

Abstract
In the last century, considerable efforts were made to understand the role of mtDNA mutations and of oxidative stress in aging. The classic mitochondrial free radical theory of aging, in which mtDNA mutations cause genotoxic oxidative stress, which in turn creates more mutations, has been a central hypothesis in the field for decades. In the last few years, however, new elements have discredited this original theory. The major source of mitochondrial DNA mutations seems to come from replication errors and failure of the repair mechanisms, and the accumulation of these mutations as observed in aged organisms appears to occur by clonal expansion and are not caused by a reactive oxygen species-dependent vicious cycle.
New hypotheses of how age-associated mitochondrial dysfunction may lead to aging are based on the role of reactive oxygen species as signaling molecules and on their role in mediating stress responses to age-dependent damage. Here, we review the changes that mtDNA undergoes during aging, and the past and most recent hypotheses linking these changes to the tissue failure observed in aging.
Graphical Abstract
Introduction
Aging is a degenerative process caused by the accumulation of cellular damage that leads to cellular dysfunction, tissue and organ failure, and death. Common features of aging include reduced tissue homeostasis and regeneration, increased oxidative stress, accelerated cellular senescence, with consequences such as decreased immunity, decreased healing and a generally higher level of risk factors for human diseases like cancer or neurodegenerative disorders [1].
The biology of aging and the exact mechanisms responsible for the aging process are still a matter of discussion and even though different theories can be identified, aging is most likely a multifactorial process. Even if still controversial [2], the prevailing explanation is the “free radical theory of aging,” first proposed by Harman in the ‘50s [3] and re-emphasized by Ames and colleagues in the ‘90s [4]. According to this theory, the major determinant of lifespan is the accumulation of tissue damage caused by cellular reactive oxygen species (ROS), which are highly unstable molecules that react with cellular macromolecules (lipids, proteins, and nucleic acids) and impair cellular functions [2, 5]. ROS are increased in aged tissues [6] and different lines of evidence corroborate the hypothesis that a decrease in metabolic rate attenuates oxidative damage and extends lifespan [6, 7]. Calorie restriction, for example, is a multi-target process that increases life span via acting on different levels: it prevents DNA damage and promotes DNA repair, it increases autophagy, decreases oxidative stress and affects mitochondrial efficiency and energy production [8].
Mitochondria are believed to have a central role in aging. They are the organelles that supply most of the energy to the cell in the form of ATP through oxidative phosphorylation (OXPHOS) conducted by the respiratory chain. Mitochondria are also involved in other tasks such as signaling, cellular differentiation, and cell death, as well as control of the cell cycle and cell growth. A drop in cellular ATP can lead to an increase in Bax, one of the first signals in the cellular apoptosis cascade, as well as impairment of ion pump function leading to membrane failure and cell death [9].
The OXPHOS is composed of four respiratory complexes (Complexes I to IV) and ATP synthase (Complex V), all located in the mitochondrial inner membrane. During aging there is a general decline in mitochondrial functions: tissues from aged animals show a decreased capacity to produce ATP, as reported in liver, heart and skeletal muscle [10, 11]. Moreover, the gross mitochondrial morphology is altered in aged tissues of mammals [4], the total number of mitochondria is lower in tissues of different ages, such as liver and muscle [12, 13], and likewise mitochondrial protein levels are decreased [14].
Mitochondria contain their own genome and most of the complexes of the electron transport chain are composed of both nuclear- and mtDNA-encoded proteins. Since the discovery of mtDNA diseases, and with the finding that mtDNA mutations can lead to mitochondrial dysfunctions, many efforts have been dedicated to the analysis of mtDNA changes and their role in aging.
Mitochondrial DNA
The human mitochondrial genome is a circular, double-stranded, supercoiled molecule present in one to several thousand of copies per cell [15]. It is maternally inherited and the copy number per cell varies according to the bioenergetic needs of the tissue. It is composed of 16569 bp and encodes for 37 genes (22 tRNAs molecules, 2 mitochondrial rRNA and 13 proteins). There are two strands called the “H-strand” (Heavy) and “L-strand” (Light) and are respectively enriched in guanines and cytosines. The majority of the mitochondrial genome is composed of coding regions, lacking introns. There is only a small non-coding portion of 1.1 kb called the D-loop (displacement loop) that is essential for mtDNA transcription and replication. All the mtDNA genes encode for components of the OXPHOS multi-subunit complexes, with the exception of Complex II, the components of which are entirely encoded by the nDNA [16].
Replication of mtDNA is independent of the cell cycle and, to date, only a few enzymes are known to participate in the process, all encoded by the nDNA and imported into the mitochondria [17]. RNA primers transcribed from mtDNA are also necessary to initiate mtDNA replication, and hence the enzymes that are part of the transcription machinery such as mitochondrial RNA polymerase (POLRMT), mitochondrial transcription factor B2 and A (TFB2M and TFAM), are essential for the replication process [15, 17, 18].
POLG (mitochondrial DNA polymerase γ, that has also a 3′–5′ exonuclease/proofreading activity), Twinkle (a DNA helicase) and mtSSB (mitochondrial single-stranded DNA-binding protein) are essential in the replication process: the helicase Twinkle is necessary to unwind double-stranded DNA into single-stranded so that POLG can synthetize DNA using as a template the ssDNA released by Twinkle while mtSSB binds the single-stranded DNA, protecting it from nucleolysis [19, 20].
mtDNA damage and repair mechanisms
The mtDNA mutation rate is believed to be ten times higher than that of nDNA [21] and multiple factors have been proposed to explain this phenomenon.
First of all, mtDNA is organized in “nucleoids,” dynamic structures of mitochondrial proteins and mtDNA. Proteins involved in mtDNA transcription and replication are localized in nucleoids, as well as other proteins involved in mtDNA packaging, including Twinkle, mtSSB and TFAM. TFAM in particular is the most abundant protein and it is important in mtDNA packaging and compaction [22–24]. Even though mtDNA is packed within these structures that serve as a protective shield, it is not as well packed as nDNA, so that it is more exposed to external genotoxic agents. Moreover, the distribution of nucleoids, and so mtDNA, inside the mitochondria can be also an important factor for the increased sensitivity of mtDNA to damage: nucleoids are membrane-associated and localized in the proximity of the mitochondrial respiratory chain that are, as mentioned above, one of the major sources of ROS in the cell and during not-replicative phases, oxidative damage represents one of the main types of DNA damage in aerobic organisms. Another important factor for the increased sensitivity of mtDNA to damage is the mtDNA replication rate: mtDNA mutations can occur during replication by the mis-incorporation of the wrong nucleotide. The most important mechanism of mtDNA in avoiding mutations during this process is the 3′–5′ exonuclease-proofreading activity of POLG. Even though the proofreading activity is very efficient, the fact that the mtDNA replication rate is higher than that of nDNA (because it is independent of cellular division), increases the possibility of mutation [19]. The most characterized repair mechanism is base excision repair (BER), but there is evidence that DNA breaks and mismatch repair can occur in mitochondria, although less frequently (reviewed by Kazak et al. [25]). Even though there is considerable redundancy among mtDNA repair proteins (with some exceptions such as NEIL1 [26], APE1, POLG and LIG3 [27, 28]), this mechanism appears to be less efficient in mtDNA compared to nDNA. This is understandable considering that degradation is not an option for damaged nDNA, while a partial number of damaged mtDNA molecules (or even entire organelles) can be eliminated without affecting mitochondrial function.
