

Abstract
The thalamus acts as a conduit for sensory and other information traveling to the cortex. In response to continuous sensory stimulation in vivo, the firing rate of thalamocortical neurons initially increases, but then within a minute firing rate decreases and T-type Ca2+ channel-dependent action potential burst firing emerges. While neuromodulatory systems could play a role in this inhibitory response, we instead report a novel and cell-autonomous inhibitory mechanism intrinsic to the thalamic relay neuron. Direct intracellular stimulation of thalamocortical neuron firing initially triggered a continuous and high rate of action potential discharge, but within a minute membrane potential (Vm) was hyperpolarized and firing rate to the same stimulus was decreased. This self-inhibition was observed across a wide variety of thalamic nuclei, and in a subset firing mode switched from tonic to bursting. The self-inhibition resisted blockers of intracellular Ca2+ signaling, Na+-K+-ATPases, and G protein-regulated inward rectifier (GIRK) channels as implicated in other neuron subtypes, but instead was in part inhibited by an ATP-sensitive K+ channel blocker. The results identify a new homeostatic mechanism within the thalamus capable of gating excitatory signals at the single-cell level.
Keywords: excitability, thalamus, KATP channel
thalamocortical (TC) relay neurons are a critical node of communication for information traveling from the peripheral sensory systems to the cerebral cortex as well as between diverse cortical and subcortical gray matter structures. In addition to the normal physiological signals, pathological signals such as those in central pain and epilepsy also disseminate via these thalamic pathways. The firing pattern of TC neurons is characterized by one of two modes, tonic or bursting, referring to the pattern of sodium action potentials (APs) (Jahnsen and Llinás 1984a; Llinás and Jahnsen 1982). The tonic firing mode consists of APs fired at a continuous and regular rate throughout a depolarizing stimulus, while burst firing mode occurs only from a hyperpolarized membrane potential (Vm) and consists of a brief cluster of APs firing at high frequencies (2–10 APs at 250-1,000 Hz) (Deschenes and Steriade 1982; Jahnsen and Llinás 1984b; Llinás and Jahnsen 1982). Burst firing is enabled by the presence of T-type Ca2+ channel currents present in TC neurons that are generated by the α-subunit Cav3.1 and that are deinactivated when Vm is hyperpolarized beyond resting levels. Cav3.1 mediates a low-threshold, slow, but transient depolarizing potential that drives burst firing in TC neurons (Anderson et al. 2005; Kim et al. 2001). Evidence also suggests that Ca2+ influx through Cav3.1 inhibits tonic firing (Anderson et al. 2005).
Recent extracellular recordings from thalamus found that visceral pain initially increases TC neuron firing rate in ventral posterior lateral (VPL) nucleus but within minutes overall firing rate decreases and firing mode switches to brief intermittent bursts (Kim et al. 2003). The concurrent emergence of burst firing during the inhibition of overall firing rate suggests the possibility that the neurons have become hyperpolarized, as this would deinactivate the Cav3.1 T-type calcium channels necessary for burst firing in TC neurons. Consistent with this notion, genetically deleting Cav3.1 abolished the burst firing (Kim et al. 2003). Importantly, deleting Cav3.1 only partially (40–50%) prevented the decrease in tonic firing rate. Although Vm of the TC neuron was not assessed, we speculate that the switch to burst firing likely results from an excitation-induced activation of K+ channels that then hyperpolarized Vm. The shunting effect of this presumed hyperpolarizing K+ current would also explain the inhibition of overall firing rate. Therefore we explored mechanisms whereby excitation might hyperpolarize and inhibit TC neurons.
In principle, Vm could hyperpolarize in response to a wide variety of known endogenous neuromodulators. Rapid transient hyperpolarization of TC neurons occurs via GABAA receptor synaptic transmission, from either the thalamic reticular nucleus (nRT) or local GABAergic neurons (Crabtree et al. 2002; Crabtree and Isaac 1998). However, this ligand-gated Cl− channel current is short-lived and only weakly hyperpolarizes Vm because of the rapid kinetics of GABAA synaptic transmission and the relatively high Cl− reversal potential (ECl ∼ −80 mV in TC neurons) (Ulrich and Huguenard 1997a). This mechanism may not be well suited to produce the strong and lasting Vm hyperpolarization needed to achieve a persistent inhibition of tonic firing rate and switch to burst firing mode. GABAB receptor synaptic transmission produces a stronger and longer-lasting hyperpolarization of Vm, because of the slower kinetics of GABAB G protein receptor signaling and the activation of K+ conductances with a more negative equilibrium potential (EK ∼ −100 mV) (Ulrich and Huguenard 1997b). However, hyperpolarizing Vm via GABAergic synaptic transmission requires continuous GABAergic neuron AP firing at high rates. Such firing can fail because of energy depletion and in some circumstances may lead to GABAergic neuron cell death. Extrasynaptic GABA can also inhibit through tonic inhibition mediated by specific GABAA receptor subunits, providing a way to couple circuit activity to local circuit inhibition (Herd et al. 2013). Yet, inactive surrounding circuits may also be inhibited by this mechanism, and therefore it lacks specificity. A cell-autonomous mechanism to activate K+ channels would overcome this limitation. Bacci et al. (2004) recently reported that cortical GABAergic interneuron firing triggers calcium-dependent cannabinoid release and autocrine activation of hyperpolarizing G protein-regulated inward rectifier (GIRK) K+ channels. However, this autocrine cannabinoid mechanism has not been described in glutamatergic neurons. Furthermore, this protective mechanism for GABAergic neurons would disinhibit glutamatergic neurons, leaving them prone to excitatory cell death. Importantly, TC neurons not only gate sensory signals but also transmit these signals through very high tonic fire rates (Kasten et al. 2007).
Therefore we hypothesized that TC neurons might possess a specialized autoinhibitory mechanism to prevent pathological signal transmission and glutamatergic neuron cell death in thalamus. This intrinsic mechanism might hyperpolarize Vm to inhibit tonic firing rate and promote burst firing as recently shown in vivo (Kim et al. 2003). We report that such mechanisms exist in TC neurons and that this function is independent of GABAergic and glutamatergic synaptic transmission, low-threshold T-type Ca2+ currents, Na+-K+-ATPases, and even GIRK K+ channels and intracellular Ca2+ signaling (Bacci et al. 2004). We also report evidence that the TC glutamatergic neuron autoinhibition occurs in part through ATP-regulated K+ channels, as recently reported in cholinergic (Allen and Brown 2004) and dopaminergic (Avshalumov et al. 2005) neurons. The results define a novel thalamic mechanism for robust, Ca2+-independent, and cell-autonomous inhibition of fast-spiking glutamatergic neurons in the central nervous system.
