

Abstract
Whole-genome sequencing (WGS) was carried out on 87 isolates of sequence type 111 (ST-111) of Pseudomonas aeruginosa collected between 2005 and 2014 from 65 patients and 12 environmental isolates from 24 hospital laboratories across the United Kingdom on an Illumina HiSeq instrument. Most isolates (73) carried VIM-2, but others carried IMP-1 or IMP-13 (5) or NDM-1 (1); one isolate had VIM-2 and IMP-18, and 7 carried no metallo-beta-lactamase (MBL) gene. Single nucleotide polymorphism analysis divided the isolates into distinct clusters; the NDM-1 isolate was an outlier, and the IMP isolates and 6/7 MBL-negative isolates clustered separately from the main set of 73 VIM-2 isolates. Within the VIM-2 set, there were at least 3 distinct clusters, including a tightly clustered set of isolates from 3 hospital laboratories consistent with an outbreak from a single introduction that was quickly brought under control and a much broader set dominated by isolates from a long-running outbreak in a London hospital likely seeded from an environmental source, requiring different control measures; isolates from 7 other hospital laboratories in London and southeast England were also included. Bayesian evolutionary analysis indicated that all the isolates shared a common ancestor dating back ∼50 years (1960s), with the main VIM-2 set separating approximately 20 to 30 years ago. Accessory gene profiling revealed blocks of genes associated with particular clusters, with some having high similarity (≥95%) to bacteriophage genes. WGS of widely found international lineages such as ST-111 provides the necessary resolution to inform epidemiological investigations and intervention policies.
INTRODUCTION
Concern over the increasing prevalence in hospitals of multiresistant bacteria, especially those that are resistant to carbapenem antibiotics, has prompted the use of typing methods that easily allow interlaboratory comparison of isolates. As a result, it has become clear that (i) high-risk clones of various Gram-negative bacteria, including Pseudomonas aeruginosa, can be found in many disparate locations, often with no obvious epidemiological links between the affected patients, and (ii) many of these clones are adept at acquiring various antibiotic resistance determinants (1, 2). These clones are commonly referred to as international lineages since they have been described in many countries across the globe. Epidemiological investigations of apparent outbreaks and interhospital spread of such lineages are confounded by the fact that they are so widely found; microbiological evidence of the same type must be considered alongside information about patient location and movement, for example, and patient location and movement information should be sufficiently robust to allow firm conclusions to be drawn. Greater resolution than that provided by conventional molecular typing methods, such as multilocus sequence typing (MLST), pulsed-field gel electrophoresis (PFGE), and variable-number tandem repeat (VNTR) analysis, is essential if we are to infer or discount potential transmission pathways with confidence. Understanding these pathways will be critical for precise public health interventions to contain these high-risk clones effectively. Whole-genome sequencing (WGS) can provide this resolution and has been used to investigate outbreaks of Klebsiella pneumoniae ST-258 producing K. pneumoniae carbapenemase (KPC) enzymes (3), the Nottingham strain of P. aeruginosa (4), and many others (e.g., see reference 5).
Carbapenemase-producing isolates of P. aeruginosa are becoming more common internationally and in the United Kingdom. Most carry blaVIM genes, but those producing other metallo-beta-lactamases (MBLs), e.g., IMP, NDM, SPM (6, 7), and non-metallo-beta-lactamases such as KPC and GES variants (8, 9) have also been described and may be locally, nationally, or regionally prevalent. Most MBL-producing P. aeruginosa strains in the United Kingdom belong to six international lineages, of which sequence type 111 (ST-111) is the most common (10). This lineage belongs to the O12 serotype, which has been associated with multidrug resistance and expansion in hospitals for decades (1, 11). It has been described in many European and Mediterranean countries, including France, Italy, Greece, the Netherlands, Spain and the Canary islands, Hungary, Scandinavia, Croatia, Austria, and Germany (6, 12–21) as well as in the United Kingdom (22, 23). It has been frequently associated with VIM-2 or less commonly with VIM-4 or IMP-type MBLs but sometimes only with noncarbapenemase beta-lactamases, such as VEB-1 and OXA extended-spectrum beta-lactamases (24, 25). A representative from outbreaks in two Colombian cities carried blaKPC and blaVIM-2 genes (26). The variety of resistance genes found in isolates of ST-111 indicates independent acquisition of resistance determinants by members of a widespread clonal lineage of P. aeruginosa (12), which may independently be selected for in hospitals in many locations.