MtDNA turnover in aging
MtDNA maintenance and mitochondrial function rely on efficient mtDNA turnover that is determined by biogenesis, dynamics, and selective autophagic removal of defective organelles. These mechanisms are essential for mtDNA stability, and their changes during the aging process may affect the mtDNA integrity [29]. There is different evidence suggesting that aging is associated with decreased mtDNA copy numbers (described in detail in the next paragraphs). One possible explanation is an age-related decreased of mitochondrial biogenesis. The expression of nuclear-encoded regulatory factors involved in mtDNA maintenance, such as AMPK, PGC-1α, or NRF-1, changes during aging. In particular, in rodent models there is reduced AMPK activation [30] as well as a reduced response to stimuli (e.g., motor exercise) that, in young animals, increases PGC1α, TFAM and NRF-1 [31]. In a recent analysis performed in our laboratory, increased activation of PGC-1α in the mitochondrial POLG-α mutator mouse muscle was sufficient to increase mitochondrial biogenesis and enhance skeletal muscle and heart function, but was not able to reduce the accumulation of mtDNA mutations [32].
Another mechanism involved in mtDNA turnover is base excision repair (BER), which is of essential importance to maintain the integrity of mtDNA. A few studies analyzed BER activity changes during the aging process [33–35] and, in mammals, it seems to be tissue-dependent. BER efficiency is increased in aged liver, kidneys, and heart of mice, while it is decreased in muscle tissue [34] and in different regions of the brain, probably owing to decreased expression of repair enzymes such as 8-OHdG glycosylase and DNA polymerase-γ [36], or to an altered process of delivering BER enzymes to the mitochondria [37].
Mitochondrial dynamics (fusion and fission processes) play a key role in mtDNA integrity because they promote the exchange of components between damaged and functionally active mitochondria. In this way, impaired mitochondria with a highly mutated/wild-type mtDNA ratio can decrease their heteroplasmy by exchanging DNA material with healthy mitochondria [38]. When these processes fail to recover damaged mitochondria, dysfunctional organelles are degraded by a process called mitophagy.
Drp1 and Fis1 (involved in mitochondrial fission) and Mfn1, Mfn2, and Opa1 (involved in fusion) are the most studied mammalian proteins involved in this process [39]. The role of these proteins in maintaining mtDNA integrity and mitochondrial function throughout the aging process is still controversial [40, 41].
On the other hand, a great deal of effort has been directed toward understanding changes of mitophagy during aging. Aged tissues show a decrease in autophagic activity, as well as in proteins related to this process, such as SIRT1, Atg5, Atg7, Beclin 1 and LC3 [42, 43]. The age-related accumulation of dysfunctional mitochondria with increased levels of mtDNA mutations can be therefore be due, in part, to defects in their removal [44, 45].
To be noted, mechanisms that prolong life span, like caloric restriction, sirtuin activation, and insulin/insulin-like growth factor inhibition are all involved in activating autophagy/mitophagy [29].
mtDNA changes in aging
Different studies support the notion that mtDNA expression decreases with age, while mutations accumulate [46]. The idea that the increased rate of mutation is relevant to the aging process dates to the 1980’s [47]. One of the first reports connecting mtDNA damage and aging was published in 1988, when Piko et al. reported an increased frequency of deletions in senescent rats and mice [48]. In 1990, Cortopassi et al. [49] showed how low levels of a common deletion (5kb between nucleotides 8470 and 13447) usually associated with mitochondrial diseases [50] were present in heart and brain of aging humans. Studies in skeletal muscle showed that the accumulation of the common deletion occurred mostly through clonal expansion of single mutation events [51]. The same deletion was also found in brains of aged humans and, in particular, the higher number of mutations was present in caudate and substantia nigra [52–54], curiously the same brain regions that appear to be the most sensitive to OXPHOS dysfunction [55, 56]. Other non-clonal deletions involving the major arc of the mtDNA (between the mtDNA origins of replication) are also increased in tissues of aged individuals [54, 57]. Other deletions involving different parts of the mtDNA (such as the conserved sequence involved in replication and transcription termination) also increase with age [54, 58]. All the first analyses of the breakpoints involved in deletion events were carried out with PCR and sequencing techniques, and with the advent and advancement of new sequencing techniques such as next generation sequence (NGS), in the last decades there has been an improvement also in the analysis of rearrangements, point mutations and SNPs. Different groups using different techniques found an accumulation of point mutations with aging (Table 1) [54, 59–63]. In differently aged tissues, both specific base substitutions and general mutations have been analyzed: Michikawa, et al. [60] reported an accumulation of a particular base substitution in the D-Loop (T414G) in fibroblasts of people above 65 years of age compared to younger controls, and the same mutation was increased in muscles of individuals older than 30 years, but not in their brains [63]. The D-Loop seems to be the mtDNA region more prone to mutations: single nucleotide variants are increased with age in this region in the striatum of aged individuals [54]. But the accumulation of mutations in other regions of the mtDNA has been reported also, for example in human colonic crypt stem cells [59].
