

Abstract
The integrin CD11b/CD18 (also known as Mac-1), which is a heterodimer of the αM (CD11b) and β2 (CD18) subunits, is critical for leukocyte adhesion and migration and for immune functions. Blocking integrin-mediated leukocyte adhesion, although beneficial in experimental models, has had limited success in treating inflammatory diseases in humans. Here, we used an alternative strategy of inhibiting leukocyte recruitment by activating CD11b/CD18 with small-molecule agonists, which we term leukadherins. These compounds increased the extent of CD11b/CD18-dependent cell adhesion of transfected cells and of primary human and mouse neutrophils, which resulted in decreased chemotaxis and transendothelial migration. Leukadherins also decreased leukocyte recruitment and reduced arterial narrowing after injury in rats. Moreover, compared to a known integrin antagonist, leukadherins better preserved kidney function in a mouse model of experimental nephritis. Leukadherins inhibited leukocyte recruitment by increasing leukocyte adhesion to the inflamed endothelium, which was reversed with a blocking antibody. Thus, we propose that pharmacological activation of CD11b/CD18 offers an alternative therapeutic approach for inflammatory diseases.
INTRODUCTION
The migration and recruitment of leukocytes is essential for their normal immune response to injury and infection and for various inflammatory and autoimmune disorders. Leukocyte functions are modulated by β2 integrins, including the highly abundant integrin CD11b/CD18 (also known as Mac-1 and CR3), which is a heterodimer of the αM (CD11b) and β2 (CD18) subunits (1-3). CD11b/CD18 is normally present in an inactive conformation in circulating leukocytes, but it is rapidly activated (4-6) to mediate leukocyte adhesion, migration, and accumulation at the sites of inflammation. Indeed, blocking CD11b/CD18 and its ligands (7-9) and ablation of the genes encoding CD11b (3) or CD18 (10) decrease the severity of inflammatory responses in many animal models; however, such blocking agents have had limited success in treating inflammatory or autoimmune diseases in humans (11, 12). This may be because complete blockade of CD11b/CD18 with antibodies is difficult owing to the availability of a large intracellular pool of CD11b/CD18 that can be mobilized to the cell surface (13, 14), or because the suppression of leukocyte recruitment with blocking agents requires >90% occupancy of active integrin receptors (15). Antibodies against β2 integrins also have unexpected side effects (16).
Here, we took an alternative approach to the treatment of inflammatory diseases that involves the activation, rather than the blockade, of CD11b/CD18. Our premise was based on the finding by Harlan and coworkers more than 15 years ago that the trapping of integrin α4β1 in a high-avidity state with an activating antibody increases cell adhesion and decreases eosinophil migration (17). Experiments with knock-in animals that express activating mutants of the integrins αLβ2 (18, 19) and α4β7 (20) provide in vivo support for this hypothesis. We asked whether small molecules, which are easily delivered in vivo and can be readily optimized for use in different mammals, could be an effective approach for activating integrins. Agonists have an additional advantage in that they need to activate only a fraction of cellular receptors to elicit a functional response in vivo (21). Whether transient activation of a fraction of native receptors in vivo, as is expected from small-molecule treatment, can have a biological effect also remains an open question. Here, in multiple physiologically relevant experimental models in different species, we showed that the severity of inflammatory diseases was reduced by the activation of CD11b/CD18 with small molecules. These findings suggest that integrin activation might be a useful pharmacological approach to the treatment of inflammatory and autoimmune diseases in humans.
RESULTS
Leukadherins are newly characterized, small-molecule agonists of CD11b/CD18
We previously used a cell-based, high-throughput screening (HTS) assay to screen a chemical library of >100,000 molecules for compounds that affected the adhesion of K562 cells that expressed CD11b/CD18 at the cell surface (K562 CD11b/CD18 cells) to fibrinogen, the physiological ligand of CD11b/CD18 (22-24). Focusing our search on compounds that increased cell adhesion (agonists), we identified a series of compounds, which we termed leukadherins, that contained a core furanyl thiazolidinone chemical structural motif that was common in all the identified compounds (22). We explored the structure-activity relationship of various substitutions of chemical residues in the central core common to leukadherins (23) and identified three compounds, leukadherin-1 (LA1), leukadherin-2 (LA2), and leukadherin-3 (LA3) (Fig. 1, A to D), which showed enhanced activity in vitro. LA1, LA2, and LA3 separately increased CD11b/CD18-dependent cell adhesion to fibrinogen with 50% effective concentration (EC50, the effective concentration for a 50% increase in adhesion) values of 4, 12, and 14 mM, respectively. Cells that did not express CD11b/CD18 did not show any substantial binding to fibrinogen. The binding of CD11b/CD18 to its ligands is mediated by divalent cations, with calcium ions (Ca2+) inhibiting the interaction (thus acting as antagonists), whereas magnesium (Mg2+) and manganese (Mn2+) ions facilitate the interaction (thus acting as agonists) (25, 26). A study described an inverse agonist of the integrin lymphocyte function-associated antigen–1 (LFA-1, also known as the αLβ2 integrin) that increased LFA-1–mediated adhesion under basal conditions but inhibited it under activating conditions (27). To evaluate whether leukadherins could similarly inhibit preactivated CD11b/CD18, we measured the binding of K562 CD11b/CD18 cells to immobilized fibrinogen in the presence of Mn2+. We found that LA1, LA2, and LA3 did not inhibit cell adhesion in the presence of the agonist Mn2+ (fig. S1), suggesting that they are true agonists. We also identified a structurally related compound, leukadherin-control (LA-C), which showed no effect on CD11b/CD18-dependent cell adhesion (Fig. 1D). Increased adhesion of CD11b/CD18-expressing cells, induced by Mn2+ (28) and LA1, LA2, or LA3, was blocked by the monoclonal antibodies (mAbs) IB4 (29) and 44a (30), which are specific for CD18 and CD11b, respectively (Fig. 1E), further confirming that these compounds mediate CD11b/CD18-dependent cell adhesion.
Fig. 1.
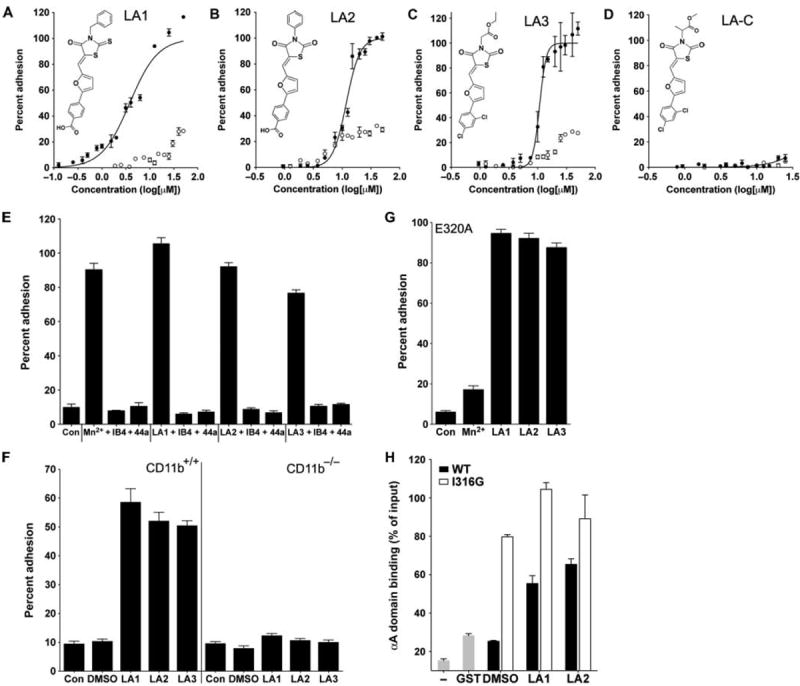
Leukadherins increase CD11b/CD18-dependent cell adhesion. (A to D) Dose-response curves showing the percentages of input K562 CD11b/CD18 cells (filled circles) and K562 cells (open circles) that adhered to immobilized fibrinogen in the presence of increasing amounts of LA1, LA2, LA3, or LA-C. The chemical structures of LA1, LA2, LA3, and LA-C are also shown. Data are means ± SEM (n = 3 to 6 replicates per data point) from one of at least three independent experiments. (E) Histograms showing the adhesion of K562 CD11b/CD18 cells to fibrinogen in response to LA1, LA2, or LA3 in the absence or presence of the blocking antibodies IB4 and 44a. Also shown is the extent of adhesion in the presence of physiologic concentrations of Ca2+ and Mg2+ (Con) and that in the presence of the known agonist Mn2+. Data are means ± SEM (n = 4 to 9 replicates per condition) from one of at least three independent experiments. (F) Histograms showing the percentage adhesion of wild-type (WT, CD11b+/+) and CD11b−/− neutrophils to immobilized fibrinogen in the absence (DMSO) or presence of LA1, LA2, or LA3 as compared to that of control cells (Con). Data are means ± SEM (n=5 replicates per condition) from one of at least three independent experiments. (G) Histograms showing the percentage of binding of K562 E320A cells to immobilized fibrinogen induced by LA1, LA2, or LA3. Also shown is the extent of K562 E320A adhesion with Ca2+ and Mg2+ ions (Con) and Mn2+. Data are means ± SEM (n = 6 replicates per condition) from one of at least three independent experiments. (H) Histograms showing the percentage binding of recombinant GST-fused αA domain constructs to immobilized fibrinogen (normalized to the amounts of input αA) in the absence of leukadherin (DMSO) or in the presence of LA1 or LA2. Also shown is the background binding obtained in the absence of any protein (−) or with the GST construct alone (GST). Data are means ± SEM (n = 3 replicates per condition) from one of at least three independent experiments.
Neutrophils contain a large, intracellular pool of CD11b/CD18 that can be mobilized to the cell surface (13, 14), which, in addition to a conformational switch in CD11b/CD18 from an inactive to an active form, helps to increase the extent of adhesion of neutrophils to the extracellular matrix. To rule out an enhancement in the abundance of CD11b/CD18 at the cell surface as a means by which LA1, LA2, and LA3 might increase cell adhesion, we measured the relative amounts of surface CD11b/CD18 on K562 cells (fig. S2) and neutrophils (fig. S3) and found that they were not increased by any of these compounds. The increase in CD11b/CD18-dependent cell adhesion stimulated by LA1, LA2, and LA3 was independent of the type of integrin ligand, because all three also increased the extent of adhesion of cells to the CD11b/CD18 ligands iC3b (fig. S4) and intercellular adhesion molecule–1 (ICAM-1) (fig. S5). Human monocytic THP-1 cells also showed a similar leukadherin-induced increase in cell adhesion, suggesting that the effects of leukadherins are independent of cell type (fig. S6). LA1, LA2, and LA3 also increased the binding of wild-type, but not CD11b−/−, neutrophils to immobilized fibrinogen (3) (Fig. 1F), further demonstrating that these compounds target CD11b/CD18. To determine whether leukadherins also affected CD11b/CD18-mediated phagocytosis, we incubated K562 CD11b/CD18 cells with iC3b-coated sheep red blood cells (EiC3bs) (31). We found that LA1, LA2, and LA3 all substantially increased the capture of EiC3bs and rosetting (the binding of multiple EiC3bs, which are much smaller than a typical human cell, to a phagocytic cell forms a rosette), suggesting that these agonists can also stimulate CD11b/CD18-mediated phagocytosis (fig. S7).
Leukadherins bind to the ligand-binding αA domain and allosterically activate CD11b/CD18
To identify the binding site of LA1, LA2, and LA3, which are predicted to bind to the ligand-binding αA (or αI) domain in CD11b/CD18 (22, 23, 32), we generated K562 cells stably expressing the mutant integrin CD11bE320A/CD18 (K562 E320A cells). The conserved residue Glu320 (E320) in the linker region that follows the activation-sensitive α7 helix in the CD11bA domain (αA domain) acts as an endogenous ligand of the von Willebrand factor type A (vWFA) domain of CD18 (βA or I domain) (33). The E320A mutation abolishes the Mn2+-mediated increase in ligand binding by CD11b/CD18; however, the stabilization of αA in a high-affinity conformation by additional activating mutations overcomes this deficit and induces ligand binding by the E320A mutant protein (34). LA1, LA2, and LA3 (but not Mn2+) selectively increased the binding of K562 E320A cells to fibrinogen (Fig. 1G), suggesting that these compounds also bind to and stabilize the αAdomain in a high-affinity conformation. To confirm this, we performed experiments with purified recombinant αA(35) and found that LA1 and LA2 increased the binding of the wild-type αA domain to immobilized ligand (Fig. 1H) to an extent similar to that observed with a mutant αA domain that contains a constitutionally activating mutation (I316G) (36). The lower solubility of LA3 compared to those of LA1 and LA2 precluded its use in this experiment.