MATERIALS AND METHODS
All protocols were approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee and comply with the policies and regulations of The Journal of Physiology's animal ethics standards as outlined by Drummond (2009). Adult (2–6 mo old) C57BL/6 mice, as well as Cav3.1-knockout mice in a C57BL/6 genetic background, were genotyped as previously described and utilized in this study (Anderson et al. 2005). Slice preparations were as previously described (Kasten et al. 2007). Mice (80 total) were deeply anesthetized with isoflurane (5%, inhaled) and decapitated, and the brain was quickly removed and placed in ice-cold, oxygenated (95% O2-5% CO2) sucrose cutting solution containing (in mM) 234 sucrose, 5 KCl, 5 MgSO4, 1 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. Coronal brain slices (230–280 μm) were cut and transferred to oxygenated (5% CO2) artificial cerebrospinal fluid (ACSF) for 40–60 min at 37°C and then stored at room temperature for 1–9 h before recording in ACSF containing (in mM) 124 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 glucose.
TC relay neurons were visually identified for recording at ×400 magnification with infrared (IR) DIC optics on an upright Olympus BX-51WI microscope (Olympus, Tokyo, Japan). More than 90% of the neurons within these thalamic nuclei are glutamatergic (see the Allen Brain Atlas, Mouse Connectivity, Transgenic Characterization, comparing the glutamatergic neuron Slc17a6-IRES-Cre reporter and GABAergic neuron Slc32a1-IRES-Cre reporter; http://www.brain-map.org). GABAergic neurons are extremely rare outside the reticular nucleus within the thalamus. These rare, typically smaller, bipolar neurons were avoided with the use of IR-guided microscopy. GABA neurons are also outliers in their electrophysiological properties, including a higher input resistance. Such a rare cell was excluded from the analysis. Recording pipettes were pulled from 1.5-mm-OD capillary tubing (A-M Systems, Carlsborg, WA) with a Flaming/Brown P-97 pipette puller (Sutter Instruments, Novato, CA) and had tip resistances of 3–5 MΩ when filled with internal solution. Internal solution contained (in mM) 135 potassium methanesulfonate, 10 HEPES, 4 KCl, 2 NaCl, 4 MgATP, 0.3 Tris-GTP, 7 phosphocreatine, 1 EGTA, 0.1 CaCl2 (pH = 7.25, osmolarity ∼ 290 mosM). In studies utilizing the calcium chelator BAPTA in the patch pipette, potassium methanesulfonate concentration was reduced to maintain osmolarity. Multiple attempts to perform perforated patch recordings in these adult TC neurons were unsuccessful.
Recordings were made with a HEKA triple EPC-10 patch-clamp amplifier utilizing PatchMaster 2 software (HEKA Instruments, Southboro, MA) at room temperature (∼22°C) or 35°C in oxygenated (5% CO2) ACSF. Current-clamp recordings were obtained from thalamic relay neurons in the centromedial (CM) nucleus and other nuclei as described in the figures. Cells were recorded only if series resistance was <15 MΩ; most demonstrated a series resistance of 7–12 MΩ. Series resistance was 80% compensated, and junction potentials were uncorrected. Data were unfiltered and sampled at 2–20 kHz. Burst firing is defined as two or more APs occurring at the beginning of a current pulse with an interspike interval of <10 ms (>100 Hz). Firing rate (spikes/s across the full 500-ms pulse) and Vm were measured from the first and last traces of the high-frequency stimulation protocol described below. To fit inset traces into limited space, APs are truncated. Statistics were assessed by Student's paired t-test for single measurements and ANOVA for repeated measurements.
Drugs were bath applied with gravity perfusion in oxygenated (95% O2-5% CO2) ACSF. All recordings were performed in the presence of AMPA/kainate antagonist DNQX (10 μM; Tocris, Ellisville, MO) and GABAA receptor antagonist picrotoxin (100 μM; Sigma, St. Louis, MO). Tetrodotoxin (TTX), ouabain, BaCl2, glibenclamide, and tolbutamide were obtained from Sigma-Aldrich.
RESULTS
Repeated firing causes thalamocortical neurons to hyperpolarize Vm and self-inhibit.
The autonomous response of individual TC neurons to prolonged AP firing was assessed with direct intracellular depolarizing current injections during whole cell patch-clamp recordings in coronal brain slices from adult mice. Current injection amplitudes were adjusted such that a 500-ms pulse elicited 5–20 spikes. These pulses were initially repeated once every 10 s, revealing a consistent AP firing rate with each stimulus. The pulses were then repeated at a higher rate of once every second for 4 min (240 depolarizations). This protocol was selected because it allowed strong stimulation of the TC neuron while allowing measurements of Vm between stimuli. We initially focused on CM neurons because of their relatively high baseline input resistance and their potential as a target of cortical seizures. Figure 1A reveals that repeated current injections cause tonic AP firing. Interestingly, when the same pulse stimulus was given at a higher rate (every 1 s), the Vm between injections gradually hyperpolarized, reaching a steady state after ∼1 min (Fig. 1A). The pattern of APs evoked also changed: early pulses caused a continuous “tonic” AP firing response (10–40 Hz), but as Vm hyperpolarized tonic firing rate decreased, eventually changing to a single AP (Fig. 1A). In CM, a low-threshold calcium spike was observed, but burst firing was most often absent, even during the rebound from a hyperpolarizing current injection firing only single spikes (Kasten and Anderson, data not shown). This contrasts with the change from tonic to burst firing (initial couple spikes firing at ≥100 Hz) observed in neurons of other thalamic nuclei [VPL and medial dorsal, central division (MDc) in Fig. 2, C and D, and Fig. 4D]. After the first firing trial 1, upon switching back to the low firing rate stimulus, Vm returned to resting (Fig. 1A, left) or even slightly depolarized values (Fig. 1A, right) in CM. In most cells, repeating the same stimulus sequence (Fig. 1A, right, trials 2–5) caused a stronger hyperpolarizing response and the accumulation of a component of persistent hyperpolarization (Fig. 1B, i–iii, and Fig. 1A, right, labeled “Early”, n = 8). These results indicate that the firing history of TC neurons determines the magnitude of the self-inhibition and Vm hyperpolarization. CM thalamic neurons challenged with this firing protocol hyperpolarized Vm by 13.6 ± 0.9 mV (P < 0.001, n = 16; Fig. 1C), and firing rate was inhibited by 62 ± 5% of baseline (P < 0.001, n = 16; Fig. 1D).