Table 1.
mtDNA changes during life. List of publications analyzing the changes in mtDNA point mutations in human aging with the age of the subjects analyzed in the studies
| Age | Tissue | Technique | mtDNA region (nucleotides) | Changes during life | Ref |
|---|---|---|---|---|---|
| 28 to 99 | Brain, blood | denaturing gradient gel electrophoresis and Cloning | D-loop, tRNAs, COI | Increased with age | [135] |
| 1 to 70 | Muscle | PCR-restriction fragment length polymorphism | MELAS and NARF related-positions | No correlation with age | [136] |
| 1–90 | Muscle, kidney, heart | PCR | 3243 | Increased with age | [137] |
| 0 to 101 | Fibroblast | denaturing gradient gel electrophoresis | D-loop | Increased with age | [60] |
| 1 to 90 | Muscle | denaturing gradient gel electrophoresis | D-loop, tRNA | Increased with age | [138] |
| 6 to 82 | Muscle | PCR-restriction fragment length polymorphism | 189 and 408 | Increased with age | [139] |
| 69 to 82 | Muscle | denaturing gradient gel electrophoresis | COII and 5 tRNAs | Increased with age | [51] |
| ≤31/≥53 | Brain | Cloning and Sequencing | COI | Increased with age | [140] |
| 0 to 92 | Skin | single strand conformation polymorphism | D-loop and COII | No correlation with age | [141] |
| 50 to 109 | Heart | Sequencing | D-loop | Increased with age | [142] |
| 19 to 95 | T cells | Heteroduplex Reference strand mediated conformation analysis | D-loop | No correlation with age | [143] |
| 10to 91 | Brain | Cloning and Sequencing | 10999–12168 | Increased with age in frontal cortex. No correlation in substantia nigra | [144] |
| 4 to 97 | Buccal cell, muscle | PCR-restriction fragment length polymorphism, Sequencing, Southern Blot | 189 | Increased with age | [145] |
| 31 | Brain | Sequencing | Whole mitochondrial genome | No correlation with age | [146] |
| 25 to 80 | Brain | Sequencing | Whole mitochondrial genome | Increased with age | [54] [82] |
The connection between the fact that mtDNA mutations are increased during aging, and the aging process itself, is still controversial and different factors have to be taken in consideration when approaching this topic. Even though mutations are accumulated in specific tissues, the ratio between mutated and wild-type mtDNA is very low, and studies of mtDNA diseases showed that pathogenic mutations have to reach at least a 70 to 90% of heteroplasmy to have an effect on mitochondrial function (the so-called “threshold effect”) [64]. MtDNA mutations accumulating with aging would not be enough to affect OXPHOS function or ATP production. Some reports, however, support the idea that mtDNA mutations in aging have an effect on respiratory function even if moderate [65]. How these mutations appear and accumulate during aging to reach a pathogenic threshold is still under investigation. Mutations can be maternally inherited, or they can originate from defects in replication or in the repair system, or they can form subsequently after exposure to mutagenic agents such as ROS or UV irradiation. A single mutation can then accumulate by clonal expansion or different mutations can appear as a result of continuous mutagenic insult. One of the main and oldest hypotheses to explain the generation of new mtDNA deletions during life is based on the controversial mitochondrial free radical theory of aging.
Recent studies by NGS showed a more general picture of the spectrum of mtDNA mutations observed during aging. Large deletions, single nucleotide variations and rearrangements at the D-loop appear to accumulate in the aging brain. However, the combined mutation load is still relatively low, approximately 3% [54, 61]. It is possible that few neurons have a large concentration of mtDNA mutations, as previously reported for dopaminergic neurons of the substantia nigra [53] and more single cell analyses would be required to addressed this question.
The mitochondrial free radical theory of aging
As mentioned above, the mitochondrial free radical theory of aging has been the most accredited theory after its first postulation. Since it has been analyzed in detail in numerous reviews [66–69], we will only briefly analyze the main characteristics and controversies. Mitochondria are an important source of ROS, in particular of superoxide anion, which is formed at Complex I and III [70, 71] of the electron transport chain in the mitochondrial inner membrane. Metabolic rate, usually estimated by measuring oxygen consumption at rest, correlates with superoxide generation in mitochondria [72]. SOD2 (superoxide dismutase-2) converts superoxide to hydrogen peroxide, which can be converted to the highly reactive hydroxyl radical in the presence of a transition metal ion (Fenton reaction) [73].
As mentioned above, many studies have suggested that mitochondrial damage plays an important role in the process of aging and that the accumulation of ROS with age is due to a decline in mitochondrial function and to reduced ROS scavenging enzymes. The mitochondrial theory of aging, first proposed by Harman in the 1970’s [74], was based on the idea that somatic mtDNA mutations impair OXPHOS Complexes (in particular Complex I and III), resulting in an increase of ROS, which in turn, damages proteins, lipids, and DNA, including mtDNA in a vicious cycle [75].
The decline in mitochondrial function concomitant with an increase in mtDNA point mutations and deletions observed in aged tissues was compatible with this theory. Moreover, different elements also fit this theory: the fact that the electron transport chain in mitochondria is an inherent source of oxygen radicals, the observation that in aged tissues there is an increased production of ROS, and the assumption that the mtDNA susceptibility to oxidative damage is higher than in nDNA.
One of the indirect proofs supporting this theory was the correlation between the levels of 8-hydroxy-2′-deoxyguanosine (8OHdG) and the levels of age-related common deletions in mtDNA [76–78]: ROS in fact facilitate the formation of oxidative DNA base damage products such as 8OHdG.
The 8-hydroxyguanine-DNA interaction leads to GC to TA transversions because 8- hydroxyguanine can sometimes pair with adenine instead of cytosine, and oxidative damage to cytosine mostly results in the formation of GC to AT transitions so that overall, GC to AT transitions and GC to TA transversions are the most commonly observed mutations resulting from oxidative DNA damage [79]. Past studies showed that mutations detected in aged mice appeared to be over 80% GC to AT transitions and GC to TA transversions [80], suggesting that most were created by oxidative DNA damage. However, in a more recent analysis, Itsara et al. showed that only a small fraction of the mtDNA mutations in aged Drosophila were GC to TA transversions, excluding oxidative stress as a major mechanism of mutation accumulation [81].
NGS analyses of human brain mtDNA also showed that the mutation spectra are comprised predominantly of transition mutations, consistent with misincorporation by DNA polymerase gamma or deamination of cytidine and adenosine as the primary mutagenic events in mtDNA. On the other hand, G -> T mutations, due to oxidative damage to DNA, did not significantly increase with age [82].
The evidence supporting the mitochondrial free radical theory of aging is thereby only indirect and in the last decades several works were published that did not validate this theory, mostly for lack of or inconclusive evidence [83]. In Drosophila, loss-of-function mutations in the mitochondrial ROS scavenging enzyme SOD or in the DNA repair enzyme Ogg1 have no influence on the somatic mtDNA mutation frequency [81].
