not be the only mechanism behind 0A activity pulsing since deletion of sda does not completely abolish pulsing. As a result, the mechanism underlying this pulsatile response remains unclear.
A hidden negative feedback loop in the phosphorelay
Our first goal was to uncover the mechanism underlying the pulsing of 0A~P. To this end, we built a detailed mathematical model of the sporulation phosphorelay network (Figure 2A) that controls 0A production and phosphorylation (see Supplemental Information). This network consists of multiple histidine kinases KinA-E, two phosphotransferases Spo0F (0F) and Spo0B (0B), and the master regulator 0A (Hoch, 1993). Among the five kinases, the cytoplasmic protein KinA is the major sporulation kinase (in our conditions-Figure S2A) and therefore we have only included this kinase in our model (Eswaramoorthy et al., 2010; Perego et al., 1989). Upon nutrient limitation, KinA auto-phosphorylates and indirectly transfers the phosphate group to 0A via the intermediate proteins 0F and 0B (Figure 2A) (Burbulys et al., 1991). The expression levels of kinA, 0F and 0A are regulated by 0A~P via direct and indirect transcriptional feedback loops. These post-translational and transcriptional interactions were described using appropriate mass-action and Hill-function type rate laws to build the model of the phosphorelay network (see Supplemental Information and Table S1).
Figure 2.

Substrate inhibition of 0F by KinA produces a negative feedback in the phosphorelay.
A. Network diagram of the sporulation phosphorelay network that controls the activity of the master regulator Spo0A (0A). The phosphorelay includes both post-translational and transcriptional regulatory interactions. Post-translationally, the kinases KinA-E (only KinA is shown) transfer phosphoryl groups to the master regulator 0A via the two phosphotransferases Spo0B (0B) and Spo0F (0F). Transcriptionally, 0A~P controls the expression of kinA, 0F and 0A forming multiple transcriptional feedback loops. Our model also includes a substrate inhibition interaction (red blunted arrow) whereby excess 0F can bind to un-phosphorylated KinA and block its auto-phosphorylation. This substrate inhibition creates a negative feedback loop wherein 0A~P activates 0F expression and 0F inhibits 0A activation by inhibiting KinA. B. Mathematical model predicts that, for a phosphorelay with substrate-inhibition of KinA by 0F (blue curve), 0A promoter activity is a non-monotonic functions of 0F concentrations and decrease ultrasensitively for [0F]>5μM. In contrast, for a phosphorelay without substrate-inhibition (orange curve), 0A promoter activity monotonically increase to saturated value. C. Predicted non-monotonic dependence of 0A activity on 0F levels is confirmed by engineering inducible 0F strain, i0FamyE (Δ0F; amyE::Phsp-0F) and measuring maximum 0A promoter activity in the at different levels of 0F induction. Gray empty circles show maximum P0A promoter activity levels achieved by individual cell lineages over 25 hours in starvation conditions at each IPTG concentration. Blue filled circles and error-bars indicate the mean and standard deviations of these measurements at each IPTG concentration. Maximum P0A promoter activity decreases at high 0F expression levels (IPTG>10μM) in agreement with the substrate-inhibition effect of 0F overexpression.
Analysis of simulations of this model showed that an increase in 0F levels leads to higher levels of 0A~P and increased expression of 0A targets (Figure 2B). However, this result contradicts previous in vivo studies, which indicate that 0F overexpression inhibits sporulation (Chapman and Piggot, 1987; Chastanet et al., 2010; Sen et al., 2011). To resolve this discrepancy, we made a change to the conventionally assumed phosphorelay architecture (Figure 2A without red arrow) and incorporated substrate inhibition of KinA by 0F into the phosphorelay model (red arrow, Figure 2A). This substrate inhibition effect was based on an in vitro study demonstrating inhibition of phosphotransfer by excess 0F (Grimshaw et al., 1998). The resulting model predicted a non-monotonic dependence of 0A~P on 0F as high 0F concentrations blocked 0A activation (Figure 2B). We verified this inhibition in an engineered strain, i0FamyE, in which the native copy of 0F was replaced by a copy of 0F expressed from an IPTG-inducible promoter (Phyperspank, or Phsp) at the amyE locus. The results indicate inhibition of 0A activity by excess of 0F induction (Figures 2C and S2A).
Since 0F expression is activated by 0A~P, the substrate inhibition of KinA by 0F results in the negative feedback loop in the phosphorelay. This feature is especially significant since negative feedback loops are known to produce adaptation-like pulsatile responses (Ma et al., 2009). In addition, the inhibition of KinA by 0F made the flux through the phosphorelay very sensitive to the ratio of KinA and 0F concentrations (Figure S2BC). As a result, any perturbation of the relative KinA/0F ratio can force the negative feedback loop to produce a pulsed 0A~P response.
Chromosomal arrangement of phosphorelay genes provides a pulse triggering perturbation
The sensitivity of the phosphorelay response to the KinA/0F ratio in our model suggested that chromosomal arrangement of kinA and 0F may affect 0A~P dynamics. On the B. subtilis chromosome 0F is located close to the origin (326°-oriC proximal) and kinA near the terminus (126°-ter proximal) of DNA replication (Figure 3A). As a result, replication of 0F precedes that of kinA and each DNA replication cycle produces a transient decrease in the kinA:0F gene dosage ratio (Figure 3A). In light of this, we proposed that this arrangement of 0F and kinA genes on the chromosome might couple the phosphorelay output to DNA replication.
Figure 3.
