

Abstract
Many cancers exhibit chromosomal rearrangements. These rearrangements can be simple, involving a single balanced fusion that preserves the proper complement of genetic information, or complex with one or more fusions that disrupt this balance. Recent technological advances have improved our ability to detect and understand these rearrangements, leading to speculation about potential causal mechanisms such as defective DNA double strand break repair and faulty DNA replication. A better understanding of these potential cancer-causing mechanisms will lead to novel therapeutic regimens to fight cancer. This review describes technological advances in methods used to detect simple and complex chromosomal rearrangements, cancers that exhibit these rearrangements, potential mechanisms for rearrangement of chromosomes, and intervention strategies designed specifically against fusion gene products and causal DNA repair/synthesis pathways.
Keywords: replication fork, DNA repair, chromosomal rearrangement, chromothripsis, oncogenesis
Introduction
Accumulation of genetic mutations can contribute to cancer development, progression, and metastasis.1 Some rearrangements are simple balanced translocations that result from a single fusion and preserve the proper complement of genetic information, although such translocations often disrupt regulation of the genes involved. Other rearrangements are complex, with multiple fusions at a single locus and do not maintain the normal complement of genetic information. Such rearrangements have the potential to cause cancer if they mutate a tumor suppressor gene or activate an oncogene2 and can also contribute to tumor heterogeneity and clonal evolution as mechanisms for metastasis and drug resistance.3,4 Thus, chromosomal rearrangements are important in cancer etiology. This review will first describe recent advancements in technologies that drove the discovery and characterization of translocations. Next, we describe cancers that exhibit simple balanced translocations or more complex rearrangements. Finally, we review potential mechanisms that lead to chromosomal rearrangements and present possible intervention strategies.
Technologies for the Discovery and Evaluation of Genomic Rearrangements
The technological advances summarized in Table 1 have enabled the detection of chromosomal rearrangements in cancer cells. Chromosomal rearrangements in cancer were first identified in the early 50s by karyotype analyses based on Giemsa (G-banding) or reverse Giemsa banding (R-banding). G-banding is a technique that produces a visible display of condensed chromosomes whereas R-banding produces bands that are complementary to Giemsa bands.5,6 This technology allowed the observation of whole chromosomes during metaphase and provided the basis for the hypothesis that tumorigenesis is a genetic disease based on the realization that alteration of chromosome structure is a frequent event in cancer. However, this technology did not effectively identify the specific chromosomal locations or structures involved in complex rearrangements.
Table 1. Techniques for identification of chromosomal rearrangements.
| Technique | Procedure | Purpose | Limitations |
|---|---|---|---|
| Giemsa (G)-banding | MSP with a banding pattern used to identify chromosomes | Identify chromosome number and structural rearrangements | Resolution < 10 Mb, not sensitive enough to detect small inversions |
| Reverse giemsa (R)-banding | MSP with a banding pattern complementary to G-banding | Identify chromosome number and structural rearrangements | Resolution ~10 Mb, not sensitive enough to detect small inversions |
| Fluorescence in situ hybridization (FISH) | Probes anneal to specific regions such as pericentromere and telomere in MSPs | Identify chromosome number, rearrangements, and specific structures such as pericentromeres and telomeres | Allows analysis of only 4 fluorochromes at one time |
| Spectral karyotyping (SKY) | Sophisticated FISH using a combination of paint probes targeting all chromosomes in a single hybridization | Identify all chromosomes and visualize complex structural alterations | Resolution 5–10 Mb, not sensitive enough to detect small inversions |
| Comparative genomic hybridization (CGH) | Hybridize test DNA to reference DNA to identify copy number variation | Identify deletions and amplifications | Resolution depends on the density of the probes, does not allow detection of inversions and balanced translocations |
| Whole genome sequencing (WGS) | Sequencing the entire genome | Analysis of sequence at the fusion site at single nucleotide resolution | Costly and requires intensive data analysis |
| High-throughput genome-wide translocation sequencing (HTGTS) and translocation capture sequencing (TC-Seq) | Sequencing applied to sites of induced DSBs in B cells | Identify large numbers of translocations | Requires skilled personnel for library preparation, costly and requires intensive data analysis |
Abbreviations: DSB, double-strand break; MSP, metaphase spread
To address this issue, the technique of fluorescence in situ hybridization (FISH) was developed in the late 70s. FISH greatly improved the resolution of standard karyotyping by providing tools for visualizing specific loci on metaphase chromosomes. In addition, FISH allowed the quantitative analysis of chromosomal alterations in interphase cells, thus providing a technique for analyzing non-dividing cells. Refinements and improvements of the technology have been ongoing and approximately 40 different applications of FISH have been reported to date.7 Although several of these applications specifically address unique biologic questions, others are widely employed for the analysis of chromosomes in cancer cells and are routinely used for the clinical evaluation of patient samples.
In general, the most commonly used methods of FISH require the generation of a locus-specific probe (LSP) targeting a gene of interest (usually an oncogene or a tumor suppressor gene) that is labeled with one fluorochrome, combined with one or more reference probes (a subcentromeric probe mapping to the same chromosome as the LSP of interest or a probe mapping to a region flanking the LSP) that is labeled with a different fluorochrome. A variety of probes can be used in different combinations depending on the specific loci of interest, allowing precise and highly detailed analysis of chromosome alterations including visualization of chromosome breakpoints, copy number alterations (gains and losses), and inversion of chromosomal regions. FISH is a cost-effective approach that has the advantages of allowing analysis at the single cell level and facilitating characterization of genomic regions that are notoriously difficult to study with other approaches (e.g., structural alterations that map to repetitive regions such as pericentromeres and telomeres).
Spectral karyotyping (SKY) is a more sophisticated FISH approach that requires access to a spectra cube (interferometer).8,9 SKY is based on the combinatorial use of paint probes targeting all chromosomes in a single hybridization, providing an exceptional ability to visualize complex structural alterations. The application of SKY to the analysis of human and murine samples has been instrumental in advancing the field of molecular cytogenetic analysis and gaining a better understanding of the complexity of chromosome alterations in cancer. It has allowed refinement of the complexity of previously known breakpoints and better characterization of cases that were difficult to resolve because of poor spreads or contracted metaphase chromosomes, highly rearranged karyotypes with numerous marker chromosomes, or subtle chromosomal aberrations.10-12 SKY has also been proven to be a powerful tool in the analysis of murine chromosomes. The use of genetically engineered mice as model systems of human cancer has fueled the need for better methods of cytogenetic analysis because of the challenges associated with studying murine chromosomes that are similar in size and are acrocentric.13 As a result, the characteristic banding pattern typically used for the identification of human chromosomes is less helpful in mice, making karyotyping very difficult. Furthermore, at the present time there are few cytogeneticists trained in karyotyping mouse chromosomes. There are more than 160 reports describing the use of SKY to characterize the karyotype of a wide range of murine models. In our experience, SKY has been an extremely valuable tool for analyzing structural alterations resulting in complex rearrangements in murine chromosomes.14,15 In one specific application, SKY has been used to resolve the chromosomes involved in the formation of dipericentrics and chromosomes with extra pericentromeres and telomeres (EPTs). The pericentromere is the region surrounding the centromere, which is a highly complex structure important for chromosome segregation during mitosis. The pericentromere is more easily detected than the centromere using FISH, hence the term dipericentric. Dipericentric chromosomes, also called dicentric chromosomes, have a pericentromere at each end of the chromosome and none in the middle. EPTs are complex structural alterations involving segmental duplications with extra centromeres and telomeres at the poles of a chromosome as well as in the middle of the chromosome.15 However, although SKY is well suited for the analysis of complex alterations it has a limited resolution of 10 Mb and is therefore not suitable for fine mapping of break points.
Comparative genomic hybridization (CGH), which was developed during the 90s, can be used to better define breakpoints.16 Conventional CGH (cCGH), originally designed for hybridization on metaphase chromosomes, makes use of differentially labeled test DNA and reference DNA that are co-hybridized with chromosomes to identify regions of copy number variation (CNV). To overcome limitations of the resolution provided by karyotyping and cCGH the technique was adapted to matrix-based hybridization protocols in array CGH (aCGH).17 A large variety of matrices is now available, offering different levels of resolution and dynamic ranges (from one to several fold changes in copy number, as frequently observed in cancer cells). aCGH, often combined with single nucleotide polymorphism (SNP) arrays, is routinely used in clinical settings to diagnose prenatal and intellectual disabilities. Several array designs are available for the cancer genome (e.g., Agilent 400K CGH/SNP and Affymetrix CytoScan and OncoScan FFPE arrays). Such arrays are used in cancer clinical genomics to stratify lymphoma patients; for example, chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), large B-cell lymphoma (DLBCL), and mantle cell lymphoma (MCL). In basic research aCGH is extensively applied to analyze complex rearrangements. For example, Carvalho and colleagues applied a custom-designed 44 k microarray to study genomic segments at the methyl CpG binding protein 2 (MECP2) and the proteolipid protein 1 (PLP1) loci that are characterized by a common genomic organization, duplication-inverted triplication-duplication.18 By mapping the junctions of breakpoints in a cohort of 9 subjects they provided evidence supporting the mechanism leading to the formation of a triplicated segment embedded within a duplication: duplication-inverted triplication-duplication complex rearrangements are formed by the combination of homology-directed break-induced replication (BIR) with microhomology-mediated BIR or nonhomologous end joining (NHEJ) that involves replication fork repair after stalling and template switching (discussed below). A major limitation of aCGH is the inability to detect balanced translocations; in addition, as the technique is based on analysis of bulk DNA (DNA isolated from a pool of cells) it has restricted sensitivity to detect mosaic alterations (more than 20% of the population of cells must present the same alteration to enable detection).