In silico docking studies with high-resolution, three-dimensional (3D) structures of the αA domain in its low- and high-affinity conformations (36-38) suggested that LA1, LA2, and LA3 preferentially bound to the open, high-affinity conformation of the αA domain, near the activation-sensitive α7 helix region, thereby allosterically stabilizing the αA domain in its high-affinity conformation (fig. S8) (23). Flow cytometric analysis showed an increase in the binding of the activation-sensitive mAb 24 to K562 CD11b/CD18 cells in the presence of LA1, confirming that LA1 activated full-length integrin on live cells (fig. S9). Björklund et al. described an agonist of CD11b/CD18, termed IMB-10, that targets the αA domain of CD11b/CD18 (32). We compared the relative affinities of LA1 and IMB-10 for CD11b/CD18 with our cell-based adhesion assay and found that LA1 showed the higher affinity (fig. S10), perhaps because of its more rotationally constrained furanyl thiazolidinone central scaffold.
Leukadherins reduce leukocyte migration by increasing the extent of CD11b/CD18-mediated cell adhesion and reducing de-adhesion of the uropod
Leukocyte chemotaxis on 2D surfaces involves sequential integrin-mediated adhesion and de-adhesion steps (39). Cells that have constitutively active mutant integrins show increased adhesion and reduced cell migration in chemotactic gradients, compared to those of their wild-type counterparts, through the trapping of the mutant integrins in a ligand-bound conformation (40, 41). To test whether the increased cell adhesion induced by leukadherins affected cell migration, we performed experiments with murine neutrophils, which undergo chemotaxis in response to a gradient of the chemoattractant peptide N-formyl-Met-Leu-Phe (fMLP) (42). Live-cell imaging showed the smooth migration of neutrophils in a physiologic buffer (Fig. 2A); however, treatment of the cells with LA1, LA2, or LA3 resulted in a substantial decrease in lateral migration and migration velocity (Fig. 2, A to C). Although the cells treated with LA1, LA2, or LA3 showed some movement toward the chemoattractant, they displayed reduced directional persistence (Fig. 2D) and reduced mean square displacement (MSD) (Fig. 2E), which was suggestive of their constrained motility, compared to the more directed motility of control cells treated with dimethyl sulfoxide (DMSO).
Fig. 2.
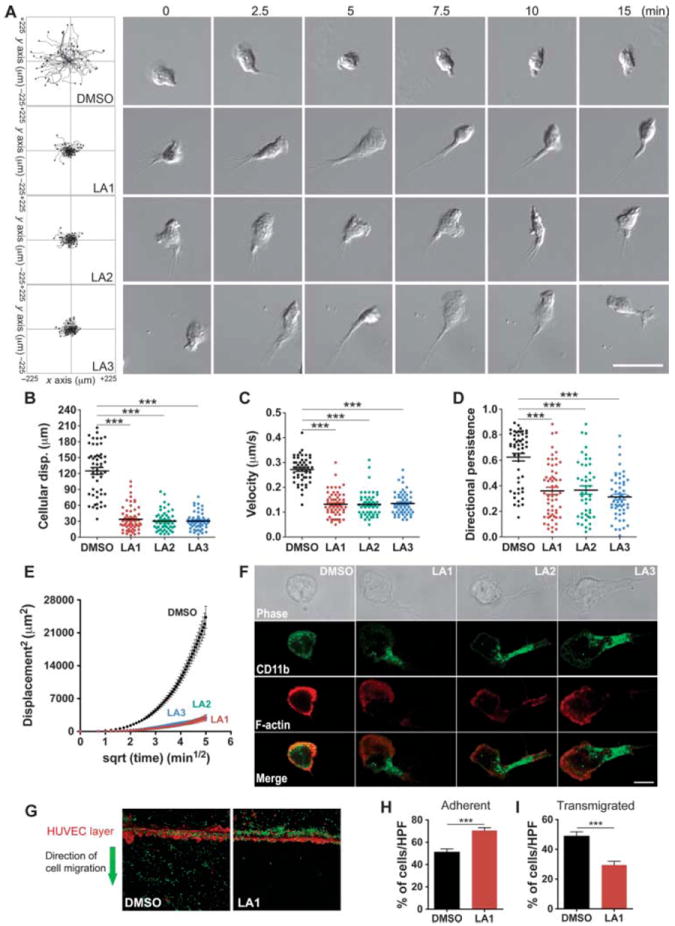
Leukadherins decrease cell migration by stabilizing integrin-substrate binding in the uropod. (A) Track plots showing the analysis of migrating WT neutrophils in Zigmond chambers in response to a gradient of fMLP in the absence of leukadherin (DMSO) or in the presence of LA1, LA2, or LA3. Data are from >50 cells per condition and from three or more independent experiments for each condition. Representative cell images at various time points from timelapse video microscopy are also shown. Scale bar, 25 μm. (B to E) Quantitative analysis of (B) the mean displacement, (C) the mean velocity, (D) the directional persistence, and (E) the mean displacement square plots shows the effects of leukadherins on cell motility. Lines indicate means ± SEM. ***P < 0.0001. (F) Fluorescence images of CD11b localization in WT neutrophils undergoing chemotaxis in response to f MLP and in the absence of leukadherins (DMSO) or in the presence of LA1, LA2, or LA3. Representative confocal and phase-contrast images of migrating neutrophils analyzed for the presence of CD11b (green) and F-actin (red) are from one of at least three independent experiments. Scale bar, 5 μm. (G to I) Transendothelial migration of THP-1 cells across a layer of HUVECs. (G) Representative confocal images (10× magnification) showing THP-1 cells (green) transmigrating across a layer of HUVECs stimulated with TNF-α (red) in response to a gradient of the chemokine MCP-1 in the absence (DMSO) or presence of LA1. Data are representative of at least five independent experiments. (H) Histogram showing the quantification of THP-1 that had adhered to the HUVECs. (I) Histograms showing the quantification of transmigrated THP-1 cells. Data are the means ± SEM. ***P < 0.0001.
In addition, unlike neutrophils undergoing chemotaxis in the absence of leukadherins, which displayed a typical flattened leading edge and a short, narrow tail, cells that migrated in the presence of LA1, LA2, or LA3 showed elongated uropods (Fig. 2A and movies S1 to S4), suggesting that a defect in cell de-adhesion was the key mechanism responsible for defective cell migration, as has been observed in cells with activating mutations of their integrins (18, 20). To investigate this, we used confocal microscopy to show that CD11b/CD18 clustered in the extended uropods of cells treated with LA1, LA2, or LA3 (Fig. 2F), suggesting that the failure to release integrin-substrate interactions in the uropod was responsible for the defective migration of these cells. Leukadherins had no effect on neutrophil migration in 3D collagen gels (fig. S11 and movies S5 and S6), supporting findings that leukocyte migration in 3D substrates is integrin-independent (43). However, leukadherins reduced the efficiency of trans-endothelial migration (TEM) by THP-1 cells across a layer of human umbilical vein endothelial cells (HUVECs) activated by tumor necrosis factor–α (TNF-α) in vitro by increasing cell adhesion to the HUVEC layer (Fig. 2, G to I). Together, these data suggest that leukadherins increase cell adhesion and reduce their lateral motility, thereby inhibiting TEM.
Leukadherins reduce recruitment of leukocytes during acute peritonitis in mice
To determine the effects of LA1, LA2, and LA3 on inflammatory responses in vivo, we used the acute, thioglycolate-induced peritonitis model in mice (3). LA1, LA2, and LA3 showed no in vitro cytotoxicity (figs. S12 and S13) at concentrations as high as 50 μM. In addition, LA1, LA2, and LA3 did not induce integrin clustering (fig. S14) or outside-in signaling (fig. S15), suggesting that they are not ligand mimics. Intraperitoneal injection of thioglycolate resulted in substantial accumulation of neutrophils in the peritoneum compared to that in mice injected with saline (Fig. 3A). Administration of LA1 30 min before injection with thioglycolate significantly reduced the amount of neutrophil accumulation by 40% (P < 0.05) compared to that in mice pretreated with vehicle, whereas LA2 reduced neutrophil accumulation by 65% (P < 0.0001), and LA3 reduced it by 55% (P < 0.05) (Fig. 3A).
Fig. 3.
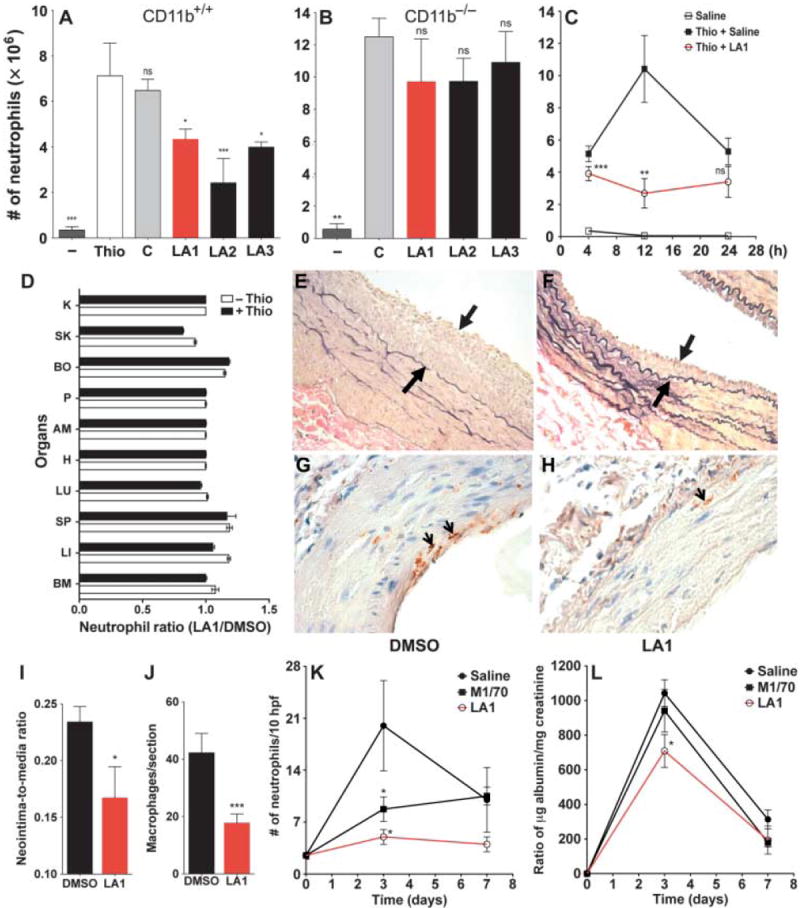
Leukadherins reduce the recruitment of leukocytes during inflammation and preserve organ function in vivo. (A and B) Bar graphs showing the total number of neutrophils in the peritoneal fluid of (A) WT and (B) CD11b−/− mice 4 hours after intraperitoneal injection of thioglycolate alone or of thioglycolate subsequent to the administration of vehicle (C), LA1, LA2, or LA3. Saline was used as a control (n = 4 to 9 mice per treatment). Data are means ± SEM. *P < 0.05; **P < 0.001; ***P < 0.0001; ns, not significant (by one-way ANOVA). (C) Graph showing the number of neutrophils in the peritoneal fluid of WT animals 4, 12, and 24 hours after thioglycolate injection in the absence (filled black square) or presence of LA1 (open red circles) (n = 3 to 4 mice per treatment). Peritoneal neutrophils in the absence of thioglycolate are also shown (open black squares). **P < 0.001; ***P < 0.0001; ns, not significant. (D) Bar graph showing the ratios of neutrophils detected in various organs 4 hours after treatment of WT mice (n = 3 mice per group) with DMSO or LA1 in the absence (−Thio) or presence of thioglycolate-induced inflammation (+Thio). BM, bone marrow; LI, liver; SP, spleen; LU, lung; H, heart; AM, abdominal muscle; P, pancreas; BO, bowel; SK, skin; and K, kidney. Data are means ± SD (n = 3 mice per treatment). (E and F) Representative photomicrographs of arteries 21 days after balloon injury in rats treated with vehicle (DMSO) or LA1. Arrows indicate neointimal thickening. (G and H) Photomicrographs of representative arteries 3 days after balloon injury in rats treated with DMSO or LA1. Arrows point to CD68+ macrophages. (I) Bar graph showing the neointima-to-media ratio as determined by morphometric analysis of the injured arteries from DMSO- or LA1-treated rats (n = 7 to 9 rats per group). Data are means ± SEM. *P < 0.05. (J) Bar graph showing quantification of macrophage infiltrates in injured arteries (3 days after injury) from rats treated with DMSO or LA1 (n = 12 mice per treatment). Data are means ± SEM. ***P < 0.0001. (K and L) The effect of agonist LA1 on ameliorating kidney injury is better than that of the antagonist M1/70. (K) Graph displaying the number of glomerular neutrophils in untreated (saline), antagonist-treated (M1/70), and LA1-treated mice at various time points (n = 3 to 4 mice per group, except at the day 0 time point, where n = 2). Data are means ± SEM. *P < 0.05. (L) Graph plotting measured proteinuria in untreated (saline), antagonist-treated (M1/70), and LA1-treated mice at various time points (n = 3 to 8 mice group, except at the day 0 time point, where n = 2). Data are means ± SEM. *P < 0.05.