Large deletions of mtDNA also accumulate with aging, and were also seen as evidence for a vicious cycle [84]. However, the rate of accumulation of age-related mtDNA deletions in patients with impaired oxidative phosphorylation does not show an accelerated accumulation of mtDNA damage [85]. Different studies also showed, that even though feeding animals with antioxidants can decrease oxidative damage and sometimes alter longevity, aging is usually not delayed [86]. Likewise, recent studies with NextGen sequencing also suggest that oxidative damage is not the major event inducing mutations of the mtDNA [54, 82]
Finally, studies on mouse models knocked in for a proofreading-deficient version of POLG-α, [87, 88] do not support this theory. The transgenic mice in which POLG-α was mutated in cardiac tissue showed no evidence of increased oxidative protein damage, or of age-dependent increase in mtDNA oxidative damage [89]. The mutator mouse, in which POLG-α-deficient protein is expressed ubiquitously, develops a premature aging phenotype, owing to a significant increase in the levels of mtDNA point mutations and deletions [87], but it does not show signs of increased oxidative damage to mitochondrial proteins, lipids, or DNA [90] while in aged mutator mice, only a very mild increase in the level of mitochondrial hydrogen peroxide is observed [91].
The Clonal expansion theory
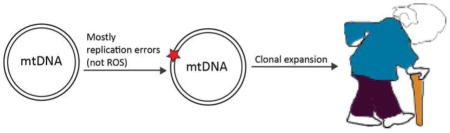
Other than the theory of the vicious cycle intrinsic to the mitochondrial free radical theory of aging, another possibility is that mtDNA mutations accumulate mostly by clonal expansion. According to this hypothesis, in some cells that carry the initial mutation, there occurs clonal expansion so that the threshold of mutated mtDNA reaches a pathogenic level with age, affecting over time the whole tissue [92]. In other words, inherited or de novo mutated mtDNA undergoes clonal expansion until the OXPHOS is impaired and the whole cell becomes deficient, while the level of mtDNA mutations overall in the tissue remains only slightly affected (Figure 1). Different studies support the hypothesis of a mosaic-like tissue: Muller-Hocker at al. showed how, in aged individuals, there is an increased number of single randomly distributed myocytes and cardiomyocytes without Cytochrome c oxidase (COX) activity [93, 94]. COX-deficient succinate dehydrogenase (SDH)-positive single neurons with high mtDNA deletions increase in aged-linked neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease [95–97]; moreover, focal respiratory chain defects accumulate to significant levels in aging human colon [59], liver, pancreas [98], stomach [99], and small intestine [92]. Through clonal expansion, both de novo and inherited mutations would increase with age (Figure 1). Supporting this theory, Ross et al. [100] analyzed a series of mouse mutants with different loads of both inherited and somatic mtDNA mutations, and reported that maternally transmitted mtDNA mutations induce mild ageing phenotypes and aggravate prematurely ageing phenotypes when associated with somatic de novo mutations in the mutator mice.
Figure 1. Proposed mechanisms to explain the role of mtDNA damage to the aging process.

mtDNA mutations can be generated during life or inherited. Both types can accumulate during life, potentially to the point where an OXPHOS defect is generated. This defect may increase superoxide (and other reactive oxygen species) generation, which in theory could further damage the mtDNA. However, experimental evidence of this vicious cycle lacks. Reactive oxygen species are more likely signals triggering protective effects. Recent studies suggest that not only postmitotic cells, but also progenitor cells seem be an important cellular target of mtDNA damage.
Different explanations for clonal expansion and mutant mtDNA accumulation have also been proposed, such as imperfect autophagocytosis [101, 102], or a replicative advantage of the mutant mtDNA compared to the wild-type [57, 103, 104], but clarification of this mechanism is still under investigation.
Although any single mutation is present at very low levels in aged tissues, it has been proposed that there could be many changes, which all combined could affect mitochondrial function. This “tip of the iceberg” model [105] was supported by quantification of base-pair substitutions and very high levels of mtDNA deletions in the elderly but, as described above, more recent and direct quantitation has not supported this hypothesis [54, 61].
Alternative role of ROS
From the most recent works, it is more apparent how the classical mitochondrial free radical theory of aging must be revisited [75]. Even though it is clear how mtDNA damage and ROS do have a role in the aging process, their correlation is still extensively under investigation. One hypothesis is that the increase in ROS is a consequence rather than a cause of aging [69] and that the role of ROS is to mediate the stress response to age-dependent damage.
ROS are not purely detrimental reactive byproducts of mitochondrial metabolism, but they are also main characters of signaling pathways that control proliferation, cell death and senescence [106]. Intracellular ROS can transiently increase after extracellular stimuli, such as growth factors, and contribute to the propagation of mitogenic and anti-apoptotic signals probably through the reversible oxidation of active sites in protein tyrosine phosphatases [107, 108]. ROS can also affect the activity of transcription factors such as NF-kB, JUN, and FOS, having a great impact on gene expression [86, 109], and they also contribute to apoptotic pathways. In particular, ROS generation in mitochondria appears to be a precise mechanism used in signaling pathways such as apoptosis, necrosis, and growth arrest [110]. Moreover, the oncogenic protein RAS increases the intracellular levels of ROS, inducing senescence [111]; likewise p21 [112] and p53 [113] activation can be regulated by ROS and can mediate apoptosis and growth arrest [114].
ROS may serve as signals to induce endogenous defenses, facilitating cellular adaptation to stresses such as hypoxia, autophagy, immune cell function and cellular differentiation [115]. Based on these observations, Hekimi and colleagues recently proposed the “gradual ROS response hypothesis” (hormesis) [69], linking the dual role of ROS as messengers and as toxic agents. They propose that ROS are early messengers in a protective stress-response pathway. With aging, the increase in cell damage induces a gradually intensifying stimulation of stress-response pathways and a consequent increased generation of ROS. As aging progresses, ROS levels reach a threshold at which their toxicity starts to contribute to cellular damage [69]. This theory would explain how also the subtle increase in mitochondrial ROS, a consequence of mtDNA mutagenesis, is sufficient to modify cellular stress response signaling. Surprisingly, the apoptosis machinery appears to be critical for ROS-related increase in longevity [116].
Support for the gradual ROS response hypothesis of aging comes from different studies in which increased oxidative stress in mitochondria of young animals protects against age-dependent loss of mitochondrial function [117] and, in some cases, increased longevity [118]. The discrepancies between the studies that correlate antioxidant administration to longevity can be approached taking account of this theory, so that it is not ROS accumulation that causes oxidative stress and consequent aging, but more an imbalance between the protective and toxic roles of ROS.
Nonetheless, it is important to take into consideration that progressive loss of mtDNA integrity can potentially affect not only the oxidative status of the cells or the production of ROS, but also a wide range of cellular processes. For example, mtDNA mutations may lead to cell dysfunction through additional mechanisms such as mediators of cell death that could be activated by misfolded mitochondria-encoded proteins [119].
From mtDNA damage to tissue failure
The discovery of mitochondrial diseases was the first proof that mutations in mtDNA lead to mitochondrial dysfunction [120]. However, in aging, how an mtDNA mutation can finally lead to tissue failure is not yet completely clarified.