Fig. 1.

Kinetics and plasticity of centromedial (CM) thalamocortical neuron autoinhibition. A: diagram of pulse protocol (top) and example of the firing response and membrane potential (Vm) shift. In all traces, a line marks the baseline resting Vm prior to and during stimulation. Left: kinetics of firing-induced Vm hyperpolarization and repolarization. Right: effects of repeated trials of the firing protocol (n = 8). Means ± SE are plotted. B: traces revealing the effects of repeated trials of the firing protocol. A second line marks the most hyperpolarized Vm achieved with repeated stimulation. Inset: repeating the protocol also potentiated the firing rate inhibition. Traces at baseline (early) and stimulated (late) Vm are shown. In the example shown, with the repeated stimulation protocol, the combined persistent and reversible components of hyperpolarization generated a very large hyperpolarization response approaching 20 mV in total. C: action potential firing hyperpolarizes Vm (n = 16). D: action potential firing inhibits firing frequency (n = 16). *P < 0.05.
Fig. 2.
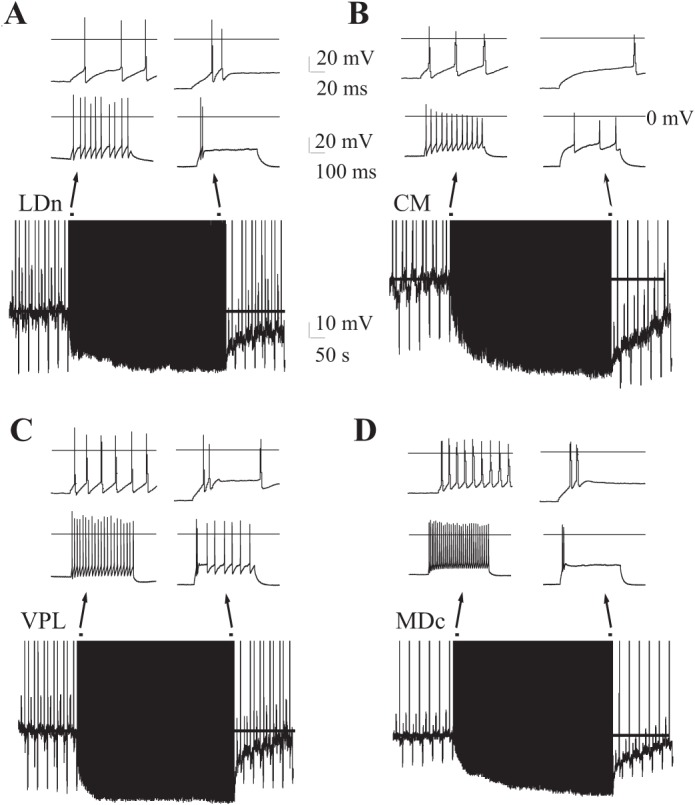
Cell-autonomous regulation of firing rate and mode in multiple thalamic nuclei. Thalamocortical neurons from multiple nuclei exhibit action potential firing-induced Vm hyperpolarization, firing rate inhibition, and firing mode switching from continuous tonic to transient with or without an initial high-frequency burst of 2 spikes at >100 Hz (see inset, top). A: lateral dorsal thalamocortical neuron (LDn) with 80-pA depolarizing current injections. B: CM thalamocortical neuron with 75-pA current injections. C: ventral posterior lateral (VPL) thalamocortical neuron with 140-pA current injections. D: medial dorsal, central division (MDc) thalamocortical neuron with 400-pA current injections. Examples are representative of 3–5 cells each. Insets: traces, early and late in firing protocol. In all traces, a line marks the baseline resting Vm prior to and during stimulation.
Fig. 4.
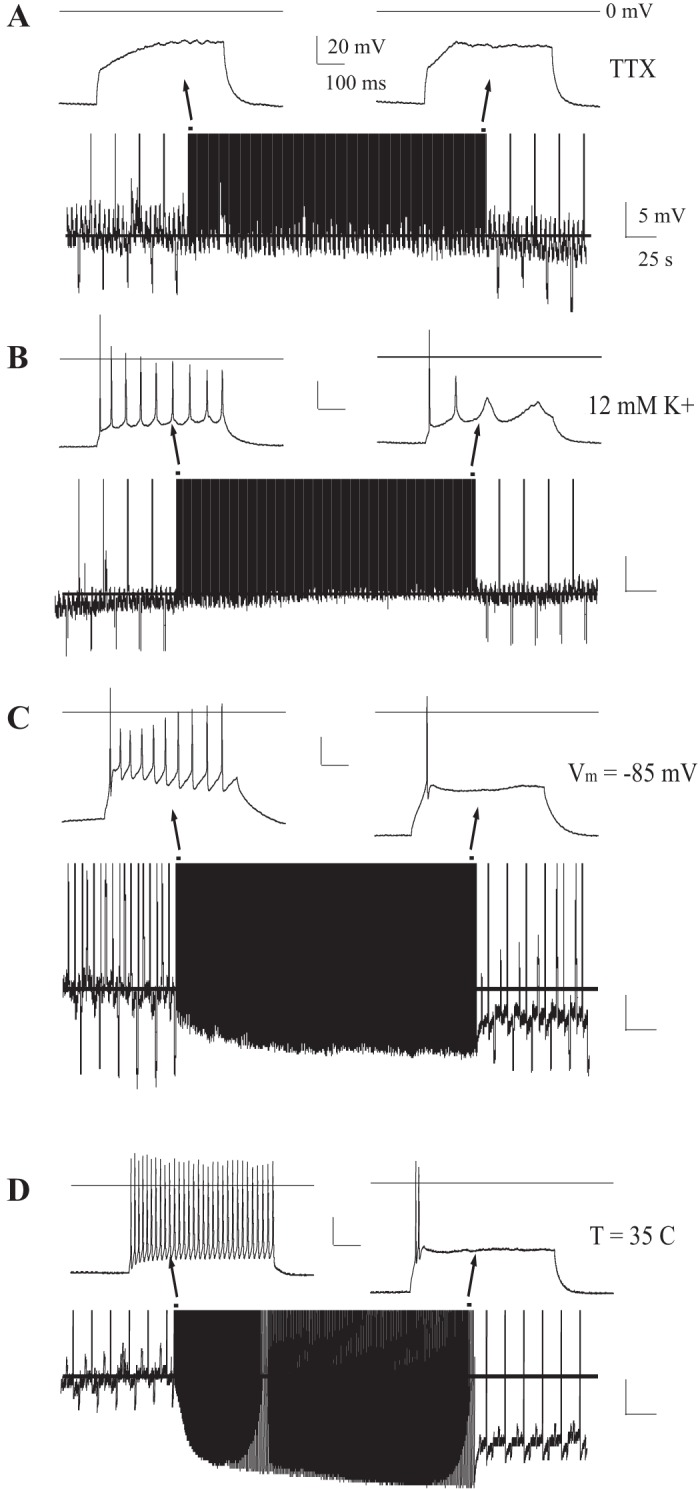
Firing-induced Vm hyperpolarization requires a transmembrane K+ gradient and voltage-gated Na+ channels. A: example CM cell (n = 3/3) with Na+ channel blockade (500 nM TTX). A broad range of depolarizing current injections failed to induce Vm hyperpolarization. B: example CM cell (n = 3/3) challenged with the firing protocol while extracellular [K+] was increased (12 mM) to shift K+ reversal potential near resting Vm. A tonic current was injected (−60 pA) to maintain Vm near resting values (−65 mV). C: example CM cell (n = 16/16) where a tonic hyperpolarizing current was injected to lower Vm ∼ −85 mV prior to and during the firing protocol. D: example MDc neuron with physiological temperatures (T = 35°C) Insets: traces, early and late in firing protocol. In all traces, a line marks the baseline resting Vm prior to and during stimulation.