We could not detect any difference in the numbers of circulating leukocytes between leukadherin-treated mice and control, vehicle-treated mice (table S1). This suggested that LA1, LA2, and LA3 did not cause leukocyte cytotoxicity in vivo, thus ruling this out as a reason for the observed reduction in the number of neutrophils in leukadherin-treated animals. We also found that LA1, LA2, and LA3 did not substantially reduce the number of neutrophils recruited to the peritoneum of thioglycolate-treated CD11b−/− mice (Fig. 3B), which showed increased neutrophil accumulation compared to that in wild-type mice, because CD11a compensates for the lack of CD11b (3). This further suggests that LA1, LA2, and LA3 target CD11b/CD18 in vivo.
In experiments with LA1 as the representative leukadherin, we also analyzed the kinetics of neutrophil recruitment. We found that the number of peritoneal neutrophils increased 4 hours after administration of thioglycolate in animals pretreated with vehicle, peaked after 12 hours, and declined thereafter (Fig. 3C). In LA1-treated animals, neutrophil accumulation was substantially reduced at 4 hours after administration of thioglycolate and stayed reduced at 12 hours, suggesting that leukadherins substantially inhibited neutrophil recruitment. We observed comparably reduced numbers of peritoneal neutrophils 24 hours after thioglycolate administration in both groups of animals. Furthermore, analysis of tissue histology of various organs and neutrophil enumeration confirmed that leukadherins did not lead to neutrophil sequestration in any organ (Fig. 3D), ruling this out as a cause of the reduced neutrophil influx into inflamed tissues. This suggests that increased neutrophil adhesion near sites of inflammation, which decreased their motility and extravasation, was the likely reason for their decreased recruitment to the peritoneum.
Leukadherins reduce neointimal thickening upon vascular injury in rats
Percutaneous transluminal coronary angioplasty (PTCA) is the most effective way to unblock occluded coronary arteries (44); however, PTCA causes denudation of the endothelial cell lining and deposition of fibrin and platelets at the site of injury. The inflammatory response to this injury induces vascular smooth muscle cell growth into the innermost layer of the vessel (neointima) and, ultimately, arterial renarrowing (restenosis). Leukocyte recruitment and infiltration, through selective binding between CD11b/CD18 expressed on the surface of leukocytes and the platelet cell surface receptor glycoprotein (GP) Ibα (45), precedes neointima formation and restenosis (46). Indeed, antibody-mediated blockade of CD11b/CD18 or loss of CD11b (for example, in CD11b−/− mice) decreases intimal thickening after angioplasty or stent implantation in experimental models (8). To further test the anti-inflammatory properties of LA1 in vivo, we used it in an arterial balloon injury model in rats (47). The extent of disease (remodeling or hyperplasia) was determined by calculating the ratio between areas of the neointima (the newly formed innermost layer) and the media (the vessel wall) at the site of injury. We administered LA1 or vehicle (DMSO) to Fischer male rats 30 min before injury and continued injections every other day for 3 weeks. Injured arteries of the LA1-treated rats developed significantly reduced neointimal thickening (Fig. 3, E and F) (neointimato- media ratio of 0.16 ± 0.02 versus 0.23 ± 0.01, P < 0.05) (Fig. 3I), whereas the control compound LA-C showed no effect (fig. S16). To determine whether leukadherin treatment leads to reduced leukocyte accumulation, which precedes vascular remodeling, we performed immunohistochemical analyses of arteries 3 days after injury. We observed a significant reduction in the number of macrophages in the arteries of LA1- treated animals (17.7 ± 3.1 versus 42.2 ± 6.7,P< 0.0001) (Fig. 3, G to J); LA3 showed similar protective effects (fig. S17). Together, these results suggest that leukadherins lead to reduced leukocyte accumulation at the site of vascular injury and a subsequent decrease in neointimal thickening.
Leukadherins are more efficient than integrin antagonists in treating inflammatory injury
To determine whether CD11b/CD18 agonists have any therapeutic advantages over antagonists, we performed a head-to-head comparison between a well-characterized CD11b/CD18 antagonist, the M1/70 antibody against CD11b (48), and LA1 in experiments with an established mouse model of kidney disease, anti–glomerular basement membrane (anti-GBM) nephritis (49). This model is characterized by neutrophil infiltration that mediates urinary protein loss, including that of albumin. Consistent with a role for CD11b in this disease, CD11b−/− mice (49) and rats treated with mAb against CD11b (50) show decreased leukocyte infiltration and protection from proteinuria relative to their wild-type counterparts. We found that induction of disease in mice led to a peak influx of neutrophils into the kidney and maximal proteinuria at day 3 (Fig. 3, K and L), whereas M1/70 substantially decreased neutrophil influx and reduced proteinuria. However, LA1 produced a maximal decrease in both the number of infiltrating neutrophils and the extent of proteinuria in treated mice, suggesting a therapeutic advantage of agonists over antagonists in this disease model.
The effects of leukadherins can be reversed by removing them from the circulation
To visualize the effects of leukadherins on leukocyte accumulation in vivo, we used transgenic Tg(mpx∷eGFP) zebrafish that express the gene encoding enhanced green fluorescent protein (eGFP) under a myeloid-specific peroxidase gene (mpx) promoter to specifically fluorescently label neutrophils (51). Tailfin transection in zebrafish larvae at 3 days post fertilization (dpf) (Fig. 4A) revealed a rapid and robust recruitment of neutrophils to the site of tissue injury (Fig. 4, B and C to E) (51). LA1 had no observable effects in uninjured larvae (Fig. 4B); however, LA1 significantly reduced the number of neutrophils at the injured zebrafish tailfin 4 hours after injury compared to that in control larvae (15.6 ± 1.7 versus 34.6 ± 4.5, P < 0.0001) (Fig. 4, C and E).
Fig. 4.
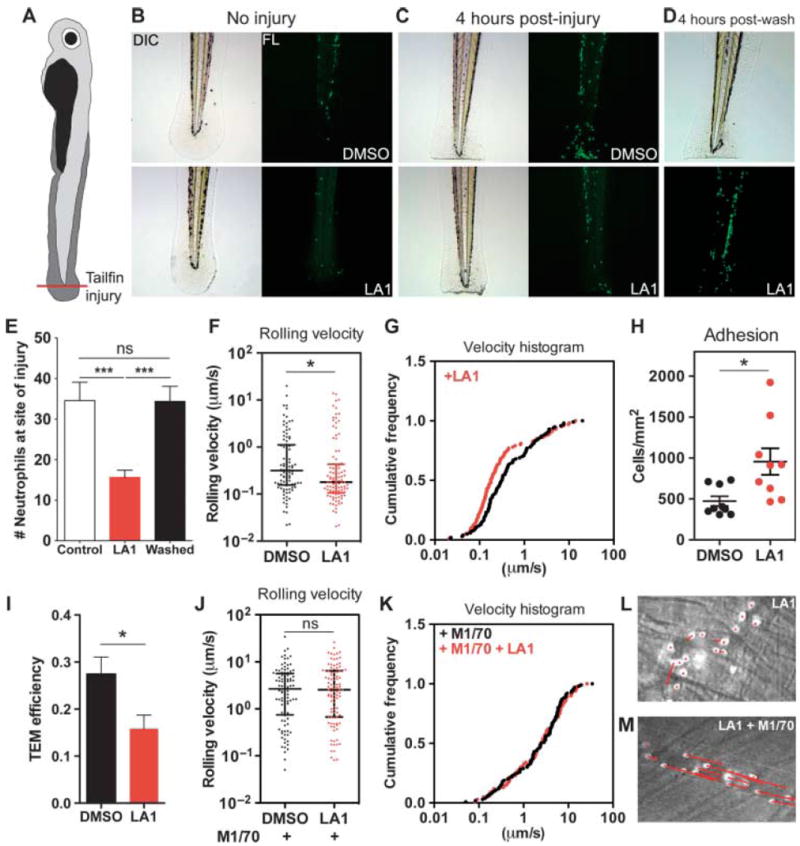
Leukadherins decrease leukocyte recruitment in vivo by increasing the extent of slow rolling and the number of adherent cells, thereby decreasing leukocyte TEM. (A) Schematic representing the zebrafish tailfin injury model. (B to D) Photomicrographs (left) and fluorescence images (right) of (B) the larvae tail without injury, (C) the tail with injury, and (D) 4 hours after the removal of LA1 from treated, injured larvae. Images show accumulation of neutrophils (green) in the injured tail. (E) Graph showing quantification of the number of neutrophils near the site of tailfin injury in zebrafish larvae treated with DMSO (Control) and LA1 (n = 12 to 16 larvae per group). Data are means ± SEM. ***P < 0.0001; ns, not significant (by one-way ANOVA). (F to M) Intravital microscopy–based determination of the effects of LA1 on leukocyte migration in vivo. (F) Graph showing the rolling velocities of individual neutrophils in the venules of TNF-α–treated mouse cremaster muscle without (DMSO) or with LA1. Lines indicate medians and 25 to 75% interquartile ranges. *P < 0.05. (G) Cumulative histograms of the rolling velocities of 100 leukocytes from DMSO-treated (black dots) and LA1-treated (red dots) animals. (H) Numbers of adherent neutrophils in venules without (DMSO) or with LA1. Data are means ± SEM. *P < 0.05. (I) Graph showing relative efficiency of neutrophil TEM (as determined by the number of transmigrated neutrophils/the number of adherent neutrophils) in the absence (DMSO) or presence of LA1. Data are means ± SEM. ***P < 0.05. (J) Rolling velocities of neutrophils in TNF-α– and M1/70-treated venules without (DMSO) or with LA1. Lines indicate median and interquartile ranges. ns, not significant. (K) Cumulative histograms of rolling velocity of 100 leukocytes from M1/70-treated mice in the absence (black dots) or presence of LA1 (red dots). Representative video micrograph images of cremaster muscle venules treated with (L) LA1 or (M) M1/70 and LA1. The lengths of the red arrows indicate neutrophil movement during a 25-s period.
Furthermore, to determine whether the effects of leukadherins in vivo were reversible, we administered LA1 to uninjured zebrafish for 4 to 8 hours, rinsed the zebrafish, induced tailfin injury, and quantified neutrophil accumulation 4 hours later. We found that the removal of LA1 led to neutrophil accumulation at the injured tailfins to an extent similar to that observed in untreated zebrafish larvae (Fig. 4, D and E). LA2 showed protective effects similar to those of LA1 (fig. S18). Fluorescence imaging of the injured whole zebrafish larvae showed no difference in total neutrophil numbers between leukadherin-treated and untreated larvae (fig. S19), which suggested that the leukadherin-mediated reduction in neutrophil accumulation was not due to a reduction in the overall number of neutrophils. This result is consistent with our experiments in mice that showed that leukadherins did not cause cytotoxicity in vivo (table S1). Together, these data demonstrate that leukadherins inhibit the accumulation of neutrophils at the sites of tissue injury and that their effects in vivo are reversible by their removal.