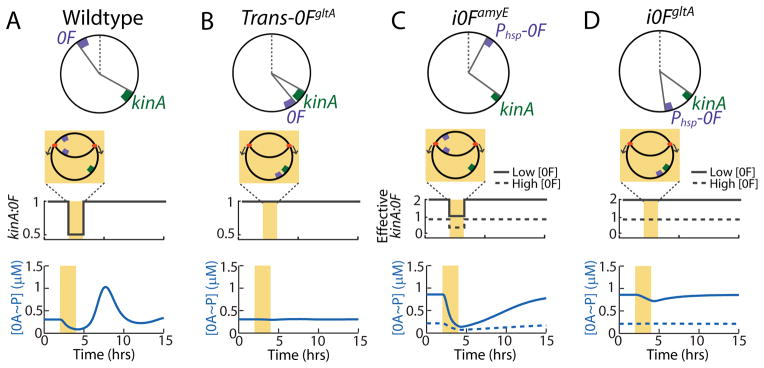
Mathematical model identifies the mechanism of 0A~P pulsing and its necessary conditions.
Top panels in (A–D) show chromosomal arrangements of 0F and kinA in (A) Wildtype (WT) B. subtilis and engineered strains: (B) Trans-0FgltA, (C) i0FamyE, (D) i0FgltA. 0F is located close to the origin of replication in WT and i0FamyE strains and close to the terminus in the Trans-0FgltA and i0FgltA. kinA is located close to the terminus in all strains. Note that 0F is expressed from the IPTG-inducible Phsp promoter, rather than the native 0A~P regulated P0F promoter in the inducible i0FamyE and i0FgltA strains. Middle panels in (A–D) show changes in kinA:0F gene dosage ratio in the WT and engineered strains. In the inducible i0FamyE and i0FgltA strains, the effective kinA:0F gene dosage depends on whether the level of 0F expression from the IPTG-inducible Phsp promoter is low (solid line) or high (dashed line). Bottom panels in (A–D) show model predictions for the response of 0A~P levels to the changes in kinA:0F ratio in WT and engineered strains. Model simulations show that the transient decrease in kinA:0F during DNA replication (yellow bar) in WT (A) inhibits the phosphorelay phosphate flux, thereby causing a decrease in 0A~P. Once DNA replication is complete, the phosphorelay produces an overshoot of 0A~P before returning to the steady state resulting in a 0A~P pulse. Model results also predict that that elimination of the transient decrease of kinA:0F in the Trans-0FgltA strain (B) abolishes 0A~P pulsing and instead the system stays at steady state. In the i0FamyE strain (C), the transient decrease in effective kinA:0F causes a decrease in 0A~P but 0A~P does not overshoot before returning to the steady state and there is no pulse. depends on the level of 0F expression. Simulations also show that elimination of the transient decrease of kinA:0F in the i0FgltA strain (D) abolishes 0A~P pulsing and instead the system stays at steady state. This 0A~P response level in i0F strains depends on whether 0F expression from the IPTG-inducible Phsp promoter is low (solid line) or high (dashed line). These results show that 0A~P pulsing is triggered by DNA replication and that both the kinA-0F chromosomal arrangement and 0A~P-0F negative feedback are essential for 0A~P pulsing.
Including a DNA replication window in our simulations, we found that this imbalance in kinA and 0F expression inhibits the phosphorelay flux and results in a decrease in 0A~P during replication (Figure 3A). Once DNA replication is completed and the kinA:0F ratio returns to one, the delayed negative feedback loop comprised of transcriptional feedback from 0A~P to 0F and the postulated substrate inhibition of KinA by 0F produces an overshoot of 0A~P above its steady state (Figure 3A). These overshoots are manifested as pulses of 0A activity occurring once per cell cycle. Thus, our model explains both the pulsing mechanism and the observed correlation between DNA replication and timing of 0A pulses (compare Figures 1C and 3A). Moreover comparison of the chromosomal locations of kinA and 0F in 45 different species of spore-forming bacteria that have both these genes showed little variation in their positions relative to the chromosomal origin (Figure S2DE and Table S2), which suggests that the proposed relative gene dosage pulsing mechanism is evolutionarily conserved.
To uncover the essential design features necessary for the pulsatile 0A~P dynamics, we tested the model response to specific network perturbations. First, we tested the effect of translocating 0F so that both kinA and 0F are close to the chromosome terminus. Simulations of this engineered Trans-0FgltA strain (Figures 3B and S3) showed that the translocation of 0F close to the terminus eliminated the transient kinA:0F decrease during DNA replication. Consequently, simulations of this modified strain showed no 0A~P pulsing and instead the system remained at steady state. In the same fashion, our model shows that elimination of the transient kinA:0F ratio decrease by translocation of kinA close to the chromosomal origin (Trans-kinAamyE) would eliminate 0A~P pulsing (Figure S3).
Next, we investigated the role of the negative feedback loop between 0A~P and 0F by testing the effect of assuming that 0F is expressed from an IPTG-inducible Phsp promoter, rather than the native 0A~P regulated P0F promoter. We tested two such inducible engineered strains: i0FamyE (Phsp-0F located close to the origin of replication - Figure 3C) and i0FgltA (Phsp-0F located close to the terminus - Figure 3D). In these inducible strains, the effective kinA:0F gene dosage was assumed to depend on the level of 0F expression. Model simulations showed that in the i0FamyE strain (Figure 3C), there is a transient decrease in effective kinA:0F. This decrease inhibits the phosphorelay phosphate flux and causes a decrease in 0A~P. However 0A~P does not overshoot before returning to the steady state and there is no pulse (Figure 3C), unlike the results for WT (Figure 3A). Simulations further showed that transient decrease of kinA:0F is eliminated by 0F translocation in the i0FgltA strain (Figure 3D) and that 0A~P stays at steady state in this case. Simulations also predicted that the 0A~P response in these inducible strains depends on the level of 0F, specifically 0A~P levels decrease with increasing 0F expression in both i0FamyE and i0FgltA (Figure 3CD).