Self-regulated firing exists in multiple thalamic nuclei.
To determine whether this self-regulatory response to firing exists in the glutamatergic neurons of other major thalamic relay nuclei, we repeated the protocol in neurons from multiple thalamic nuclei. A lateral dorsal (LD) TC neuron with 80-pA depolarizing current injection hyperpolarized Vm from −70 mV to −81 mV, inhibited firing rate from 20 Hz to 4 Hz, and switched from tonic to burst firing (Fig. 2A). A CM TC neuron with 75-pA current injection hyperpolarized Vm from −65 mV to −82 mV, inhibited firing rate from 24 Hz to 6 Hz, but failed to burst (Fig. 2B). A VPL TC neuron with 140-pA current injection hyperpolarized Vm from −70 to −84 mV, inhibited firing rate from 42 Hz to 16 Hz, and developed an initial burst response to the pulse (Fig. 2C). A MDc TC neuron with 400-pA current injections hyperpolarized Vm from −66 mV to −82 mV, inhibited firing rate from 62 Hz to 4 Hz, and fired an initial burst (Fig. 2D). A subset of neurons developed a persistent hyperpolarization of Vm after the stimulus trial shown (Fig. 2A). These studies reveal that self-regulated firing is pervasive among thalamic nuclei.
Firing-induced hyperpolarization requires Na+ but not Ca2+ entry.
T-type Ca2+ channels are necessary for thalamic neuron burst firing. While the function of thalamic burst firing is largely unknown, one study has found it necessary to stabilize sleep (Anderson et al. 2005). To test whether T-type Ca2+ channels contribute to this autoinhibitory response in vitro, we recorded thalamic neurons in mice with genetic deletion of exons 11–13 in CACN1G, which encodes the major TC neuron T-type Ca2+ channel subunit, Cav3.1 (Anderson et al. 2005). This targeted gene knockout abolishes T-type Ca2+ currents and burst firing in TC neurons (Anderson et al. 2005; Kasten et al. 2007). Despite the absence of burst firing, firing-induced hyperpolarization of Vm was preserved [11.2 ± 2.5 mV (n = 8) vs. 13.6 ± 0.9 mV in control (ctrl) (n = 16), P > 0.2; Fig. 3, A and C]. Consistent with the studies in vivo (Kim et al. 2003), loss of Cav3.1 did partially attenuate firing rate inhibition [45.5 ± 4.1% (n = 8) vs. 61.7 ± 5.1% in ctrl (n = 16), P < 0.05; Fig. 3D].
Fig. 3.
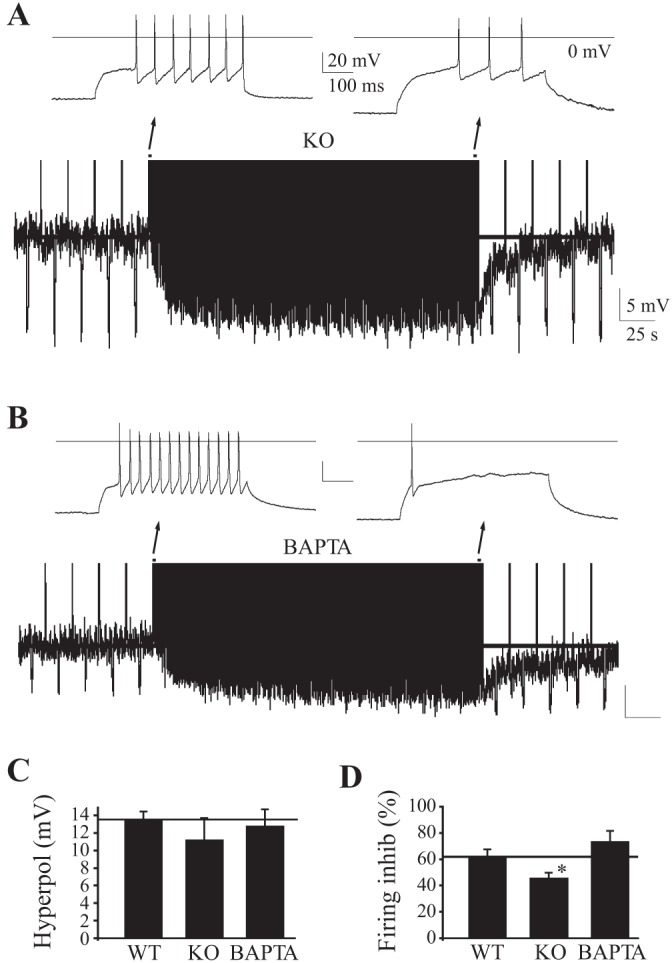
Ca2+ signaling is not required for firing-induced Vm hyperpolarization in thalamocortical neurons. A: CM thalamocortical neurons from mice lacking Cav3.1 T-type Ca2+ channels (KO). B: CM thalamocortical neuron with chelation of intracellular Ca2+ with pipette BAPTA (15 mM). C and D: effects of chelating intracellular Ca2+ with pipette BAPTA. WT, wild-type control. *P < 0.05. Insets: traces, early and late in firing protocol. In all traces, a line marks the baseline resting Vm prior to and during stimulation.