Leukadherins reduce the extravasation of leukocytes in vivo
Finally, to determine the mechanism of action of leukadherins in vivo, we performed intravital microscopic analysis of mouse cremaster muscle. CD11b/CD18 binds to ICAM-1 on the surface of TNF-α–activated endothelium to mediate slow rolling and arrest of leukocytes (52). We found that LA1 substantially decreased the rolling velocity of leukocytes in TNF-α–stimulated venules (Fig. 4, F and G), which led to a substantial increase in the number of adherent cells (Fig. 4H); however, the number of transmigrated cells adjacent to the vessel in LA1-treated mice was similar to that in the DMSOtreated animals (fig. S20), suggesting that LA1 substantially reduced the overall efficiency of leukocyte TEM (Fig. 4I). Injection of M1/70, which blocks CD11b, reversed the effects of LA1 on leukocyte rolling (Fig. 4, J to M), confirming that the effects of LA1 were CD11b/CD18-specific. Thus, these in vivo measurements mirror the in vitro data on the effects of leukadherins on leukocyte motility.
DISCUSSION
Leukocyte infiltration is a common finding in most inflammatory diseases, and the leukocytic integrin CD11b/CD18 plays an important role in this process (1, 2). CD11b/CD18-mediated leukocyte rolling and firm adhesion to the vascular wall lead to leukocyte transmigration across the vasculature and accumulation in the tissue as part of a multistep infiltration process (Fig. 5) (52). Current approaches that treat leukocyte infiltration are focused on blocking the adhesion of leukocytic integrins to their respective ligands (“anti-adhesion” therapy) (53, 54). Antibodies against integrins, such as the M1/70 mAb, block the binding of integrins to ligands found on the vascular wall, thus reducing the infiltration of leukocytes into the tissue (Fig. 5). Although such strategies have proven beneficial in certain animal models, many agents that block the binding of integrins to their ligands have failed in clinical trials (11, 53), had substantial side effects, or have had to be withdrawn from the market (55).
Fig. 5.
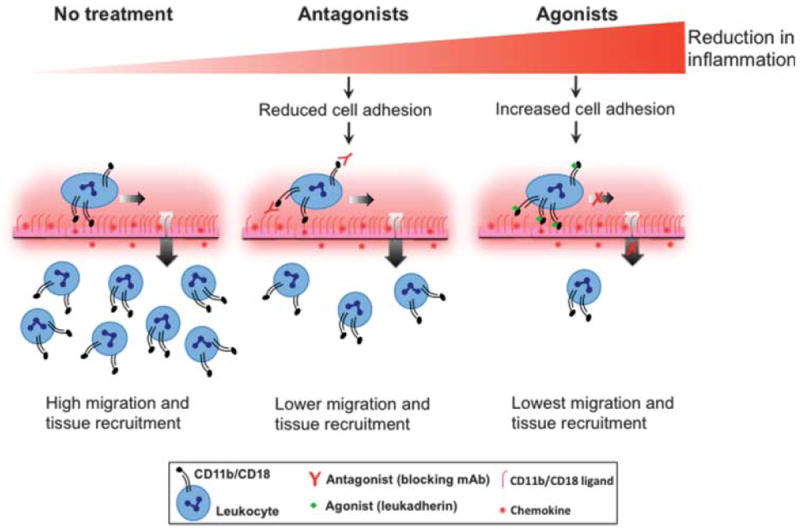
Schematic showing how integrin antagonists and agonists differ in their ability to reduce inflammatory disease. Integrin antagonists (central panel), such as blocking mAbs, prevent leukocyte adhesion to the inflamed endothelium, thereby reducing leukocyte migration and tissue recruitment as compared to the untreated situation (left panel). On the other hand, integrin agonists (such as leukadherins), promote the adhesion of leukocytes, which reduces their lateral and transendothelial migration and leads to an even greater decrease in the tissue recruitment of leukocytes. Thus, small-molecule integrin agonists (such as our prototype compounds, leukadherins) represent an alternative strategy for modulating leukocyte recruitment and inflammatory diseases.
Our data demonstrate that an alternative approach to inhibiting leukocyte migration by enhancing the activation of integrins with small molecules is highly effective in reducing leukocyte infiltration and subsequent inflammation in vivo (Fig. 5). We used a cell-based HTS assay (22, 24) to identify and optimize small-molecule agonists of CD11b/CD18, which we have termed leukadherins. The leukadherin compounds LA1 to LA3 have similar chemical structures, and they bind to the ligand-binding αA domain and convert CD11b/CD18 into its active conformation. We showed that leukadherins, but not the structurally similar compound LA-C, promoted CD11b/CD18-dependent cell adhesion and decreased leukocyte motility, which led to a substantial reduction in leukocyte TEM and recruitment into tissues.
In addition, we found that leukadherins had a higher affinity for CD11b/CD18 than did a CD11b/CD18 agonist (32), perhaps because of their more rotationally constrained furanyl thiazolidinone central ring structure. Furthermore, through experiments with various disease models and multiple animal species, we demonstrated that these CD11b/CD18 agonists reduced inflammation in physiologically relevant settings, suggesting that they are viable therapeutic leads for future optimization. Our data also reveal that when directly compared with a known antagonist, CD11b/CD18 agonists better preserve organ function upon inflammatory injury (Fig. 5). Finally, the in vivo effects of leukadherins were transient and were reversed by removing them from the circulation. Our results suggest that integrin-specific, small-molecule agonists represent an effective pharmacological approach for the treatment of inflammatory and autoimmune diseases.
Our results are consistent with previous studies that showed that integrin activation leads to increased cell adhesion and decreased migration (17-20, 27, 32). Additionally, knock-in mice expressing constitutively active mutants of the integrins αLβ2 (18, 19) and α4β7 (20) in the germline show reduced recruitment of inflammatory cells because of an increase in cellular adhesion and a reduction in cell migration through constitutively active integrins. However, unlike the knock-in animals, in which all of the mutant integrin receptors are expressed in a constitutively active form, whether activation of a fraction of wild-type receptors (as is expected upon treatment with a small-molecule agonist) could have a similar phenotype in vivo and could reduce inflammation in physiologically relevant disease models was not known. Not all of the studies of knock-in animals with active integrin mutants have presented the same response in vivo. Notably, homozygous knock-ins carrying an activating D759A mutation in the cytoplasmic tail of the integrin β1 subunit show no obvious phenotype and no functional changes under physiological conditions (56). Additionally, keratinocytes from these knock-in mice show comparable adhesion, spreading, and migration in vitro to those of cells expressing wild-type integrins. On the other hand, introduction of active mutants of the integrin α2 subunit in mice results in a marked reduction (in the case of leukocytes) or a complete loss (in the case of platelets) of activated α2 on the cell surface during thrombopoiesis, and the expression of active integrin α2 on progenitor cells has no effect on their migration in vivo (57). It is possible in this case that some compensatory mechanisms during animal development overcame the adhesion and migration deficiencies of the introduced mutations. In that respect, we believe that the chemical-biological approach highlighted in this study, in which the function of endogenous, wild-type protein is perturbed by a specific agonist, represents away forward to analyze the effects of integrin activation on cellular functions in vivo.
A potential concern with the use of integrin agonists as therapeutics in vivo is that increased adhesion of inflammatory cells to the vascular endothelium may harm the vascular cells, leading to vascular damage and leakage. However, we have not observed any signs of vascular injury in the various experimental models that we have studied, perhaps because such an increase in leukocyte adhesion is only transient. Previous studies with knock-in animals that express constitutively active mutants of the integrins αLβ2 (18, 19) and α4β7 (20) have also not reported any signs of vascular injury. We analyzed animals that received compounds for more than 3 months, and we found no systemic signs of vascular injury or leakage—no signs of edema or systemic vascular compromise. Whereas these observations mitigate the concern of systemic vascular toxicity associated with integrin agonists, detailed future studies will be needed to fully address some of these concerns. In conclusion, we showed that CD11b/CD18 agonists increased the adhesion and decreased the motility of leukocytes, which resulted in reduced leukocyte TEM, recruitment into the tissues, and inflammation. We suggest that integrin-specific, small-molecule agonists represent an effective pharmacological approach for the treatment of inflammatory and autoimmune diseases.
MATERIALS AND METHODS
Reagents and antibodies
The 44a mAb against CD11b [an immunoglobulin G (IgG) 2a (IgG2a) isotype] (30) and the heterodimer-specific IB4 mAb against CD18 (IgG2a) (29, 58) were from the American Type Culture Collection (ATCC). M1/70, a rat mAb against mouse CD11b (IgG2b) (48), was from the mAb core at University of California, San Francisco. We obtained mAb 24 (IgG1) (59) from Abcam, and the isotype control antibodies clone X40 (IgG1) and clone X39 (IgG2a), fluorescein isothiocyanate (FITC)–conjugated mAb A85-1 (rat anti-mouse IgG1), FITC-conjugated R19-15 (rat anti-mouse IgG2a), FITC-conjugated goat antibody against mouse immunoglobulin, rat antibody against mouse GR-1, GR-1–FITC, and phycoerythrin (PE)–conjugated rat antibody against mouse Mac-1 were from BD Pharmingen. Human fibrinogen (depleted of plasminogen, von Willebrand factor, and fibronectin) was from Enzyme Research Laboratories, bovine serum albumin (BSA) was from Sigma, recombinant human ICAM-1–Fc was from R&D Systems, and iC3b was from Calbiochem. MaxiSorp and High Bind 384-well plates were obtained from Nalgene and Corning, respectively. Nonfat milk was obtained from Bio-Rad. The cell quantitation reagent MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)- 2H-tetrazolium salt] was from Promega. Polymerase chain reaction (PCR) assay reagents, aswell as restriction and modification enzymes, were obtained from New England Biolabs Inc. Glutathione beads were purchased from Sigma. All cell culture reagents were from Invitrogen Corp. and Mediatech. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals Inc. The antibiotic G418was purchased from Invivogen. The sheep antibody against rabbit GBM was a gift from S. Shankland and J. Pippin.
Mice
The C57BL/6J (B6) wild-type and the B6 CD11b−/− (Jax 3991) (3) GFP mice were purchased from The Jackson Laboratory. Lys-eGFP mice have been described previously (60). The wild-type Fischer 344 rats were purchased from Harlan Laboratories. Animal care and procedures were approved by the Institutional Animal Care and Use Committee and were performed in accordance with institutional guidelines.
Cell lines
K562 cells (ATCC) stably transfected with plasmid encoding wild-type integrin CD11b/CD18 (K562 CD11b/CD18 cells) have been described previously (22, 31). The mutant integrin subunit CD11bE320A has been described previously (33). K562 cells stably transfected with mutant integrin CD11bE320A/CD18 (K562 E320A cells) were generated according to previously published protocols (22, 31). All cell lines were maintained in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% heat-inactivated FBS, penicillin and streptomycin (50 U/ml each), and G418 (0.5 mg/ml). THP-1 cells (ATCC) were maintained in RPMI 1640 supplemented with 10% heat-inactivated FBS and β-mercaptoethanol (50 mM) according to the manufacturer’s instructions.
Cell adhesion assays
Cell adhesion assays with immobilized ligands were performed as previously described (22). Assays with all cell lines were performed in an identical fashion. A stock solution of the leukadherin family of small-molecule agonists was prepared by dissolving the compounds in DMSO at a concentration of 2 to 10 mM. The final concentration of DMSO in the assay was ~1%. Assays were performed in three to six replicate wells. Data are from one of at least three independent experiments. The HTS assay to identify previously uncharacterized agonists with a library of >100,000 small molecules was performed as previously described (22, 61).
Neutrophil adhesion assay
Neutrophils from 8- to 10-week-old wild-type and CD11b−/− B6 mice were isolated from thioglycolate-stimulated peritonea as previously described (62). Cells were suspended in serum-free IMDM and incubated with leukadherins in ligand-coated wells in a 384-well plate for 10 min at 37°C. The assay plates were then gently inverted and kept in the inverted position for 30 min at room temperature to dislodge the nonadherent cells. The remaining adherent cells were quantified by imaging microscopy as previously described (22, 63). Assays were performed in triplicate wells. Data reported are from one of at least three independent experiments.