In this study, we undertook single nucleotide polymorphism (SNP) phylogenetic analysis, Bayesian evolutionary analysis sampling trees (BEAST), and accessory genome analysis of the whole-genome sequences of 87 P. aeruginosa ST-111 isolates from the United Kingdom collected over a 9-year period with the aim of exploiting the extra resolution provided by WGS to determine to what extent spread within and between hospitals has occurred during the emergence of this ST as the most common MBL-producing clone of P. aeruginosa in the United Kingdom.
MATERIALS AND METHODS
P. aeruginosa isolates.
We studied 87 P. aeruginosa isolates that had been submitted to Public Health England (PHE) Antimicrobial Resistance and Healthcare Associated Infections (AMRHAI) Reference Unit from 24 hospital laboratories in the United Kingdom between April 2005 and April 2014. The isolates were from 65 patients and included six pairs of isolates each from a single patient and 12 associated isolates from the hospital environment. They also included four screening isolates, received at the same time as one another, from feces from an unknown patient(s). They were from 24 hospital laboratories across the United Kingdom, including Wales and Scotland, but most were from hospitals in the London area (11 hospital laboratories). Representatives were described by patient (P1 to P65), hospital laboratory (by region of the United Kingdom and unique number within that region), as discussed previously (10), MBL if present (e.g., VIM-2), and date (month.year) of collection; if the date of collection was unavailable, date of receipt was used. Environmental and screening isolates were given as E (E1 to E12) and S (S1 to S4) numbers, respectively.
The study set included representatives from 54% (65/120) of patients and from 83% (24/29) of the hospital trusts from which isolates of this type had been submitted over the 2005 to 2014 period. The remaining isolates were excluded because they were either no longer available in our archives (pre-2010 isolates from 32 patients) or because WGS failed to provide sufficient depth of coverage (mean depth of 15-fold or greater) for them to be included. The 87 isolates were from a variety of sources, the most common of which were sputum (18), urine (15), blood (10), wound (8), feces (6), and skin (4), with a wide spectrum of clinical details given by the sending laboratories, including screening, chest infection, pneumonia, pyrexia, sepsis/bacteremia, abscess, and necrotizing fasciitis.
Conventional typing and resistance characterization.
The 87 isolates had been typed as part of reference investigations by VNTR analysis at nine loci (27) and shared the profile 11,3,4,3,2,2,x,4,x (where x is variable), which corresponds to ST-111 (10, 22). In some cases, PFGE of SpeI-digested genomic DNA had also been carried out to offer additional resolution between isolates sharing highly similar VNTR types (27).
Carbapenemase genes were sought by PCR as described previously (28, 29), usually following interpretative reading of antibiograms. Carbapenemase gene alleles were determined from the WGS data as described in Antibiotic Resistance Gene Profiling.
Whole-genome sequencing.
Genomic DNA was prepared using a GeneJET genomic DNA purification kit (Thermo Fisher Scientific, United Kingdom) and sequenced on a HiSeq instrument (Illumina, United Kingdom) on Rapid Run mode using the TruSeq Rapid PE Cluster v2 kit and the TruSeq Rapid SBS v2 kit (200 cycles) by the PHE Genomic Services Unit.
The library was prepared using Nextera XT DNA sample preparation kits (Illumina, Cambridge, United Kingdom) following the manufacturer's protocol. The average fragment size and the concentration of clusterable material were determined as recommended by Illumina to quality check the library generated. The determination of the average fragment size of the libraries generated was performed using the DNA high-sensitivity reagent kit and DNA extended-range LabChip prepared according to the manufacturer's protocol and loaded onto a LabChip GX instrument (PerkinElmer, United Kingdom). The concentration of clusterable material for loading onto the HiSeq was determined by quantitative PCR (qPCR)-based quantification, using the KAPA universal library quantification kit (KAPA Biosystems, London, United Kingdom) following the manufacturer's protocol and real-time PCR cycle instrument setting. The library was clustered onto a HiSeq flow cell using the TruSeq Rapid Duo sample loading kit on an Illumina cBot instrument (Illumina) before being loaded onto the HiSeq instrument.
Maximum-likelihood tree construction.