TC neurons possess an assortment of high-threshold Ca2+ channels that can regulate firing (Budde et al. 1998; Coulter et al. 1989; Crunelli et al. 1989; Kasten et al. 2007). To test whether increases of intracellular Ca2+ are necessary for firing-induced hyperpolarization and inhibition, we included the rapid Ca2+ chelator BAPTA (15 mM) in the pipette solution to block increases of intracellular Ca2+ triggered by repetitive firing. BAPTA not only failed to block firing-induced hyperpolarization [12.6 ± 1.5 mV (n = 9) vs. 13.6 ± 0.9 in ctrl (n = 16), P > 0.2; Fig. 3, B and C] but also failed to block autoinhibition [22.6 ± 2.6 Hz before vs. 6.2 ± 2.0 Hz after (n = 9) inhibition of 73.0 ± 9.1% vs. 61.7 ± 5.1% in ctrl (n = 9), P > 0.2; Fig. 3, B and C]. Thus Ca2+-mediated pathways are unnecessary for firing-induced Vm hyperpolarization or autoinhibition.
Bacci et al. (2004) found that depolarization alone, independent of Na+ APs, hyperpolarizes Vm in cortical GABAergic interneurons via Ca2+-induced cannabinoid signaling. However, the Ca2+ independence of TC neuron autoinhibition suggested a different mechanism. Consistent with this suggestion, unlike these cortical GABAergic interneurons, blocking voltage-gated Na+ channels and APs with TTX (500 nM) abolished the depolarization-induced hyperpolarizing of TC glutamatergic neurons (Fig. 4A; n = 3).
Firing hyperpolarizes Vm via a K+ conductance and self-inhibits via shunting.
Firing not only hyperpolarized Vm but also markedly decreased input resistance (931 ± 96 MΩ vs. 547 ± 90 MΩ, P < 0.01, n = 6), suggesting the possible activation of K+ conductances to hyperpolarize Vm and inhibit firing rate. However, TC neurons also display a large nonlinear membrane conductance; hyperpolarizing Vm from rest opens both hyperpolarization-activated, nonselective cation conductances (Ih) and inwardly rectifying K+ channels that reduce input resistance (Kasten et al. 2007; Meuth et al. 2006). To establish that firing activates a K+ conductance to hyperpolarize Vm, we raised extracellular K+ concentration ([K+]) from 3.5 to 12 mM, shifting the reversal potential for K+ (EK) from −102 to −63 mV. Shifting EK toward resting Vm blocked the firing-induced Vm hyperpolarization but preserved firing-induced inhibition (Fig. 4B; n = 3), revealing the shunting effects of the firing-activated K+ conductance. The findings suggest that firing activates a K+ conductance to hyperpolarize Vm and inhibit firing. Also consistent with this suggestion, prior hyperpolarization of Vm below resting values to −85 mV (EK is −102 mV) with tonic negative current injections failed to occlude the firing-induced inhibition of evoked firing rate [71.0 ± 12.0% (n = 7) vs. 61.7 ± 5.1% in ctrl (n = 16), P > 0.2; Fig. 4C]. In fact, even from this relatively hyperpolarized potential, repeated firing further hyperpolarized Vm beyond −85 mV [ΔVm: 4.3 ± 1.9 mV (n = 7) vs. 13.6 ± 0.9 mV in ctrl (n = 16), P < 0.01; Fig. 4C] toward EK. To confirm that this mechanism functions under physiological conditions, we increased bath temperature to T = 35°C and repeated the same firing protocol. Under these conditions, the firing protocol robustly hyperpolarized Vm and inhibited firing rate [Fig. 4D; MDc (n = 3) and CM (n = 4)]. Additionally, the persistent hyperpolarization of Vm developed within the first stimulus challenge (Fig. 4D) rather than after multiple challenges at room temperature (Fig. 1B).
The GIRK inhibitor barium (100 μM; Fig. 5A), which blocks firing-induced hyperpolarization of GABAergic neurons in cortex (Bacci et al. 2004), failed to block firing-induced Vm hyperpolarization in TC neurons.
Fig. 5.

Autoinhibition resists G protein-regulated inward rectifier (GIRK) K+ channel and Na+-K+-ATPase blockers. A: effects of GIRK K+ channel blocker Ba2+ (100 μM, n = 7) on firing-induced Vm hyperpolarization and firing rate inhibition; ctrl, control. B: effects of Na+-K+-ATPase blocker ouabain (250 nM, n = 5/5) on firing-induced Vm hyperpolarization and firing rate inhibition. Studies in CM. *P < 0.05. Insets: traces, early and late in firing protocol. In all traces, a line marks the baseline resting Vm prior to and during stimulation.
The Na+-K+-ATPase inhibitor ouabain, previously shown to inhibit the hyperpolarizing response of hippocampal neurons to glutamate and calyx of Held terminals to firing (Kim et al. 2007; Thompson and Prince 1986), actually enhanced firing-induced Vm hyperpolarization and firing rate autoinhibition (Fig. 5B). The results indicate that TC neurons autoregulate firing via mechanisms distinct from cortical [low threshold spike (LTS)] GABAergic neurons and the calyx of Held synaptic terminus.
Firing autoregulation in thalamocortical neurons is inhibited by KATP channel blockers.
Activating KATP channels with diazoxide inhibited tonic firing in TC neurons, but blockers of these channels had minimal effects during a brief (1 s) excitatory stimulus (Kasten et al. 2007). We speculated that KATP channels might instead contribute during periods of prolonged AP firing.