Next-generation sequencing (NGS) approaches, including whole genome sequencing (WGS), promise to further advance our understanding of complex chromosomal alterations by precise mapping of breakpoints and junctions, thus enabling unprecedented sensitivity and resolution of structural and mutational changes. Improvements in WGS have allowed the discovery of complex chromosomal rearrangements in solid tumors.19 Furthermore, although these translocations were initially thought to be simple and balanced, deep sequencing has revealed that some are in fact quite complicated. WGS has revealed genomic architecture that is susceptible to complicated rearrangements and has narrowed down the sequence flanking the genomic alterations so that breakpoints can be identified. The single-nucleotide resolution of WGS enables mapping of chromosome regions too small for analysis by FISH or aCGH. A combination of WGS, SNP arrays, and FISH approaches were used in the discovery of the phenomenon of chromothripsis (or chromosome shattering), which shows a complicated pattern of multiple fusions at a single location.20 In the past few years WGS has been instrumental in revealing new causative mechanisms of chromosomal breaks and rearrangements.
High-throughput genome-wide translocation sequencing (HTGTS) and translocation capture sequencing (TC-Seq) were applied to identify translocation junctions in B cells upon induction of DNA double-strand breaks (DSBs) at specific locations in the genome.21,22 These studies identified a large number of translocations, revealing a marked association between transcription start sites and translocation targets. The majority of translocation junctions were formed through end joining with short microhomologies. Although most cells repair induced DSBs by rejoining the ends without causing major genomic rearrangements, a significant fraction of cells join the induced DSB ends to endogenous DSBs elsewhere in the genome, creating intra- and interchromosomal rearrangements. Translocations occur more frequently on the chromosome carrying the induced break and preferentially target transcribed chromosomal regions, even up to 50 Mb away.
The application of NGS sequencing to study chromosome alterations in cancer opens new and exciting directions of study but has certain challenges. Although the price of sequencing is decreasing dramatically, the cost of sequencing a mammalian genome remains high when considering that analysis of several samples is usually required to address biological questions. Moreover, the analytical tools essential for WGS analysis in cancer samples are not yet fully developed. For example, in the case of chromothripsis, automated methods designed to detect or annotate high throughput sequencing data are not available. To address these limitations, several new technologies are being developed. One such method is ShatterProof, which uses structural variation calls (translocations, copy number variations, short insertions, and loss of heterozygosity) and performs robust statistical tests to accurately predict the presence and location of chromothriptic events.23 Another limitation of applying WGS studies to cancer cells is the need for high coverage (> 30 × ) to allow characterization of clonal heterogeneity by tumor biopsy. Despite these limitations, advances in technology have greatly improved our ability to identify and characterize chromosomal rearrangements, thus presenting the opportunity to address causal factors.
The technologies described above have been instrumental for the mapping of genomic alterations, especially in the cancer genome where chromosomal alterations characterize many solid tumors and hematologic malignancies. As a result, specialized databases compiling a detailed inventory of chromosomal alterations have been developed. The most comprehensive of these are the Mitelman Database (http://cgap.nci.nih.gov/Chromosomes/Mitelman), which contains all published chromosome aberrations in neoplastic disorders with clinical features of more than 64,000 cancers, and the Catalogue of Somatic Mutations in Cancer (COSMIC) database,24 which currently reports the characterization of 327 types of gene fusions identified in more than 60,000 cancer cases (accessed May 5, 2014). Data available at this time indicate that gene fusions occur in approximately 15% of all solid cancer cases analyzed, which is less frequent than the reported rate in hematological malignancies (16.2%). Chromosomal rearrangements might be found in more tumors as high throughput NGS sequencing becomes more widespread and affordable.
Simple and Complex Rearrangements Found in Cancers
The presence of simple and balanced rearrangements (summarized in Table 2) can increase cancer risk. Simple rearrangements have no or limited alterations at the fusion ends whereas balanced rearrangements fuse chromosomes without losing genetic information. Their role in cancer etiology was first described in 1960 with the discovery of the Philadelphia chromosome in patients with various forms of leukemia.25,26 The Philadelphia chromosome arises from a head-to-tail fusion of the breakpoint cluster region (BCR) with the gene encoding the c-abl (ABL1) proto-oncogene tyrosine kinase to generate the BCR-ABL fusion transcript. The BCR-ABL fusion generates a constitutively active tyrosine kinase that can transform cells and inhibit apoptosis induced by a variety of agents.27 Activation of protein kinases is seen in other gene fusions. For example, anaplastic lymphoma kinase (ALK) can be activated by an inversion that fuses the ALK gene with the echinoderm microtubule-associated protein like 4 (EML4) gene. This rearrangement occurs in 2–5% of non-small cell lung cancers (NSCLCs).28 C-ros oncogene 1 (ROS1) receptor tyrosine kinase (conserved with ALK) is also activated in approximately 1% of NSCLCs and in other cancers (cholangiocarcinoma, glioblastoma multiforme, gastric adenocarcinoma) after fusion with a variety of genes.29-31 In addition to the Philadelphia chromosome, a variety of other balanced translocations are commonly observed in hematologic malignancies.19 For example, 10% of all cases of acute myeloid leukemia (AML) exhibit one of the following three fusions: (1) the N-terminal DNA-binding domain of AML1 with the Eight-Twenty One oncoprotein (ETO); (2) the promyelocytic leukemia (PML) gene with the retinoic acid receptor α (RARA); and 3) the mixed lineage leukemia (MLL) gene with one of several partners.32 The AML-ETO fusion causes aberrant recruitment of epigenetic modifiers, thus affecting normal myelomonocytic development, whereas PML-RARA affects both nuclear receptor signaling and PML body assembly. In addition, the transactivation domains of the forkhead box (FOX) gene family fuse with the DNA binding domains of a variety of genes to produce a fusion transcript that encodes an unregulated transcription factor. Such fusions include paired box (PAX) with FOXO133-35 and MLL with either FOXO3 or FOXO4. These translocations have been found in alveolar rhabdomyosarcoma and leukemia.36,37 Furthermore, PAX3-FOXO1 is associated with an aggressive phenotype and poor prognosis.38 The presence of fusion proteins that activate similar oncogenic pathways suggests that cancers of different molecular and cellular origin share common pathogenetic mechanisms determined by the transcriptional effector properties of the forkhead protein subfamily.39 Thus, simple and balanced chromosomal translocations can result in fusion proteins that enable cancer development.
Table 2. Examples of simple balanced rearrangements found in cancers.
| Fusion partners | Breakpoint | Cancer | Defect | Targeted therapy |
|---|---|---|---|---|
| Philadelphia chromosome: breakpoint cluster region with c-abl (BCR-ABL1) | t(9;22)(q34;q11) | CML, ALL, AML | Tyrosine kinase activation Balanced |
Imatinib |
| Anaplastic lymphoma kinase gene with echinoderm microtubule-associated protein like 4 (ALK-EML4) | Inv(2)(p21;p23) | NSCLC | Tyrosine kinase activation Balanced |
Crizotinib |
| c-ros oncogene 1 (ROS1) with multiple genes | NSCLC, cholangiocarcinoma, glioblastoma multiforme, gastric adenocarcinoma |
Tyrosine kinase activation Balanced and non-balanced |
Crizotinib | |
| AML1/ETO | t(8;21)(q22;q22) | AML | Aberrant recruitment of epigenetic modifiers affecting normal myelomonocytic development Balanced |
General chemotherapy (cytarabine and anthracycline) |
| Promyelocytic leukemia with retinoic acid receptor α (PML-RARA) | t(15;17)(q22;q21) | AML | Nuclear receptor signaling and PML body assembly Balanced |
ATRA and arsenic oxide (AS203) |
| Mixed lineage leukemia (MLL)-unclassified partners | AML | Four types of unclassified fusion partners: (1) nuclear proteins, (2) cytoplasmic proteins, (3) histone acetyltransferases, (4) septins Balanced |
ATRA | |
| Paired box with Forkhead box (PAX3-FOXO1) |
t(2;13)(q36;q14) | ARMS | Transcriptional activation Balanced |
Thapsigargin |
| PAX7-FOXO1 | t(1;13)(p36;q14) | ARMS | Transcriptional regulation Balanced and non-balanced |
Targeting downstream pathways |
| FOXO3-MLL | t(6;11)(q21;q23) | Leukemia and ARMS | Transcriptional regulation Usually balanced |
ATRA |
| FOXO4-MLL | t(X;11)(q13;q23) | Leukemia and ARMS | Transcriptional regulation Usually balanced |
ATRA |
| FOXP1-PAX5 | t(3;9)(p13;p13) | Lymphoblastic lymphoma | Transcriptional regulation Balanced and non-balanced |
None |
Abbreviations: ALL, acute lymphoblastic leukemia; AML, acute myelogenous leukemia; ARMS, alveolar rhabdomyosarcoma; ATRA, all-trans-retinoic acid; CML, chronic myelogenous leukemia; NSCLC, non-small cell lung cancer.