Thus far our phosphorelay model only included one kinase, KinA, as its deletion (ΔkinA) has most drastic effect on sporulation efficiency under most sporulation conditions (Figure S2A) (LeDeaux et al., 1995). However, in certain conditions (S7 minimal media), KinB is known to act as the major kinase. To explore the role of KinB we extended our model to include KinB. Notably the location of kinB is about 60 degrees further from oriC than 0F (Figure S3A). As a result, similar to kinA there is a transient period during which gene dosage of 0F exceeds that of kinB resulting in the inhibition of kinase and reduction in 0A activity. According to our mathematical model, upon kinB replication relief of this inhibition can be sufficient to trigger a 0A~P pulse (Figure S3E). The lower amplitudes of 0A~P pulses triggered by KinB relative to KinA are consistent with the results of Levine et al. (Levine et al., 2012) who reported 0A-activity pulsing in ΔkinA strain. This prediction also explains the reduced sporulation efficiency of this strain (LeDeaux et al., 1995). Overall these results show that KinB can partially compensate for KinA and trigger pulsing of 0A activity by relying on the same design features as the kinA:0F pulsing mechanism.
Based on these results we constructed a simplified model of the network that can capture the negative feedback based 0A~P pulsing mechanism. This model used KinA concentration as a time-varying signal and had just three variables: 0A~P, 0F and an auxiliary variable to account for a delay in transcriptional activation of 0F by 0A~P (Figure S4A and Supplemental Information). This minimal model also demonstrated that a change in the kinA:0F ratio together with ultrasensitive repression of 0A~P by 0F and delayed activation of 0F transcription are necessary and sufficient to produce the observed pulses of 0A~P (Fig. S4BC). Elimination of either of these ingredients destroyed the pulsing (Figure S4D–G). Notably, introduction of the positive feedback into the model to mimic the effect of transcriptional activation of kinA and spo0A by 0A~P enhanced the ultrasensitivity of the repression of 0A phosphorylation by 0F and increased the pulse amplitudes resulting from changes in KinA:0F dosage ratio (Figure S4D–G). However in agreement with the observations of Levine et al, this positive feedback is not essential for pulsing (Levine et al., 2012).
Altogether our model results suggest that two design features of the phosphorelay are crucial for the pulsatile response of 0A~P during starvation: (i) negative feedback between 0A~P and 0F and (ii) transient gene dosage imbalance between kinA and 0F resulting from the chromosomal arrangement of these genes. Consequently, our model predicts that disrupting these key features would abolish 0A~P pulsing and thereby affect sporulation (Figure 3B–D, S3 and S4).
Experimental tests confirm the role of gene-dose imbalance and negative feedback in 0A~P pulsing
We tested our modeling predictions by engineering two sets of B. subtilis strains. The first set of strains was engineered to examine how the chromosomal arrangement of kinA and 0F and the resulting transient gene dosage imbalance affects 0A~P pulsing (Figure 4A–E). In the first strain, Trans-0FgltA (Figure 4B), we eliminated the transient imbalance in the kinA:0F ratio by translocation of the 0F gene to the gltA locus close to the terminus. As a control, we engineered a second strain (Trans-0FamyE; Figure 4C) by moving the 0F gene to amyE locus near the origin so that the kinA:0F imbalance is retained despite the translocation. Because the entire 0F gene with the upstream region containing regulatory sequences was translocated in both strains, only the relative gene dosage during replication was perturbed whereas the negative feedback regulation remained intact. As predicted by the model (Figure 3B), pulsing was abolished in the Trans-0FgltA strain but not in the Trans-0FamyE, which exhibited pulsing similar to the WT cells (Figure 4F–H). The lack of pulsing in the Trans-0FgltA resulted solely from the change in chromosomal position of 0F and not from reduced 0F expression. Specifically, we measured the activity of P0F at amyE and gltA loci and found that it displays the same pulsatile behavior with similar expression levels (Figure S1).
Figure 4.

Experimental observation confirm that 0A~P pulsing depends on kinA-0F chromosomal arrangement and transcriptional feedback from 0A~P to 0F negative feedback. A–E Chromosomal arrangements of 0F and kinA in Wildtype B. subtilis (A), Trans-0FgltA (B), Trans-0FamyE (C), Trans-kinAgltA (D) and Trans-kinAamyE (E) strains. Blue and orange bars mark strains that have transient kinA:0F imbalance and negative feedback respectively. F–J Measurements of P0A-yfp activity in single cells during starvation. 0A~P pulses seen in Wildtype B. subtilis cells (F) are abolished by the translocation of 0F to the gltA locus in the Trans-0FgltA strain (G) but not by the translocation of 0F to the amyE locus in the Trans-0FamyE strain (H). 0A~P pulses are also abolished by the translocation of kinA to the amyE locus in the Trans-kinAgltA strain (J) but not by the translocation of kinA to the gltA locus in the Trans-0FgltA strain (I). Note that P0A-yfp promoter activity level does not increase in the Trans-0FgltA strain. Vertical dashed lines indicate cell divisions. K–O Chromosomal arrangements of 0F and kinA in iTrans-0F (K), i0FamyE (L, N) and i0FgltA strains (M, O). Note that both i0F strains lack the 0A~P-0F negative feedback. P–T Measurements of P0A-yfp activity in single cells during starvation. Addition of an IPTG inducible copy of 0F near the origin in the iTrans-0F strain (P) recovers the transient kinA:0F imbalance lost in Trans-0FgltA strain and rescues the 0A~P pulses seen in wildtype B. subtilis cells. 0A activity pulsing is greatly decreased in the i0FamyE (Q, S) and i0FgltA strains (R, T) which lack the negative feedback. 0A activity in the i0FamyE (Q) strain fluctuates due to transient changes in kinA:Phsp-0F but does not pulse (Figure S5). If 0F expression is low (at 5μM IPTG; Q, R), both i0F strains accumulate high 0A activity levels. High level expression of 0F (at 20μM IPTG; S, T) blocks 0A activation in both i0F strains similar to Trans-0FgltA.