FASTQ reads were trimmed using Trimmomatic 0.32 (30) with the following parameters: LEADING, 30; TRAILING, 30; SLIDINGWINDOW, 10:30; and MINLEN, 50. Subsequently, they were mapped to the Liverpool epidemic strain of ST-146 LESB58 chromosome (GenBank accession number NC_011770.1) using bwa-mem 0.7.5a (31) and variants determined using GATK 2.6.5 (32). Variants were parsed to retain high-quality SNPs based on the following conditions: a depth of coverage (DP) of ≥5, an AD ratio (ratio between variant base and alternative bases) of ≥0.8, a mapping quality (MQ) of ≥30, a ratio of reads with MQ0 to total number of reads of ≤0.05, and a distance to nearest SNP of >10. All positions that fulfilled these criteria in >0.9 of the samples were joined to produce a multiple FASTA format file where the sequence for each strain consists of the concatenated variants. This file was used as an input to generate a maximum-likelihood (ML) tree using Randomized Axelerated Maximum Likelihood (RAxML) (33) as implemented on the CIPRES portal (34) with the following parameters: –m (substitution model), GTRCAT; –b (bootstrap random number seed), 12345 -#; number of runs, 100; and –c (number of categories), 25.
BEAST analysis.
BEAST analysis (http://beast.bio.ed.ac.uk/), which has previously been successfully used for the investigation of the evolution of isolates of P. aeruginosa from patients with cystic fibrosis (35), produces time-measured phylogenies, allowing estimation of the time of divergence of different clades and is therefore a useful additional tool for analyzing sequences from isolates obtained over an extended period. The FASTA file describing SNPs in multiple samples was imported into BEAUti 1.8 and used to generate a BEAST 1.8 compatible xml file using the following parameters: Hasegawa-Kishino-Yano (HKY) substitution model, gamma site heterogeneity model, lognormal relaxed clock, coalescent constant size tree prior, UPGMA starting tree model, and 5 million Markov Chain Monte Carlo (MCMC) chain length (36). To account for invariant sites, a constantPatterns block listing the counts for the number of each invariant base was added to the xml file. The output from BEAST was summarized using TreeAnnotator (burn-in of 5,000 samples) and displayed using FigTree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree).
Accessory gene profiling.
Genome sequences were assembled using SPAdes v3.0.0 (37), and genes were predicted using Prokka v1.7 (38) with default parameters. All genes were processed to determine a nonredundant representative set using an iterative BLAST procedure whereby a novel gene was added to the set when its self-BLAST bit score was greater than 2× the bit score of BLASTs against all other genes in the set (39). Genes present in all isolate sequences (core genes) were removed from the set and the presence/absence of each accessory gene was plotted alongside the isolates on a phylogenetic tree.
In a secondary approach, contigs were assembled using VelvetOptimizer (http://bioinformatics.net.au/software.velvetoptimiser.shtml). Open reading frames were found with Prodigal (http://prodigal.ornl.gov), and the amino acid sequences clustered with CD-HIT (http://cd-hit.org) were found using an 85% similarity threshold. The presence or absence of genes not found in all isolates was shown in a matrix, and those found in particular clusters were identified by BLAST searches.
Antibiotic resistance gene profiling.
Trimmed reads from each genome were mapped against a database of reference sequences for resistance genes using Bowtie 2 (http://bowtie-bio.sourceforge.net/bowtie2), and binary alignment map (BAM) and variant calling format (VCF) files were produced with SAMtools (http://samtools.sourceforge.net). VCF generated thereby was parsed using a simple script that extracts, for each reference sequence with aligned reads, sites covered by >5 reads and the number of nucleotide alterations with respect to the reference sequence to calculate nucleotide identities. The presence of resistance genes was determined with a stringent threshold of inclusion of >90% identity over the length of the reference sequence whereas beta-lactamase variants were determined with 100% identity. The reference database used in this study was obtained from the Comprehensive Antibiotic Resistance Database (http://arpcard.mcmaster.ca) and from the NCBI nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore) using the accession numbers described in the supplementary data of Zankari et al. (40).
Nucleotide sequence accession numbers.
Sequence files were deposited in the European Nucleotide Archive under study number ERP010395 and accession numbers ERS716506 to ERS716591.
RESULTS
Conventional analysis.
The 87 P. aeruginosa isolates examined included representatives of ST-111 referred from United Kingdom hospitals over a 9-year period, starting in 2005 when it was uncommon to find MBLs in this species in the United Kingdom, and therefore provided a unique opportunity to investigate the relative roles of clonal spread versus independent introductions/acquisitions, as this lineage emerged as the dominant host carrying carbapenemases. The total numbers of patients and hospital laboratories for each year from which we received these isolates is shown in Fig. 1. A summary of the data for the 87 isolates included in this study is provided in Table S1 in the supplemental material.