More complex rearrangements are also found in cancer. These rearrangements have multiple joins at the fusions and can involve more than two chromosomes, and result in a change in genetic content as well as a change in the chromosomal linear structure. Complex genomic rearrangements have been reported in lymphoma.40-45 Complex chromosomal translocations leading to gene fusions are also found in solid cancers,19 including whole arm translocations and isochromosomes in head and neck squamous cell carcinoma,46 complex genomic rearrangements including inversions in pancreatic cancer,47 and rearrangements including tandem duplications in breast cancers.48,49 Palindrome structures were also found in cells derived from colon cancer, breast cancer, and embryonal rhabdomyosarcoma, and in primary medulloblastomas.50 Furthermore, rearrangements that have undergone chromothripsis have been identified, with tens to hundreds of fusions mapping to a single chromosomal location.20,51,52 Chromothripsis has been detected in as many as 3% of cancers (and more might be found with wider application of WGS) and occurs in a wide range of cancers including leukemia, medulloblastoma, melanoma, glioma, sarcoma, bone cancer, colorectal cancer, renal cancer, and thyroid cancer.53 It appears that chromosomal disruption and pulverization might be linked to the generation of micronuclei as a consequence of mitotic errors.54 The discovery and understanding of these complex rearrangements is continuing to advance as technology improves yet the causal mechanisms are still not understood. Defects in the repair of DNA DSB repair and faulty DNA synthesis have been proposed as mechanisms underlying these rearrangements.40
Double Strand Break Repair Pathways that Influence Chromosomal Rearrangements
Multiple pathways are responsible for DNA DSB repair, including nonhomologous end joining (NHEJ), homologous recombination (HR), and break-induced replication (BIR). These pathways are important for maintaining chromosomal integrity. Conversely, their faulty application has the potential to cause chromosomal rearrangements (Fig. 1).
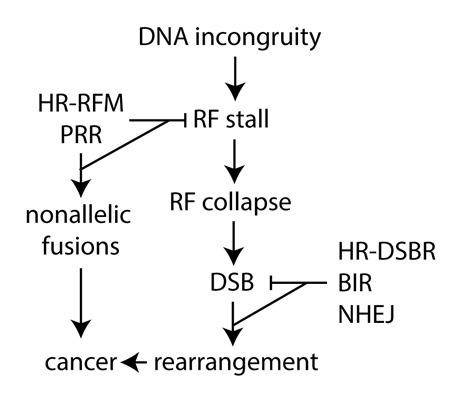
Figure 1. Replication fork maintenance and double strand break repair can either suppress or cause chromosomal rearrangements. BIR, break-induced replication; DSB, double-strand break; DSBR, double strand break repair; HR, homologous recombination; RF, replication fork; NHEJ, nonhomologous end joining; PRR; post replication repair; RFM, replication fork maintenance .
The NHEJ pathway repairs DSBs without a homologous DNA template; therefore, it is the predominate repair pathway during G1 but also can be involved during S/G2.55 NHEJ components include the KU heterodimer composed of KU70 and KU80, which binds to DNA ends together with the kinase DNA-PKCS. The XRCC4/DNA ligase IV heterodimer subsequently ligates the DNA ends. Loss of any of these proteins results in a severe DSB repair defect that causes hypersensitivity to clastogenic agents and defective V(D)J recombination during lymphocyte development.56,57 NHEJ-deficient cells are predisposed to chromosomal alterations and telomere joining events that result in chromosome fusions.58,59 In contrast, NHEJ itself can also rearrange chromosomes by joining noncontiguous ends, as occurs in Fanconi anemia cells.60,61 This is a particularly interesting find, because the Fanconi anemia pathway maintains replication forks in association with HR.62 Similarly, NHEJ enables chromosomal translocations in cells mutant for breast cancer 1 (BRCA1), breast cancer 2 (BRCA2), and ataxia telangiectasia mutated (ATM),63,64 which are all tumor suppressors important for HR. Theoretically, NHEJ-mediated joining of chromosome ends after telomere deletion would result in dicentric products of two chromosomes. Similarly, NHEJ could join the ends of sister chromatids to generate a palindromic chromosome. Thus, NHEJ repairs DSBs to suppress rearrangements but can also inappropriately join ends to yield rearrangements.
There are also alternative pathways to the classic KU-dependent NHEJ that have the potential to rearrange chromosomes.65 This alternative-NHEJ is poorly understood, but is known to be error prone because it frequently changes the DNA ends found at the joins.66,67 Furthermore, alternative-NHEJ can utilize microhomology to imprecisely join ends (microhomology-mediated end joining or MMEJ). Microhomologies are often found at the fusion joins of translocations in human cancer cells, implicating MMEJ as an enabler of cancer development and progression.68 Thus, alternative end joining pathways also have the potential to rearrange chromosomes.
Homologous recombination pathways repair DSBs using a homologous template for fidelity, and HR is therefore restricted to S/G2 phases of the cell cycle. The RecA recombinase RAD51 forms a filament on 3′ single DNA strands to induce annealing to the sister chromatid template.69 The recombination mediator BRCA2 assists in the formation and stability of the RAD51 filament. The RecQ helicase Bloom Syndrome mutated (BLM)70,71 regulates unwanted recombination that occurs through the dissolution of Holliday junctions.72-75 Both BRCA2 and BLM suppress cancer, thereby demonstrating that deficient or unregulated HR is mutagenic. Expression of mutant RAD51 (K133A) in mammalian cells causes a large number of chromosomal rearrangements, some of which are highly complex.15 The K133A mutation prevents binding of ATP to the highly conserved Walker A motif and disables the ability of RAD51 to induce topological changes in duplex DNA in an ATP-dependent manner. This mutant is highly toxic when expressed in proliferating cells. In addition, normally functioning HR can also rearrange chromosomes if the invading strand anneals to a homologous but nonallelic substrate, which creates a potential problem with repeated sequences.
Break-induced replication repairs one-ended DSBs or gaps during replication.76,77 A single strand at the DSB or gap invades a complementary strand to initiate DNA synthesis that has the potential to proceed to the end of the chromosome, causing loss of heterozygosity. BIR in yeast is dependent on Rad52 and the Pol32 subunit of DNA polymerase delta, demonstrating its dependence on DNA synthesis. In mammals, the Pol32 ortholog, POLD3, is required for cell cycle progression and processive DNA synthesis in cells undergoing replicative stress induced by cyclin E overexpression. Cyclin E overexpression results in BIR-mediated, POLD3-dependent segmental duplications.78 POLD3-mediated BIR facilitates genomic amplifications of up to 200 kb. Thus, BIR can also cause chromosomal rearrangements.
DNA Synthesis Pathways that Influence Chromosomal Rearrangements
As inappropriate NHEJ, HR, and BIR can result in chromosomal rearrangement, pathways that suppress DSB generation during DNA synthesis would decrease the need to engage these potentially mutagenic pathways (Fig. 1). DSB formation can occur when a replication fork encounters an incongruity in DNA such as secondary structure, damage, a protein adduct, or a transcription bubble.79 These impediments have the potential to stall or collapse replication forks. Whereas a stalled fork is simply a temporary pause, a collapsed fork has lost the replisome and can lead to fork failure or “floundering” if not properly resolved.40 A myriad of proteins can process unresolved collapsed forks with the potential formation of intermediate structures such a regressed fork (chickenfoot), a hemicatenane, and a single-strand gap.73,80-82 These structures are susceptible to formation of one-ended DSBs that will become two-ended DSBs at the point where forks converge, thus invoking DNA repair pathways. To avoid fork collapse, the nascent strand could simply switch templates to the complementary sister chromatid to bypass the DNA incongruity. In such circumstances, the lesion or alternative structure is bypassed but not corrected. Lesion bypass mechanisms appear to be important for replication fork maintenance but are not well understood.
It is possible that components of the HR pathway facilitate lesion bypass. In yeast, HR suppresses blocked replication forks through a template exchange mechanism83 and in mammals a RAD51-dependent pathway responds to stalled replication forks without a DSB.84 RAD51 was also purified from the nascent replication strand before formation of DSBs.85 Furthermore, the RAD51 K133A mutant shows impaired replication fork restart.15 RAD51 overexpression and enhanced RAD51 filament stability also rescue nascent strand degradation in FANCD2-mutant cells.62 Moreover, after nucleotide depletion, BRCA2 protects nascent replication strands from degradation and enables replication fork restart.62,86,87 BLM similarly enables replication fork recovery. Moreover, RAD51, BRCA2, and BRCA1 minimize long-tract gene conversion in response to stalled forks, but not DSBs.88 The response of a RAD51-mediated pathway to stalled replication forks is consistent with a potential template switch mechanism.
Postreplication repair (PRR) is a lesion bypass pathway that suppresses broken replication forks to maintain genome stability. The term PRR is actually a misnomer because the lesion is not repaired but bypassed, and this pathway may also be called lesion bypass or DNA damage tolerance. There are two branches to PRR that are best understood in yeast.89,90 The first branch is translesion synthesis (TLS), which bypasses lesions simply by exchanging a high fidelity replication polymerase with a translesion synthesis polymerase. TLS is induced by monoubiquitination of PCNA lysine 164 (K164) by the E2/E3 ubiquitinase RAD6/RAD18. A translesion polymerase then replaces the processive polymerase to bypass the lesion. Some TLS polymerases have low stringency and introduce mismatches at the base lesion, and as a consequence are error prone.91 TLS is conserved from yeast to mammals. The second branch of PRR is error-free postreplication repair (EF-PRR), which bypasses the lesion through a template switch. EF-PRR is induced by monoubiquitination of PCNA lysine K164 by RAD6/RAD18 followed by polyubiquitination of the previously monoubiquitinated K164 by the E2/E3 ubiquitinase UBC13-MMS2/RAD5. EF-PRR is poorly understood in yeast and even more elusive in higher eukaryotes. However, RAD6/RAD18 and UBC13-MMS2 are conserved from yeast to mammals suggesting conservation of function. In addition, mammals possess two functional yeast RAD5 orthologs: helicase-like transcription factor (HLTF) and SNF2 histone-linker PHD-finger RING-finger helicase (SHPRH). Both HLTF and SHPRH mediate PCNA polyubiquitination and suppress chromosomal alterations92,93; however, they are not redundant because they impart lesion specificity.94 Furthermore, HLTF has DNA translocase activity that reverses regressed forks95 and can form a D loop independent of RAD51 and RAD54,96 both of which suggest a mechanism for strand switching. Thus, EF-PRR in mammals likely suppresses replication fork anomalies to minimize fork collapse.