Following the same line of thought, we constructed two Trans-kinA strains by chromosomally translocating kinA to perturb the transient kinA:0F imbalance. In Trans-kinAgltA (Figure 4D) and Trans-kinAamyE (Figure 4E) we moved the kinA gene to gltA locus near the terminus and the amyE locus near the origin respectively. Translocation of kinA close to the origin in Trans-kinAamyE eliminated the transient imbalance in the kinA:0F ratio whereas the kinA:0F imbalance was retained in Trans-kinAgltA. Similar to the Trans-0F strains the negative feedback regulation remained intact in the Trans-kinA strains. As predicted by our model, Trans-kinAgltA exhibited pulsing similar to the WT cells whereas pulsing was abolished in the Trans-kinAamyE strain (Figure 4IJ). However in contrast to the non-pulsing Trans-0FgltA strain, the non-pulsing Trans-kinAamyE displayed a high level of 0A activity. This difference can be attributed to the compensatory effect of other kinases (such as KinB) or to the increased expression of kinA from the amyE locus. Together, the results from these translocation experiments confirm the prediction that transient kinA:0F gene dosage imbalance is necessary for 0A pulse generation.
To further establish the role of transient imbalance in 0F to kinA expression rates, we constructed a rescue strain, iTrans-0F (Figure 4K). In addition to 0F translocation to the terminus (Trans-0FgltA), we integrated an additional IPTG-inducible copy of 0F close to the chromosome origin to recover the transient imbalance in 0F to kinA expression rates during replication. In the absence of IPTG, this strain acted like the non-responsive Trans-0FgltA strain and showed no 0A~P pulsing. However, at 5μM IPTG the sporulation in the iTrans-0F strain was restored similar to the WT strain (Figure 4P). Thus, we concluded that the transient imbalance in kinA and 0F expression resulting from their chromosomal locations acts as an essential trigger for 0A~P pulses.
To test the role of negative feedback between 0A~P and 0F in pulse generation, we created a second set of strains, i0FamyE and i0FgltA (Figure 4L-O), in which 0F is expressed from the IPTG-inducible Phsp promoter, rather than the native 0A~P regulated promoter. According to our simulations (Figure 3CD), even though 0A~P pulsing in these strains would be disrupted, cells could still accumulate high levels of 0A~P if 0F expression was below the inhibitory range determined in Figure 2C. Indeed, we found that in i0FamyE and i0FgltA strains, the 0A promoter activity increased gradually over time to levels comparable to that in WT at 5μM IPTG (low 0F expression; Figure 4QR). In contrast, at 20μM IPTG (high 0F expression; Figure 4ST) there was no significant increase in 0A promoter. We also found that, consistent with our model predictions (Figure 3C), 0A promoter activity in i0FamyE fluctuates (Figure 4M) due to transient changes in kinA:0F expression ratios in this strain. However, these fluctuations did not resemble the adaptation type pulsatile responses of the WT and Trans-0FamyE strains. In fact, unlike the WT and Trans-0FamyE strains both i0FamyE and i0FgltA strains showed no statistically significant difference between the peak 0A activity during a cell-cycle and the 0A activity at the end of the cell-cycle (Figure S5). This led us to conclude that 0A activity does not pulse in these strains, thereby confirming our prediction that the negative feedback in the phosphorelay is essential for producing pulses of 0A~P in response to starvation.
Lack of 0A~P pulsing leads to sporulation defects
Notably, the lack of 0A~P pulsing in the kinA translocation strain Trans-kinAamyE and the inducible i0FamyE and i0FgltA strains (Figure S5) did not prevent them from producing spores (for i0F strains this is the case only at 5μM IPTG when 0F expression was low; Figure 5A). Thus, 0A~P pulsing was not essential for sporulation, but we hypothesized that it was necessary for ensuring that the threshold level of 0A~P activity required for asymmetric septation or σF activation would only be reached in the cells with two complete chromosomes. Accordingly, we predicted that strains with non-pulsatile accumulation of 0A~P would exhibit an increased frequency of defective sporulation phenotypes resulting from untimely 0A activation.
Figure 5.
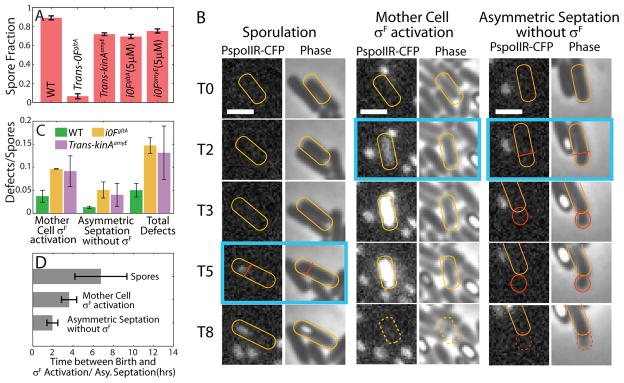
Loss of coordination of sporulation program with DNA replication in non-pulsing strains could lead to sporulation defects. A. Fraction of cells that have formed spores by 25hrs into starvation in different strains. Bars and error-bars respectively show the means and standard deviation of spore fraction calculated using 3 independent measurements for each condition.. B. Phase-contrast and fluorescence microscopy (PspoIIR-cfp) images from a time-lapse experiment showing the difference in timing of SpoIIR activation/Asymmetric septation in sporulation and sporulation defects. T0 represents the time of birth for each indicated cell (yellow outline). T2, T3 etc. indicate time after birth in hours. Time-point of SpoIIR activation/Asymmetric septation in each case is marked by blue box. Asymmetric septation and σF activation happen late in the cell-cycle (T5) during normal sporulation as compared to the sporulation defect cases. Early activation of σF in the whole cell at T2 results in cell death. Early asymmetric septation at T2 produce a small daughter cell (red outline) which dies without activating σF in the forespore. Scale Bars: 2μm. C. Quantification of number of defects per spore produced over 30hrs in starvation conditions by the pulsing WT strain (green bars) and the non-pulsing strains i0FgltA (yellow bars) and Trans-kinAamyE (purple bars). Error-bars indicate the standard deviation of 3 independent measurements. The defects/spore ratio is significantly higher for non-pulsing strains. D. Time difference between birth and SpoIIR (a σF reporter) activation/Asymmetric septation in cell-cycles that produce spores and those that end in lysis due to sporulation defects. SpoIIR activation/Asymmetric septation happens significantly earlier in cell-cycles that end sporulation defects.