Consistent with this notion, the KATP channel blocker tolbutamide (100 μM) markedly decreased the firing-induced Vm hyperpolarization and self-inhibition (Fig. 6, A–C). Tolbutamide reduced the ΔVm hyperpolarization to 6.7 ± 1.2 mV [vs. 13.6 ± 0.9 mV in ctrl (n = 9 each), P < 0.001] and diminished autoinhibition to 23.8 ± 3.2% [vs. 61.7 ± 5.1% in ctrl (n = 9 each), P < 0.001].
Fig. 6.

KATP channels mediate the self-regulation of firing in CM thalamocortical neurons. A: effects of KATP channel blocker tolbutamide (100 μM) on firing-induced Vm hyperpolarization and firing rate inhibition. The same example cell is shown at baseline and with tolbutamide. B and C: Vm hyperpolarization (P < 0.001) and firing rate inhibition (P < 0.001) with and without tolbutamide. *P < 0.05. D: overlay of traces from steady-state, firing-induced hyperpolarization protocol in a CM thalamocortical neuron. Traces are before (red) and after (black) treatment with glibenclamide (30 μM). In all traces, a line marks the baseline resting Vm prior to and during stimulation.
Another KATP channel inhibitor (glibenclamide, 30 μM, n = 3 of 3, example in Fig. 6D) had similar effects. The small residual hyperpolarization and inhibition (Fig. 6B) could involve compensatory activation of Ca2+-activated K+ currents (Kasten et al. 2007), Na+-activated K+ currents (Bhattacharjee et al. 2002, 2003; Kim and McCormick 1989), or the Na+-K+-ATPase (Kim et al. 2007; Thompson and Prince 1986). The results provide evidence that KATP channels mediate a major component of the firing-induced hyperpolarization and autoinhibition of glutamatergic TC neurons.
DISCUSSION
This study reveals that TC glutamatergic neurons self-inhibit. While firing-induced autoinhibition and Vm hyperpolarization have been observed in several other neuronal subtypes, including superior colliculus neurons, GABAergic neurons, the synaptic terminus of the calyx of Held, and others (Bacci et al. 2004; Kim et al. 2007; Kim and McCormick 1998; Kubota and Saito 1991; Llinás and Lopez-Barneo 1988), this study identifies this mechanism in relay neurons of the thalamus. Importantly, self-inhibition could occur even at relatively low firing rates and displayed plasticity. KATP channels have been functionally identified in cholinergic neurons (Allen and Brown 2004) and were shown to regulate baseline tonic firing in dopaminergic neurons (Avshalumov et al. 2005). However, this channel subtype had not been previously implicated in the regulation of induced glutamatergic neuron AP firing rate. The finding of plasticity of this mechanism is also novel; the reversible (and a component of slowly reversible or irreversible) homeostatic response to firing was potentiated by prior neuronal activity, revealing a new form of plasticity of membrane excitability. KATP-dependent hyperpolarization provides an important link between energy metabolism and neuronal firing within the thalamus that could mediate homeostatic responses to the excessive discharges that impinge on thalamus in conditions such as epilepsy, peripheral pain syndromes, and possibly also certain psychiatric diseases (reviewed in Jeanmonod et al. 1996). This energy-dependent regulatory mechanism of thalamic sensory transmission may also explain changes in sensory transmission that occur across the sleep-wake cycle as energy is depleted and replenished.
KATP channels play important role in inhibiting thalamic output.
Multiple neuromodulators and ion channels are capable of regulating Vm and firing in TC relay neurons. The nonselective cation and two-pore K+ channels shape resting Vm and regulate the firing response of TC neurons (Meuth et al. 2006). Strong GABAergic input from nRT neurons generates both a GABAA channel-mediated Cl− conductance and a GABAB receptor-mediated G protein-activated inward-rectifying K+ channel conductance (Cox et al. 1997; Ulrich and Huguenard 1996) to inhibit firing. Finally, cholinergic stimulation is capable of altering firing of TC neurons (Varela and Sherman 2007).
We previously performed a comprehensive analysis of the ion channels that regulate firing in TC neurons and identified specific K+ channels (Kv1 and Kv3.2 voltage-gated, SK Ca2+-activated, and Kir2.2-like leak channels regulate components of tonic firing) and Ca2+ channels (largely T type and N type) that regulate elements of the firing response profile (Kasten et al. 2007). The present findings extend this characterization to the domain of prolonged tonic firing. While KATP channels are strongly expressed in thalamus (Dunn-Meynell et al. 1998; Gehlert et al. 1991), our initial studies found that KATP channel blockers had a relatively small contribution to the firing response during a brief 1-s stimulus (Kasten et al. 2007). By contrast, these channels have a profound inhibitory effect during prolonged tonic firing.
The finding that even very low firing rates (as low as 10 Hz; Fig. 1C) induce autoinhibition indicates that this mechanism could have effects even during physiological conditions and normal behavioral states. Importantly, KATP-mediated inhibition of TC neurons is independent of extrinsic synaptic inputs and occurs at the single-cell level, permitting highly focused regulation of neuronal circuit transmission within the thalamus. This would be an ideal mechanism to limit the transmission of signals generated from a limited peripheral somatosensory pain field or a cortical or subcortical seizure focus. Kim et al. (2003) showed that nociceptive stimuli cause an initial increase of tonic firing rate of TC neurons in the somatosensory thalamus that then decreases and switches to Cav3.1-dependent burst firing; however, Cav3.1 T-type Ca2+ channel-dependent burst firing cannot occur without Vm hyperpolarization to deinactivate the channel. We show that tonic firing alone is sufficient to hyperpolarize Vm and that this response is mediated in large part by the activation of KATP channels. As observed in vivo (Kim et al. 2003), we found that a relatively smaller portion of the in vitro autoinhibition was lost when Cav3.1 T-type Ca2+ channels were deleted (∼30–40%; Fig. 3D), whereas a much greater portion of this autoinhibition was lost when KATP channels were inhibited (∼75%; Fig. 6C).
KATP channels are adaptable modulators of neuronal function.