Template switch pathways have been mostly studied in bacteria and yeast by observing the switch between nonallelic repeats that result in a chromosomal alteration. In bacteria, a template switch was observed between inverted97,98 and direct99 repeats and found to be independent of the recA recombinase. In yeast, a template switch between repeats occurred during BIR.100 Faulty template switching was also observed between inverted repeats during replication that required RAD59, but not RAD51.101 In fission102 and budding103,104 yeast, replication-based pathways without a DSB fuse inverted repeats to generate palindromic chromosomes. In fission yeast, HR was found to be primarily responsible for fusing repeats after replication forks were experimentally induced to stall at a reporter. In budding yeast, a genetically undefined pathway was found to be responsible for spontaneous inverted repeat fusion and this process was suppressed by HR and EF-PRR. Thus, template switch-mediated repeat fusion is observed in bacteria and yeast, although the causal pathways for spontaneous fusion are not fully understood.
Mammalian cells are potentially vulnerable to template switch-mediated rearrangements because of the high number of repeats found in the genome. Approximately 11% of the human genome is composed of 300-bp Alu repeat elements and an additional 5% includes larger repeat regions called low copy number repeats (LCRs).105 Furthermore, pericentromeres, centromeres, and telomeres are composed of repeats that are dispersed in the chromosomal arms.106-109 These repeats are potential substrates for rearrangements.105 Replication forks are prone to stalling in areas dense with repeats such as telomeres,110 tRNA genes,111,112 and triplet repeats.113,114 Inverted repeats can form hairpins that have the potential to stall replication forks and cause chromosomal rearrangements in yeast115 and stimulate PCNA polyubiquitination in human cell-free extracts.116 Rearrangements are found at palindromic structures in humans,117 suggesting that they originate from a homology-based mechanism combined with defects in DSB repair20 or DNA synthesis.118-120 DNA repeats pose a particular problem for replication forks because they can form cruciforms, hairpins, triplex H-DNAs, left handed Z-DNAs, and slipped strand S-DNAs.121 Repeat-induced chromosomal rearrangements are common. Copy number variation is a spontaneous change in the number of DNA segments that occurs between repeats.44,120 Moreover, monozygotic twins display different DNA CNV profiles, demonstrating plasticity involving repeats.122 CNV is a common event that is important for murine123 and primate124,125 evolution. Palindromic chromosomes are also found in human cells50,117 and the involvement of repeats at the rearrangement joins suggests a DNA synthesis mechanism such as faulty template switch or fork stalling and template switching.40,45 The etiology of these structural variations is not known, but could involve defects in DSB repair or DNA synthesis.
Recently, we described two template switch pathways in wild type mouse embryonic stem cells that fused inverted repeats to generate unstable multipericentric chromosomes.14 The level of sequence identity within the repeats determined pathway choice: exposure to γ-radiation enhanced the fusion of identical but not mismatched repeats whereas exposure to UV light had the opposite affect. Thus, genotoxic exposure delineated two pathways that fuse repeats based on sequence identity. Even though these are distinct pathways, both appear to fuse repeats as a consequence of replication fork stalling rather than DSB repair since fusion of both identical and mismatched repeats was spontaneous (a DSB was not induced) and enhanced by hydroxyurea-induced replication fork stalling (hydroxyurea depletes nucleotides to stall forks).
We hypothesized that HR enabled identical repeat fusion because HR repairs damage caused by hydroxyurea and γ-radiation but not UV light84,126 and only identical repeats contain sufficient homology for HR.127 Repeat fusion was tested in BLM-deficient cells which exhibit unregulated HR.128 We found that BLM-deficient cells exhibited enhanced fusion of identical, but not mismatched, repeats and that reducing the level of either RAD51 or BRCA2 substantially reduced identical repeat fusion in these cells.14 The existence of a BLM-regulated pathway that depends on RAD51 and BRCA2 and fuses identical, but not mismatched, repeats is consistent with HR.
We hypothesized that EF-PRR fuses mismatched repeats because EF-PRR, as measured by PCNA ubiquitination, processes damage induced by UV light but not by γ-radiation.93,129 Repeat fusion was tested in RAD18-mutant cells130 which do not efficiently ubiquitinate PCNA14 and should therefore be defective in EF-PRR. We found that lack of RAD18 slightly decreased identical repeat fusion, possibly reflecting a subtle role in HR.131-133 In contrast, loss of RAD18 almost completely ablated mismatch repeat fusion implicating a role for EF-PRR.14 Moreover, mutation of a nonprocessive 3′→5′ exonuclease, TREX2, ablated mismatch repeat fusion but enhanced identical repeat fusion, clearly delineating these pathways. TREX2 appears to be epistatic with RAD18 with regard to PCNA ubiquitination and both are important for replication fork maintenance. These observations are consistent with an epistatic relationship for RAD18 and TREX2 mediating mismatch repeat fusion through EF-PRR.
Both the BLM-regulated fusion of identical repeats and the RAD18/TREX2-mediated fusion of mismatched repeats generate very complicated chromosomal rearrangements that include dipericentrics and EPTs.14 We propose that a DNA synthesis pathway causes dipericentrics as a result of a template switch that bypasses a hairpin followed by replication to the telomere. Subsequent breakage-fusion-bridge cycles cause further alterations as chromosomes segregate during mitosis. Interestingly, using a combination of SKY and LSPs on interphase nuclei we showed that a repeat fusion reporter was amplified, mobile, and located in micronuclei. Thus, template switch mechanisms have the potential to generate extrachromosomal double minutes that enhance cancer development and progression and cause resistance to chemotherapeutic agents.134 These repeat fusion pathways could influence cancer etiology and drug effectiveness.
Targets for Cancer Therapy
Simple balanced chromosomal rearrangements generate gene fusions that can be exploited for cancer therapy. Cancers with rearrangements that activate kinases might be susceptible to small molecule inhibitors,135 for example the tyrosine kinase inhibitors imatinib and crizotinib are for cancers possessing the BCR-ABL fusion protein-positive cancers136 and for EML4-ALK and ROS1-positive NSCLC,28,137 respectively. Another example is the use of all-trans-retinoic acid (ATRA) for PML patients with the PML/RARA fusion. Retinoic acids are key players in myeloid differentiation and act through their agonistic nuclear receptors (RAR α/RXR) to modulate the expression of target genes. The PML-RARA fusion generates an oncoprotein that blocks granulocytic differentiation, and ATRA is thought to modulate cellular differentiation that is dependent on PML-RARα proteolysis.138 Patients that become resistant to ATRA are treated with arsenic trioxide (As2O3), which exerts its therapeutic effect by promoting degradation of the PML/RARA protein that drives the growth of acute promyelocytic leukemia cells.139 ATRA is also able to overcome the differentiation block induced by MLL chimeric proteins in acute promyelocytic leukemia and therefore constitutes the standard of care for induction therapy in this disease. In addition to kinase inhibitors, thapsigargin, a sarco/endoplasmic reticulum Ca2+-ATPases (SERCA) inhibitor, targets the PAX3-FOXO1 fusion. Thapsigargin activates AKT to alter cytosolic calcium levels by blocking the ability to pump calcium into the sarcoplasmic and endoplasmic reticulum, thereby inhibiting the fusion of autophagosomes with lysosomes and ultimately causing endoplasmic reticulum stress and cell death.140,141 Thus, the fusion products from simple balanced translocations represent good therapeutic targets for the treatment of cancer.
In addition to targeting fusion proteins, future cancer therapies could attack the pathway that compensates for the primary DNA repair defect that initially caused the cancer. The compensatory pathway is an attractive target because it would attack the tumor on multiple fronts. The first front would take advantage of synthetic sickness/lethality.142 A drug that targets the compensatory pathway would enhance sensitivity to commonly used cytotoxic drugs that cause DNA damage. Such a drug would be especially effective if the compensatory pathway is adept at bypassing the cytotoxic drug-induced lesions.143 In addition, this class of drug would have a good therapeutic index for cases where only the cancer, but not the patient, is mutant for the primary pathway as a result of loss of heterozygosity. The second front would prohibit further genome modification.144 The compensatory pathway is often mutagenic because it is overused and under-regulated in an attempt to atone for the primary defect. In addition, lesion bypass pathways such as TLS are often error prone.143 Thus, a drug that targets the compensatory pathway would suppress mutations that might otherwise enhance cancer development and metastasis and lead to drug resistance. These novel drugs would be especially attractive when lesion bypass mechanisms compensate for DNA repair defects because such mechanisms are nonessential for cell and organism viability,145,146 unlike NHEJ56,147-149 and HR.150,151 Thus, a drug that attacks a compensatory pathway has the potential to both enhance sensitivity to genotoxic therapeutics and reduce further mutations.
Summary
Chromosomal rearrangements frequently found in cancers are proposed to facilitate cancer development, progression, metastasis, and drug resistance. Cytogenetics and NGS have revealed simple and complex chromosomal rearrangements in many cancer cells with more expected as throughput increases. Knowledge of such rearrangements will enable drug development. Many drugs that target fusion proteins generated from simple rearrangements are currently used in treatment of cancers. Novel drug strategies could be developed that attack compensatory pathways to enhance the efficacy of current cytotoxic drugs through synthetic sickness/lethality and suppress continued rearrangements that would otherwise facilitate cancer progression, metastasis, and drug resistance. Thus, a better understanding of cancer-causing chromosomal rearrangements will enable the development of novel anticancer regimens.