As previously mentioned, mutations have to reach a pathogenic threshold in order to have an effect on mitochondrial function [64], and the mutations accumulated in aged tissues are significant but still very low compared, for example, to mitochondrial diseases. A recent hypothesis is that either acute damage or clonally expanded mutations affect the pool of stem cells in different tissues, affecting in particular their regenerative properties (Figure 1).
Mathematical models and in vivo analysis of animal models suggest that the principal causes of mtDNA mutations are replication errors arising during early development, and that their accumulation throughout life is mainly due to clonal expansion [51, 98, 121]. Considering that one of the characteristics of aging is diminished somatic stem cell function [122] and that in mitotic tissues the stem cells are the main long-lived cells, different analyses have been performed in order to investigate a possible role for mtDNA mutations in stem cell aging [123]. One of the first clues was reported by Taylor and colleagues [59]: they described the accumulation of mtDNA mutations in colonic crypt stem cells deficient in cytochrome c oxidase activity and an age-related increase in their number. mtDNA mutations in human colon have been associated with a decrease in crypt cell number and with a defect in cell proliferation [124].
McDonald et al. [99] showed that mtDNA mutations are present in stem cells within normal human gastric epithelium and are passed on to the differentiated progeny. Also, patches of hepatocytes deficient in cytochrome c oxidase activity contain clonal mtDNA mutations, suggesting their origin from a stem cell [98].
More evidence supporting the role of mtDNA mutations in the stem cell theory of aging come in fact from the analysis of animal models. In the mutator mouse, for example, neural stem cells and hematopoietic stem cells are dysfunctional from embryogenesis, suggesting a causal role of mtDNA accumulation: neural stem cells are reduced and have a decreased ability to self-renew, while hematopoietic stem cells show abnormal lineage differentiation [125, 126]. Another mouse model of aging, in which mtDNA in deleted ubiquitously by a restriction enzyme targeted to mitochondria, shows muscle wasting owing to a decline in muscle satellite cells, which decreases the muscle’s capacity to regenerate and repair during aging [127].
Why stem cells are more affected by mtDNA damage still remains elusive. It is still unclear whether mtDNA damage directly reduces the somatic stem cell pool or only their renewal capacities, and if mitochondrial impairment occurs intrinsically in the stem cells or indirectly owing to a systemic effect. Stem cells contain, in general, fewer mitochondria, as their quiescent state requires less energy and relies more on glycolysis than oxidative metabolism [128, 129]. Therefore, it is possible that their sensitivity to the same amount of mtDNA damage is increased compared to differentiated cells that contain a larger amount of mitochondria. On the other hand, mtDNA damage may not affect the quiescent cells themselves but it could impair the first steps of differentiation, when cells rely more on oxidative metabolism to satisfy their increased need for ATP [130]. Moreover, the stem cell renewal capacity could be affected because of altered redox status to which they are particularly sensitive [131, 132].
Interestingly, in the mutator mouse, N-acetyl-L-cysteine treatment in embryogenesis rescued the stem cell abnormalities, supporting this hypothesis that ROS/redox subtle changes are the link between mtDNA mutations and tissue failure [125].
Another challenging piece of information that has been studied to solve the puzzle of the biology of aging is how a focal impairment of mitochondrial function can then spread throughout the tissue. The “reductive hotspot hypothesis,” for example, proposes that cells harboring mtDNA mutations are toxic to the surrounding tissue. This theory is based on the assumption that cells with OXPHOS defects can reduce molecular oxygen through the plasma membrane redox system, producing extracellular superoxide and increasing oxidative burden to the surrounding tissue [105, 133]. Other theories, such as the activation of the NLRP3 inflammasome, have been proposed: mtDNA damage is suggested to modify the response of the immune system with the production of ROS, linking mtDNA mutations to another typical sign of aging [44, 91].
Conclusions
Since the discovery of a link between mtDNA mutations, mitochondrial dysfunction and mitochondrial diseases, great efforts have been made to investigate the role of mtDNA mutations in aging. The accumulation of mtDNA deletions and point mutations, together with the increase of ROS in aging tissues, led naturally to the mitochondrial free radical theory of aging. If at the beginning this original theory was very appealing owing to numerous examples of indirect evidence linking mtDNA, ROS, mitochondrial dysfunction and genotoxicity, it has become apparent that it needs to be revised.
With the advance of sequencing techniques, laser capture microdissection analysis of single dysfunctional cells, and the creation of animal models of aging, new evidence disproves the main player in the theory: the origin of mtDNA mutations seems to be inherited or to come from errors in mtDNA replication or from failure of repair mechanisms other than from oxidative damage. The cause of the accumulation of mutated mtDNA molecules observed with age seems to be their clonal expansion, and not continuous exposure to genotoxic agents, as proposed by the “vicious cycle” theory. Moreover, the initial excitement in proposing antioxidants as aging-delaying molecules mostly failed2 to give any conclusive output, resulting in disappointment in the field and raising more questions and doubts than answers and conclusions. New emerging theories propose ROS as tightly regulated stress signal molecules, and thus as a physiological response rather than a cause of aging even though the exact signaling pathways need still to be characterized [134].
Still, it is possible that ROS damage occurs to other macromolecules, such as lipids and proteins, with consequences to the aging process. As mentioned above, changes in ROS signaling also could have functional relevance to the aging process [134]. We believe that if the Free Radical theory of aging is to remain alive, it should move away from the mtDNA as the key target. However, even if not caused by oxidative damage, the role of mtDNA mutations in aging is likely still important. For example, how can focal, clonal-expanded accumulation of mtDNA mutations lead to the general tissue failure observed in aging? This and several related questions still need explanation. The recent theories about the involvement of stem cells in the general tissue failure observed in aging and the impact of mtDNA mutations in stem cell function are promising new venues.
Highlights.
Mitochondrial dysfunction increases with aging
Mitochondrial DNA (mtDNA) mutations accumulate with aging
Features of age-related mtDNA mutations suggest that they were not created by oxidative stress
The vicious cycle theory for mtDNA damage does not have strong experimental support
ROS is likely to play an important signaling role associated with mitochondrial stress during aging
Acknowledgments
Our work is supported by the US National Institutes of Health Grants 5R01EY010804, 1R01AG036871, 1R01NS079965, 1R21ES025673, the Muscular Dystrophy Association, the United Mitochondrial Disease Foundation and the JM fund for mitochondrial disease research.
