

ABSTRACT
The CRISPR-Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated genes) system provides prokaryotic cells with an adaptive and heritable immune response to foreign genetic elements, such as viruses, plasmids, and transposons. It is present in the majority of Archaea and almost half of species of Bacteria. Porphyromonas gingivalis is an important human pathogen that has been proven to be an etiological agent of periodontitis and has been linked to systemic conditions, such as rheumatoid arthritis and cardiovascular disease. At least 95% of clinical strains of P. gingivalis carry CRISPR arrays, suggesting that these arrays play an important function in vivo. Here we show that all four CRISPR arrays present in the P. gingivalis W83 genome are transcribed. For one of the arrays, we demonstrate in vivo activity against double-stranded DNA constructs containing protospacer sequences accompanied at the 3′ end by an NGG protospacer-adjacent motif (PAM). Most of the 44 spacers present in the genome of P. gingivalis W83 share no significant similarity with any known sequences, although 4 spacers are similar to sequences from bacteria found in the oral cavity and the gastrointestinal tract. Four spacers match genomic sequences of the host; however, none of these is flanked at its 3′ terminus by the appropriate PAM element.
IMPORTANCE The CRISPR-Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated genes) system is a unique system that provides prokaryotic cells with an adaptive and heritable immunity. In this report, we show that the CRISPR-Cas system of P. gingivalis, an important human pathogen associated with periodontitis and possibly also other conditions, such as rheumatoid arthritis and cardiovascular disease, is active and provides protection from foreign genetic elements. Importantly, the data presented here may be useful for better understanding the communication between cells in larger bacterial communities and, consequently, the process of disease development and progression.
INTRODUCTION
Prokaryotes are well known for their ability to respond dynamically to the changing environment by means of genetic alterations (1). In addition, they have developed more sophisticated defense mechanisms against foreign nucleic acids, including masking, mutating, or downregulating phage receptors (2, 3); interference with phage/plasmid DNA transfer (4, 5); digestion of nonself DNA by restriction-modification systems (6); and highly specific degradation of foreign nucleic acids in a sequence-dependent manner by the CRISPR-Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated genes) system. CRISPRs were first discovered in 1987 (7), but their function was elucidated only recently (8). They have been identified in more than 80% of Archaea species and about 45% of Bacteria species (9). The CRISPR-Cas system is the only adaptive and heritable prokaryotic immune system identified to date (10).
Structurally, a CRISPR-Cas genetic element consists of an array of repeats interspaced with relatively short DNA stretches, called spacers, with a set of cas genes in close proximity (11). The CRISPR array is located downstream of the leader sequence, which plays an important role in acquisition of new spacers as well as transcription of the CRISPR array. Spacers are short sequences originating from foreign (or sometimes self) nucleic acids that serve as a memory bank of past infections and events involving introduction of nucleic acids to the bacterial cell; their presence enables complementarity-based recognition of nucleic acids and their subsequent degradation. Sequences complementary to the spacers in the target nucleic acids are called protospacers. The exact mechanism of CRISPR-Cas-mediated nucleic acid degradation varies between species. Based on the presence/absence of particular Cas proteins encoded in the genome, CRISPR-Cas systems are divided into three major types (types I to III) and 11 subtypes (12). Cas1 and Cas2 proteins are present in all active systems. The signature protein of type I CRISPR-Cas systems is Cas3, whose DNase activity is responsible for degradation of target DNA (13). In type II systems, the signature protein is Cas9, which contributes to processing of the long precursor transcript, called the pre-crRNA, degradation of the targeted nucleic acid (14), and spacer acquisition (15). The cleavage of pre-crRNA requires trans-activating small crRNA (tracrRNA), base pairing with the repeat fragment of pre-crRNA, and the housekeeping protein RNase III (16). Finally, Cas10 is the signature protein of type III CRISPR-Cas systems, which are further divided into subtypes III-A and III-B; subtype III-A was shown to degrade both DNA and RNA (17, 18), whereas subtype III-B is RNA specific (19). A detailed description of all CRISPR-Cas types is provided in a recent review (10).
Periodontitis is the most prevalent infectious inflammatory disease of humankind; up to 47% of the adult population suffers from this disease (20, 21). One of the key players in development of periodontitis is the Gram-negative anaerobic bacterium Porphyromonas gingivalis (22). Importantly, P. gingivalis infection has also been associated with other conditions, including rheumatoid arthritis, cardiovascular disease, and aspiration pneumonia (23–25). In silico analysis of three publicly available genomic sequences of P. gingivalis strains (W83, TDC60, and ATCC 33277), as well as clinical strains, revealed that at least four different CRISPR regions (30, 36.1, 36.2, and 37) and two sets of accompanying cas genes are present in the P. gingivalis genome (26), including one belonging to type I-C (neighboring CRISPR 30) and the other belonging to type III-B (neighboring CRISPR 37).
The RNA produced during transcription of a CRISPR region (pre-crRNA) is processed by Cas proteins to yield short crRNA molecules containing spacer sequences, which serve as guides for subsequent CRISPR-Cas-mediated degradation of nucleic acids (16, 27). In type I and III CRISPR-Cas systems, pre-crRNA primary processing is performed by one of the following Cas endonucleases: Cas6, Cas6b, Cas6e, Cas6f, or Cas5d. All of these enzymes cleave repeat sequences at a single position, producing crRNAs containing the complete spacer sequence and fragments of the repeat sequence at both ends (called 5′ and 3′ handles). In most of the characterized type I and III systems, the 5′ handle is 8 nucleotides (nt) long, whereas the 3′ handle contains the rest of the repeat sequence (19, 27–31). Some exceptions exist, however, such as the type I-C system, in which the 5′ handle is 11 nt long (30). Another example is the Synechocystis sp. system, with a 5′ handle of 13 nt (32). In some bacterial species, crRNA is further trimmed during maturation. In Staphylococcus epidermidis (which has a type III-A CRISPR-Cas system), this process yields two mature crRNA species, of 43 nt and 37 nt, by 3′-side trimming of crRNA (33). In Streptococcus pyogenes (with a type II system), crRNAs are cleaved from the 5′ side, yielding 39- to 42-nt mature crRNAs (16). In Pyrococcus furiosus (with at least seven CRISPR loci and cas genes characteristic of type I-A, I-B, and III-B systems), crRNAs are cleaved from the 3′ side, yielding 45-nt and 39-nt mature crRNAs (19). In type I and III CRISPR-Cas systems, trimming involves only the 3′ end, and the original 5′ handle is preserved (19, 33). However, transcript maturation is not the only means of crRNA generation. In Neisseria spp., in addition to regular type II processing, the crRNAs are also transcribed separately from promoters embedded within repeat sequences and then trimmed (34).
Alignment of protospacer flanking sequences in genetic elements targeted by type I and II CRISPR-Cas systems led to the identification of conserved sequence motifs, called protospacer-adjacent motifs (PAMs), which are required for spacer uptake and for CRISPR interference (35, 36). The presence of PAMs allows for easy discrimination between foreign DNA and genomic CRISPR loci. The data obtained so far suggest that type I systems require a PAM at the 3′ end of the protospacer (defining the protospacer as a sequence complementary to but not identical to the crRNA), whereas type II systems require a PAM at the 5′ end of the protospacer (10, 35, 36). Type III CRISPR-Cas systems seem not to require PAM elements for sequence recognition. Instead, the lack of complementarity between the 5′ handle of the mature crRNA molecule and the target sequence enables the cleavage process and prevents the system from cleaving its own CRISPR array (37).