To determine whether pulsatile 0A activation plays a role in preventing faulty sporulation, we counted the frequency of defects in the pulsing WT and non-pulsing Trans-kinAamyE and i0FgltA strains. We specifically focused on two types of sporulation defects (Figure 5B): (i) Asymmetric septation without activation of σF in the forespore and (ii) Activation of σF in the mother cell before asymmetric septation. We found that asymmetric septation without activation of σF causes cells to bud off a small daughter cell that lacks DNA and dies soon after division (Figure 5B). On the other hand, activation of σF in the mother cell causes cell death (Figure 5B). Thus, both types of defects affect the ability of cells to efficiently produce spores. Counting the number of such abnormalities, we found that the frequency of defects per spore produced over 30hrs in starvation conditions was about three-fold higher in the i0FgltA strain (14.7% ± 1.7%; 3 independent measurements >250 spores each) and Trans-kinAamyE strain (13.1% ± 5.8%; 3 independent measurements >100 spores each) relative to the WT strain (5.0% ± 1.5%; 3 independent measurements, >250 spores each) (Figure 5C). Therefore, we find that 0A~P pulsing plays a key role in preventing defective sporulation.
Next we examined whether the higher frequency of defects/spore ratio in the non-pulsing i0FgltA strain results from lack of proper coordination of sporulation with the cell-cycle. To test this idea, we used time-lapse microscopy data for the i0FgltA strain to compute the time of cell-fate decisions both in cell-cycles that successfully produce spores and those that end in defective sporulation. The time of cell-fate decision was defined as the time from the start of the cell-cycle to the time of PspoIIR-cfp (a σF reporter) activation in the cases of normal sporulation and the mother cell σF activation defect. For the defect of asymmetric septation without σF activation, the ime of cell-fate decision was defined as the time from the start of the cell-cycle to the time of asymmetric septation. As shown in Figure 5B and 5D, cell-cycles that end in sporulation defects reach cell-fate decisions early in the cell-cycle (2–3hrs after the start of the cell-cycle). We note that unlike rich medium conditions, cell-cycle durations in starvation conditions are typically 5–6 hours long. As DNA replication is incomplete early in the cell-cycle, these early cell-fate decisions appear to arise from the attempt to execute the sporulation program without two complete chromosomes. In contrast, in cell-cycles that successfully produce a spore, the timing of cell-fate decisions is typically late in the cell-cycle (>4hrs after the start of the cell-cycle; Figure 5B, D), after the completion of DNA replication. Thus we concluded that activation of 0A and commitment to sporulation too early in the cell-cycle, before the completion of DNA replication, is responsible for both sporulation defects. Moreover, since these defects occur at a higher frequency in the non-pulsing i0FgltA strain, these results show that the 0A~P pulsing plays a key role in preventing sporulation defects because of its ability to ensure proper coordination of the sporulation program with DNA replication.
Discussion
Taken together, our results reveal how B. subtilis cells are able to couple cell-fate decisions to DNA replication. Using an ultrasensitive, delayed negative feedback loop to detect the transient imbalance of gene dosage resulting from directional chromosome replication allows B. subtilis to use DNA replication itself as the trigger for 0A activation, thereby ensuring that these two do not temporally conflict with each other. Moreover, the pulsatile activation of 0A during every cell cycle offers B. subtilis cells an opportunity to evaluate their starvation level and decide between sporulation and continued vegetative growth on a cell-cycle by cell-cycle basis.
One of the key design features that underlies the pulsatile 0A~P dynamics appears to be a negative feedback loop, which are known to be one of the few network motifs capable of generating adaptation-like pulsatile responses (Ma et al., 2009). The crucial component that creates this negative feedback is the substrate inhibition of KinA by 0F through the formation of a dead-end complex that blocks KinA autophosphorylation. This substrate inhibition effect has been demonstrated previously (Grimshaw et al., 1998), but has received little attention in mathematical modeling studies. This effect is however essential for explaining the inhibitory effect of 0F overexpression on sporulation (Chapman and Piggot, 1987; Chastanet et al., 2010; Sen et al., 2011). Our results demonstrate that this negative feedback based on the substrate inhibition of KinA by 0F plays a critical role in coupling 0A~P pulsing to DNA replication. Alternative explanations for 0A~P pulsing suggested by earlier studies invoke either the 0A~P-AbrB-Spo0E negative feedback loop (Schultz et al., 2009) or the inhibition of KinA by Sda (Veening et al., 2009). However, a pulsing mechanism based on the 0A~P-AbrB-Spo0E negative feedback loop is unlikely since it cannot explain our observations of the cell-cycle coupling of pulses (Figure 1D). In addition, a recent study has shown the Spo0E deletion does not affect pulsing (Levine et al., 2012). On the other hand, the cell-cycle dependent oscillations of Sda provides a viable explanation for the DNA replication coupled 0A~P pulses. However, our results showing the lack of pulsing in i0FgltA strain where 0A~P-0F negative feedback is perturbed (Figure 3D), suggests that substrate inhibition feedback that we propose here plays the key role in controlling 0A~P dynamics.
Notably, dead-end complex based substrate inhibition mechanisms have been previously postulated to act as a source of ultrasensitivity in the response of bacterial two-component systems and sigma factor regulation (Clarkson et al., 2004; Igoshin et al., 2008; Igoshin et al., 2006). Our results reveal that these mechanisms can also result in ultrasensitivity in the ratio of the two genes involved in dead-end complex substrate inhibition – specifically the KinA:0F ratio. In the case of sporulation, this ratio sensitive response forms the basis of the coupling of 0A~P pulsing to DNA replication by a gene dosage mechanism. More generally, however, this ratio-sensitive response provides a unique mechanism for the integration of different environmental signals - a feature that may be relevant to a wide variety systems that employ phosphorelay networks (Appleby et al., 1996).