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Long YC, et al. The biochemistry and cell biology of aging: metabolic regulation through mitochondrial signaling. Am J Physiol Endocrinol Metab. 2014;306(6):E581–91. doi: 10.1152/ajpendo.00665.2013. [DOI] [PubMed] [Google Scholar]
- 2.Edrey YH, Salmon AB. Revisiting an age-old question regarding oxidative stress. Free Radic Biol Med. 2014 doi: 10.1016/j.freeradbiomed.2014.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 4.Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91(23):10771–8. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller FL, et al. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43(4):477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 6.Hamilton ML, et al. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci USA. 2001;98(18):10469–74. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med. 2002;33(1):37–44. doi: 10.1016/s0891-5849(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 8.Szafranski K, Mekhail K. The fine line between lifespan extension and shortening in response to caloric restriction. Nucleus. 2014;5(1) doi: 10.4161/nucl.27929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006;11(4):473–85. doi: 10.1007/s10495-006-5881-9. [DOI] [PubMed] [Google Scholar]
- 10.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292(2):C670–86. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 11.Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123(3):951–7. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526(Pt 1):203–10. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Short KR, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102(15):5618–23. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ojaimi J, et al. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech Ageing Dev. 1999;111(1):39–47. doi: 10.1016/s0047-6374(99)00071-8. [DOI] [PubMed] [Google Scholar]
- 15.Takamatsu C, et al. Regulation of mitochondrial D-loops by transcription factor A and single-stranded DNA-binding protein. EMBO Rep. 2002;3(5):451–6. doi: 10.1093/embo-reports/kvf099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull. 2013;106:135–59. doi: 10.1093/bmb/ldt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–99. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 18.Metodiev MD, et al. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 2009;9(4):386–97. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 20.Korhonen JA, et al. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004;23(12):2423–9. doi: 10.1038/sj.emboj.7600257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Prog Neurobiol. 2011;94(2):166–200. doi: 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrido N, et al. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell. 2003;14(4):1583–96. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ekstrand MI, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13(9):935–44. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 24.Kukat C, et al. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A. 2011;108(33):13534–9. doi: 10.1073/pnas.1109263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kazak L, Reyes A, Holt IJ. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol. 2012;13(10):659–71. doi: 10.1038/nrm3439. [DOI] [PubMed] [Google Scholar]
- 26.Vartanian V, et al. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci U S A. 2006;103(6):1864–9. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sengupta S, et al. Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-l-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene. 2011;30(4):482–93. doi: 10.1038/onc.2010.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holt IJ. Mitochondrial DNA replication and repair: all a flap. Trends Biochem Sci. 2009;34(7):358–65. doi: 10.1016/j.tibs.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Gaziev AI, Abdullaev S, Podlutsky A. Mitochondrial function and mitochondrial DNA maintenance with advancing age. Biogerontology. 2014 doi: 10.1007/s10522-014-9515-2. [DOI] [PubMed] [Google Scholar]
- 30.Reznick RM, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5(2):151–6. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derbre F, et al. Age associated low mitochondrial biogenesis may be explained by lack of response of PGC-1alpha to exercise training. Age (Dordr) 2012;34(3):669–79. doi: 10.1007/s11357-011-9264-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dillon LM, et al. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum Mol Genet. 2012;21(10):2288–97. doi: 10.1093/hmg/dds049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imam SZ, et al. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27(8):1129–36. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Szczesny B, Tann AW, Mitra S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: Susceptibility of skeletal muscles to oxidative injury. Mech Ageing Dev. 2010;131(5):330–7. doi: 10.1016/j.mad.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garreau-Balandier I, et al. A comprehensive approach to determining BER capacities and their change with aging in Drosophila melanogaster mitochondria by oligonucleotide microarray. FEBS Lett. 2014;588(9):1673–9. doi: 10.1016/j.febslet.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Chen D, et al. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J Neurochem. 2002;81(6):1273–84. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- 37.Gredilla R, Bohr VA, Stevnsner T. Mitochondrial DNA repair and association with aging--an update. Exp Gerontol. 2010;45(7–8):478–88. doi: 10.1016/j.exger.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–87. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 39.Hoppins S. The regulation of mitochondrial dynamics. Curr Opin Cell Biol. 2014;29C:46–52. doi: 10.1016/j.ceb.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 40.Figge MT, Osiewacz HD, Reichert AS. Quality control of mitochondria during aging: is there a good and a bad side of mitochondrial dynamics? Bioessays. 2013;35(4):314–22. doi: 10.1002/bies.201200125. [DOI] [PubMed] [Google Scholar]
- 41.Weber TA, Reichert AS. Impaired quality control of mitochondria: aging from a new perspective. Exp Gerontol. 2010;45(7–8):503–11. doi: 10.1016/j.exger.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 42.Kaushik S, et al. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012;13(3):258–65. doi: 10.1038/embor.2011.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donati A, et al. Age-related changes in the autophagic proteolysis of rat isolated liver cells: effects of antiaging dietary restrictions. J Gerontol A Biol Sci Med Sci. 2001;56(9):B375–83. doi: 10.1093/gerona/56.9.b375. [DOI] [PubMed] [Google Scholar]
- 44.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–12. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 46.Manczak M, et al. Time-course of mitochondrial gene expressions in mice brains: implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J Neurochem. 2005;92(3):494–504. doi: 10.1111/j.1471-4159.2004.02884.x. [DOI] [PubMed] [Google Scholar]
- 47.Linnane AW, et al. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1(8639):642–5. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 48.Piko L, Hougham AJ, Bulpitt KJ. Studies of sequence heterogeneity of mitochondrial DNA from rat and mouse tissues: evidence for an increased frequency of deletions/additions with aging. Mech Ageing Dev. 1988;43(3):279–93. doi: 10.1016/0047-6374(88)90037-1. [DOI] [PubMed] [Google Scholar]
- 49.Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990;18(23):6927–33. doi: 10.1093/nar/18.23.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeviani M, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988;38(9):1339–46. doi: 10.1212/wnl.38.9.1339. [DOI] [PubMed] [Google Scholar]
- 51.Fayet G, et al. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12(5):484–93. doi: 10.1016/s0960-8966(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 52.Soong NW, et al. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat Genet. 1992;2(4):318–23. doi: 10.1038/ng1292-318. [DOI] [PubMed] [Google Scholar]