For the present study, we assessed the activity of the CRISPR-Cas modules in P. gingivalis strain W83. The results clearly showed that all CRISPR loci are transcribed and that at least some transcripts are processed to form crRNAs. Furthermore, the CRISPR-Cas system was able to mediate degradation of plasmids containing elements complementary to the spacers. Functional analysis indicated that protospacer flanking sequences are important for recognition of the target sequence.
MATERIALS AND METHODS
In silico analysis.
CRISPR regions present in P. gingivalis strain W83 were identified using CRISPRfinder and the CRISPR database (9, 38). The cas genes were previously annotated by Watanabe et al. (26). Individual spacer sequences present in four identified CRISPR arrays were used to search for potential protospacer sequences by using CRISPRTarget (39). Consensus repeat sequences for each of the identified CRISPR arrays were used to search the CRISPRmap database to identify structural motifs and sequence families (40).
Bacterial culture.
P. gingivalis W83 was grown anaerobically in tryptic soy broth (Fluka, Switzerland) supplemented with 0.5% yeast extract (Bioshop, Canada), l-cysteine (0.5 mg/ml; Bioshop, Canada), menadione (0.5 μg/ml; ICN Biomedicals), and hemin (5 μg/ml; ICN Biomedicals). For agar plates (henceforth called blood agar plates), medium was supplemented with 5% sheep blood and 1.5% agar. Tetracycline (1 μg/ml) and gentamicin (150 μg/ml) were added if required. All cultures were processed in an MACS500 anaerobic chamber (Don Whitley Scientific Limited, Frederick, MD) in an atmosphere of 80% N2, 10% CO2, and 10% H2. Bacterial stocks were stored at −80°C in storage medium (culture medium supplemented with glycerol). For preparation of liquid cultures, bacterial stocks were first cultured on blood agar plates, and selected colonies were used to inoculate the broth (seed cultures). After overnight growth, the required volume of fresh broth was inoculated with a seed culture to an optical density at 550 nm (OD550) of 0.1 and grown anaerobically at 37°C until the desired OD550 was reached. Escherichia coli strain S17-1 was purchased from Leibniz-Institut DSMZ. All E. coli strains were grown aerobically in LB medium (Bioshop, Canada) with shaking. If required, ampicillin (100 μg/ml) was added.
Purification of nucleic acids.
Total RNA of P. gingivalis was extracted using Tri reagent (Sigma-Aldrich, Poland) as indicated by the manufacturer, with modifications. Briefly, 10 ml of P. gingivalis culture was centrifuged at 3,000 × g for 30 min, and the pellet was resuspended in 1 ml of Tri reagent. After 5 min of incubation at room temperature, 0.2 ml of chloroform was added, and the sample was shaken vigorously and incubated for 10 min at room temperature. The resulting mixture was centrifuged (12,000 × g for 15 min at 4°C). The aqueous phase was transferred to a new tube, and RNA was precipitated with 0.5 ml of 2-propanol (16 h, −20°C). Subsequently, RNA was pelleted by centrifugation (12,000 × g for 10 min at 4°C) and washed with 1 ml of 75% ethanol. Dried RNA was resuspended in 50 μl of sterile, nuclease-free water (Sigma-Aldrich, Poland). Isolated RNA was quantified using a NanoDrop spectrophotometer (Thermo Scientific) and stored at −80°C.
Plasmids from E. coli and P. gingivalis were purified using a GeneJET plasmid miniprep kit (Thermo Scientific) as indicated by the manufacturer.
P. gingivalis genomic DNA was purified using a genomic minikit (A&A Biotechnology, Poland) according to a protocol provided by the manufacturer.
Northern blot analysis.
P. gingivalis total RNA was isolated from bacteria after 24 h of culture to an OD600 between 1.6 and 1.7. Samples were treated with Turbo DNase (Life Technologies) according to the manufacturer's instructions. Total RNA (15 μg) was separated in a 15% polyacrylamide gel (19:1 acrylamide-bis) supplemented with 8 M urea. The gel was prerun at 180 V for 30 min. Samples were separated at 100 V until they completely entered the gel and afterwards at 180 V. RNAs were transferred to a nylon membrane (Immobilon-Ny+; Millipore) by use of a Trans-Blot SD semidry transfer cell (Bio-Rad) and were fixed with a Hoefer UVC 500 UV cross-linker (Hoefer) set to 70,000 μJ/cm2. Following cross-linking, the membrane was prehybridized in Perfect Hyb hybridization buffer (Sigma-Aldrich, Poland) for 5 min at 42°C. Furthermore, the hybridization buffer was supplemented with the biotinylated probes listed in Table 1. The overall scheme of annealing places for probes is presented in Fig. 1B. The membrane was incubated overnight at 42°C in a hybridization oven (Micro-4; Hybaid, United Kingdom). The signal was visualized using a biotin chromogenic detection kit (Thermo Scientific) as indicated by the manufacturer. A small RNA marker was purchased from Abnova (Taiwan). In addition, two RNA fragments obtained by in vitro transcription, with lengths of 30 and 50 bases, were used. In vitro transcription was performed using an mMESSAGE mMACHINE T7 kit (Life Technologies) according to the manufacturer's instructions. Two oligonucleotides (5′-AAT AAA GCA GAT TGG GAA AAT CCT ATA CCC TAT AGT GAG TCG TAT TA-3′ and 5′-TAT CGT GCT GTA AAT AAA GCA GAT TGG GAA AAT CCT ATA TGT ACT TTC CCT ATA GTG AGT CGT ATT A-3′) containing the T7 promoter sequence were used as templates. Both fragments encoded transcripts recognized by the probe CRISPR 30 spacer 20+ in order to allow detection on a Northern blot membrane.
TABLE 1.
Probes for Northern blot detection of crRNA
| Targeted CRISPR and region | Probe sequence |
|---|---|
| CRISPR 36.1 | |
| Repeat + | GTTGGATCTACCCTCTATTCGAAGGGTACACACAAC-biotin |
| Repeat − | GTTGTGTGTACCCTTCGAATAGAGGGTAGATCCAAC-biotin |
| Spacer 4+ | TCAAGGGTCGGGTTCTTGACCTTACCTCCA-biotin |
| Spacer 4− | TGGAGGTAAGGTCAAGAACCCGACCCTTGA-biotin |
| CRISPR 36.2 | |
| Repeat + | Biotin-GTTGTCTCCACCCTTCTAACTAAGGGTATTCCCAAC |
| Repeat − | Biotin-GTTGGGAATACCCTTAGTTAGAAGGGTGGAGACAAC |
| Spacer 4+ | Biotin-GCAATCACAAAAACTTATAACGATGCGTTT |
| Spacer 4− | Biotin-AAACGCATCGTTATAAGTTTTTGTGATTGC |
| CRISPR 37 | |
| Repeat + | Biotin-GTCTTAATAGCCTTACGGACTGTGTATGTATAGTGAG |
| Repeat − | Biotin-CTCACTATACATACACAGTCCGTAAGGCTATTAAGAC |
| Spacer 4+ | Biotin-TGCAGGGAGTTGGTTCAGCAAAAACCCCGCTGTC |
| Spacer 4− | Biotin-GACAGCGGGGTTTTTGCTGAACCAACTCCCTGCA |
| CRISPR 30 | |
| Repeat + | GTTTTAATTCCTGTATGGTGCAATTGAAAT-biotin |
| Repeat − | ATTTCAATTGCACCATACAGGAATTAAAAC-biotin |
| Spacer 1+ | Biotin-CTCCCGGGTTGGGCAGCACGGCTTTGAGGAATTGG |
| Spacer 1− | Biotin- CCAATTCCTCAAAGCCGTGCTGCCCAACCCGGGAG |
| Spacer 4+ | GATTCTCTATATGTTCAGTTCAATACAATGCTGAAA-biotin |
| Spacer 4− | TTTCAGCATTGTATTGAACTGAACATATAGAGAATC-biotin |
| Spacer 7+ | Biotin-CTATAAAAGAAATTCCAAGAGAGCATGATTCTGAG |
| Spacer 7− | Biotin- CTCAGAATCATGCTCTCTTGGAATTTCTTTTATAG |
| Spacer 20+ | AATAAAGCAGATTGGGAAAATCCTATATGTACTTT-biotin |
| Spacer 20− | AAAGTACATATAGGATTTTCCCAATCTGCTTTATT-biotin |
FIG 1.