The sensitivity of phosphorelay response to the KinA:0F ratio and as a result to their gene dosages also suggests a unique mechanism for growth rate based control of sporulation. In nutrient rich conditions, for cells with a generation time of less than the time required replication, new rounds of replication begin before the previous round terminates, resulting in multifork replication (Wang and Levin, 2009). As a result of multifork replication, cells have multiple copies of the origin-proximal chromosomal region and a single copy of the terminus-proximal region. Consequently the relative gene dosage of origin-proximal and terminus-proximal genes increases at high growth rate (Wang and Levin, 2009). Considering that 0F and kinA are origin-proximal and terminus-proximal respectively, this implies that the low KinA:0F ratio at high growth rates could prevent 0A activation sporulation in nutrient rich conditions. A similar origin-terminus relative gene dosage mechanism has been shown to regulate histidine metabolism in response to growth rate in Salmonella typhimurium (Blanc-Potard et al., 1999). Our results suggest that in Bacillus subtilis, the kinA:0F relative gene dosage sensitive phosphorelay may act similarly and function as a growth rate dependent decision mechanism although this requires further investigation.
Our results also show that to enable the pulsatile 0A~P response strategy, the design of the sporulation network exploits a universal feature of bacterial physiology: the transient imbalance of gene dosage between origin-proximal and terminus-proximal genes during chromosome replication. It is well-known that chromosomal location of genes affects their expression (Block et al., 2012; Klumpp et al., 2009) and that clustering of genes facilitates their co-regulation (Lathe et al., 2000). Recent studies have also revealed that origin proximal location of genes can be used to detect DNA replication stress and trigger bacterial competence (Slager et al., 2014). Our results add to this growing repertoire of functional roles for chromosomal arrangement of regulatory genes and provide a simple cell-cycle coupling mechanism that could very well be employed in a wide range of other microbial species and stress response mechanisms.
Experimental Procedures
Strain construction
All strains were derived from Bacillus subtilis PY79 (Youngman et al., 1984) using standard molecular biology techniques. Details of the strain construction, strain genotypes and primer sequences and vectors used are provided in the Table S3 in the Supplemental Information.
Culture preparation and Microscopy
Culture preparation
For imaging, B. subtilis culture was started from an overnight LB agar plate containing appropriate antibiotics (final concentrations: 5 μg/ml chloramphenicol, 5 μg/ml neomycin, 5μg/ml erythromycin, 5μg/ml phleomycin and 100 μg/ml spectinomycin). Strains containing multiple resistance genes were grown on a combination of no more than three antibiotics at a time. Cells were resuspended in casein hydrolysate (CH) medium (Sterlini and Mandelstam, 1969) and grown at 37°C with shaking. After reaching OD 1.8–2.0, cells were washed once and resuspended in 0.5 volume of Resuspension Medium (RM) (Sterlini and Mandelstam, 1969). The resuspended cells were grown at 37°C for 1 hour, then diluted 15-fold and applied onto a 1.5% low-melting agarose pad made with RM-MOPS medium with desired IPTG or glucose concentration, if necessary. The pads were covered, left to air-dry for 1 hour at 37°C and placed into a coverslip-bottom Willco dish for imaging.
Time-Lapse Microscopy
Differentiation of B. subtilis microcolonies was monitored with fluorescence time-lapse microscopy at 37°C with an Olympus IX-81 inverted microscope with a motorized stage (ASI) and an incubation chamber. Image acquisition was set to every 20 min with a Hamamatsu ORCA-ER camera. Custom Visual Basic software in combination with the Image Pro Plus (Media Cybernetics) was used to automate image acquisition and microscope control.
Image Analysis
A combination of Schnitzcells software (http://cell.caltech.edu/schnitzcells), custom written MATLAB programs, MicrobeTracker tool (Sliusarenko et al., 2011) and freely available ImageJ plugins (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2014) was used to analyze microscopy data as described below.
Data Analysis
Quantification of Promoter Activity
The measurements of promoter activities for P0A-cfp/yfp promoters (Figures 1C and 4) and the P0F-yfp promoter (Figure S1) refer to rate of protein production calculated from fluorescence time-lapse data. These promoter activities were calculated by using an explicit finite-difference method described previously (Levine et al., 2012; Rosenfeld et al., 2005). Briefly, the promoter activity PF(t) was determined from fluorescence time-series data for mean cell fluorescence (M(t)) and cell-length (L(t)) using the following equation:
Here Δt is the time difference between successive frames (20 minutes). μ(t) and γ are the protein dilution and degradation rates respectively. We used γ=0.1 hr−1 since in our conditions, fluorescent proteins CFP and YFP are stable. μ(t), the cell-growth rate at every frame was calculated using the following equation:
See Supplemental Information for a detailed description of the Promoter Activity quantification procedure.
Quantification and characterization of promoter activity pulses
The time to reach the maximum promoter activity from the start of the cell-cycle and was used to calculate the Tp period (Figure 1D).
To differentiate pulsing and non-pulsing strains (Figure S5), the promoter activity in each cell-cycle was quantified at three time points: at the start of the cell-cycle, at the point during the cell-cycle where the promoter activity reaches its maximum value and at the end of the cell-cycle (Figure S5AB). To determine whether increase in 0A promoter activity during each cell cycle occurs in a pulsatile manner we compared ΔMax, the difference between Peak promoter activity and promoter activity at the start of the cell cycle, and ΔEnd, the difference between the promoter activities at the start and end of the cell-cycles. In each strain, we aggregate the data for ΔMax and ΔEnd at each cell cycle and compare their distributions with a two sample t-test. Statistical significance of the observed differences in ΔMax and ΔEnd is used to detect whether pronounced pulsing can be detected for each strain.