Chemotaxis assay and time-lapse video microscopy
Neutrophil chemotaxis on 2D surfaces was performed with Zigmond chambers (Neuro Probe) as described previously (42, 64) on acid-cleaned glass or fibrinogen-coated glass coverslips. Cell migration was studied in a gradient of f MLP (Sigma), which was generated by placing buffer alone in one well of the chamber and f MLP (10 μM) in the other well, in the absence or presence of leukadherins (15 μM). Cell migration was recorded at 5- to 30-s intervals for a period of 25 min with a Nikon Eclipse 90i or a Leica DMI16000 deconvolution microscope. Images from the Nikon microscope were acquired with a Nikon DS camera with a PLAN APO 20× differential interference contrast (DIC) microscope objective and captured with Nikon Imaging software. Leica DIC images were acquired with an HCX PL APO 40×/0.75 numerical aperture (NA) objective with a DCF360FX camera driven by LAS-AF software. Analysis of neutrophil migration was performed with the motile population that had moved more than 10 μm (64) with ImageJ software [National Institutes of Health (NIH)] using manual cell tracking by the Ibidi chemotaxis and migration tool plug-in for ImageJ. Motility parameters, such as migration velocity, the total cellular displacement (distance from the origin), and directional persistence, were analyzed for individual cell tracks with the Ibidi chemotaxis plug-in for ImageJ. At least 50 cells under each condition from at least three independent experiments were quantified.
Neutrophil migration in 3D collagen gels
Wild-type B6 neutrophils were suspended in a collagen gel solution (BD Biosciences) with a final collagen concentration of 1.6 mg/ml in the presence of vehicle (1% DMSO) or LA1 (15 μM) and cast in custom-built migration chambers with a thickness of 0.5 to 1 mm as described previously (65). Final neutrophil concentrations in the assay were 1 × 106 cells/ml of gel. After polymerization of the collagen fibers at 37°C for 30 min, gels were overlaid with 50 ml of culture medium containing fMLP (10 μm) and subsequently imaged with a Leica DMI16000 deconvolution microscope with DIC. Cell migration was recorded at 60-s intervals for a period of 45 min and was captured with an HC PL FLUOTAR 20×/0.5 NA objective with a DCF360FX camera and Leica LAS-AF software. Cell motility data were derived by constructing individual cell tracks with the manual tracking plug-in tool for ImageJ. Analysis of individual cell tracks was performed to determine migration velocity, the total cellular displacement (distance from the origin), and directional persistence with the chemotaxis plug-in tool for ImageJ. We quantified at least 40 independent cells under each condition from three independent experiments.
Imaging TEM of cells across a layer of activated HUVECs
To measure the effects of leukadherins on the TEM capacity of THP-1 cells, we performed vertical collagen gel invasion assays as described previously (66). Briefly, we prepared collagen gel chambers containing the chemokine monocyte chemotactic protein–1 (MCP-1, also known as CCL2) (200 ng/ml, Sigma) as described earlier. HUVECs (1 × 105, Millipore) were incubated with a cell-permeable red fluorescent dye (CellTracker Red CMTPX, Invitrogen) and then seeded on top of the collagen gels. After 16 hours in culture, THP-1 cells (2 × 104 cells) incubated with a cellpermeable green fluorescent dye (CellTracker Green CMFDA, Invitrogen) were placed on top of a layer of HUVECs that were activated with TNF-α (10 μg/ml) for 4 hours, and their adhesion to HUVECs and TEM across the HUVEC layer were determined with a Leica TCS SP5 confocal microscope or a Leica DMI16000 deconvolution microscope with an HCX PL APO 10×/1.3 NA objective driven by Leica LAS-AF software. At least 25 images were taken for each condition, and we determined the numbers of cells that adhered to the HUVEC layer and those that completely migrated through the HUVEC layer by manual counting.
Immunofluorescence microscopy
To examine the localization of CD11b/CD18 and polymerized actin (F-actin) in migrating neutrophils, we stimulated 1 × 104 cells with f MLP (10 μm) in serum-free RPMI 1640 on glass coverslips for 15 min at 37°C in the absence or presence of leukadherins (15 μM). The cells were fixed, permeabilized with 0.1% Triton X-100, and incubated with the M1/70 mAb against CD11b followed by Alexa Fluor 488–conjugated goat antibody against mouse Ig (Invitrogen) and rhodamine-labeled phalloidin (Invitrogen). A z series of fluorescence images was recorded with a Leica TCS SP5 confocal microscope and an HCX PL APO 63×/1.4 NA objective and with Leica LAS-AF software. The images presented are from a z stack projection of 15 confocal sections from the basal to the apical side of the cell (stack z spacing, 0.29 μm). Images presented are representative of at least 20 cells analyzed for each condition from at least two independent experiments. To examine clustering of CD11b/CD18 on the cell surface, we suspended 1 × 104 K562 CD11b/CD18 cells in serum-free IMDM and incubated the cells without or with fibrinogen (100 μg) for 3 hours at 37°C, as described previously (67), in the absence or presence of leukadherins (15 μM). The cells were fixed in suspension and incubated with the IB4 mAb, which is specific for CD11b/CD18, followed by Alexa Fluor 488–conjugated goat antibody against mouse Ig (Sigma). Fluorescence images were recorded with a Leica DMI16000 deconvolution microscope and an HCX PL APO 63×/1.3 NA objective with a DCF360FX camera and with Leica LAS-AF software. The CD11b/CD18 clusters were analyzed in ImageJ, and a 3D representation of fluorescence intensity was also generated in ImageJ. The images presented are representative of at least 20 cells analyzed for each condition from at least three independent experiments.
Purification of recombinant CD11b A domain (αA domain)
Recombinant human αA domains were constructed and purified according to published protocols (68). Briefly, the αA domain in its inactive conformation was generated by cloning and expressing protein fragments spanning residues Gly111 to Gly321 (321WT) (forward primer, 5′-ggttccgcgtggatccgagaacctgtactttcaaggaggatccaacctacggcag- 3′; reverse primer, 5′- gaattcccggggatccaccctcgatcgcaaagat-3′) with the Infusion Cloning Kit (Clontech) into the Bam HI site in the vector pGEX-2T according to the manufacturer’s protocol. The αA domain in its active conformation was generated by replacing Ile316 with glycine [I316G (36)] using the forward primer 5′-ggttccgcgtggatccgagaacctgtactttcaaggaggttttcaggaatgt-3′ and the reverse primer 5′-atatccccgggattaaccctcgatcgcaaagcccttctc-3′. The insert was digested with Bam HI and Sma I and inserted into the pGEX-2T vector that was digested with Bam HI and Sma I. All constructs were confirmed by direct DNA sequencing. All recombinant proteins were expressed as glutathione S-transferase (GST) fusion proteins in Escherichia coli and purified by affinity chromatography (glutathione beads, Sigma) according to the manufacturer’s instructions. Purified protein preparations were dialyzed against tris-buffered saline (TBS) [20 mm tris-HCl (pH 7.5), 150 mm NaCl], subsequently concentrated with Amicon-10 columns (Millipore), and stored at −80°C. Purity was confirmed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis.
αA domain ligand-binding assay
MaxiSorp 96-well plates were coated overnight with fibrinogen (1 μg per well) in 10 mM phosphate-buffered saline (PBS, pH 7.4) and blocked with 1% BSA in PBS. Binding of purified, GST-tagged αA domain (50 μl per well of a 5 μg/ml solution) to immobilized fibrinogen was performed in TBS-based assay buffer (TBS containing 0.1% BSA, 1 mM MgCl2, 1 mM CaCl2, and 0.05% Tween 20) (TBS-Ca/Mg buffer) for 1 hour at room temperature. The αA domain was also added to uncoated wells on the plate to estimate the maximum amount of protein that could be captured and detected in each well for data normalization. Unbound αA domain was removed by washing the wells twice with TBS-Ca/Mg buffer. Subsequently, the amount of bound protein was determined by incubation with horseradish peroxidase–conjugated antibody against GST (GE, 1:2000 dilution) for 1 hour. Unbound antibody was removed by washing the wells twice with TBS-Ca/Mg buffer. Detection of bound protein was performed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate kit (Vector Labs) according to the manufacturer’s protocol. Absorbance was read with a SpectraMax M5 spectrophotometer (Molecular Devices). Absorbance values were normalized such that the mean absorbance from the input αA domain wells was set at 100%, and the results are presented as the percentage of the total input amounts of the wild-type αA domain. Assays were performed in triplicate wells, and the data shown are from one of at least three independent experiments.
Flow cytometry
Flow cytometric analysis of K562 cells and human neutrophils for the surface expression of CD11b/CD18 was performed as previously described (69, 70). Briefly, cells were suspended in the assay buffer [TBS containing 1 mM each of Ca2+ and Mg2+ ions (TBS++) and 0.1% BSA]. Cells (5 × 105) were incubated with primary mAb [a 1:100 dilution of IB4 or 44a ascites, or of mAb 24 (15 μg/ml)] in the absence or presence of leukadherin (25 to 50 μM) in 100 ml of TBS++ on ice (except for mAb 24 and the isotype control antibody, for which incubations were performed at 37°C in TBS++) for 30 min. Subsequently, the cells were washed three times with the assay buffer and incubated with allophycocyanin (APC)–conjugated goat antibody against mouse Ig (Invitrogen) for 20 min at 4°C. Cells were washed twice with the assay buffer and analyzed with a FACS-Calibur flow cytometer (BD Biosciences), counting at least 10,000 events (cells). Data were analyzed with CellQuest software (BD Biosciences). Assays were performed in triplicate and the data shown are from one of at least three independent experiments. Human neutrophils contain an intracellular pool of CD11b/CD18 that is rapidly brought to the cell surface upon activation (13, 14, 71). To determine whether leukadherins had any effect on the mobilization of the intracellular pools of CD11b/CD18, we purified human neutrophils in a quiescent state from whole blood and kept the cells on ice, as previously described (72). For analysis, cells were suspended in the assay buffer [Hanks’ balanced salt solution (HBSS) containing 1 mM each of CaCl2 and MgCl2 (HBSS-Ca/Mg) and 0.1% human serum albumin (HSA)]. Cells (5 × 105) were incubated with the mAbs described earlier in the absence or presence of LA1, LA2, or LA3 (25 μM) for 30 min. The cells were then washed three times with assay buffer, labeled with secondary antibody (APC-conjugated goat antibody against mouse Ig), and analyzed by flow cytometry. As a positive control, neutrophils were activated by incubating the cells with lipopolysaccharide (100 ng/ml) or phorbol myristate acetate (20 nM) (72, 73) at 37°C in assay buffer, after which they were labeled and analyzed as described earlier.
Phagocytosis assay with complement iC3b-coated sheep erythrocytes (EiC3bs)
Sheep erythrocytes coated with complement iC3b were prepared and used in the phagocytosis assay as described previously (31). Coated erythrocytes (EiC3bs) were diluted to a concentration of 1.5 × 107 to 6 × 107 cells/ml. K562 CD11b/CD18 cells were washed twice in TBS and resuspended to 1 × 106/ml, of which 40 μl (4 × 104 cells) was incubated in suspension with EiC3bs (1.2 × 106) in a total volume of 100 μl at 37°C for 25 min in the presence of 1 mM each of CaCl2 and MgCl2 (in the absence or presence of 50 to 100 μM LA1, LA2, or LA3), in 1 mM MnCl2, or in 10 mM EDTA. Binding was detected by visually analyzing the formation of rosettes [the binding of multiple erythrocytes (EiC3bs) to individual K562 cells] by phase-contrast microscopy, as has been described previously (31). For scoring, only those K562 cells that were bound to ≥3 EiC3bs were scored as positive, and >200 cells were examined in multiple fields under each condition. Binding results, showing the percentages of all cells showing rosettes in a field, are reported as histograms representing the mean ± SEM of triplicate experiments; the data shown are from one of at least three independent experiments.
Cell viability assays
Cell viability assays were performed with commercially available reagents and kits. Briefly, 1 × 104 K562 CD11b/CD18 cells or wild-type B6 neutrophils were incubated in each well of a 96-well plate (Corning) with increasing amounts of the indicated compounds, and the number of viable cellswas determined with the MTS reagent (Promega), according to the manufacturers’ instructions, after 4 hours (neutrophils) or 24 hours (K562 cells) of incubation. A SpectraMaxM5 spectrophotometer was used to read the assay plates. Data are representative of at least two independent experiments.