(A) Locations of CRISPR arrays and cas genes in the P. gingivalis chromosome. Protein annotation and CRISPR array nomenclature are presented according to the system of Watanabe et al. (26). The name of each CRISPR contains the length of a single repeat (and a consecutive number, if there are other arrays with the same repeat length). Arrows indicate the predicted direction of gene transcription. Blocks representing overlapping sequences are shifted upwards. Regions of the genome not related to the CRISPR-Cas system are omitted. Positions of CRISPR regions in the genome are shown below the axis. (B) Scheme for analysis of the CRISPR arrays. Probes used in Northern blots are marked with thin arrows above and below the CRISPR arrays. The arrow at the end of a given CRISPR array indicates the determined direction of transcription. Self-targeting spacers are marked with bold frames.
Construction of plasmids containing protospacers.
The shuttle plasmid pT-COW encodes a TetQ protein (responsible for tetracycline resistance), replicates both in E. coli and in P. gingivalis, and can be delivered efficiently into P. gingivalis cells via conjugation with E. coli S17-1 (41). The shuttle plasmid pT-COW was kindly provided by Don R. Demuth (University of Louisville, School of Dentistry). The plasmid was digested with the HindIII and SalI restriction enzymes (Thermo Scientific) and gel purified. The DNA fragments listed in Table 2 were synthesized (Genomed, Poland) and annealed to form double-stranded DNAs (dsDNAs) with overhanging ends compatible with the linear plasmid mentioned above. The annealing procedure was performed as follows. Oligonucleotides (100 μmol) (Table 2) dissolved in Tris-EDTA (TE) buffer were mixed in appropriate pairs at a 1:1 molar ratio. Samples were incubated for 3 min at 95°C and cooled at room temperature. Resulting double-stranded DNAs (50 μmol) were diluted 100-fold. Two microliters of diluted DNA (∼1 pmol) was mixed with 50 ng of linear plasmid, ligated with T4 DNA ligase (Thermo Scientific) as recommended by the manufacturer, and used to transform chemically competent E. coli DH5α cells. The cells were plated on LB agar with ampicillin (100 μg/ml). Resulting bacterial colonies were checked by sequencing, and clones containing appropriate inserts were selected.
TABLE 2.
Oligonucleotides used for construction of plasmids containing protospacers
| Plasmid | Oligonucleotide sequencea |
|---|---|
| sp4_AAA/TTT | AGCTaaaTTTCAGCATTGTATTGAACTGAACATATAGAGAATCaaa |
| TCGAtttGATTCTCTATATGTTCAGTTCAATACAATGCTGAAAttt | |
| sp4 CCT/TTT | AGCTcctTTTCAGCATTGTATTGAACTGAACATATAGAGAATCaaa |
| TCGAtttGATTCTCTATATGTTCAGTTCAATACAATGCTGAAAagg | |
| sp4 AAA/AGG | AGCTaaaTTTCAGCATTGTATTGAACTGAACATATAGAGAATCagg |
| TCGAcctGATTCTCTATATGTTCAGTTCAATACAATGCTGAAAttt | |
| sp5 AAA/TTT | AGCTaaaAAAGTTTTAAGATTAGCAAACATTTTACCATCTTGTaaa |
| TCGAtttACAAGATGGTAAAATGTTTGCTAATCTTAAAACTTTttt | |
| sp5 CCT/TTT | AGCTcctAAAGTTTTAAGATTAGCAAACATTTTACCATCTTGTaaa |
| TCGAtttACAAGATGGTAAAATGTTTGCTAATCTTAAAACTTTagg | |
| sp5 AAA/AGG | AGCTaaaAAAGTTTTAAGATTAGCAAACATTTTACCATCTTGTagg |
| TCGAcctACAAGATGGTAAAATGTTTGCTAATCTTAAAACTTTttt | |
| sp4scr AAA/TTT | AGCTaaaAGACCTTGCAAGTATATTTGAAACGACTTATTGATAaaa |
| TCGAtttTATCAATAAGTCGTTTCAAATATACTTGCAAGGTCTttt | |
| sp4scr CCT/TTT | AGCTcctAGACCTTGCAAGTATATTTGAAACGACTTATTGATAaaa |
| TCGAtttTATCAATAAGTCGTTTCAAATATACTTGCAAGGTCTagg | |
| sp4scr AAA/AGG | AGCTaaaAGACCTTGCAAGTATATTTGAAACGACTTATTGATAagg |
| TCGAcctTATCAATAAGTCGTTTCAAATATACTTGCAAGGTCTttt | |
| RNA random seq. | GATCCGCGTCTCCTTGCGGGTAGATCGCCGACCGCAGAG |
| GATCCTCTGCGGTCGGCGATCTACCCGCAAGGAGACGCG | |
| RNA C30/sp4 (+/−) | GATCCGATTCTCTATATGTTCAGTTCAATACAATGCTGAAAG |
| GATCCTTTCAGCATTGTATTGAACTGAACATATAGAGAATCG |
Lowercase letters indicate PAM sequences.
Assessment of RNA degradation by the CRISPR-Cas system.
In order to determine whether RNA constitutes a substrate for the CRISPR 30/Cas system, a quantitative reverse transcription-PCR (RT-PCR) assay with a reporter sequence was designed. Briefly, a synthetic reporter cassette was prepared that carried annealing sites for a fluorescent probe and primers. A BglII restriction site was introduced between primer/probe annealing sites. Conjugation of the pT-COW plasmid in P. gingivalis results in transcription of the plasmid DNA, probably due to the presence of a cryptic promoter site. A synthetic reporter cassette was cloned into the pT-COW plasmid by use of BamHI and SalI restriction sites, using T4 DNA ligase (Thermo Scientific) according to the manufacturer's instructions. The resulting plasmid was recovered in E. coli DH5α bacteria and sequenced (Genomed, Poland).
Synthetic protospacers (Genomed, Poland) (Table 2) were introduced into the plasmid through the BglII restriction site by ligation with T4 DNA ligase (Thermo Scientific). In order to prevent self-ligation of the plasmid, the ligation reaction was done in the presence of 1 U of the BglII restriction enzyme. Resulting plasmids were recovered in E. coli DH5α bacteria and sequenced (Genomed, Poland). Each tested protospacer was introduced in both orientations, while a random sequence was introduced as a control.