While KATP channels activate primarily in response to low intracellular [ATP], a broad variability in the [ATP] sensitivity of KATP channels has been observed in different systems due largely to differences in subunit composition and splice variants (Inagaki et al. 1995, 1996; Sakura et al. 1999). Furthermore, KATP channel protein is strongly upregulated in response to repeated neuronal activity caused by picrotoxin seizure kindling (Jiang et al. 2004). In addition to direct regulation via intracellular [ATP] and [ADP], KATP channels are also activated by mitochondrial release of peroxide, as demonstrated in heart and vascular tissue and more recently in brain (Avshalumov et al. 2005; Avshalumov and Rice 2003; Bao et al. 2005; Ichinari et al. 1996; Thompson et al. 1998). Peroxide-induced oxidation of KATP channels (an irreversible modification requiring protein turnover to clear) increases the [ATP] needed to inhibit channels in excised patches causing activity even at baseline [ATP] (Ichinari et al. 1996). Such mechanisms (peroxide modification, increased protein expression, or increased surface recruitment) could potentially explain the plasticity (including the tonic irreversible Vm hyperpolarization) observed with repeated stimulation (Fig. 1A, right, and Fig. 1B).
Other channels contributing to firing-induced inhibition.
Blocking KATP channels nearly eliminated firing-induced autoinhibition (∼75%) but only partially inhibited firing-induced hyperpolarization (∼50%). Several other channels were considered as potential explanations for this residual firing-induced hyperpolarization, including slow afterhyperpolarization (sAHP) K+ channels, Ca2+-activated K+ channels, and Na+-activated K+ channels. TC neurons possess a wide range of Ca2+ channels, which can regulate firing through small- and large-conductance K+ channels (SK and BK Ca2+-activated K+ channels) as well as the sAHP Ca2+-activated K+ channel (Kasten et al. 2007; Zhang et al. 2009); however, these channels generally provide feedback inhibition on a much faster timescale (ms to s) to hyperpolarize and inhibit firing. Significantly, complete chelation of intracellular Ca2+ with BAPTA produced no alteration in firing-induced Vm hyperpolarization or autoinhibition, suggesting that Ca2+-dependent pathways are not required. Na+-activated K+ (KNa+) channels are activated in response to high concentrations of intracellular Na+, and activity of these channels can be enhanced by decreases of intracellular [ATP] (Bhattacharjee et al. 2003; Schwindt et al. 1989; Tamsett et al. 2009; Yang et al. 2007). KNa+ channels have been implicated in firing-induced hyperpolarization, and mRNA for these channels is present in thalamus (Bhattacharjee et al. 2002, 2003; Kim and McCormick 1998; Zhang et al. 2010). Activation of KNa+ channels could explain the residual hyperpolarization that occurs in the presence of KATP channel blockers and could also explain the paradoxical increase of Vm hyperpolarization and firing rate inhibition found when the Na+-K+-ATPase was blocked (Fig. 5B). The enlarged response would be consistent with a greatly enhanced increase of intracellular [Na+] and KNa+ channel activation. An alternative non-ion channel-based mechanism that should also be considered in future studies is feedback inhibitory glioneuronal transmission.
Functional implications.
The cell-autonomous hyperpolarization of TC neurons described here will supplement the well-described feedback inhibition induced by GABAergic neurons of the nRT (Cox et al. 1997; Crabtree et al. 2002; Crabtree and Isaac 1998; Destexhe et al. 1998; Ulrich and Huguenard 1997a, 1997b). However, this cell-autonomous, self-regulatory mechanism in thalamus is distinct from GABAergic inhibition, operating on a much slower timescale and demonstrating plasticity following prior bouts of high neuronal activity. This new mechanism could overcome the failure of inhibition that might occur when energy is depleted or death occurs in GABAergic neurons and would permit the regulation of thalamic signal transmission at the single-cell level.
In conclusion, this study demonstrates a novel form of self-regulation in TC neurons that likely occurs through KATP channels. Enhancing KATP channel activity may hold promise for treating several neurological and psychiatric disorders (epilepsy, schizophrenia, Parkinson's disease, depression, and neurogenic pain) associated with excessive excitatory discharge of thalamus (Jeanmonod et al. 1996).
GRANTS
This work was supported in part by National Institute of Neurological Disorders and Stroke Grant R01 NS-057444 (M. P. Anderson), the Nancy Lurie Marks Family Foundation (M. P. Anderson), Autism Speaks/NAAR (M. P. Anderson), and Beth Israel Deaconess Medical Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.R.K. and M.P.A. conception and design of research; M.R.K. performed experiments; M.R.K. and M.P.A. analyzed data; M.R.K. and M.P.A. interpreted results of experiments; M.R.K. and M.P.A. prepared figures; M.R.K. and M.P.A. drafted manuscript; M.R.K. and M.P.A. edited and revised manuscript; M.R.K. and M.P.A. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of M. R. Kasten: Department of Rehabilitation Medicine, University of Washington, Seattle, WA 98104 (e-mail: mkasten@uw.edu).