Western blotting analysis
K562 CD11b/CD18 cells were incubated with LA1, LA2, or LA3 (15 μM) or fibrinogen (200 μg) in serum-free medium for 1 hour at 37°C. Cell lysates were resolved on a 10% SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane (Thermo Scientific) by means of established protocols. Membranes were incubated with a 1:1000 dilution of an antibody against phosphorylated extracellular signal–regulated kinase 1/2 (ERK1/2) (Thr202/Tyr204, Cell Signaling), stripped with Reblot mild stripping solution (Millipore), and then incubated, first with an antibody against total ERK1/2 (Cell Signaling) and then with an antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cell Signaling), and developed according to the manufacturer’s instructions (Thermo Scientific). Data presented are representative of at least three independent experiments.
Blood cell count
Complete peripheral blood leukocyte counts from the different mice were quantified by the University of Miami mouse pathology core by flow cytometry.
In vivo peritonitis model
Thioglycolate-induced peritonitis in 8- to 10-week-old wild-type B6 and CD11b−/− B6 mice was performed as previously described (62). Leukadherins were administered 30 min before intraperitoneal injection with 3% thioglycolate. LA1 and LA2 (200 μl of a 50 μM solution in saline) were administered intravenously, whereas LA3 was administered intraperitoneally (500 μl of a 50 μM solution in saline). To evaluate peritoneal neutrophil recruitment, we euthanized mice at 4, 12, or 24 hours after thioglycolate injection; the peritoneal lavage was collected, and the number of emigrated neutrophils was quantified by flow cytometric analysis for cells expressing both GR-1 and Mac-1 on the surface, as described previously (18).
Balloon-induced arterial injury in rats
All surgeries were performed under isoflurane anesthesia (Baxter). Compounds were administered intramuscularly (1 ml of a 50 μM solution in saline). Balloon injury in the right iliac artery was inflicted with a 2F Fogarty catheter (Baxter) adapted to a custom angiographic kit (Boston Scientific, Scimed) (47). An aortotomy in the abdominal aorta was made to insert a catheter to the level of the right iliac artery. The balloon was inflated to 1.5 to 1.6 atm and retracted to the arteriotomy site three times. The aortic excision was repaired with eight sutures. The abdominal cavity was closed by planes with an interrupted suture pattern. Arterial specimens were collected 3 to 30 days after injury, fixed in 4% formalin–PBS (Sigma-Aldrich) for 5 min, and analyzed by histology and immunostaining.
Anti-GBM nephritis
Experimental anti-GBM nephritis was induced in wild-type B6 mice (n = 4 mice per group) by intraperitoneal injection of sheep antibody against rabbit GBM (0.5 ml per 20 g body weight per day for 2 consecutive days) as previously described (74, 75). One group of animals was treated with leukadherins by daily intraperitoneal injection of LA1 (500 μl of a 50 μM solution in saline) starting at 2 hours before induction of nephritis and continuing until the end of the experiment. A group of animals was treated with a known antagonist, the blocking antibody M1/70, as described previously (76). Briefly, M1/70 (100 μg per injection) in a saline solution was injected intraperitoneally every other day starting at 2 hours before induction of nephritis and continuing until the end of the experiment. Urine collections were performed every 24 hours and analyzed for evidence of proteinuria on SDS-PAGE gels with BSA standards, as described previously (77). The amounts of creatinine were determined with a creatinine assay kit (Cayman Chemical) according to the manufacturer’s instructions. Mice were killed on days 0, 3, and 7, and renal biopsies were obtained from each animal. Tissue sections were fixed with 4% paraformaldehyde and were used for histochemical analyses and leukocyte enumeration.
Evaluation of neutrophil sequestration
To evaluate whether leukadherins induced tissue sequestration of neutrophils, we fixed various organs from wild-type B6 mice (n = 3 mice per group) with formalin and stained them with hematoxylin and eosin (H&E). The numbers of neutrophils in untreated and LA1-treated animals were quantified from four random fields for each specimen at either 40× or 1000× magnification in a blinded fashion. Bone marrow was isolated as described previously (78). Briefly, mice were euthanized, and the femurs and tibia from both hind legs were removed and freed of soft tissue. The extreme ends of the bones were cut off, and a solution of RPMI 1640 containing 10% FBS was forced through the bone with a 27-gauge syringe needle. Cell clumps were dispersed, passed through an 80-μm filter, and collected by centrifugation. Red blood cells (RBCs) were removed by hypotonic lysis buffer followed by two washes with HBSS buffer containing 0.1% BSA. Neutrophil numbers were determined by flow cytometry as described earlier.
Histology and immunostaining
Elastica van Gieson staining was used for histochemical analysis to evaluate the formation of neointima. Morphometric analysis was performed in a blinded fashion with NIH ImageJ. Immunostaining with antibody against rat CD68 (1:50 dilution, AbD Serotec) was used to detect macrophages in the tissue. To examine renal histology in mice from experiments involving anti-GBM nephritis, we stained kidney sections with H&E stain or periodic acid and Schiff’s reagent. The number of infiltrating neutrophils was enumerated by immunostaining with antibody against GR-1 according to the manufacturer’s instructions. The cells were counted by the same operator in a blinded fashion.
Intravital microscopy
Mice were given an intrascrotal injection of TNF-α (500 ng, PeproTech) in 0.25 ml of saline 3 hours before cremaster muscle exteriorization. Some animals also received intravenous injections of the blocking mAb M1/70 (30 μg per mouse) in 0.1 ml of saline and intraperitoneal injections of LA1 (100 μM) or DMSO in 0.5 ml of saline 30 min before injection with TNF-α. Mice were anesthetized with an intraperitoneal injection of ketamine (125 mg/kg), xylazine (12.5 mg/kg), and atropine sulfate (0.025 mg/kg) and placed on a 38°C heating pad. After tracheal intubation and cannulation of one carotid artery, the cremaster was exteriorized, pinned to the stage, and superfused with thermocontrolled bicarbonate-buffered saline (131.9 mM NaCl, 18 mM NaHCO3, 4.7 mM KCl, 2.0 mM CaCl2, and 1.2 mM MgCl2) equilibrated with 5% CO2 in N2. Cremaster muscles were illuminated with stroboscopic flash epi-illumination (DPS-1, Rapp OptoElectronic) and halogen transillumination. Microscopic observations were made on postcapillary venules with a diameter of between 20 and 40 mm by means of an intravital microscope (Axioskop; Carl Zeiss MicroImaging Inc.) with a saline immersion objective (SW 40/0.75). A charge-coupled device (CCD) camera (model SIT66, DAGE-MTI) was used for recording. In a limited analysis, cells adjacent to the venules were counted to determine the number of transmigrated neutrophils. The surface area (S) was calculated for each vessel as S = πdIν, where d is the diameter of the vessel and Iν is the length of the vessel. Adherent leukocytes were defined as those cells that were stationary for more than 30 s.
Zebrafish tailfin injury assays
Transgenic Tg(mpx∷eGFP) zebrafish (51) were maintained according to standard protocols (79). Tailfin injury in larvae 3 days after fertilization was performed as described previously (51). Larvae were anesthetized by immersion in E3 with 4.2% tricane, and tails were completely transected with a sterile microdissection scalpel, in accordance with the approved protocols, and were recovered at the indicated time points. Zebrafish larvae (3 dpf) were treated with the appropriate compounds as described (80). Briefly, small-molecule compounds were administered by immersing the larvae in a solution of the compounds in E3. The final concentration of DMSO was kept at <1%. For the assessment of the inflammatory response, injured larvae were analyzed 4 hours after injury. For the post-wash assay, uninjured larvae incubated with the compounds in E3 for 4 to 8 hours were washed in E3 and injured. Larvae were analyzed with a Leica DMI6000B microscope and a Hamamatsu Orca-3CCD camera with Volocity. Excitation was performed with the laser set at 488 nm, and the images were analyzed with Volocity. The numbers of fluorescent neutrophils at sites of inflammation were counted by eye in a blinded fashion.
Statistical analysis
Data were analyzed with GraphPad Prism and compared with the Mann-Whitney test and the Student’s t test, where appropriate, or by one-way analysis of variance (ANOVA) with post hoc analysis, when comparing two or more groups. P < 0.05 was considered statistically significant.
Computational modeling
To model the binding of leukadherins to the active, open conformation of the αA domain, we performed a series of computational studies as previously described (81). To identify possible ligand-binding modes, we applied an induced-fit docking (IFD) procedure implemented in the Schrodinger software suite as previously described (23). The optimized receptor structures after IFD were then used to dock the agonists LA1 and LA2 with Schrodinger Glide and the SP scoring function. Next, the best-scoring structures for LA1 and LA2 in their Z configuration were further optimized in molecular dynamics (MD) simulations, which were performed as multistep protocols with several minimization and simulation steps preceding the production MD run. Simulations were performed with the MD package Desmond by DEShaw Research (82) at 300 and 325 K (NPT ensemble) with the Simple Point Charge (SPC) water model (cubic box of 10 Å around the receptor) on an IBM E-server 1350 cluster (36 nodes of eight Xeon 2.3-GHz cores and 12 gigabytes of memory). The final simulation times were 12 ns, in which the reported poses remained stable. The data presented show the poses of LA1 and LA2 after 1.2 ns of production simulation.
Supplementary Material
Movie S1. Neutrophil chemotaxis in 2D in the presence of DMSO (control).
Movie S2. Neutrophil chemotaxis in 2D in the presence of LA1.
Movie S3. Neutrophil chemotaxis in 2D in the presence of LA2.
Movie S4. Neutrophil chemotaxis in 2D in the presence of LA3.
Movie S5. Neutrophil migration in 3D networks in vitro in the presence of DMSO (control).
Movie S6. Neutrophil migration in 3D networks in vitro in the presence of LA1.
Movie S7. Intravital microscopy in TNF-α–treated cremaster muscle with LA1.
Movie S8. Intravitalmicroscopy inTNF-α–treated cremastermuscle withLA1 and themAbM1/70.
www.sciencesignaling.org/cgi/content/full/4/189/ra57/DC1
Fig. S1. Leukadherins are true agonists and do not inhibit cell adhesion in the presence of the agonist Mn2+.
Fig. S2. Leukadherins do not affect the surface abundance of CD11b/CD18 on K562 cells.
Fig. S3. Leukadherins do not mobilize CD11b/CD18 from internal pools or affect the amount of CD11b/CD18 on the surface of human neutrophils.
Fig. S4. Leukadherins increase the adhesion of CD11b/CD18-expressing cells to iC3b.
Fig. S5. Leukadherin-dependent CD11b/CD18 activation is independent of ligand type.
Fig. S6. Leukadherin-dependent activation of CD11b/CD18 occurs in THP-1 cells.
Fig. S7. Leukadherins increase the extent of binding of iC3b-coated RBCs by K562 cells.
Fig. S8. Ribbon diagrams showing computational models for the binding of LA1 and LA2 in an activation-sensitive region of the CD11b A domain.
Fig. S9. Leukadherins activate full-length CD11b/CD18 on live K562 cells.
Fig. S10. Leukadherins have a higher affinity than does IMB-10 for CD11b/CD18.
Fig. S11. Leukadherins do not affect neutrophil migration in 3D gels in vitro.
Fig. S12. Leukadherins do not cause cytotoxicity in vitro.
Fig. S13. Leukadherins do not cause neutrophil cytotoxicity in vitro.
Fig. S14. Leukadherins do not induce integrin clustering or outside-in signaling.
Fig. S15. Leukadherins do not induce CD11b/CD18-mediated outside-in signaling.
Fig. S16. The control compound LA-C has no effect on neointimal thickening upon balloon injury in wild-type rats.
Fig. S17. LA3 substantially reduces neointimal thickening after balloon injury in rats.
Fig. S18. LA2 prevents neutrophil recruitment to injured tissue in a reversible manner.
Fig. S19. Leukadherins do not lead to loss of neutrophil numbers in zebrafish larvae.
Fig. S20. Leukadherins reduce the number of transmigrated cells in vivo.
Table S1. White blood cell counts in mouse whole-blood samples.