The plasmids were introduced into P. gingivalis via conjugation with E. coli strain S17-1, as described below. The obtained bacterial colonies were collected with a sterile loop and used for total RNA extraction with an RNA extraction kit (Bio Basic, Canada) according to the manufacturer's instructions. RNA samples were digested with Turbo DNase (Life Technologies, Poland) as indicated by the manufacturer and repurified using an RNA extraction kit (Bio Basic, Canada). Subsequently, samples were divided into two sets. In one set, RNA was reverse transcribed with a High Capacity cDNA reverse transcription kit (Life Technologies, Poland) according to the manufacturer's protocol. In the second (control) set, no reverse transcriptase was added. The obtained cDNAs were analyzed quantitatively by real-time PCR for the presence of the reporter sequence, using 1× TaqMan universal master mix II, no AmpErase UNG (Life Technologies, Poland), 900 nM (each) primers (5′-AAA CCT CGT TGG AAG CGT GT-3′ and 5′-CTG TGG AAA ACC TTT GGC ATC-3′), and 200 nM specific probe labeled with 6-carboxyfluorescein (FAM) and 6-carboxytetramethylrhodamine (TAMRA) (5′-FAM-ATG TTA TTC AGT GCT TTG GTC CTC GTG AT-TAMRA-3′). Rox was used as a reference dye. The reaction was monitored on a model 7500 Fast real-time PCR machine (Applied Biosystems) with the following settings: 2 min at 50°C, 10 min at 92°C, and 40 cycles of 15 s at 92°C and 1 min at 60°C.
Conjugal transfer of plasmids.
E. coli strain S17-1 was used as a donor strain to introduce the pT-COW plasmid and its derivatives into P. gingivalis by conjugation (42). Plasmids carrying spacer sequences (Table 2) were transformed into chemically competent E. coli S17-1 cells. Multiple colonies from each transformation plate were scraped using a sterile loop and mixed with P. gingivalis W83 cells prepared in the same manner on blood agar without antibiotics. The plates were incubated anaerobically at 37°C overnight. Following the incubation, cocultures were collected with a sterile loop and plated onto blood agar plates with tetracycline (1 μg/ml) and gentamicin (150 μg/ml). The plates were incubated for 7 to 10 days under anaerobic conditions at 37°C, and bacterial colonies were counted following incubation.
5′ rapid amplification of cDNA ends (5′ RACE).
Total RNA was extracted from P. gingivalis W83 by using Tri reagent (Sigma-Aldrich, Poland), digested with Turbo DNase (Life Technologies, Poland) as indicated by the manufacturer, and repurified using an RNA extraction kit (Bio Basic, Canada). Reverse transcription was conducted using a primer complementary to a fragment of the 3rd spacer (5′-CGG TCT ATC TCG TAG CGT TC-3′) by using a High Capacity cDNA reverse transcription kit (Life Technologies, Poland) according to the manufacturer's protocol. The remaining RNA was digested with RNase H and RNase T1 (both from Thermo Scientific) in 50 μl RNase H buffer (both added at 1 U per 10 μl of reverse transcription reaction mixture). The resulting pool of cDNAs was purified using a GeneJET PCR purification kit (Thermo Scientific), and a poly(C) tail was added using terminal deoxynucleotidyltransferase (Thermo Scientific) according to the manufacturer's protocol. The obtained cDNAs were PCR amplified using primers complementary to the 2nd primer and the poly(C) tail (5′-GAG GAG ATA GGA CTC GCG CT-3′ and 5′-GGC CAC GCG TCG ACT AGT ACG GGG GGG GGG GGG IIG-3′, respectively). Amplification products were cloned into the pTZ57R/T plasmid by using an InsTAclone PCR cloning kit (Thermo Fisher Scientific) and then sequenced.
RESULTS
CRISPR elements in the P. gingivalis genome.
There are four CRISPR regions in the genome of P. gingivalis W83, two of which are accompanied by a cluster of cas genes (9, 26, 43). The genomic organization of these elements is presented in Fig. 1.
Ribonucleases cleaving pre-crRNAs into crRNAs recognize the direct repeat sequences, and even slight modifications of these elements may prevent cleavage (44, 45). Analysis of the P. gingivalis CRISPR regions revealed that the repeats are well conserved within each of the four arrays, yet their sequences differ between arrays (Table 3). CRISPRs 36.1 and 36.2 exhibit the highest level of conservation: all repeats within these arrays are identical. Two other CRISPR regions are slightly less conserved. In CRISPR 37, there is a single-nucleotide difference in the middle of the penultimate repeat, and in CRISPR 30, there is a single-nucleotide variation at the end of the last repeat. Furthermore, the repeats of CRISPRs 36.1 and 36.2 share 72% identity (26 of 36 nt are identical) (Table 3). The CRISPRmap analysis assigned CRISPRs 36.1 and 36.2 into superclass F, without identifying the sequence family or structural motif. CRISPR 30 was assigned into superclass A in family sequence 2, without identifying the structural motif. For CRISPR 37 of superclass E, no family sequence was assigned, but it was assigned to structural motif 4.
TABLE 3.
Basic characteristics of CRISPR arrays in the P. gingivalis W83 genome
| CRISPR array | Repeat consensus sequence | No. of spacers | Spacer length (nt) | Putative associated CRISPR-Cas type |
|---|---|---|---|---|
| 36.1 | GTTGGATCTACCCTCTATTCGAAGGGTACACACAAC | 7 | 30 | Unknown |
| 36.2 | GTTGTCTCCACCCTTCTAACTAAGGGTATTCCCAAC | 7 | 30 | Unknown |
| 37 | GTCTTAATAGCCTTACGGACTGTGTATGTATAGTGAG | 7 | 34–38 | III-B |
| 30 | GTTTTAATTCCTGTATGGTGCAATTGAAAT | 23 | 34–37 | I-C |
Spacer sequences in CRISPR arrays are essential for recognition of the target nucleic acids. These elements are introduced into the genome during an adaptive response to endogenous or exogenous nucleic acids. The origin of P. gingivalis W83 spacers remains largely unknown; most of them are not similar to any known sequence, but four of them exhibit significant degrees of similarity to the bacterium's own genomic sequence. The first leader-proximal spacer of CRISPR 37 matches an intergenic region in the P. gingivalis W83 genome (36/36 nt; positions 975677 to 975712). The first two leader-proximal spacers of CRISPR 30 share 100% identity with two different regions of the gene encoding saccharopine dehydrogenase (35/35 nt; positions 728442 to 728476 and 728638 to 728672). Finally, the last spacer of CRISPR 30 matches a gene encoding an outer membrane efflux protein (positions 729776 to 729811); however, in this case, the identity is not complete (34 of 36 nt). Moreover, none of the four identified protospacers in the P. gingivalis genome is neighbored by an identified PAM. Visualization of the self-targeting spacer alignment is presented in Fig. S1 in the supplemental material. CRISPRTarget analysis performed for all spacers from the four CRISPR arrays resulted in hits from other bacterial species that can be found in the oral cavity and gastrointestinal tract. Hits were noted for spacers 1, 2, 5, and 11 of CRISPR 30. Spacers 1 and 2 show homology to sequences encoding saccharopine dehydrogenases (Prevotella spp. and Bacteroides spp.). Spacer 5 shows homology to sequences encoding the beta subunit of acetyl coenzyme A (acetyl-CoA) carboxylase (Streptococcus agalactiae) and PtrB protease (Flavobacteria spp.). The results for spacer 11 indicate homology to sequences encoding an ABC transporter permease (Butyrivibrio spp.) and sequences that are not associated with open reading frames (Leuconostoc inhae and Bacillus cereus). However, the differences between all of these spacers and identified sequences are considerable, ranging from 3 to 8 nt.