- 53.Kraytsberg Y, et al. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38(5):518–20. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 54.Williams SL, et al. Somatic mtDNA mutation spectra in the aging human putamen. PLoS Genet. 2013;9(12):el003990. doi: 10.1371/journal.pgen.1003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pickrell AM, et al. The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J Neurosci. 2011;31(27):9895–904. doi: 10.1523/JNEUROSCI.6223-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinto M, Pickrell AM, Moraes CT. Regional susceptibilities to mitochondrial dysfunctions in the CNS. Biol Chem. 2012;393(4):275–81. doi: 10.1515/hsz-2011-0236. [DOI] [PubMed] [Google Scholar]
- 57.Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet. 2009;18(6):1028–36. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pham XH, et al. Conserved sequence box II directs transcription termination and primer formation in mitochondria. J Biol Chem. 2006;281(34):24647–52. doi: 10.1074/jbc.M602429200. [DOI] [PubMed] [Google Scholar]
- 59.Taylor RW, et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J Clin Invest. 2003;112(9):1351–60. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michikawa Y, et al. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science. 1999;286(5440):774–9. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 61.Sevini F, et al. mtDNA mutations in human aging and longevity: Controversies and new perspectives opened by high-throughput technologies. Exp Gerontol. 2014 doi: 10.1016/j.exger.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 62.Brierley EJ, et al. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol. 1998;43(2):217–23. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 63.Murdock DG, Christacos NC, Wallace DC. The age-related accumulation of a mitochondrial DNA control region mutation in muscle, but not brain, detected by a sensitive PNA-directed PCR clamping based method. Nucleic Acids Res. 2000;28(21):4350–5. doi: 10.1093/nar/28.21.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rossignol R, et al. Mitochondrial threshold effects. Biochem J. 2003;370(Pt 3):751–62. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trounce I, Byrne E, Marzuki S. Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet. 1989;1(8639):637–9. doi: 10.1016/s0140-6736(89)92143-0. [DOI] [PubMed] [Google Scholar]
- 66.Barja G. Updating the mitochondrial free radical theory of aging: an integrated view, key aspects, and confounding concepts. Antioxid Redox Signal. 2013;19(12):1420–45. doi: 10.1089/ars.2012.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liochev SI. Reactive oxygen species and the free radical theory of aging. Free Radic Biol Med. 2013;60:1–4. doi: 10.1016/j.freeradbiomed.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 68.Gomez-Cabrera MC, et al. Mitochondria as sources and targets of damage in cellular aging. Clin Chem Lab Med. 2012;50(8):1287–95. doi: 10.1515/cclm-2011-0795. [DOI] [PubMed] [Google Scholar]
- 69.Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21(10):569–76. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237(2):408–14. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 72.Sohal RS, Mockett RJ, Orr WC. Mechanisms of aging: an appraisal of the oxidative stress hypothesis. Free Radic Biol Med. 2002;33(5):575–86. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- 73.Inoue M, et al. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr Med Chem. 2003;10(23):2495–505. doi: 10.2174/0929867033456477. [DOI] [PubMed] [Google Scholar]
- 74.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20(4):145–7. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 75.Lagouge M, Larsson NG. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J Intern Med. 2013;273(6):529–43. doi: 10.1111/joim.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85(17):6465–7. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beckman KB, Ames BN. Endogenous oxidative damage of mtDNA. Mutat Res. 1999;424(1–2):51–8. doi: 10.1016/s0027-5107(99)00007-x. [DOI] [PubMed] [Google Scholar]
- 78.Hamilton ML, et al. A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA. Nucleic Acids Res. 2001;29(10):2117–26. doi: 10.1093/nar/29.10.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang D, Kreutzer DA, Essigmann JM. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res. 1998;400(1–2):99–115. doi: 10.1016/s0027-5107(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 80.Vermulst M, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39(4):540–3. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 81.Itsara LS, et al. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014;10(2):el003974. doi: 10.1371/journal.pgen.1003974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kennedy SR, et al. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9(9):el003794. doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gruber J, Schaffer S, Halliwell B. The mitochondrial free radical theory of ageing-where do we stand? Front Biosci. 2008;13:6554–79. doi: 10.2741/3174. [DOI] [PubMed] [Google Scholar]
- 84.Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic Biol Med. 1990;8(6):523–39. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 85.Tengan CH, et al. Oxidative phosphorylation dysfunction does not increase the rate of accumulation of age-related mtDNA deletions in skeletal muscle. Mutat Res. 1997;379(1):1–11. doi: 10.1016/s0027-5107(97)00076-6. [DOI] [PubMed] [Google Scholar]
- 86.de Magalhaes JP, Church GM. Cells discover fire: employing reactive oxygen species in development and consequences for aging. Exp Gerontol. 2006;41(1):1–10. doi: 10.1016/j.exger.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 87.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–23. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 88.Zhang D, et al. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69(2):151–61. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]
- 89.Mott JL, et al. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat Res. 2001;474(1–2):35–45. doi: 10.1016/s0027-5107(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 90.Thompson LV. Oxidative stress, mitochondria and mtDNA-mutator mice. Exp Gerontol. 2006;41(12):1220–2. doi: 10.1016/j.exger.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Logan A, et al. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell. 2014 doi: 10.1111/acel.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gutierrez-Gonzalez L, et al. Analysis of the clonal architecture of the human small intestinal epithelium establishes a common stem cell for all lineages and reveals a mechanism for the fixation and spread of mutations. J Pathol. 2009;217(4):489–96. doi: 10.1002/path.2502. [DOI] [PubMed] [Google Scholar]
- 93.Muller-Hocker J. Cytochrome-c-oxidase deficient cardiomyocytes in the human heart--an age-related phenomenon. A histochemical ultracytochemical study. Am J Pathol. 1989;134(5):1167–73. [PMC free article] [PubMed] [Google Scholar]
- 94.Muller-Hocker J. Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: an age-related alteration. J Neurol Sci. 1990;100(1–2):14–21. doi: 10.1016/0022-510x(90)90006-9. [DOI] [PubMed] [Google Scholar]
- 95.Cottrell DA, et al. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology. 2001;57(2):260–4. doi: 10.1212/wnl.57.2.260. [DOI] [PubMed] [Google Scholar]
- 96.Bender A, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38(5):515–7. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 97.Pinto M, Moraes CT. Mitochondrial genome changes and neurodegenerative diseases. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbadis.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fellous TG, et al. Locating the stem cell niche and tracing hepatocyte lineages in human liver. Hepatology. 2009;49(5):1655–63. doi: 10.1002/hep.22791. [DOI] [PubMed] [Google Scholar]