Glossary
Abbreviations:
- aCGH
array comparative genomic hybridization
- ALK
anaplastic lymphoma kinase
- ALL
acute lymphoblastic leukemia
- AML
acute myelogenous leukemia
- ATM
ataxia telangiectasia mutated
- ATRA
all-trans-retinoic acid
- BCR
breakpoint cluster region
- BIR
break-induced replication
- BLM
Bloom Syndrome mutated
- BRCA
breast cancer
- CGH
comparative genomic hybridization
- cCGH
conventional CGH
- CML
chronic myelogenous leukemia
- CNV
copy number variation
- DSB
DNA double strand break
- EF-PRR
error free-postreplication repair
- EML4
echinoderm microtubule-associated protein like 4
- EPT
extra pericentromeres and telomere
- ETO
Eight-Twenty One oncoprotein
- FISH
fluorescence in situ hybridization
- FOX
forkhead box protein
- HLTF
helicase-like transcription factor
- HTGTS
high-throughput genome-wide translocation sequencing
- HR
homologous recombination
- LCR
low copy number repeat
- LSP
locus specific probe
- MLL
mixed lineage leukemia
- NGS
next-generation sequencing
- NHEJ
nonhomologous end joining
- NSCLC
non-small cell lung cancer
- PAX
paired box
- PRR
post replication repair
- PML
promyelocytic leukemia
- RARA
retinoic acid receptor alpha
- ROS1
c-ros oncogene 1
- SKY
spectral karyotyping
- SNP
single nucleotide polymorphism
- TLS
translesion synthesis
- WGS
whole genome sequencing
Acknowledgments
This work was supported by the following grants from the NIH: 2P01AG017242–12 and 1 RO1 ES022054–01 to PH and an ACS grant RSG-11–021–01-CNE to CM. We would also like to thank the CTRC (CA054174).
References
- 1.Meyerson M, Pellman D. . Cancer genomes evolve by pulverizing single chromosomes. Cell 2011; 144:9 - 10; http://dx.doi.org/ 10.1016/j.cell.2010.12.025; PMID: 21215363 [DOI] [PubMed] [Google Scholar]
- 2.Rowley JD. . Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243:290 - 3; http://dx.doi.org/ 10.1038/243290a0; PMID: 4126434 [DOI] [PubMed] [Google Scholar]
- 3.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. . Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46:225 - 33; http://dx.doi.org/ 10.1038/ng.2891; PMID: 24487277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klco JM, Spencer DH, Miller CA, Griffith M, Lamprecht TL, O’Laughlin M, Fronick C, Magrini V, Demeter RT, Fulton RS, et al. . Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell 2014; 25:379 - 92; http://dx.doi.org/ 10.1016/j.ccr.2014.01.031; PMID: 24613412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caspersson T, Zech L, Modest EJ, Foley GE, Wagh U, Simonsson E. . Chemical differentiation with fluorescent alkylating agents in Vicia faba metaphase chromosomes. Exp Cell Res 1969; 58:128 - 40; http://dx.doi.org/ 10.1016/0014-4827(69)90123-2; PMID: 5404060 [DOI] [PubMed] [Google Scholar]
- 6.Caspersson T, Hultén M, Lindsten J, Zech L. . Distinction between extra G-like chromosomes by quinacrine mustard fluorescence analysis. Exp Cell Res 1970; 63:240 - 3; http://dx.doi.org/ 10.1016/0014-4827(70)90363-0; PMID: 4250490 [DOI] [PubMed] [Google Scholar]
- 7.Volpi EV, Bridger JM. . FISH glossary: an overview of the fluorescence in situ hybridization technique. Biotechniques 2008; 45:385 - 6, 388, 390 passim; http://dx.doi.org/ 10.2144/000112811; PMID: 18855767 [DOI] [PubMed] [Google Scholar]
- 8.Schröck E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, Ning Y, Ledbetter DH, Bar-Am I, Soenksen D, et al. . Multicolor spectral karyotyping of human chromosomes. Science 1996; 273:494 - 7; http://dx.doi.org/ 10.1126/science.273.5274.494; PMID: 8662537 [DOI] [PubMed] [Google Scholar]
- 9.Liyanage M, Coleman A, du Manoir S, Veldman T, McCormack S, Dickson RB, Barlow C, Wynshaw-Boris A, Janz S, Wienberg J, et al. . Multicolour spectral karyotyping of mouse chromosomes. Nat Genet 1996; 14:312 - 5; http://dx.doi.org/ 10.1038/ng1196-312; PMID: 8896561 [DOI] [PubMed] [Google Scholar]
- 10.Veldman T, Vignon C, Schröck E, Rowley JD, Ried T. . Hidden chromosome abnormalities in haematological malignancies detected by multicolour spectral karyotyping. Nat Genet 1997; 15:406 - 10; http://dx.doi.org/ 10.1038/ng0497-406; PMID: 9090389 [DOI] [PubMed] [Google Scholar]
- 11.Coleman AE, Schröck E, Weaver Z, du Manoir S, Yang F, Ferguson-Smith MA, Ried T, Janz S. . Previously hidden chromosome aberrations in T(12;15)-positive BALB/c plasmacytomas uncovered by multicolor spectral karyotyping. Cancer Res 1997; 57:4585 - 92; PMID: 9377573 [PubMed] [Google Scholar]
- 12.Rao PH, Cigudosa JC, Ning Y, Calasanz MJ, Iida S, Tagawa S, Michaeli J, Klein B, Dalla-Favera R, Jhanwar SC, et al. . Multicolor spectral karyotyping identifies new recurring breakpoints and translocations in multiple myeloma. Blood 1998; 92:1743 - 8; PMID: 9716604 [PubMed] [Google Scholar]
- 13.Dorritie K, Montagna C, Difilippantonio MJ, Ried T. . Advanced molecular cytogenetics in human and mouse. Expert Rev Mol Diagn 2004; 4:663 - 76; http://dx.doi.org/ 10.1586/14737159.4.5.663; PMID: 15347260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu L, Kim TM, Son MY, Kim SA, Holland CL, Tateishi S, Kim DH, Yew PR, Montagna C, Dumitrache LC, et al. . Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes. Nature 2013; 501:569 - 72; http://dx.doi.org/ 10.1038/nature12500; PMID: 24013173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim TM, Ko JH, Hu L, Kim SA, Bishop AJ, Vijg J, Montagna C, Hasty P. . RAD51 mutants cause replication defects and chromosomal instability. Mol Cell Biol 2012; 32:3663 - 80; http://dx.doi.org/ 10.1128/MCB.00406-12; PMID: 22778135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D. . Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992; 258:818 - 21; http://dx.doi.org/ 10.1126/science.1359641; PMID: 1359641 [DOI] [PubMed] [Google Scholar]
- 17.Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Döhner H, Cremer T, Lichter P. . Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 1997; 20:399 - 407; http://dx.doi.org/; PMID: 9408757 [DOI] [PubMed] [Google Scholar]
- 18.Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, McCall A, Pivnick EK, Hines-Dowell S, Seaver LH, et al. . Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet 2011; 43:1074 - 81; http://dx.doi.org/ 10.1038/ng.944; PMID: 21964572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitelman F, Johansson B, Mertens F. . The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007; 7:233 - 45; http://dx.doi.org/ 10.1038/nrc2091; PMID: 17361217 [DOI] [PubMed] [Google Scholar]
- 20.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. . Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144:27 - 40; http://dx.doi.org/ 10.1016/j.cell.2010.11.055; PMID: 21215367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. . Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 2011; 147:95 - 106; http://dx.doi.org/ 10.1016/j.cell.2011.07.048; PMID: 21962510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. . Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 2011; 147:107 - 19; http://dx.doi.org/ 10.1016/j.cell.2011.07.049; PMID: 21962511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Govind SK, Zia A, Hennings-Yeomans PH, Watson JD, Fraser M, Anghel C, Wyatt AW, van der Kwast T, Collins CC, McPherson JD, et al. . ShatterProof: operational detection and quantification of chromothripsis. BMC Bioinformatics 2014; 15:78; http://dx.doi.org/ 10.1186/1471-2105-15-78; PMID: 24646301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. . COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011; 39:D945 - 50; http://dx.doi.org/ 10.1093/nar/gkq929; PMID: 20952405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nowell PC, Hungerford DA. . Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst 1960; 25:85 - 109; PMID: 14427847 [PubMed] [Google Scholar]
- 26.Kurzrock R, Gutterman JU, Talpaz M. . The molecular genetics of Philadelphia chromosome-positive leukemias. N Engl J Med 1988; 319:990 - 8; http://dx.doi.org/ 10.1056/NEJM198810133191506; PMID: 3047582 [DOI] [PubMed] [Google Scholar]
- 27.Ren R. . Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer 2005; 5:172 - 83; http://dx.doi.org/ 10.1038/nrc1567; PMID: 15719031 [DOI] [PubMed] [Google Scholar]
- 28.Gridelli C, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardiello F. . ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat Rev 2014; 40:300 - 6; http://dx.doi.org/ 10.1016/j.ctrv.2013.07.002; PMID: 23931927 [DOI] [PubMed] [Google Scholar]
- 29.Chin LP, Soo RA, Soong R, Ou SH. . Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol 2012; 7:1625 - 30; http://dx.doi.org/ 10.1097/JTO.0b013e31826baf83; PMID: 23070242 [DOI] [PubMed] [Google Scholar]
- 30.Lee J, Lee SE, Kang SY, Do IG, Lee S, Ha SY, Cho J, Kang WK, Jang J, Ou SH, et al. . Identification of ROS1 rearrangement in gastric adenocarcinoma. Cancer 2013; 119:1627 - 35; http://dx.doi.org/ 10.1002/cncr.27967; PMID: 23400546 [DOI] [PubMed] [Google Scholar]
- 31.Choi CM. . Overview of ALK and ROS1 Rearranged Lung Cancer. Tuberc Respir Dis (Seoul) 2013; 75:236 - 7; http://dx.doi.org/ 10.4046/trd.2013.75.6.236; PMID: 24416053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cerveira N, Bizarro S, Teixeira MR. . MLL-SEPTIN gene fusions in hematological malignancies. Biol Chem 2011; 392:713 - 24; http://dx.doi.org/ 10.1515/BC.2011.072; PMID: 21714766 [DOI] [PubMed] [Google Scholar]