Descriptions for Movies S1 to S8
References
Acknowledgments
We thank H. Tannoury, J. Y. Park, and J. Rosa for their generous help and discussions with the cell-based assays; T. D. Y. Chung, S. Vasile, E. Sergienko, and the staff of Conrad Prebys Center for Chemical Genomics at the Burnham Institute for Medical Research; and C. Shamu and the staff of the Institute for Chemistry and Chemical Biology at Harvard Medical School for their generous support in the implementation of the HTS assay. We thank S. Shankland and J. Pippin for the gift of sheep antibody against rabbit GBM. We thank M. A. Arnaout, P. Mundel, and D. Stoub for helpful discussions. We also thank Y.Wei and D. Mateu for help with the rat balloon injury model and immunostaining procedures. Funding: This work was supported in part by K01- HL096413 (to R.I.V.-P.) and with resources from the Miller School of Medicine, the Katz Family Fund, and the Center for Computational Science at the University of Miami.
Footnotes
Author contributions: D.M. and M.H.F. contributed equally to this work and performed most of the experiments in this study, with technical assistance from C.J.B. and M.D. C.W. performed the anti-GBM nephritis experiment; Y.K. performed the intravital microscopy experiment; K.M.B. and G.L. performed the zebrafish experiments; D.H., A.N., and P.R. conducted the histological analyses; R.I.V.-P. conducted the balloon injury experiment; L.F.M., S.S., D.T., R.I.V.-P., K.L., J.R., and V.G. designed the experiments; L.F.M., D.T., K.L., and J.R. provided input in the preparation of the manuscript; and D.M., M.H.F., J.R., and V.G. co-wrote the paper.
Competing interests: V.G. is an inventor of pending patents related to the study, and V.G. and the University of Miami have the potential for financial benefit from their future commercialization. The pending patents have been licensed to Adhaere Pharmaceuticals Inc., a company cofounded by V.G. The authors have no additional financial interests.
REFERENCES AND NOTES
- 1.Arnaout MA. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood. 1990;75:1037–1050. [PubMed] [Google Scholar]
- 2.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 3.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 4.Altieri DC, Edgington TS. The saturable high affinity association of factor X to ADP-stimulated monocytes defines a novel function of the Mac-1 receptor. J Biol Chem. 1988;263:7007–7015. [PubMed] [Google Scholar]
- 5.Ginsberg MH, Du X, Plow EF. Inside-out integrin signalling. Curr Opin Cell Biol. 1992;4:766–771. doi: 10.1016/0955-0674(92)90099-x. [DOI] [PubMed] [Google Scholar]
- 6.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 7.Jaeschke H, Farhood A, Bautista AP, Spolarics Z, Spitzer JJ, Smith CW. Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia-reperfusion injury in rat liver. Hepatology. 1993;17:915–923. [PubMed] [Google Scholar]
- 8.Rogers C, Edelman ER, Simon DI. A mAb to the β2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc Natl Acad Sci U S A. 1998;95:10134–10139. doi: 10.1073/pnas.95.17.10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson I, Gillinov AM, Curtis WE, DiNatale J, Burch RM, Gardner TJ, Cameron DE. Inhibition of neutrophil adherence improves postischemic ventricular performance of the neonatal heart. Circulation. 1993;88:II372–II379. [PubMed] [Google Scholar]
- 10.Wilson RW, Ballantyne CM, Smith CW, Montgomery C, Bradley A, O’Brien WE, Beaudet AL. Gene targeting yields a CD18-mutant mouse for study of inflammation. J Immunol. 1993;151:1571–1578. [PubMed] [Google Scholar]
- 11.Dove A. CD18 trials disappoint again. Nat Biotechnol. 2000;18:817–818. doi: 10.1038/78412. [DOI] [PubMed] [Google Scholar]
- 12.Harlan JM, Winn RK. Leukocyte-endothelial interactions: Clinical trials of anti-adhesion therapy. Crit Care Med. 2002;30:S214–S219. doi: 10.1097/00003246-200205001-00007. [DOI] [PubMed] [Google Scholar]
- 13.Berger M, O’Shea J, Cross AS, Folks TM, Chused TM, Brown EJ, Frank MM. Human neutrophils increase expression of C3bi as well as C3b receptors upon activation. J Clin Invest. 1984;74:1566–1571. doi: 10.1172/JCI111572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes BJ, Hollers JC, Crockett-Torabi E, Smith CW. Recruitment of CD11b/CD18 to the neutrophil surface and adherence-dependent cell locomotion. J Clin Invest. 1992;90:1687–1696. doi: 10.1172/JCI116041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lum AF, Green CE, Lee GR, Staunton DE, Simon SI. Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. J Biol Chem. 2002;277:20660–20670. doi: 10.1074/jbc.M202223200. [DOI] [PubMed] [Google Scholar]
- 16.Molloy ES, Calabrese LH. Therapy: Targeted but not trouble-free: Efalizumab and PML. Nat Rev Rheumatol. 2009;5:418–419. doi: 10.1038/nrrheum.2009.142. [DOI] [PubMed] [Google Scholar]
- 17.Kuijpers TW, Mul EP, Blom M, Kovach NL, Gaeta FC, Tollefson V, Elices MJ, Harlan JM. Freezing adhesion molecules in a state of high-avidity binding blocks eosinophil migration. J Exp Med. 1993;178:279–284. doi: 10.1084/jem.178.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semmrich M, Smith A, Feterowski C, Beer S, Engelhardt B, Busch DH, Bartsch B, Laschinger M, Hogg N, Pfeffer K, Holzmann B. Importance of integrin LFA-1 deactivation for the generation of immune responses. J Exp Med. 2005;201:1987–1998. doi: 10.1084/jem.20041850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park EJ, Peixoto A, Imai Y, Goodarzi A, Cheng G, Carman CV, von Andrian UH, Shimaoka M. Distinct roles for LFA-1 affinity regulation during T-cell adhesion, diapedesis, and interstitial migration in lymph nodes. Blood. 2010;115:1572–1581. doi: 10.1182/blood-2009-08-237917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park EJ, Mora JR, Carman CV, Chen J, Sasaki Y, Cheng G, von Andrian UH, Shimaoka M. Aberrant activation of integrin α4β7 suppresses lymphocyte migration to the gut. J Clin Invest. 2007;117:2526–2538. doi: 10.1172/JCI31570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diamond MS, Springer TA. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J Cell Biol. 1993;120:545–556. doi: 10.1083/jcb.120.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JY, Arnaout MA, Gupta V. A simple, no-wash cell adhesion–based high-throughput assay for the discovery of small-molecule regulators of the integrin CD11b/CD18. J Biomol Screen. 2007;12:406–417. doi: 10.1177/1087057106299162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faridi MH, Maiguel D, Barth CJ, Stoub D, Day R, Schürer S, Gupta V. Identification of novel agonists of the integrin CD11b/CD18. Bioorg Med Chem Lett. 2009;19:6902–6906. doi: 10.1016/j.bmcl.2009.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faridi MH, Maiguel D, Brown BT, Suyama E, Barth CJ, Hedrick M, Vasile S, Sergienko E, Schürer S, Gupta V. High-throughput screening based identification of small molecule antagonists of integrin CD11b/CD18 ligand binding. Biochem Biophys Res Commun. 2010;394:194–199. doi: 10.1016/j.bbrc.2010.02.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ajroud K, Sugimori T, Goldmann WH, Fathallah DM, Xiong JP, Arnaout MA. Binding affinity of metal ions to the CD11b A-domain is regulated by integrin activation and ligands. J Biol Chem. 2004;279:25483–25488. doi: 10.1074/jbc.M402901200. [DOI] [PubMed] [Google Scholar]
- 26.Michishita M, Videm V, Arnaout MA. A novel divalent cation-binding site in the A domain of the β2 integrin CR3 (CD11b/CD18) is essential for ligand binding. Cell. 1993;72:857–867. doi: 10.1016/0092-8674(93)90575-b. [DOI] [PubMed] [Google Scholar]
- 27.Yang W, Carman CV, Kim M, Salas A, Shimaoka M, Springer TA. A small molecule agonist of an integrin, αLβ2. J Biol Chem. 2006;281:37904–37912. doi: 10.1074/jbc.M606888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altieri DC. Occupancy of CD11b/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J Immunol. 1991;147:1891–1898. [PubMed] [Google Scholar]
- 29.Hogg N, Stewart MP, Scarth SL, Newton R, Shaw JM, Law SK, Klein N. A novel leukocyte adhesion deficiency caused by expressed but nonfunctional β2 integrins Mac-1 and LFA-1. J Clin Invest. 1999;103:97–106. doi: 10.1172/JCI3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnaout MA, Todd RF, III, Dana N, Melamed J, Schlossman SF, Colten HR. Inhibition of phagocytosis of complement C3- or immunoglobulin G-coated particles and of C3bi binding by monoclonal antibodies to a monocyte-granulocyte membrane glycoprotein (Mol) J Clin Invest. 1983;72:171–179. doi: 10.1172/JCI110955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta V, Gylling A, Alonso JL, Sugimori T, Ianakiev P, Xiong JP, Arnaout MA. The β-tail domain (βTD) regulates physiologic ligand binding to integrin CD11b/CD18. Blood. 2007;109:3513–3520. doi: 10.1182/blood-2005-11-056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Björklund M, Aitio O, Stefanidakis M, Suojanen J, Salo T, Sorsa T, Koivunen E. Stabilization of the activated αMβ2 integrin by a small molecule inhibits leukocyte migration and recruitment. Biochemistry. 2006;45:2862–2871. doi: 10.1021/bi052238b. [DOI] [PubMed] [Google Scholar]
- 33.Alonso JL, Essafi M, Xiong JP, Stehle T, Arnaout MA. Does the integrin αA domain act as a ligand for its βA domain? Curr Biol. 2002;12:R340–R342. doi: 10.1016/s0960-9822(02)00852-7. [DOI] [PubMed] [Google Scholar]
- 34.Shimaoka M, Lu C, Salas A, Xiao T, Takagi J, Springer TA. Stabilizing the integrin αM inserted domain in alternative conformations with a range of engineered disulfide bonds. Proc Natl Acad Sci U S A. 2002;99:16737–16741. doi: 10.1073/pnas.252633099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R, Rieu P, Griffith DL, Scott D, Arnaout MA. Two functional states of the CD11b A-domain: Correlations with key features of two Mn2+-complexed crystal structures. J Cell Biol. 1998;143:1523–1534. doi: 10.1083/jcb.143.6.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong JP, Li R, Essafi M, Stehle T, Arnaout MA. An isoleucine-based allosteric switch controls affinity and shape shifting in integrin CD11b A-domain. J Biol Chem. 2000;275:38762–38767. doi: 10.1074/jbc.C000563200. [DOI] [PubMed] [Google Scholar]
- 37.Lee JO, Bankston LA, Arnaout MA, Liddington RC. Two conformations of the integrin A-domain (I-domain): A pathway for activation? Structure. 1995;3:1333–1340. doi: 10.1016/s0969-2126(01)00271-4. [DOI] [PubMed] [Google Scholar]
- 38.Lee JO, Rieu P, Arnaout MA, Liddington R. Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18) Cell. 1995;80:631–638. doi: 10.1016/0092-8674(95)90517-0. [DOI] [PubMed] [Google Scholar]
- 39.Liu L, Schwartz BR, Lin N, Winn RK, Harlan JM. Requirement for RhoA kinase activation in leukocyte de-adhesion. J Immunol. 2002;169:2330–2336. doi: 10.4049/jimmunol.169.5.2330. [DOI] [PubMed] [Google Scholar]
- 40.Huttenlocher A, Ginsberg MH, Horwitz AF. Modulation of cell migration by integrinmediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. 1996;134:1551–1562. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- 42.Zigmond SH. Orientation chamber in chemotaxis. Methods Enzymol. 1988;162:65–72. doi: 10.1016/0076-6879(88)62064-7. [DOI] [PubMed] [Google Scholar]
- 43.Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fässler R, Sixt M. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008;453:51–55. doi: 10.1038/nature06887. [DOI] [PubMed] [Google Scholar]