CRISPR loci are transcriptionally active.
Northern blotting using biotinylated probes specific to repeats and spacers in the CRISPR cassette was used to detect transcripts containing CRISPR elements (Fig. 2). This analysis revealed that CRISPRs 36.2, 37, and 30 are transcribed in the same direction (consistent with the transcription direction of cas genes), whereas CRISPR 36.1 is transcribed in the opposite direction. For some probes, however (CRISPR 37 spacer 4 and CRISPR 36.2 repeat), bands were present for both possible directions of transcription. This may indicate bidirectional transcription of crRNAs.
FIG 2.

Northern analysis of CRISPR array transcription. Total RNA of P. gingivalis W83 was separated in a 15% polyacrylamide gel containing 8 M urea; RNAs were transferred to nylon membranes by electroblotting and analyzed using biotinylated synthetic DNA probes specific to repeat and spacer sequences. “+” probes have a direction consistent with the direction of cas gene transcription (identical for all). “−” probes are complementary to “+” probes. Arrows to the right of each gel indicate the expected sizes of crRNA processing products, as follows: crRNA, mature crRNA; 1×, single repeat-spacer unit; and 2×, double repeat-spacer unit.
Analysis of total bacterial RNA with probes specific to both spacers and repeats revealed the presence of a distinct product of ∼70 bases, corresponding in size to an RNA molecule containing a single spacer-repeat tandem, in all four CRISPR arrays (expected lengths, 66 nt for CRISPRs 36.1 and 36.2, 64 to 67 nt for CRISPR 30, and 71 to 75 nt for CRISPR 37). Additional bands present for each CRISPR array, with lengths exceeding 100 nt, correspond to double tandem units consisting of two spacers and two repeats. The results obtained for CRISPR 30 suggest that crRNA molecules originating from different spacers may be processed differently. The signal for spacer 1 was weak, but an RNA fragment of about 70 nt (which corresponds in length to the single repeat-spacer unit) could be observed. For spacer 4, in addition to the 70-nt RNA fragment, three additional RNA fragments were present (∼60 nt, slightly larger than 40 nt, and slightly smaller than 40 nt). For spacer 7, there was an ∼70-nt fragment accompanied by two smaller RNA molecules, which were slightly larger and slightly smaller than 40 nt. In this case, the 60-nt product was not visible. For spacer 20, a single crRNA was visible, although no additional bands were detected. All probes specific to the repeats and two probes specific to the spacers (CRISPR 36.1 spacer 4 and CRISPR 30 spacer 4) recognized products of pre-crRNA scission that were approximately 70 nt long. The probe specific to the CRISPR 36.1 repeat recognized RNA molecules smaller than 70 nt.
To determine the transcription start site for the CRISPR 30 array, we used 5′ RACE. Most of the RNAs originating from the CRISPR 30 region reached only the first repeat of the array (exactly 8 nt from its 3′ end). RT-PCR analysis revealed the presence of longer CRISPR 30 transcripts, stretching at least 84 nt beyond the first repeat (data not shown). The 5′ RACE technique confirmed the scission of pre-crRNA in the region of the first repeat sequence, resulting in a product with an 8-nt 5′ handle, but unfortunately, it failed to reveal the transcription start site.
Specificity and activity of the CRISPR 30 machinery.
No known phages infect P. gingivalis. Therefore, to evaluate the in vivo activity, specificity, and effectiveness of the P. gingivalis CRISPR-Cas system, we constructed artificial mobile genetic elements (plasmids) based on the shuttle vector pT-COW (37). To assess whether the CRISPR-Cas system is active in P. gingivalis, a single CRISPR cassette (CRISPR 30) was selected for detailed analysis. This decision was motivated by the fact that two of the CRISPR loci (CRISPR 36.1 and CRISPR 36.2) lack cas genes and the third locus (CRISPR 37) is of type III-B, which targets RNA rather than DNA.
To assess whether the CRISPR-Cas system is able to specifically degrade dsDNA, pT-COW-derived plasmids carrying sequences complementary to crRNA (originating from CRISPR 30) flanked by various adjacent sequences were delivered into P. gingivalis by conjugation. These sequences included spacers 4 and 5 (counting from the leader end of the array), as well as a scrambled spacer with the nucleotide content of spacer 4, but with a randomized sequence to avoid similarity to any spacer of the studied array. Each of the aforementioned protospacers was prepared in three variants: with the predicted PAM at the 5′ end, with the predicted PAM at the 3′ end, and with no PAM at all. The results clearly show that dsDNA may be a target of the CRISPR 30/Cas module and that bacteria conjugated with plasmids carrying sequences complementary to crRNAs with the appropriate flanking regions were not able to survive in the presence of tetracycline (Fig. 3).
FIG 3.
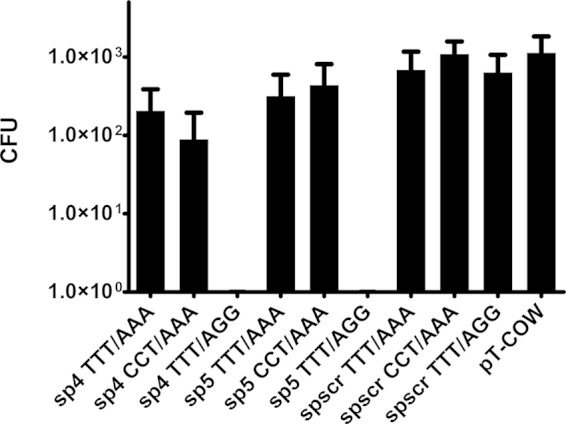
Protospacer-adjacent sequences are important for CRISPR-Cas-mediated target degradation. The plasmid pT-COW and its derivatives were delivered to P. gingivalis W83 via conjugation. The name of each plasmid contains the number of the spacer introduced into the plasmid and the three-nucleotide 5′ and 3′ flanks of the protospacer. The significance of the observed differences between samples and the control plasmid was analyzed using Student's t test. All experiments were repeated three times, and results are expressed as means ± standard deviations (SD).
The choice of sequences adjacent to the protospacer selected for testing was based on data reported in the literature. CRISPR arrays can be grouped into at least 12 clusters according to repeat sequence similarity (46). CRISPR 30 belongs to cluster 1; therefore, the CRISPR 30/Cas system is predicted to recognize the NGG sequence as a PAM (35). In the aforementioned publication, the direction of transcription of CRISPR regions was not verified in vivo, so it is possible that the exact PAM sequence is CCN at the 5′ end of the protospacer or NGG at the 3′ end. For the no-PAM variant, the sequences 5′ TTT and 3′ AAA were chosen because no single-base trinucleotides are expected to trigger interference (Table 4).
TABLE 4.