- 33.Bennicelli JL, Edwards RH, Barr FG. . Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma. Proc Natl Acad Sci U S A 1996; 93:5455 - 9; http://dx.doi.org/ 10.1073/pnas.93.11.5455; PMID: 8643596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, Barr FG. . Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res 1994; 54:2869 - 72; PMID: 8187070 [PubMed] [Google Scholar]
- 35.Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, et al. . Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007; 446:758 - 64; http://dx.doi.org/ 10.1038/nature05690; PMID: 17344859 [DOI] [PubMed] [Google Scholar]
- 36.Myatt SS, Lam EW. . The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 2007; 7:847 - 59; http://dx.doi.org/ 10.1038/nrc2223; PMID: 17943136 [DOI] [PubMed] [Google Scholar]
- 37.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ 3rd, Emanuel BS, Rovera G, Barr FG. . Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet 1993; 5:230 - 5; http://dx.doi.org/ 10.1038/ng1193-230; PMID: 8275086 [DOI] [PubMed] [Google Scholar]
- 38.Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, Concordet JP, Thway K, Oberlin O, Pritchard-Jones K, et al. . PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol 2012; 30:1670 - 7; http://dx.doi.org/ 10.1200/JCO.2011.38.5591; PMID: 22454413 [DOI] [PubMed] [Google Scholar]
- 39.So CW, Cleary ML. . Common mechanism for oncogenic activation of MLL by forkhead family proteins. Blood 2003; 101:633 - 9; http://dx.doi.org/ 10.1182/blood-2002-06-1785; PMID: 12393557 [DOI] [PubMed] [Google Scholar]
- 40.Carr AM, Paek AL, Weinert T. . DNA replication: failures and inverted fusions. Semin Cell Dev Biol 2011; 22:866 - 74; http://dx.doi.org/ 10.1016/j.semcdb.2011.10.008; PMID: 22020070 [DOI] [PubMed] [Google Scholar]
- 41.Liu P, Carvalho CM, Hastings PJ, Lupski JR. . Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev 2012; 22:211 - 20; http://dx.doi.org/ 10.1016/j.gde.2012.02.012; PMID: 22440479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang F, Carvalho CM, Lupski JR. . Complex human chromosomal and genomic rearrangements. Trends Genet 2009; 25:298 - 307; http://dx.doi.org/ 10.1016/j.tig.2009.05.005; PMID: 19560228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. . The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 2009; 41:849 - 53; http://dx.doi.org/ 10.1038/ng.399; PMID: 19543269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. . Mechanisms of change in gene copy number. Nat Rev Genet 2009; 10:551 - 64; http://dx.doi.org/ 10.1038/nrg2593; PMID: 19597530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colnaghi R, Carpenter G, Volker M, O’Driscoll M. . The consequences of structural genomic alterations in humans: genomic disorders, genomic instability and cancer. Semin Cell Dev Biol 2011; 22:875 - 85; http://dx.doi.org/ 10.1016/j.semcdb.2011.07.010; PMID: 21802523 [DOI] [PubMed] [Google Scholar]
- 46.Martínez JG, Pérez-Escuredo J, Castro-Santos P, Marcos CA, Pendás JL, Fraga MF, Hermsen MA. . Hypomethylation of LINE-1, and not centromeric SAT-α, is associated with centromeric instability in head and neck squamous cell carcinoma. Cell Oncol (Dordr) 2012; 35:259 - 67; http://dx.doi.org/ 10.1007/s13402-012-0085-5; PMID: 22718136 [DOI] [PubMed] [Google Scholar]
- 47.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, Morsberger LA, Latimer C, McLaren S, Lin ML, et al. . The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010; 467:1109 - 13; http://dx.doi.org/ 10.1038/nature09460; PMID: 20981101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. . Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009; 462:1005 - 10; http://dx.doi.org/ 10.1038/nature08645; PMID: 20033038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McBride DJ, Etemadmoghadam D, Cooke SL, Alsop K, George J, Butler A, Cho J, Galappaththige D, Greenman C, Howarth KD, et al. . Tandem duplication of chromosomal segments is common in ovarian and breast cancer genomes. J Pathol 2012; 227:446 - 55; http://dx.doi.org/ 10.1002/path.4042; PMID: 22514011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka H, Bergstrom DA, Yao MC, Tapscott SJ. . Widespread and nonrandom distribution of DNA palindromes in cancer cells provides a structural platform for subsequent gene amplification. Nat Genet 2005; 37:320 - 7; http://dx.doi.org/ 10.1038/ng1515; PMID: 15711546 [DOI] [PubMed] [Google Scholar]
- 51.Zhang CZ, Leibowitz ML, Pellman D. . Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev 2013; 27:2513 - 30; http://dx.doi.org/ 10.1101/gad.229559.113; PMID: 24298051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korbel JO, Campbell PJ. . Criteria for inference of chromothripsis in cancer genomes. Cell 2013; 152:1226 - 36; http://dx.doi.org/ 10.1016/j.cell.2013.02.023; PMID: 23498933 [DOI] [PubMed] [Google Scholar]
- 53.Maher CA, Wilson RK. . Chromothripsis and human disease: piecing together the shattering process. Cell 2012; 148:29 - 32; http://dx.doi.org/ 10.1016/j.cell.2012.01.006; PMID: 22265399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. . DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012; 482:53 - 8; http://dx.doi.org/ 10.1038/nature10802; PMID: 22258507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burma S, Chen BP, Chen DJ. . Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006; 5:1042 - 8; http://dx.doi.org/ 10.1016/j.dnarep.2006.05.026; PMID: 16822724 [DOI] [PubMed] [Google Scholar]
- 56.Zhu C, Bogue MA, Lim DS, Hasty P, Roth DB. . Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell 1996; 86:379 - 89; http://dx.doi.org/ 10.1016/S0092-8674(00)80111-7; PMID: 8756720 [DOI] [PubMed] [Google Scholar]
- 57.Lim DS, Vogel H, Willerford DM, Sands AT, Platt KA, Hasty P. . Analysis of ku80-mutant mice and cells with deficient levels of p53. Mol Cell Biol 2000; 20:3772 - 80; http://dx.doi.org/ 10.1128/MCB.20.11.3772-3780.2000; PMID: 10805721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH. . DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc Natl Acad Sci U S A 1999; 96:14899 - 904; http://dx.doi.org/ 10.1073/pnas.96.26.14899; PMID: 10611310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Choi YJ, Hanes MA, Marple T, Vogel H, Hasty P. . Deleting Ku70 is milder than deleting Ku80 in p53-mutant mice and cells. Oncogene 2009; 28:1875 - 8; http://dx.doi.org/ 10.1038/onc.2009.57; PMID: 19330025 [DOI] [PubMed] [Google Scholar]
- 60.Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. . Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010; 329:219 - 23; http://dx.doi.org/ 10.1126/science.1192277; PMID: 20538911 [DOI] [PubMed] [Google Scholar]
- 61.Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. . Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 2010; 39:25 - 35; http://dx.doi.org/ 10.1016/j.molcel.2010.06.026; PMID: 20598602 [DOI] [PubMed] [Google Scholar]
- 62.Schlacher K, Wu H, Jasin M. . A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106 - 16; http://dx.doi.org/ 10.1016/j.ccr.2012.05.015; PMID: 22789542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. . 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243 - 54; http://dx.doi.org/ 10.1016/j.cell.2010.03.012; PMID: 20362325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel AG, Sarkaria JN, Kaufmann SH. . Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A 2011; 108:3406 - 11; http://dx.doi.org/ 10.1073/pnas.1013715108; PMID: 21300883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bunting SF, Nussenzweig A. . End-joining, translocations and cancer. Nat Rev Cancer 2013; 13:443 - 54; http://dx.doi.org/ 10.1038/nrc3537; PMID: 23760025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bennardo N, Cheng A, Huang N, Stark JM. . Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 2008; 4:e1000110; http://dx.doi.org/ 10.1371/journal.pgen.1000110; PMID: 18584027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bennardo N, Gunn A, Cheng A, Hasty P, Stark JM. . Limiting the persistence of a chromosome break diminishes its mutagenic potential. PLoS Genet 2009; 5:e1000683; http://dx.doi.org/ 10.1371/journal.pgen.1000683; PMID: 19834534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McVey M, Lee SE. . MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet 2008; 24:529 - 38; http://dx.doi.org/ 10.1016/j.tig.2008.08.007; PMID: 18809224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.San Filippo J, Sung P, Klein H. . Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229 - 57; http://dx.doi.org/ 10.1146/annurev.biochem.77.061306.125255; PMID: 18275380 [DOI] [PubMed] [Google Scholar]
- 70.Bachrati CZ, Hickson ID. . RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J 2003; 374:577 - 606; http://dx.doi.org/ 10.1042/BJ20030491; PMID: 12803543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. . The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell 1995; 83:655 - 66; http://dx.doi.org/ 10.1016/0092-8674(95)90105-1; PMID: 7585968 [DOI] [PubMed] [Google Scholar]
- 72.Wu L, Hickson ID. . The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003; 426:870 - 4; http://dx.doi.org/ 10.1038/nature02253; PMID: 14685245 [DOI] [PubMed] [Google Scholar]