- 44.Stroupe KT, Morrison DA, Hlatky MA, Barnett PG, Cao L, Lyttle C, Hynes DM, Henderson WG Investigators of Veterans Affairs Cooperative Studies Program #385 (AWESOME: Angina With Extremely Serious Operative Mortality Evaluation) Cost-effectiveness of coronary artery bypass grafts versus percutaneous coronary intervention for revascularization of high-risk patients. Circulation. 2006;114:1251–1257. doi: 10.1161/CIRCULATIONAHA.105.570838. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, Zago AC, Lopez J, Andre P, Plow E, Simon DI. Leukocyte engagement of platelet glycoprotein Ibα via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. 2005;112:2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315. [DOI] [PubMed] [Google Scholar]
- 46.Simon DI, Dhen Z, Seifert P, Edelman ER, Ballantyne CM, Rogers C. Decreased neointimal formation in Mac-1−/− mice reveals a role for inflammation in vascular repair after angioplasty. J Clin Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gabeler EE, van Hillegersberg R, Statius van Eps RG, Sluiter W, Gussenhoven EJ, Mulder P, van Urk H. A comparison of balloon injury models of endovascular lesions in rat arteries. BMC Cardiovasc Disord. 2002;2:16. doi: 10.1186/1471-2261-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Springer T, Galfré G, Secher DS, Milstein C. Mac-1: A macrophage differentiation antigen identified by monoclonal antibody. Eur J Immunol. 1979;9:301–306. doi: 10.1002/eji.1830090410. [DOI] [PubMed] [Google Scholar]
- 49.Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, Cotran RS, Mayadas TN. A role for Mac-1 (CDIIb/CD18) in immune complex–stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcγ receptor–dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186:1853–1863. doi: 10.1084/jem.186.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Vriese AS, Endlich K, Elger M, Lameire NH, Atkins RC, Lan HY, Rupin A, Kriz W, Steinhausen MW. The role of selectins in glomerular leukocyte recruitment in rat anti-glomerular basement membrane glomerulonephritis. J Am Soc Nephrol. 1999;10:2510–2517. doi: 10.1681/ASN.V10122510. [DOI] [PubMed] [Google Scholar]
- 51.Renshaw SA, Loynes CA, Trushell DM, Elworthy S, Ingham PW, Whyte MK. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108:3976–3978. doi: 10.1182/blood-2006-05-024075. [DOI] [PubMed] [Google Scholar]
- 52.Dunne JL, Collins RG, Beaudet AL, Ballantyne CM, Ley K. Mac-1, but not LFA-1, uses intercellular adhesion molecule-1 to mediate slow leukocyte rolling in TNF-α-induced inflammation. J Immunol. 2003;171:6105–6111. doi: 10.4049/jimmunol.171.11.6105. [DOI] [PubMed] [Google Scholar]
- 53.Yonekawa K, Harlan JM. Targeting leukocyte integrins in human diseases. J Leukoc Biol. 2005;77:129–140. doi: 10.1189/jlb.0804460. [DOI] [PubMed] [Google Scholar]
- 54.Cox D, Brennan M, Moran N. Integrins as therapeutic targets: Lessons and opportunities. Nat Rev Drug Discov. 2010;9:804–820. doi: 10.1038/nrd3266. [DOI] [PubMed] [Google Scholar]
- 55.Allison M. PML problems loom for Rituxan. Nat Biotechnol. 2010;28:105–106. doi: 10.1038/nbt0210-105. [DOI] [PubMed] [Google Scholar]
- 56.Czuchra A, Meyer H, Legate KR, Brakebusch C, Fässler R. Genetic analysis of β1 integrin “activation motifs” in mice. J Cell Biol. 2006;174:889–899. doi: 10.1083/jcb.200604060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou Z, Schmaier AA, Cheng L, Mericko P, Dickeson SK, Stricker TP, Santoro SA, Kahn ML. Negative regulation of activated α2 integrins during thrombopoiesis. Blood. 2009;113:6428–6439. doi: 10.1182/blood-2008-08-175356. [DOI] [PubMed] [Google Scholar]
- 58.Wright SD, Rao PE, Van Voorhis WC, Craigmyle LS, Iida K, Talle MA, Westberg EF, Goldstein G, Silverstein SC. Identification of the C3bi receptor of human monocytes and macrophages by using monoclonal antibodies. Proc Natl Acad Sci U S A. 1983;80:5699–5703. doi: 10.1073/pnas.80.18.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dransfield I, Hogg N. Regulated expression of Mg2+ binding epitope on leukocyte integrin α subunits. EMBO J. 1989;8:3759–3765. doi: 10.1002/j.1460-2075.1989.tb08552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- 61.Gupta V. PubChem BioAssay. AID; 2009. HTS identification of compounds that enhance the binding of CD11b/CD18 to fibrinogen via a luminescence assay; p. 1499. [Google Scholar]
- 62.Chen LY, Shieh JJ, Lin B, Pan CJ, Gao JL, Murphy PM, Roe TF, Moses S, Ward JM, Lee EJ, Westphal H, Mansfield BC, Chou JY. Impaired glucose homeostasis, neutrophil trafficking and function in mice lacking the glucose-6-phosphate transporter. Hum Mol Genet. 2003;12:2547–2558. doi: 10.1093/hmg/ddg263. [DOI] [PubMed] [Google Scholar]
- 63.Bergmeier W, Goerge T, Wang HW, Crittenden JR, Baldwin AC, Cifuni SM, Housman DE, Graybiel AM, Wagner DD. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest. 2007;117:1699–1707. doi: 10.1172/JCI30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szczur K, Xu H, Atkinson S, Zheng Y, Filippi MD. Rho GTPase CDC42 regulates directionality and random movement via distinct MAPK pathways in neutrophils. Blood. 2006;108:4205–4213. doi: 10.1182/blood-2006-03-013789. [DOI] [PubMed] [Google Scholar]
- 65.Friedl P, Bröcker EB. Reconstructing leukocyte migration in 3D extracellular matrix by time-lapse videomicroscopy and computer-assisted tracking. Methods Mol Biol. 2004;239:77–90. doi: 10.1385/1-59259-435-2:77. [DOI] [PubMed] [Google Scholar]
- 66.Yoon CH, Hur J, Park KW, Kim JH, Lee CS, Oh IY, Kim TY, Cho HJ, Kang HJ, Chae IH, Yang HK, Oh BH, Park YB, Kim HS. Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: The role of angiogenic cytokines and matrix metalloproteinases. Circulation. 2005;112:1618–1627. doi: 10.1161/CIRCULATIONAHA.104.503433. [DOI] [PubMed] [Google Scholar]
- 67.Pluskota E, Soloviev DA, Szpak D, Weber C, Plow EF. Neutrophil apoptosis: Selective regulation by different ligands of integrin αMβ2. J Immunol. 2008;181:3609–3619. doi: 10.4049/jimmunol.181.5.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li R, Arnaout MA. Functional analysis of the β2 integrins. Methods Mol Biol. 1999;129:105–124. doi: 10.1385/1-59259-249-X:105. [DOI] [PubMed] [Google Scholar]
- 69.Lu C, Ferzly M, Takagi J, Springer TA. Epitope mapping of antibodies to the C-terminal region of the integrin β2 subunit reveals regions that become exposed upon receptor activation. J Immunol. 2001;166:5629–5637. doi: 10.4049/jimmunol.166.9.5629. [DOI] [PubMed] [Google Scholar]
- 70.Xiong JP, Stehle T, Goodman SL, Arnaout MA. New insights into the structural basis of integrin activation. Blood. 2003;102:1155–1159. doi: 10.1182/blood-2003-01-0334. [DOI] [PubMed] [Google Scholar]
- 71.Todd RF, III, Arnaout MA, Rosin RE, Crowley CA, Peters WA, Babior BM. Subcellular localization of the large subunit of Mo1 (Mo1α; formerly gp 110), a surface glycoprotein associated with neutrophil adhesion. J Clin Invest. 1984;74:1280–1290. doi: 10.1172/JCI111538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989;83:2008–2017. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Griffin JD, Spertini O, Ernst TJ, Belvin MP, Levine HB, Kanakura Y, Tedder TF. Granulocyte-macrophage colony-stimulating factor and other cytokines regulate surface expression of the leukocyte adhesion molecule-1 on human neutrophils, monocytes, and their precursors. J Immunol. 1990;145:576–584. [PubMed] [Google Scholar]
- 74.Ophascharoensuk V, Fero ML, Hughes J, Roberts JM, Shankland SJ. The cyclin-dependent kinase inhibitor p27Kip1 safeguards against inflammatory injury. Nat Med. 1998;4:575–580. doi: 10.1038/nm0598-575. [DOI] [PubMed] [Google Scholar]
- 75.Ophascharoensuk V, Pippin JW, Gordon KL, Shankland SJ, Couser WG, Johnson RJ. Role of intrinsic renal cells versus infiltrating cells in glomerular crescent formation. Kidney Int. 1998;54:416–425. doi: 10.1046/j.1523-1755.1998.00003.x. [DOI] [PubMed] [Google Scholar]
- 76.Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8368. doi: 10.1073/pnas.0911378107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K, Reiser J, Mundel P. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boxio R, Bossenmeyer-Pourié C, Steinckwich N, Dournon C, Nüsse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol. 2004;75:604–611. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- 79.Nüsslein-Volhard C. Zebrafish: A Practical Approach. Oxford Univ. Press; Oxford, UK: 2002. [Google Scholar]
- 80.Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]
- 81.Faridi MH, Maiguel D, Barth CJ, Stoub D, Day R, Schürer S, Gupta V. Identification of novel agonists of the integrin CD11b/CD18. Bioorg Med Chem Lett. 2009;19:6902–6906. doi: 10.1016/j.bmcl.2009.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kevin JB, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossváry I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Scalable algorithms for MD simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06); Tampa, Florida. 11 to 17 November 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Neutrophil chemotaxis in 2D in the presence of DMSO (control).
Movie S2. Neutrophil chemotaxis in 2D in the presence of LA1.
Movie S3. Neutrophil chemotaxis in 2D in the presence of LA2.
Movie S4. Neutrophil chemotaxis in 2D in the presence of LA3.
Movie S5. Neutrophil migration in 3D networks in vitro in the presence of DMSO (control).
Movie S6. Neutrophil migration in 3D networks in vitro in the presence of LA1.
Movie S7. Intravital microscopy in TNF-α–treated cremaster muscle with LA1.
Movie S8. Intravitalmicroscopy inTNF-α–treated cremastermuscle withLA1 and themAbM1/70.
www.sciencesignaling.org/cgi/content/full/4/189/ra57/DC1
Fig. S1. Leukadherins are true agonists and do not inhibit cell adhesion in the presence of the agonist Mn2+.
Fig. S2. Leukadherins do not affect the surface abundance of CD11b/CD18 on K562 cells.
Fig. S3. Leukadherins do not mobilize CD11b/CD18 from internal pools or affect the amount of CD11b/CD18 on the surface of human neutrophils.
Fig. S4. Leukadherins increase the adhesion of CD11b/CD18-expressing cells to iC3b.
Fig. S5. Leukadherin-dependent CD11b/CD18 activation is independent of ligand type.
Fig. S6. Leukadherin-dependent activation of CD11b/CD18 occurs in THP-1 cells.
Fig. S7. Leukadherins increase the extent of binding of iC3b-coated RBCs by K562 cells.
Fig. S8. Ribbon diagrams showing computational models for the binding of LA1 and LA2 in an activation-sensitive region of the CD11b A domain.
Fig. S9. Leukadherins activate full-length CD11b/CD18 on live K562 cells.
Fig. S10. Leukadherins have a higher affinity than does IMB-10 for CD11b/CD18.
Fig. S11. Leukadherins do not affect neutrophil migration in 3D gels in vitro.
Fig. S12. Leukadherins do not cause cytotoxicity in vitro.
Fig. S13. Leukadherins do not cause neutrophil cytotoxicity in vitro.
Fig. S14. Leukadherins do not induce integrin clustering or outside-in signaling.
Fig. S15. Leukadherins do not induce CD11b/CD18-mediated outside-in signaling.
Fig. S16. The control compound LA-C has no effect on neointimal thickening upon balloon injury in wild-type rats.
Fig. S17. LA3 substantially reduces neointimal thickening after balloon injury in rats.
Fig. S18. LA2 prevents neutrophil recruitment to injured tissue in a reversible manner.
Fig. S19. Leukadherins do not lead to loss of neutrophil numbers in zebrafish larvae.
Fig. S20. Leukadherins reduce the number of transmigrated cells in vivo.
Table S1. White blood cell counts in mouse whole-blood samples.
Descriptions for Movies S1 to S8
References