Sequences of protospacer regions of pT-COW derivatives used for conjugation assaya
| Plasmid | 5′-end-adjacent sequence | Protospacer sequence (5′–3′) | 3′-end-adjacent sequence |
|---|---|---|---|
| sp4_TTT/AAA | TATTCGATTT | GAT TCT CTA TAT GTT CAG TTC AAT ACA ATG CTG AAA | AAAAGCTGGC |
| sp4 CCT/AAA | TATTCGACCT | GAT TCT CTA TAT GTT CAG TTC AAT ACA ATG CTG AAA | AAAAGCTGGC |
| sp4 TTT/AGG | TATTCGATTT | GAT TCT CTA TAT GTT CAG TTC AAT ACA ATG CTG AAA | AGGAGCTGGC |
| sp5 TTT/AAA | TATTCGATTT | ACA AGA TGG TAA AAT GTT TGC TAA TCT TAA AAC TTT | AAAAGCTGGC |
| sp5 CCT/AAA | TATTCGACCT | ACA AGA TGG TAA AAT GTT TGC TAA TCT TAA AAC TTT | AAAAGCTGGC |
| sp5 TTT/AGG | TATTCGATTT | ACA AGA TGG TAA AAT GTT TGC TAA TCT TAA AAC TTT | AGGAGCTGGC |
| sp4scr TTT/AAA | TATTCGATTT | TAT CAA TAA GTC GTT TCA AAT ATA CTT GCA AGG TCT | AAAAGCTGGC |
| sp4scr CCT/AAA | TATTCGACCT | TAT CAA TAA GTC GTT TCA AAT ATA CTT GCA AGG TCT | AAAAGCTGGC |
| sp4scr TTT/AGG | TATTCGATTT | TAT CAA TAA GTC GTT TCA AAT ATA CTT GCA AGG TCT | AGGAGCTGGC |
Nucleotides in bold indicate PAM sequences.
Delivery of plasmids containing protospacers from the CRISPR 30 array reduced the number of surviving bacteria for variants containing the proper protospacer sequence flanked by a putative PAM sequence at the 3′ end, namely, sp4 TTT/AGG (P < 0.05) and sp5 TTT/AGG (P < 0.05) (Fig. 3). For all other variants, including a randomized spacer with a PAM sequence at the 3′ end, no statistically significant difference was observed relative to the parental plasmid.
Some CRISPR-Cas modules are also able to process RNA targets; these include CRISPR-Cas type III-B, which was detected in the genome of P. gingivalis W83. To exclude the possibility of CRISPR 30 involvement in type III-B immunity, we tested its spacers by RNA interference. Because the crRNA-mediated RNA cleavage of type III-B is not dependent on the presence of PAMs (47), no alternative protospacer-adjacent sequences were introduced. Plasmids designed for expression of the reporter RNA, containing spacer 4 from CRISPR 30 in both orientations, were introduced into P. gingivalis by conjugation, and then the levels of the produced RNA were assessed by quantitative RT-PCR (Fig. 4A and B). The levels of RNA containing potential targets for the CRISPR 30/Cas system were not affected compared to that of the control random RNA.
FIG 4.
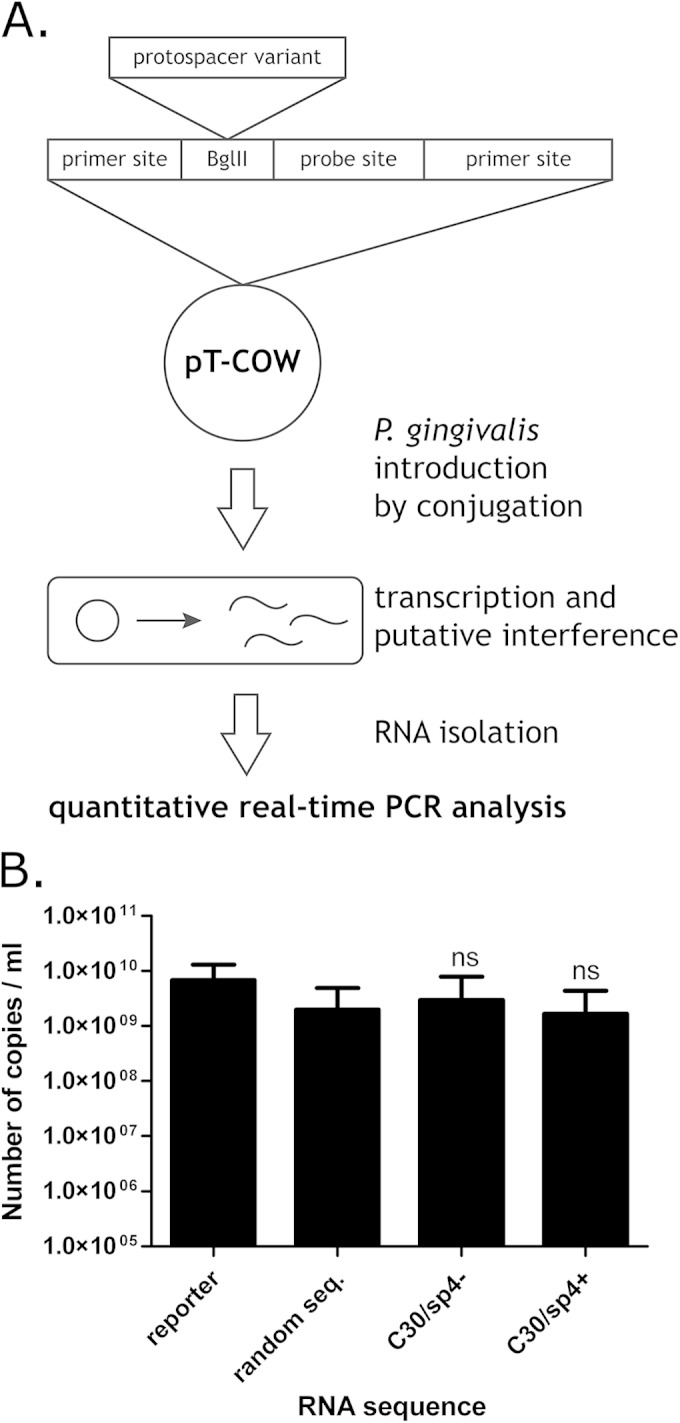
(A) Scheme of CRISPR 30 RNA interference experiment. The pT-COW plasmid was supplemented with a synthetic reporter sequence containing a protospacer variant together with a nucleotide probe annealing site flanked by PCR priming sites. Introduction of the modified plasmid into P. gingivalis results in transcription of the reporter cassette, which is a potential target for CRISPR RNA interference. This interference results in a decrease of transcript levels, which can be detected by quantitative RT-PCR analysis. (B) Degradation of RNA by the CRISPR 30/Cas system. The graph shows numbers of RNA copies in bacteria conjugated with plasmids carrying the reporter cassette with protospacers. Reporter, plasmid carrying the reporter cassette; random seq., plasmid carrying the reporter cassette with a random sequence inserted into the region targeted by the real-time PCR primers and probe; C30/sp4−, plasmid carrying the reporter cassette with a sequence identical to that of the corresponding crRNA inserted into the region targeted by the real-time PCR primers and probe; C30/sp4+, plasmid carrying the reporter cassette with a sequence complementary to that of the corresponding crRNA inserted into the region targeted by the real-time PCR primers and probe. The significance of the observed differences between samples and positive-control samples was analyzed using Student's t test. ns, not significant (P > 0.05). All experiments were repeated three times, and the results are expressed as means ± SD.
DISCUSSION
The presence of CRISPR genetic elements in the majority of clinical P. gingivalis strains and their structural conservation (26) suggest that they play an essential role in bacterial survival. To date, however, no phages able to infect P. gingivalis have been identified, and this species has no known naturally occurring plasmids (48, 49). Hence, the main function of this system remains unconfirmed. A recent report on P. gingivalis DNA-exchange mechanisms revealed that this bacterium is naturally competent and that DNA uptake is not sensitive to the DNA source or modification status (50). Given that extracellular DNA is a common constituent of bacterial biofilms (51–53), the CRISPR-Cas system may have evolved as a protective system that verifies and selects DNA molecules entering bacterial cells, helping to regulate horizontal gene transfer. The results of CRISPRTarget analysis, which revealed some degree of similarity between specific spacers of CRISPR 30 and sequences from bacteria found in the oral cavity and gastrointestinal tract, support this hypothesis.