REFERENCES
- Allen TG, Brown DA. Modulation of the excitability of cholinergic forebrain neurons by KATP channels. J Physiol 554: 353–370, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci USA 102: 1743–1748, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avshalumov MV, Chen BT, Koós T, Tepper JM, Rice ME. Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP-sensitive potassium channels. J Neurosci 25: 4222–4231, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avshalumov MV, Rice ME. Activation of ATP-sensitive K+ (KATP) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc Natl Acad Sci USA 1000: 11729–11734, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA. Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 431: 321–326, 2004. [DOI] [PubMed] [Google Scholar]
- Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci 26: 10029–10040, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. J Comp Neurol 454: 241–254, 2002. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Joiner WJ, Wu M, Yang Y, Sigworth FJ, Kaczmarek LK. Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J Neurosci 23: 11681–11691, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T, Munsch T, Pape HC. Distribution of L-type calcium channels in rat thalamic neurons. Eur J Neurosci 10: 586–597, 1998. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Calcium currents in rat thalamocortical neurons: kinetic properties of the transient, low-threshold current. J Physiol 414: 587–604, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Nucleus reticularis neurons mediate diverse inhibitory effects in thalamus. Proc Natl Acad Sci USA 94: 8854–8859, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JW, Collingridge GL, Isaac JT. A new intrathalamic pathway linking modality-related nuclei in the dorsal thalamus. Nat Neurosci 1: 389–394, 1998. [DOI] [PubMed] [Google Scholar]
- Crabtree JW, Isaac JT. New intrathalamic pathways allowing modality-related and cross-modality switching in the dorsal thalamus. J Neurosci 22: 8754–8761, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Lightowler S, Pollard CE. A T-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J Physiol 413: 543–561, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes M, Steriade M. Thalamic bursting mechanism: an inward slow current revealed by membrane hyperpolarization. Brain Res 239: 289–293, 1982. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M. Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells. J Neurophysiol 79: 999–1016, 1998. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol 587: 713–719, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K+ channel in rat brain. Brain Res 814: 41–54, 1998. [DOI] [PubMed] [Google Scholar]
- Gehlert DR, Gackenheimer SL, Mais DE, Robertson DW. Quantitative autoradiography of the binding sites for [125I]iodoglyburide, a novel high-affinity ligand for ATP-sensitive potassium channels in rat brain. J Pharmacol Exp Ther 257: 901–907, 1991. [PubMed] [Google Scholar]
- Herd MB, Brown AR, Lambert JJ, Belelli D. Extrasynaptic GABAA receptors couple presynaptic activity to postsynaptic inhibition in the somatosensory thalamus. J Neurosci 33: 14850–14868, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinari K, Kakei M, Matsuoka T, Nakashima H, Tanaka H. Direct activation of the ATP-sensitive potassium channel by oxygen free radicals in guinea-pig ventricular cells: its potentiation by MgADP. J Mol Cell Cardiol 28: 1867–1877, 1996. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron 16: 1011–1017, 1996. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP 4th, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270: 1166–1170, 1995. [DOI] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol 349: 205–226, 1984a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Ionic basis for the electroresponsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. J Physiol 349: 227–247, 1984b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmonod D, Magnin M, Morel A. Low-threshold calcium spike bursts in the human thalamus. Common physiopathology for sensory, motor and limbic positive symptoms. Brain 119: 363–375, 1996. [DOI] [PubMed] [Google Scholar]
- Jiang K, Shui Q, Xia Z, Yu Z. Changes in the gene and protein expression of KATP channel subunits in the hippocampus of rats subjected to picrotoxin-induced kindling. Mol Brain Res 128: 83–89, 2004. [DOI] [PubMed] [Google Scholar]
- Kasten MR, Rudy B, Anderson MP. Differential regulation of action potential firing in adult murine thalamocortical neurons by Kv3.2, Kv1, and SK potassium and N-type calcium channels. J Physiol 584: 565–582, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Park D, Choi S, Lee T, Sun M, Kim C, Shin HS. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science 302: 117–119, 2003. [DOI] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking a1G T-type Ca2+ channels. Neuron 31: 35–45, 2001. [DOI] [PubMed] [Google Scholar]
- Kim JH, Sizov I, Dobretsov M, von Gersdorff H. Presynaptic Ca2+ buffers control the strength of a fast post-tetanic hyperpolarization mediated by the alpha3 Na+/K+-ATPase. Nat Neurosci 10: 196–205, 2007. [DOI] [PubMed] [Google Scholar]
- Kim U, McCormick DA. Functional and ionic properties of a slow afterhyperpolarization in ferret perigeniculate neurons in vitro. J Neurophysiol 80: 1222–1235, 1998. [DOI] [PubMed] [Google Scholar]
- Kubota M, Saito N. Sodium- and calcium-dependent conductances of neurons in the zebra finch hyperstriatum ventrale pars caudale in vitro. J Physiol 440: 131–142, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Jahnsen H. Electrophysiology of mammalian thalamic neurons in vitro. Nature 297: 406–408, 1982. [DOI] [PubMed] [Google Scholar]
- Llinás R, Lopez-Barneo R. Electrophysiology of mammalian tectal neurons in vitro. II. Long-term adaptation. J Neurophysiol 60: 869–878, 1988. [DOI] [PubMed] [Google Scholar]
- Meuth SG, Kanyshkova T, Meuth P, Landgraf P, Munsch T, Ludwig A, Hofmann F, Pape HC, Budde T. Membrane resting potential of thalamocortical relay neurons is shaped by the interaction among TASK3 and HCN2 channels. J Neurophysiol 96: 1517–1529, 2006. [DOI] [PubMed] [Google Scholar]
- Sakura H, Trapp S, Liss B, Ashcroft FM. Altered functional properties of KATP channel conferred by a novel splice variant of SUR1. J Physiol 521: 337–350, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Crill WE. Long-lasting reduction of excitability by a sodium-dependent potassium current in neocortical neurons. J Physiol 61: 233–244, 1989. [DOI] [PubMed] [Google Scholar]
- Tamsett TJ, Picchione KE, Bhattacharjee A. NAD+ activates KNa channels in dorsal root ganglion neurons. J Neurosci 29: 5127–5134, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson GW, Horackova M, Armour JA. Sensitivity of canine intrinsic cardiac neurons to H2O2 and hydroxyl radical. Am J Physiol Heart Circ Physiol 275: H1434–H1440, 1998. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Prince DA. Activation of electrogenic sodium pump in hippocampal CA1 neurons following glutamate-induced depolarization. J Neurophysiol 56: 507–522, 1986. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. Gamma-aminobutyric acid type B receptor-dependent burst-firing in thalamic neurons: a dynamic clamp study. Proc Natl Acad Sci USA 93: 13245–13249, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci 17: 2348–2354, 1997a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. GABAA-receptor-mediated rebound burst firing and burst shunting in thalamus. J Neurophysiol 78: 1748–1751, 1997b. [DOI] [PubMed] [Google Scholar]
- Varela C, Sherman SM. Differences in response to muscarinic activation between first and higher order thalamic relays. J Neurophysiol 98: 3538–3547, 2007. [DOI] [PubMed] [Google Scholar]
- Yang B, Desai R, Kaczmarek LK. Slack and Slick KNa channels regulate the accuracy of timing of auditory neurons. J Neurosci 27: 2617–2627, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kolaj M, Renaud LP. Ca2+-dependent and Na+-dependent K+ conductances contribute to a slow AHP in thalamic paraventricular nucleus neurons: a novel target for orexin receptors. J Neurophysiol 104: 2052–2062, 2010. [DOI] [PubMed] [Google Scholar]
- Zhang L, Renaud LP, Kolaj M. Properties of a T-type Ca2+ channel-activated slow afterhyperpolarization in thalamic paraventricular nucleus and other thalamic midline neurons. J Neurophysiol 101: 2741–2750, 2009. [DOI] [PubMed] [Google Scholar]