- 73.Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. . Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 2005; 19:339 - 50; http://dx.doi.org/ 10.1101/gad.322605; PMID: 15687257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ira G, Malkova A, Liberi G, Foiani M, Haber JE. . Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 2003; 115:401 - 11; http://dx.doi.org/ 10.1016/S0092-8674(03)00886-9; PMID: 14622595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raynard S, Bussen W, Sung P. . A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J Biol Chem 2006; 281:13861 - 4; http://dx.doi.org/ 10.1074/jbc.C600051200; PMID: 16595695 [DOI] [PubMed] [Google Scholar]
- 76.Llorente B, Smith CE, Symington LS. . Break-induced replication: what is it and what is it for?. Cell Cycle 2008; 7:859 - 64; http://dx.doi.org/ 10.4161/cc.7.7.5613; PMID: 18414031 [DOI] [PubMed] [Google Scholar]
- 77.Malkova A, Ira G. . Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev 2013; 23:271 - 9; http://dx.doi.org/ 10.1016/j.gde.2013.05.007; PMID: 23790415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. . Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014; 343:88 - 91; http://dx.doi.org/ 10.1126/science.1243211; PMID: 24310611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Branzei D, Foiani M. . Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 2010; 11:208 - 19; http://dx.doi.org/ 10.1038/nrm2852; PMID: 20177396 [DOI] [PubMed] [Google Scholar]
- 80.Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M. . Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell 2005; 17:153 - 9; http://dx.doi.org/ 10.1016/j.molcel.2004.11.032; PMID: 15629726 [DOI] [PubMed] [Google Scholar]
- 81.Olavarrieta L, Martínez-Robles ML, Sogo JM, Stasiak A, Hernández P, Krimer DB, Schvartzman JB. . Supercoiling, knotting and replication fork reversal in partially replicated plasmids. Nucleic Acids Res 2002; 30:656 - 66; http://dx.doi.org/ 10.1093/nar/30.3.656; PMID: 11809877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sogo JM, Lopes M, Foiani M. . Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002; 297:599 - 602; http://dx.doi.org/ 10.1126/science.1074023; PMID: 12142537 [DOI] [PubMed] [Google Scholar]
- 83.Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Fréon K, Murray JM, Carr AM, Baldacci G. . Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 2010; 39:346 - 59; http://dx.doi.org/ 10.1016/j.molcel.2010.07.015; PMID: 20705238 [DOI] [PubMed] [Google Scholar]
- 84.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. . Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492 - 502; http://dx.doi.org/ 10.1016/j.molcel.2010.01.021; PMID: 20188668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. . Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 2011; 25:1320 - 7; http://dx.doi.org/ 10.1101/gad.2053211; PMID: 21685366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. . Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529 - 42; http://dx.doi.org/ 10.1016/j.cell.2011.03.041; PMID: 21565612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim TM, Son MY, Dodds S, Hu L, Hasty P. . Deletion of BRCA2 exon 27 causes defects in response to both stalled and collapsed replication forks. Mutat Res 2014; In press http://dx.doi.org/ 10.1016/j.mrfmmm.2014.06.003 [DOI] [PubMed] [Google Scholar]
- 88.Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, Scully R. . BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 2014; 510:556 - 9; http://dx.doi.org/ 10.1038/nature13295; PMID: 24776801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ulrich HD. . Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst) 2009; 8:461 - 9; http://dx.doi.org/ 10.1016/j.dnarep.2009.01.006; PMID: 19217833 [DOI] [PubMed] [Google Scholar]
- 90.Lee KY, Myung K. . PCNA modifications for regulation of post-replication repair pathways. Mol Cells 2008; 26:5 - 11; PMID: 18525240 [PMC free article] [PubMed] [Google Scholar]
- 91.Makridakis NM, Reichardt JK. . Translesion DNA polymerases and cancer. Front Genet 2012; 3:174; http://dx.doi.org/ 10.3389/fgene.2012.00174; PMID: 22973298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Motegi A, Sood R, Moinova H, Markowitz SD, Liu PP, Myung K. . Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J Cell Biol 2006; 175:703 - 8; http://dx.doi.org/ 10.1083/jcb.200606145; PMID: 17130289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Motegi A, Liaw HJ, Lee KY, Roest HP, Maas A, Wu X, Moinova H, Markowitz SD, Ding H, Hoeijmakers JH, et al. . Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc Natl Acad Sci U S A 2008; 105:12411 - 6; http://dx.doi.org/ 10.1073/pnas.0805685105; PMID: 18719106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin JR, Zeman MK, Chen JY, Yee MC, Cimprich KA. . SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell 2011; 42:237 - 49; http://dx.doi.org/ 10.1016/j.molcel.2011.02.026; PMID: 21396873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Blastyák A, Hajdú I, Unk I, Haracska L. . Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol 2010; 30:684 - 93; http://dx.doi.org/ 10.1128/MCB.00863-09; PMID: 19948885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Burkovics P, Sebesta M, Balogh D, Haracska L, Krejci L. . Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res 2014; 42:1711 - 20; http://dx.doi.org/ 10.1093/nar/gkt1040; PMID: 24198246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bi X, Liu LF. . DNA rearrangement mediated by inverted repeats. Proc Natl Acad Sci U S A 1996; 93:819 - 23; http://dx.doi.org/ 10.1073/pnas.93.2.819; PMID: 8570641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahmed A, Podemski L. . Observations on template switching during DNA replication through long inverted repeats. Gene 1998; 223:187 - 94; http://dx.doi.org/ 10.1016/S0378-1119(98)00159-0; PMID: 9858727 [DOI] [PubMed] [Google Scholar]
- 99.Goldfless SJ, Morag AS, Belisle KA, Sutera VA Jr., Lovett ST. . DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol Cell 2006; 21:595 - 604; http://dx.doi.org/ 10.1016/j.molcel.2006.01.025; PMID: 16507358 [DOI] [PubMed] [Google Scholar]
- 100.Smith CE, Llorente B, Symington LS. . Template switching during break-induced replication. Nature 2007; 447:102 - 5; http://dx.doi.org/ 10.1038/nature05723; PMID: 17410126 [DOI] [PubMed] [Google Scholar]
- 101.Mott C, Symington LS. . RAD51-independent inverted-repeat recombination by a strand-annealing mechanism. DNA Repair (Amst) 2011; 10:408 - 15; http://dx.doi.org/ 10.1016/j.dnarep.2011.01.007; PMID: 21317047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mizuno K, Lambert S, Baldacci G, Murray JM, Carr AM. . Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev 2009; 23:2876 - 86; http://dx.doi.org/ 10.1101/gad.1863009; PMID: 20008937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paek AL, Kaochar S, Jones H, Elezaby A, Shanks L, Weinert T. . Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev 2009; 23:2861 - 75; http://dx.doi.org/ 10.1101/gad.1862709; PMID: 20008936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paek AL, Jones H, Kaochar S, Weinert T. . The role of replication bypass pathways in dicentric chromosome formation in budding yeast. Genetics 2010; 186:1161 - 73; http://dx.doi.org/ 10.1534/genetics.110.122663; PMID: 20837992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Richard GF, Kerrest A, Dujon B. . Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev 2008; 72:686 - 727; http://dx.doi.org/ 10.1128/MMBR.00011-08; PMID: 19052325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin KW, Yan J. . Endings in the middle: current knowledge of interstitial telomeric sequences. Mutat Res 2008; 658:95 - 110; http://dx.doi.org/ 10.1016/j.mrrev.2007.08.006; PMID: 17921045 [DOI] [PubMed] [Google Scholar]
- 107.Kuznetsova I, Podgornaya O, Ferguson-Smith MA. . High-resolution organization of mouse centromeric and pericentromeric DNA. Cytogenet Genome Res 2006; 112:248 - 55; http://dx.doi.org/ 10.1159/000089878; PMID: 16484780 [DOI] [PubMed] [Google Scholar]
- 108.Podgornaya O, Gavrilova E, Stephanova V, Demin S, Komissarov A. . Large tandem repeats make up the chromosome bar code: a hypothesis. Adv Protein Chem Struct Biol 2013; 90:1 - 30; http://dx.doi.org/ 10.1016/B978-0-12-410523-2.00001-8; PMID: 23582200 [DOI] [PubMed] [Google Scholar]
- 109.Bolzán AD. . Chromosomal aberrations involving telomeres and interstitial telomeric sequences. Mutagenesis 2012; 27:1 - 15; http://dx.doi.org/ 10.1093/mutage/ger052; PMID: 21857006 [DOI] [PubMed] [Google Scholar]
- 110.Makovets S, Herskowitz I, Blackburn EH. . Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol Cell Biol 2004; 24:4019 - 31; http://dx.doi.org/ 10.1128/MCB.24.9.4019-4031.2004; PMID: 15082794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Deshpande AM, Newlon CS. . DNA replication fork pause sites dependent on transcription. Science 1996; 272:1030 - 3; http://dx.doi.org/ 10.1126/science.272.5264.1030; PMID: 8638128 [DOI] [PubMed] [Google Scholar]
- 112.Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA. . The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 2003; 12:1525 - 36; http://dx.doi.org/ 10.1016/S1097-2765(03)00456-8; PMID: 14690605 [DOI] [PubMed] [Google Scholar]