Two different sets of cas genes, characteristic of the CRISPR-Cas I-C and III-B systems, are present in the P. gingivalis W83 genome (12, 26). In both of these systems, the primary processing of pre-crRNA results in formation of immature crRNAs containing a 5′ handle (11 nt in type I-C and 8 nt in type III-B) derived from the repeat sequence, with the rest of the repeat sequence at the 3′ end (27–30). In type III-B, secondary processing is performed by an unknown nuclease that trims the 3′ end. Four CRISPR arrays are present in the P. gingivalis W83 genome (9, 43). Here we showed that all these regions are transcribed and processed to form single repeat-spacer crRNA units, consistent with previous reports (54). Furthermore, some of these units seem to be transcribed bidirectionally. Such RNA species were also identified in Sulfolobus solfataricus (55). It was hypothesized that these small cRNAs can neutralize crRNAs in the absence of invading nucleic acids. Another proposed explanation for this phenomenon is that transcription in the reverse orientation relative to that of spacer elements is specific to organisms with a relaxed transcription start site rather than a feature of the CRISPR-Cas system itself (31). Employment of probes specific for spacers and repeat sequences allowed visualization of repeat-spacer tandem units for CRISPR 37 (spacer 4) and CRISPR 30 (spacers 4 and 7). However, we also observed smaller-than-predicted products. For CRISPR 37, which belongs to type III-B, the obtained band pattern fits the results available for P. furiosus (66- to 72-nt intermediate bands and two mature crRNA populations, of 39 nt and 45 nt) (19, 47). In contrast, the band pattern for spacers 4 and 7 of CRISPR 30 fits the data available for type I-C, in which the mature form of crRNA correlates in size with a single repeat-spacer unit (30). The presence of additional bands may be the result of unspecific cleavage; notably, the band pattern for spacer 20, which lacks the additional bands, fits the I-C system model.
Formation of crRNA itself does not define its function and activity; therefore, an effort was made to confirm the functionality of the CRISPR-Cas system in P. gingivalis. The CRISPR 30 cassette was selected for detailed characterization due to its proximity to the cas gene cluster and confirmed crRNA transcription. The results confirmed that the system is functional: introduction of the properly flanked protospacer to foreign DNA resulted in its specific degradation. The protein composition of the products of the adjacent cas gene cassette and the structure of the repeats follow the pattern typical for a type I-C system. However, to exclude the possibility that CRISPR 30 crRNAs target RNA, we verified the specificity of the system for the RNA substrate. It is important that cmr genes present in the genome of P. gingivalis W83 may cooperate with another CRISPR array; alternatively, the cmr module may not be functional (the genome of P. gingivalis W83 lacks the cmr1 gene, whose product is essential for the activity of the Cmr protein complex [19]). As expected, we observed no CRISPR 30-mediated degradation of RNA.
Based on their repeat sequences, CRISPR arrays were allocated into 12 clusters (46), and P. gingivalis CRISPR 30 was assigned to cluster 1. In silico analysis suggested that in this cluster, the preferred PAM sequence is NGG; however, previous work showed that this rule is not absolute (35, 56, 57). Our results indicate that NGG is an active PAM when it is located at the 3′ end of a protospacer (Fig. 3).
The presence of self-targeting spacers may represent the aftermath of an autoimmune event. This idea is supported by the fact that none of the protospacers within the P. gingivalis W83 genome are flanked by experimentally determined PAM sequences, which are required for interference. Mutations within PAMs are among the ways that bacteria avoid CRISPR-Cas-based autoimmunity (58).
To summarize, we showed here that all four CRISPR regions present in the P. gingivalis W83 genome are transcribed and that at least one of them is active against dsDNA in vivo. The recognition of the protospacer is mediated by the presence of PAM elements. Together, the results of this and previous studies suggest that the P. gingivalis CRISPR-Cas system is highly efficient and may play an important role in protection against foreign DNA or regulation of physiological processes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Science Center, Poland (grants 2011/01/D/NZ6/00269 and N N302 654640 to K. Pyrc and M. Bochtler, respectively). J. Potempa acknowledges support by grants from the NIDCR (grants DE 09761 and DE 022597) and the European Commission (grant FP7-HEALTH-F3-2012-306029 “TRIGGER”). The Faculty of Biochemistry, Biophysics and Biotechnology of Jagiellonian University is a beneficiary of structural funds from the European Union (grant POIG.02.01.00-12-064/08 [molecular biotechnology for health]). The Faculty of Biochemistry, Biophysics and Biotechnology of Jagiellonian University is a partner of the Leading National Research Center (KNOW), supported by the Ministry of Science and Higher Education.
We thank Anna Golda for her technical support.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We declare that we have no competing interests.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00261-15.
REFERENCES
- 1.Bryant J, Chewapreecha C, Bentley SD. 2012. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol 7:1283–1296. doi: 10.2217/fmb.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyman P, Abedon ST. 2010. Bacteriophage host range and bacterial resistance. Adv Appl Microbiol 70:217–248. doi: 10.1016/S0065-2164(10)70007-1. [DOI] [PubMed] [Google Scholar]
- 3.Labrie SJ, Samson JE, Moineau S. 2010. Bacteriophage resistance mechanisms. Nat Rev Microbiol 8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- 4.Forde A, Fitzgerald GF. 1999. Bacteriophage defence systems in lactic acid bacteria. Antonie Van Leeuwenhoek 76:89–113. doi: 10.1023/A:1002027321171. [DOI] [PubMed] [Google Scholar]
- 5.McGrath S, Fitzgerald GF, van Sinderen D. 2002. Identification and characterization of phage-resistance genes in temperate lactococcal bacteriophages. Mol Microbiol 43:509–520. doi: 10.1046/j.1365-2958.2002.02763.x. [DOI] [PubMed] [Google Scholar]
- 6.Pingoud A, Fuxreiter M, Pingoud V, Wende W. 2005. Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci 62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PubMed] [Google Scholar]
- 7.Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. 1987. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 9.Grissa I, Vergnaud G, Pourcel C. 2007. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ, van der Oost J. 2012. The CRISPRs, they are a-changin': how prokaryotes generate adaptive immunity. Annu Rev Genet 46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 11.Jansen R, Embden JD, Gaastra W, Schouls LM. 2002. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 12.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beloglazova N, Petit P, Flick R, Brown G, Savchenko A, Yakunin AF. 2011. Structure and activity of the Cas3 HD nuclease MJ0384, an effector enzyme of the CRISPR interference. EMBO J 30:4616–4627. doi: 10.1038/emboj.2011.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, Marraffini LA. 2015. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 519:199–202. doi: 10.1038/nature14245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marraffini LA, Sontheimer EJ. 2008. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamulaitis G, Kazlauskiene M, Manakova E, Venclovas C, Nwokeoji AO, Dickman MJ, Horvath P, Siksnys V. 2014. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell 56:506–517. doi: 10.1016/j.molcel.2014.09.027. [DOI] [PubMed] [Google Scholar]