Identification of DNA replication periods using DnaN foci
To identify DNA replication windows in time-lapse experiments (Figure 1BC) we expressed a fluorescent DnaN-YFP fusion protein from the IPTG inducible Phyper-spank promoter. During DNA replication, the DnaN forms subcellular foci (Su'etsugu and Errington, 2011; Veening et al., 2009). Using the DnaN-YFP fusion protein, these foci can be detected as diffraction-limited spots. We used the SpotFinderF MATLAB program (Sliusarenko et al., 2011) with a signal-to-background ratio of 40 as the minimum peak height to identify DnaN-YFP foci and active DNA replication periods. DNA replication periods identified in this way were used to calculate the difference between the start of the cell-cycle and the end of DNA replication or Tr period (Figure 1D) for each cell-cycle in the time-lapse data.
Estimation of the spore fraction
To get an estimate of the sporulation efficiency of the various strains in Figure 3 we calculated a spore fraction for each strain after 25 hrs in starvation conditions (see Figure 5A). The spore fraction was calculated by dividing the number of cells that had formed phase-bright spores by the total number of cells. Three independent measurements were made for each strain with more than 200 cells counted in each instance to calculate the mean and standard deviations of spore fractions.
Quantification of sporulation defects
We manually counted the number of both types of sporulation defects (Asymmetric septation without σF activation and Activation of σF in the mother cell) in the time-lapse images of pulsing WT strain (harboring the same integrated reporters and GltA knockout as i0FgltA strain) and of the non-pulsing i0FgltA (at 5μM IPTG) and Trans-kinAamyE strains over 30hrs in starvation conditions. We also counted the total number of spores produced over those 30hrs to calculate the frequency of defects produced for both strains per spore (Figure 5B). Two-sample t-test was used to determine the statistical significance of the observed differences.
To determine the timing of cell-fate decision in sporulation defects and normal sporulation (Figure 5C) we used time-lapse microscopy data from the non-pulsing i0FgltA strain. The timing of cell-fate decision was defined as the time from the start of the cell-cycle to the time of PSpoIIR-cfp (a σF reporter) activation in the cases of normal sporulation and mother cell σF activation defect. For the case of asymmetric septation without σF activation defect, the time of cell-fate decision was defined as the time from the start of the cell-cycle to the time of asymmetric septation.
Supplementary Material
Highlights.
The sporulation network detects transient gene dosage imbalance during replication
Negative feedback leads to pulsatile activation of sporulation master regulator
Pulsed response couples sporulation and DNA replication
Gene translocations eliminating pulses lead to sporulation defects
Acknowledgments
Authors thank Carla Vidal for her help with strain construction. This work is supported by NSF grants MCB-1244135 (to OAI), EAGER-1450867 (to GS), MCB-1244423 (to MF), NIH NIGMS grant R01 GM088428 (to GS) and HHMI International Student Fellowship to JN.
Footnotes
Author Contributions
JN, AK, GMS and OAI designed the research. JN developed the mathematical model. AK and DL constructed the strains and performed the experiments. JN and AK analyzed the data. All authors wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appleby JL, Parkinson JS, Bourret RB. Signal transduction via the multi-step phosphorelay: not necessarily a road less traveled. Cell. 1996;86:845–848. doi: 10.1016/s0092-8674(00)80158-0. [DOI] [PubMed] [Google Scholar]
- Blanc-Potard AB, Figueroa-Bossi N, Bossi L. Histidine operon deattenuation in dnaA mutants of Salmonella typhimurium correlates with a decrease in the gene dosage ratio between tRNA(His) and histidine biosynthetic loci. J Bacteriol. 1999;181:2938–2941. doi: 10.1128/jb.181.9.2938-2941.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block DH, Hussein R, Liang LW, Lim HN. Regulatory consequences of gene translocation in bacteria. Nucleic Acids Res. 2012;40:8979–8992. doi: 10.1093/nar/gks694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulys D, Trach KA, Hoch JA. Initiation of sporulation in B. subtilis is controlled by a multicomponent phosphorelay. Cell. 1991;64:545–552. doi: 10.1016/0092-8674(91)90238-t. [DOI] [PubMed] [Google Scholar]
- Chapman JW, Piggot PJ. Analysis of the inhibition of sporulation of Bacillus subtilis caused by increasing the number of copies of the spo0F gene. J Gen Microbiol. 1987;133:2079–2088. doi: 10.1099/00221287-133-8-2079. [DOI] [PubMed] [Google Scholar]
- Chastanet A, Vitkup D, Yuan GC, Norman TM, Liu JS, Losick RM. Broadly heterogeneous activation of the master regulator for sporulation in Bacillus subtilis. Proc Natl Acad Sci U S A. 2010;107:8486–8491. doi: 10.1073/pnas.1002499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson J, Shu JC, Harris DA, Campbell ID, Yudkin MD. Fluorescence and kinetic analysis of the SpoIIAB phosphorylation reaction, a key regulator of sporulation in Bacillus subtilis. Biochemistry. 2004;43:3120–3128. doi: 10.1021/bi036014+. [DOI] [PubMed] [Google Scholar]
- Doncic A, Falleur-Fettig M, Skotheim JM. Distinct Interactions Select and Maintain a Specific Cell Fate. Molecular Cell. 2011;43:528–539. doi: 10.1016/j.molcel.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington J. Regulation of endospore formation in Bacillus subtilis. Nat Rev Microbiol. 2003;1:117–126. doi: 10.1038/nrmicro750. [DOI] [PubMed] [Google Scholar]
- Eswaramoorthy P, Duan D, Dinh J, Dravis A, Devi SN, Fujita M. The threshold level of the sensor histidine kinase KinA governs entry into sporulation in Bacillus subtilis. J Bacteriol. 2010;192:3870–3882. doi: 10.1128/JB.00466-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M, Losick R. Evidence that entry into sporulation in Bacillus subtilis is governed by a gradual increase in the level and activity of the master regulator Spo0A. Genes Dev. 2005;19:2236–2244. doi: 10.1101/gad.1335705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimshaw CE, Huang S, Hanstein CG, Strauch MA, Burbulys D, Wang L, Hoch JA, Whiteley JM. Synergistic kinetic interactions between components of the phosphorelay controlling sporulation in Bacillus subtilis. Biochemistry. 1998;37:1365–1375. doi: 10.1021/bi971917m. [DOI] [PubMed] [Google Scholar]