- 113.Sundararajan R, Freudenreich CH. . Expanded CAG/CTG repeat DNA induces a checkpoint response that impacts cell proliferation in Saccharomyces cerevisiae. PLoS Genet 2011; 7:e1001339; http://dx.doi.org/ 10.1371/journal.pgen.1001339; PMID: 21437275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM. . Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol 2009; 16:226 - 8; http://dx.doi.org/ 10.1038/nsmb.1527; PMID: 19136957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. . Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci U S A 2008; 105:9936 - 41; http://dx.doi.org/ 10.1073/pnas.0804510105; PMID: 18632578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schmutz V, Wagner J, Janel-Bintz R, Fuchs RP, Cordonnier AM. . Requirements for PCNA monoubiquitination in human cell-free extracts. DNA Repair (Amst) 2007; 6:1726 - 31; http://dx.doi.org/ 10.1016/j.dnarep.2007.06.003; PMID: 17669698 [DOI] [PubMed] [Google Scholar]
- 117.Hsiao MC, Piotrowski A, Alexander J, Callens T, Fu C, Mikhail FM, Claes KB, Messiaen L. . Palindrome-mediated and replication-dependent pathogenic structural rearrangements within the NF1 gene. Hum Mutat 2014; 35:891 - 8; http://dx.doi.org/ 10.1002/humu.22569; PMID: 24760680 [DOI] [PubMed] [Google Scholar]
- 118.Liu P, Erez A, Nagamani SC, Dhar SU, Kołodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. . Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 2011; 146:889 - 903; http://dx.doi.org/ 10.1016/j.cell.2011.07.042; PMID: 21925314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee JA, Carvalho CM, Lupski JR. . A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 2007; 131:1235 - 47; http://dx.doi.org/ 10.1016/j.cell.2007.11.037; PMID: 18160035 [DOI] [PubMed] [Google Scholar]
- 120.Hastings PJ, Ira G, Lupski JR. . A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 2009; 5:e1000327; http://dx.doi.org/ 10.1371/journal.pgen.1000327; PMID: 19180184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mirkin EV, Mirkin SM. . Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 2007; 71:13 - 35; http://dx.doi.org/ 10.1128/MMBR.00030-06; PMID: 17347517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bruder CE, Piotrowski A, Gijsbers AA, Andersson R, Erickson S, Diaz de Ståhl T, Menzel U, Sandgren J, von Tell D, Poplawski A, et al. . Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet 2008; 82:763 - 71; http://dx.doi.org/ 10.1016/j.ajhg.2007.12.011; PMID: 18304490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cheung J, Wilson MD, Zhang J, Khaja R, MacDonald JR, Heng HH, Koop BF, Scherer SW. . Recent segmental and gene duplications in the mouse genome. Genome Biol 2003; 4:R47; http://dx.doi.org/ 10.1186/gb-2003-4-8-r47; PMID: 12914656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bailey JA, Eichler EE. . Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet 2006; 7:552 - 64; http://dx.doi.org/ 10.1038/nrg1895; PMID: 16770338 [DOI] [PubMed] [Google Scholar]
- 125.Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. . Genetic evidence for complex speciation of humans and chimpanzees. Nature 2006; 441:1103 - 8; http://dx.doi.org/ 10.1038/nature04789; PMID: 16710306 [DOI] [PubMed] [Google Scholar]
- 126.Morimatsu M, Donoho G, Hasty P. . Cells deleted for Brca2 COOH terminus exhibit hypersensitivity to gamma-radiation and premature senescence. Cancer Res 1998; 58:3441 - 7; PMID: 9699678 [PubMed] [Google Scholar]
- 127.Waldman AS, Liskay RM. . Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol Cell Biol 1988; 8:5350 - 7; PMID: 2854196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A. . Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet 2000; 26:424 - 9; http://dx.doi.org/ 10.1038/82548; PMID: 11101838 [DOI] [PubMed] [Google Scholar]
- 129.Kannouche PL, Wing J, Lehmann AR. . Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 2004; 14:491 - 500; http://dx.doi.org/ 10.1016/S1097-2765(04)00259-X; PMID: 15149598 [DOI] [PubMed] [Google Scholar]
- 130.Tateishi S, Niwa H, Miyazaki J, Fujimoto S, Inoue H, Yamaizumi M. . Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol Cell Biol 2003; 23:474 - 81; http://dx.doi.org/ 10.1128/MCB.23.2.474-481.2003; PMID: 12509447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Helchowski CM, Skow LF, Roberts KH, Chute CL, Canman CE. . A small ubiquitin binding domain inhibits ubiquitin-dependent protein recruitment to DNA repair foci. Cell Cycle 2013; 12:3749 - 58; http://dx.doi.org/ 10.4161/cc.26640; PMID: 24107634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Huang J, Huen MS, Kim H, Leung CC, Glover JN, Yu X, Chen J. . RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat Cell Biol 2009; 11:592 - 603; http://dx.doi.org/ 10.1038/ncb1865; PMID: 19396164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saberi A, Hochegger H, Szuts D, Lan L, Yasui A, Sale JE, Taniguchi Y, Murakawa Y, Zeng W, Yokomori K, et al. . RAD18 and poly(ADP-ribose) polymerase independently suppress the access of nonhomologous end joining to double-strand breaks and facilitate homologous recombination-mediated repair. Mol Cell Biol 2007; 27:2562 - 71; http://dx.doi.org/ 10.1128/MCB.01243-06; PMID: 17242200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shimizu N. . Extrachromosomal double minutes and chromosomal homogeneously staining regions as probes for chromosome research. Cytogenet Genome Res 2009; 124:312 - 26; http://dx.doi.org/ 10.1159/000218135; PMID: 19556783 [DOI] [PubMed] [Google Scholar]
- 135.Shaw AT, Hsu PP, Awad MM, Engelman JA. . Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer 2013; 13:772 - 87; http://dx.doi.org/ 10.1038/nrc3612; PMID: 24132104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Goldman JM, Melo JV. . Chronic myeloid leukemia--advances in biology and new approaches to treatment. N Engl J Med 2003; 349:1451 - 64; http://dx.doi.org/ 10.1056/NEJMra020777; PMID: 14534339 [DOI] [PubMed] [Google Scholar]
- 137.Reungwetwattana T, Dy GK. . Targeted therapies in development for non-small cell lung cancer. J Carcinog 2013; 12:22; http://dx.doi.org/ 10.4103/1477-3163.123972; PMID: 24574860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jing Y. . The PML-RARalpha fusion protein and targeted therapy for acute promyelocytic leukemia. Leuk Lymphoma 2004; 45:639 - 48; http://dx.doi.org/ 10.1080/10428190310001609933; PMID: 15160934 [DOI] [PubMed] [Google Scholar]
- 139.Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand-Breitenbach V, Jeanne M, et al. . Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 2010; 328:240 - 3; http://dx.doi.org/ 10.1126/science.1183424; PMID: 20378816 [DOI] [PubMed] [Google Scholar]
- 140.Jothi M, Mal M, Keller C, Mal AK. . Small molecule inhibition of PAX3-FOXO1 through AKT activation suppresses malignant phenotypes of alveolar rhabdomyosarcoma. Mol Cancer Ther 2013; 12:2663 - 74; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0277; PMID: 24107448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ganley IG, Wong PM, Gammoh N, Jiang X. . Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell 2011; 42:731 - 43; http://dx.doi.org/ 10.1016/j.molcel.2011.04.024; PMID: 21700220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ashworth A. . A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 2008; 26:3785 - 90; http://dx.doi.org/ 10.1200/JCO.2008.16.0812; PMID: 18591545 [DOI] [PubMed] [Google Scholar]
- 143.Salehan MR, Morse HR. . DNA damage repair and tolerance: a role in chemotherapeutic drug resistance. Br J Biomed Sci 2013; 70:31 - 40; PMID: 23617096 [DOI] [PubMed] [Google Scholar]
- 144.Rosenberg SM, Queitsch C. . Medicine. Combating evolution to fight disease. Science 2014; 343:1088 - 9; http://dx.doi.org/ 10.1126/science.1247472; PMID: 24604189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dumitrache LC, Hu L, Son MY, Li H, Wesevich A, Scully R, Stark J, Hasty P. . Trex2 enables spontaneous sister chromatid exchanges without facilitating DNA double-strand break repair. Genetics 2011; 188:787 - 97; http://dx.doi.org/ 10.1534/genetics.111.129833; PMID: 21546543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Parra D, Manils J, Castellana B, Viña-Vilaseca A, Morán-Salvador E, Vázquez-Villoldo N, Tarancón G, Borràs M, Sancho S, Benito C, et al. . Increased Susceptibility to Skin Carcinogenesis in TREX2 Knockout Mice. Cancer Res 2009; 69:6676 - 84; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1208; PMID: 19654293 [DOI] [PubMed] [Google Scholar]
- 147.Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. . Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci U S A 1999; 96:10770 - 5; http://dx.doi.org/ 10.1073/pnas.96.19.10770; PMID: 10485901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Holcomb VB, Rodier F, Choi Y, Busuttil RA, Vogel H, Vijg J, Campisi J, Hasty P. . Ku80 deletion suppresses spontaneous tumors and induces a p53-mediated DNA damage response. Cancer Res 2008; 68:9497 - 502; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2085; PMID: 19010925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li G, Nelsen C, Hendrickson EA. . Ku86 is essential in human somatic cells. Proc Natl Acad Sci U S A 2002; 99:832 - 7; http://dx.doi.org/ 10.1073/pnas.022649699; PMID: 11792868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lim DS, Hasty P. . A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol 1996; 16:7133 - 43; PMID: 8943369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. . Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. [see comments] Nature 1997; 386:804 - 10; http://dx.doi.org/ 10.1038/386804a0; PMID: 9126738 [DOI] [PubMed] [Google Scholar]