- 19.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. 2009. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cobb CM, Williams KB, Gerkovitch MM. 2009. Is the prevalence of periodontitis in the USA in decline? Periodontol 2000 50:13–24. doi: 10.1111/j.1600-0757.2008.00284.x. [DOI] [PubMed] [Google Scholar]
- 21.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ, Beck J, Douglass G, Page R. 2012. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J Dent Res 91:914–920. doi: 10.1177/0022034512457373. [DOI] [PubMed] [Google Scholar]
- 22.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL. 1998. Microbial complexes in subgingival plaque. J Clin Periodontol 25:134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 23.de Pablo P, Chapple IL, Buckley CD, Dietrich T. 2009. Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol 5:218–224. doi: 10.1038/nrrheum.2009.28. [DOI] [PubMed] [Google Scholar]
- 24.Friedewald VE, Kornman KS, Beck JD, Genco R, Goldfine A, Libby P, Offenbacher S, Ridker PM, Van Dyke TE, Roberts WC, American Journal of Cardiology, Journal of Periodontology. 2009. The American Journal of Cardiology and Journal of Periodontology editors' consensus: periodontitis and atherosclerotic cardiovascular disease. Am J Cardiol 104:59–68. doi: 10.1016/j.amjcard.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Terpenning MS, Taylor GW, Lopatin DE, Kerr CK, Dominguez BL, Loesche WJ. 2001. Aspiration pneumonia: dental and oral risk factors in an older veteran population. J Am Geriatr Soc 49:557–563. doi: 10.1046/j.1532-5415.2001.49113.x. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe T, Nozawa T, Aikawa C, Amano A, Maruyama F, Nakagawa I. 2013. CRISPR regulation of intraspecies diversification by limiting IS transposition and intercellular recombination. Genome Biol Evol 5:1099–1114. doi: 10.1093/gbe/evt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carte J, Wang R, Li H, Terns RM, Terns MP. 2008. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. 2010. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, Ke A. 2012. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure 20:1574–1584. doi: 10.1016/j.str.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L. 2012. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res 40:9887–9896. doi: 10.1093/nar/gks737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scholz I, Lange SJ, Hein S, Hess WR, Backofen R. 2013. CRISPR-Cas systems in the cyanobacterium Synechocystis sp. PCC6803 exhibit distinct processing pathways involving at least two Cas6 and a Cmr2 protein. PLoS One 8:e56470. doi: 10.1371/journal.pone.0056470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hatoum-Aslan A, Maniv I, Marraffini LA. 2011. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc Natl Acad Sci U S A 108:21218–21222. doi: 10.1073/pnas.1112832108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ. 2013. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 36.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marraffini LA, Sontheimer EJ. 2010. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463:568–571. doi: 10.1038/nature08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biswas A, Gagnon JN, Brouns SJ, Fineran PC, Brown CM. 2013. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol 10:817–827. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lange SJ, Alkhnbashi OS, Rose D, Will S, Backofen R. 2013. CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res 41:8034–8044. doi: 10.1093/nar/gkt606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gardner RG, Russell JB, Wilson DB, Wang GR, Shoemaker NB. 1996. Use of a modified Bacteroides-Prevotella shuttle vector to transfer a reconstructed beta-1,4-d-endoglucanase gene into Bacteroides uniformis and Prevotella ruminicola B(1)4. Appl Environ Microbiol 62:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bélanger M, Rodrigues P, Progulske-Fox A. 2007. Genetic manipulation of Porphyromonas gingivalis. Curr Protoc Microbiol Chapter 13:Unit13C.12. doi: 10.1002/9780471729259.mc13c02s05. [DOI] [PubMed] [Google Scholar]
- 43.Nelson KE, Fleischmann RD, DeBoy RT, Paulsen IT, Fouts DE, Eisen JA, Daugherty SC, Dodson RJ, Durkin AS, Gwinn M, Haft DH, Kolonay JF, Nelson WC, Mason T, Tallon L, Gray J, Granger D, Tettelin H, Dong H, Galvin JL, Duncan MJ, Dewhirst FE, Fraser CM. 2003. Complete genome sequence of the oral pathogenic bacterium Porphyromonas gingivalis strain W83. J Bacteriol 185:5591–5601. doi: 10.1128/JB.185.18.5591-5601.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang R, Preamplume G, Terns MP, Terns RM, Li H. 2011. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure 19:257–264. doi: 10.1016/j.str.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sternberg SH, Haurwitz RE, Doudna JA. 2012. Mechanism of substrate selection by a highly specific CRISPR endoribonuclease. RNA 18:661–672. doi: 10.1261/rna.030882.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kunin V, Sorek R, Hugenholtz P. 2007. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol 8:R61. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, Resch AM, Glover CV, Graveley BR, Terns RM, Terns MP. 2012. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell 45:292–302. doi: 10.1016/j.molcel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cugini C, Klepac-Ceraj V, Rackaityte E, Riggs JE, Davey ME. 3 April 2013. Porphyromonas gingivalis: keeping the pathos out of the biont. J Oral Microbiol doi: 10.3402/jom.v5i0.19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tribble GD, Kerr JE, Wang BY. 2013. Genetic diversity in the oral pathogen Porphyromonas gingivalis: molecular mechanisms and biological consequences. Future Microbiol 8:607–620. doi: 10.2217/fmb.13.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tribble GD, Rigney TW, Dao DH, Wong CT, Kerr JE, Taylor BE, Pacha S, Kaplan HB. 2012. Natural competence is a major mechanism for horizontal DNA transfer in the oral pathogen Porphyromonas gingivalis. mBio 3:e00231-11. doi: 10.1128/mBio.00231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harmsen M, Lappann M, Knøchel S, Molin S. 2010. Role of extracellular DNA during biofilm formation by Listeria monocytogenes. Appl Environ Microbiol 76:2271–2279. doi: 10.1128/AEM.02361-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas VC, Hiromasa Y, Harms N, Thurlow L, Tomich J, Hancock LE. 2009. A fratricidal mechanism is responsible for eDNA release and contributes to biofilm development of Enterococcus faecalis. Mol Microbiol 72:1022–1036. doi: 10.1111/j.1365-2958.2009.06703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vilain S, Pretorius JM, Theron J, Brözel VS. 2009. DNA as an adhesin: Bacillus cereus requires extracellular DNA to form biofilms. Appl Environ Microbiol 75:2861–2868. doi: 10.1128/AEM.01317-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phillips P, Progulske-Fox A, Grieshaber S, Grieshaber N. 2014. Expression of Porphyromonas gingivalis small RNA in response to hemin availability identified using microarray and RNA-seq analysis. FEMS Microbiol Lett 351:202–208. doi: 10.1111/1574-6968.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lillestøl RK, Shah SA, Brügger K, Redder P, Phan H, Christiansen J, Garrett RA. 2009. CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol Microbiol 72:259–272. doi: 10.1111/j.1365-2958.2009.06641.x. [DOI] [PubMed] [Google Scholar]
- 56.Westra ER, van Erp PB, Künne T, Wong SP, Staals RH, Seegers CL, Bollen S, Jore MM, Semenova E, Severinov K, de Vos WM, Dame RT, de Vries R, Brouns SJ, van der Oost J. 2012. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol Cell 46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swarts DC, Mosterd C, van Passel MW, Brouns SJ. 2012. CRISPR interference directs strand specific spacer acquisition. PLoS One 7:e35888. doi: 10.1371/journal.pone.0035888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. 2010. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet 26:335–340. doi: 10.1016/j.tig.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.