- Hauser PM, Errington J. Characterization of cell cycle events during the onset of sporulation in Bacillus subtilis. J Bacteriol. 1995;177:3923–3931. doi: 10.1128/jb.177.14.3923-3931.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D, Dworkin J. Recent progress in Bacillus subtilis sporulation. FEMS Microbiol Rev. 2012;36:131–148. doi: 10.1111/j.1574-6976.2011.00310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch JA. Regulation of the phosphorelay and the initiation of sporulation in Bacillus subtilis. Annu Rev Microbiol. 1993;47:441–465. doi: 10.1146/annurev.mi.47.100193.002301. [DOI] [PubMed] [Google Scholar]
- Igoshin OA, Alves R, Savageau MA. Hysteretic and graded responses in bacterial two-component signal transduction. Mol Microbiol. 2008;68:1196–1215. doi: 10.1111/j.1365-2958.2008.06221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoshin OA, Price CW, Savageau MA. Signalling network with a bistable hysteretic switch controls developmental activation of the sigma transcription factor in Bacillus subtilis. Mol Microbiol. 2006;61:165–184. doi: 10.1111/j.1365-2958.2006.05212.x. [DOI] [PubMed] [Google Scholar]
- Klumpp S, Zhang Z, Hwa T. Growth rate-dependent global effects on gene expression in bacteria. Cell. 2009;139:1366–1375. doi: 10.1016/j.cell.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchina A, Espinar L, Ca Gcaron Atay T, Balbin AO, Zhang F, Alvarado A, Garcia-Ojalvo J, Suel GM. Temporal competition between differentiation programs determines cell fate choice. Mol Syst Biol. 2011a;7:557. doi: 10.1038/msb.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchina A, Espinar L, Garcia-Ojalvo J, Suel GM. Reversible and Noisy Progression towards a Commitment Point Enables Adaptable and Reliable Cellular Decision-Making. PLoS Comput Biol. 2011b;7:e1002273. doi: 10.1371/journal.pcbi.1002273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathe WC, Snel B, Bork P. Gene context conservation of a higher order than operons. Trends Biochem Sci. 2000;25:474–479. doi: 10.1016/s0968-0004(00)01663-7. [DOI] [PubMed] [Google Scholar]
- LeDeaux JR, Yu N, Grossman AD. Different roles for KinA, KinB, and KinC in the initiation of sporulation in Bacillus subtilis. J Bacteriol. 1995;177:861–863. doi: 10.1128/jb.177.3.861-863.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Fontes ME, Dworkin J, Elowitz MB. Pulsed feedback defers cellular differentiation. PLoS Biol. 2012;10:e1001252. doi: 10.1371/journal.pbio.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009;138:760–773. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narula J, Devi SN, Fujita M, Igoshin OA. Ultrasensitivity of the Bacillus subtilis sporulation decision. P Natl Acad Sci USA. 2012;109:E3513–E3522. doi: 10.1073/pnas.1213974109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M, Cole SP, Burbulys D, Trach K, Hoch JA. Characterization of the gene for a protein kinase which phosphorylates the sporulation-regulatory proteins Spo0A and Spo0F of Bacillus subtilis. J Bacteriol. 1989;171:6187–6196. doi: 10.1128/jb.171.11.6187-6196.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Gene regulation at the single-cell level. Science. 2005;307:1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- Schultz D, Wolynes PG, Jacob EB, Onuchic JN. Deciding fate in adverse times: sporulation and competence in Bacillus subtilis. Proc Natl Acad Sci U S A. 2009;106:21027–21034. doi: 10.1073/pnas.0912185106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Garcia-Ojalvo J, Elowitz MB. Dynamical consequences of bandpass feedback loops in a bacterial phosphorelay. PLoS One. 2011;6:e25102. doi: 10.1371/journal.pone.0025102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager J, Kjos M, Attaiech L, Veening JW. Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell. 2014;157:395–406. doi: 10.1016/j.cell.2014.01.068. [DOI] [PubMed] [Google Scholar]
- Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Molecular Microbiology. 2011;80:612–627. doi: 10.1111/j.1365-2958.2011.07579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterlini JM, Mandelstam J. Commitment to sporulation in Bacillus subtilis and its relationship to development of actinomycin resistance. Biochem J. 1969;113:29–37. doi: 10.1042/bj1130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su'etsugu M, Errington J. The replicase sliding clamp dynamically accumulates behind progressing replication forks in Bacillus subtilis cells. Mol Cell. 2011;41:720–732. doi: 10.1016/j.molcel.2011.02.024. [DOI] [PubMed] [Google Scholar]
- Toettcher JE, Loewer A, Ostheimer GJ, Yaffe MB, Tidor B, Lahav G. Distinct mechanisms act in concert to mediate cell cycle arrest. Proc Natl Acad Sci U S A. 2009;106:785–790. doi: 10.1073/pnas.0806196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JW, Murray H, Errington J. A mechanism for cell cycle regulation of sporulation initiation in Bacillus subtilis. Genes Dev. 2009;23:1959–1970. doi: 10.1101/gad.528209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veening JW, Stewart EJ, Berngruber TW, Taddei F, Kuipers OP, Hamoen LW. Bet-hedging and epigenetic inheritance in bacterial cell development. Proc Natl Acad Sci U S A. 2008;105:4393–4398. doi: 10.1073/pnas.0700463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JD, Levin PA. Metabolism, cell growth and the bacterial cell cycle. Nat Rev Microbiol. 2009;7:822–827. doi: 10.1038/nrmicro2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman P, Perkins JB, Losick R. Construction of a cloning site near one end of Tn917 into which foreign DNA may be inserted without affecting transposition in Bacillus subtilis or expression of the transposon-borne erm gene. Plasmid. 1984;12:1–9. doi: 10.1016/0147-619x(84)90061-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.