- 99.McDonald SA, et al. Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology. 2008;134(2):500–10. doi: 10.1053/j.gastro.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 100.Ross JM, et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature. 2013;501(7467):412–5. doi: 10.1038/nature12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269(8):1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 102.de Grey AD. A proposed refinement of the mitochondrial free radical theory of aging. Bioessays. 1997;19(2):161–6. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- 103.Diaz F, et al. Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res. 2002;30(21):4626–33. doi: 10.1093/nar/gkf602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoneda M, et al. Marked replicative advantage of human mtDNA carrying a point mutation that causes the MELAS encephalomyopathy. Proc Natl Acad Sci U S A. 1992;89(23):11164–8. doi: 10.1073/pnas.89.23.11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de Grey AD. The reductive hotspot hypothesis: an update. Arch Biochem Biophys. 2000;373(1):295–301. doi: 10.1006/abbi.1999.1509. [DOI] [PubMed] [Google Scholar]
- 106.de Magalhaes JP, Church G. Cells discover fire: Employing reactive oxygen species in development and consequences for aging. Experimental Gerontology. 2006;41(1):1–10. doi: 10.1016/j.exger.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 107.Berner YN, Stern F. Energy restriction controls aging through neuroendocrine signal transduction. Ageing Res Rev. 2004;3(2):189–98. doi: 10.1016/j.arr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 108.Chiarugi P, Cirri P. Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem Sci. 2003;28(9):509–14. doi: 10.1016/S0968-0004(03)00174-9. [DOI] [PubMed] [Google Scholar]
- 109.Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28(3):463–99. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 110.Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84(2–3):131–41. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- 111.Lee AC, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274(12):7936–40. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- 112.Macip S, et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002;21(9):2180–8. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Macip S, et al. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol. 2003;23(23):8576–85. doi: 10.1128/MCB.23.23.8576-8585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 115.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48(2):158–67. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell. 2014;157(4):897–909. doi: 10.1016/j.cell.2014.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J Biol Chem. 2008;283(38):26217–27. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8(12):el000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mott JL, Zhang D, Zassenhaus HP. Mitochondrial DNA mutations, apoptosis, and the misfolded protein response. Rejuvenation Res. 2005;8(4):216–26. doi: 10.1089/rej.2005.8.216. [DOI] [PubMed] [Google Scholar]
- 120.Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med. 2012;44(1):41–59. doi: 10.3109/07853890.2011.598547. [DOI] [PubMed] [Google Scholar]
- 121.Elson JL, et al. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68(3):802–6. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8(9):703–13. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 123.Baines HL, Turnbull DM, Greaves LC. Human stem cell aging: do mitochondrial DNA mutations have a causal role? Aging Cell. 2014;13(2):201–5. doi: 10.1111/acel.12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nooteboom M, et al. Age-associated mitochondrial DNA mutations lead to small but significant changes in cell proliferation and apoptosis in human colonic crypts. Aging Cell. 2010;9(1):96–9. doi: 10.1111/j.1474-9726.2009.00531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ahlqvist KJ, et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 2012;15(1):100–9. doi: 10.1016/j.cmet.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 126.Norddahl GL, et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8(5):499–510. doi: 10.1016/j.stem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 127.Wang X, et al. Transient systemic mtDNA damage leads to muscle wasting by reducing the satellite cell pool. Hum Mol Genet. 2013;22(19):3976–86. doi: 10.1093/hmg/ddt251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Oburoglu L, et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell. 2014;15(2):169–84. doi: 10.1016/j.stem.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 129.Forsberg K, Di Giovanni S. Cross Talk between Cellular Redox Status, Metabolism, and p53 in Neural Stem Cell Biology. Neuroscientist. 2014;20(4):326–342. doi: 10.1177/1073858413514634. [DOI] [PubMed] [Google Scholar]
- 130.Inoue S, et al. Mitochondrial respiration defects modulate differentiation but not proliferation of hematopoietic stem and progenitor cells. FEBS Lett. 2010;584(15):3402–9. doi: 10.1016/j.febslet.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 131.Simsek T, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380–90. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shao L, et al. Reactive oxygen species and hematopoietic stem cell senescence. Int J Hematol. 2011;94(1):24–32. doi: 10.1007/s12185-011-0872-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.de Grey AD. The reductive hotspot hypothesis of mammalian aging: membrane metabolism magnifies mutant mitochondrial mischief. Eur J Biochem. 2002;269(8):2003–9. doi: 10.1046/j.1432-1033.2002.02868.x. [DOI] [PubMed] [Google Scholar]
- 134.Vina J, et al. The free radical theory of aging revisited: the cell signaling disruption theory of aging. Antioxid Redox Signal. 2013;19(8):779–87. doi: 10.1089/ars.2012.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jazin EE, et al. Human brain contains high levels of heteroplasmy in the noncoding regions of mitochondrial DNA. Proc Natl Acad Sci U S A. 1996;93(22):12382–7. doi: 10.1073/pnas.93.22.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pallotti F, et al. Evidence that specific mtDNA point mutations may not accumulate in skeletal muscle during normal human aging. Am J Hum Genet. 1996;59(3):591–602. [PMC free article] [PubMed] [Google Scholar]
- 137.Liu VW, Zhang C, Nagley P. Mutations in mitochondrial DNA accumulate differentially in three different human tissues during ageing. Nucleic Acids Res. 1998;26(5):1268–75. doi: 10.1093/nar/26.5.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang Y, et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc Natl Acad Sci U S A. 2001;98(7):4022–7. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Del Bo R, et al. Evidence and age-related distribution of mtDNA D-loop point mutations in skeletal muscle from healthy subjects and mitochondrial patients. J Neurol Sci. 2002;202(1–2):85–91. doi: 10.1016/s0022-510x(02)00247-2. [DOI] [PubMed] [Google Scholar]
- 140.Lin MT, et al. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet. 2002;11(2):133–45. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 141.Gerhard GS, et al. Mitochondrial DNA mutation analysis in human skin fibroblasts from fetal, young, and old donors. Mech Ageing Dev. 2002;123(2–3):155–66. doi: 10.1016/s0047-6374(01)00328-1. [DOI] [PubMed] [Google Scholar]
- 142.Nekhaeva E, et al. Clonally expanded mtDNA point mutations are abundant in individual cells of human tissues. Proc Natl Acad Sci U S A. 2002;99(8):5521–6. doi: 10.1073/pnas.072670199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ross OA, et al. Mitochondrial DNA damage in lymphocytes: a role in immunosenescence? Exp Gerontol. 2002;37(2–3):329–40. doi: 10.1016/s0531-5565(01)00200-5. [DOI] [PubMed] [Google Scholar]
- 144.Simon DK, et al. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiol Aging. 2004;25(1):71–81. doi: 10.1016/s0197-4580(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 145.Theves C, et al. Detection and quantification of the age-related point mutation A189G in the human mitochondrial DNA. J Forensic Sci. 2006;51(4):865–73. doi: 10.1111/j.1556-4029.2006.00163.x. [DOI] [PubMed] [Google Scholar]
- 146.Reeve AK, et al. The low abundance of clonally expanded mitochondrial DNA point mutations in aged substantia nigra neurons. Aging Cell. 2009;8(4):496–8. doi: 10.1111/j.1474-9726.2009.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]