FIG 1.

Charts showing numbers of patients (shown by metallo-beta-lactamase status in panel a and additionally with number of hospital laboratories [requestors] in panel b) from which representatives of ST-111 were received between 2005 and April 2014. Patients are included only once (by year when the first isolate was received), but requestors are included for every year that they submitted isolates from new patients. “Not tested” applies to isolates subjected only to typing that are no longer available in our archives; all but one was from hospital London_17, and it is assumed that they are highly likely to be VIM positive.
Most of the isolates (74/87, 85%) produced VIM-2 carbapenemase, but some had IMP variants (5) (IMP-1, IMP-13, or IMP-18) or NDM-1 (1) enzymes; no carbapenemase gene was detected in seven isolates. One isolate (P19, London_7, VIM-2, 05.10) had a VIM-2 and an IMP-18 gene; this was confirmed from a single colony pick.
Most of the isolates (57/87; 66%) shared an identical profile of 11,3,4,3,2,2,5,4,7 by VNTR analysis. Of the rest, most (18/28) differed only by a single repeat unit at the ninth, most discriminatory locus (11,3,4,3,2,2,5,4,6/8); these were distributed among multiple hospitals and appeared to show no clear pattern, although all the IMP-only producers had a profile of 11,3,4,3,2,2,5,4,6. Ten isolates (from patients P42, P52, and P55 and environmental isolates E10 to E14) from a single laboratory (London_26) (one patient also attended London_28), that were related in time and space, shared a consistent profile of 11,3,4,3,2,2,6,4,7; these were the only isolates in the WGS set with this profile and clustered together by all methods tested. One isolate (from patient P19) had a profile of 11,3,4,3,2,2,4,4,7. Finally the NDM-1 producer had the most distant VNTR profile from the others (11,3,4,3,2,2,6,4,13), differing from the most common profile at locus 7 (by one repeat unit) and locus 9 (by 6 repeat units). Phylogenetic analysis clearly showed that this single NDM producer was an outlier compared with the remainder of the set, with an average of 1,377 SNPs from the other isolates, and it was removed from the analysis.
Phylogenetic analysis of whole-genome sequences.
Among the remaining set of 86 isolates analyzed, there were between 0 and 548 high-quality (as determined by the criteria described in the methods) SNPs identified between any two isolates with a total of 21,278 variable positions. These were included in an alignment used to generate a maximum-likelihood tree (Fig. 2; see also Table S2 in the supplemental material). The number of SNPs quoted in the alignment is unlikely to be absolute, since SNP positions of low quality or with insufficient coverage in greater than 10% of the isolates were not included in the analysis. The five IMP-only producers from two geographically distant hospital laboratories (IMP set) separated by approximately 5 years were relatively diverse, having up to 46 SNPs between them; surprisingly, isolates from different hospitals separated by a period of 4 years (from patients P1 and P6) were the most similar, differing by only 5 SNPs. All IMP-only isolates were in clades that were phylogenetically distinct from those where the VIM producers were found (Fig. 2). The isolate producing VIM and IMP (from patient P19) from a third hospital laboratory (London_7) clustered apart from the other VIM or IMP producers; this isolate had between 111 and 491 SNPs from any other isolate by our criteria. Similarly, most of the MBL-negative isolates clustered in separate branches from the VIM producers and showed greater diversity than the IMP producers. Among the VIM producers, pairs of isolates were included from six patients (P8, P42, P43, P51, P52, and P54) separated in time from between 3 days (P43) and 4 years (P8), although most were collected within a month of each other. Five pairs of isolates were clustered closely, supporting the validity of the analysis (with 0 SNPs detected between isolates of four pairs and with 1 SNP found in a fifth pair); the greatest difference (18 SNPs) was observed between the pair of isolates that had the greatest time difference between them (isolates from P8 separated by 4 years and isolated in different hospitals) (Fig. 2). The length of time between these isolates suggests carriage in this patient (41).
FIG 2.

Maximum-likelihood tree estimation using single nucleotide polymorphism (SNP) analysis of representatives of ST-111 from United Kingdom hospitals collected between 2005 and 2014. Isolates are labeled by patient (P1 to P64 and S1 to S4), hospital, metallo-beta-lactamase gene (where present), and date (month.year) of collection. Environmental isolates are labeled E1 to E12, followed by this information. Two isolates from patient P8, separated by 4 years, are marked with stars; other patient pairs are marked with triangles, circles, squares, diamonds, or hexagons. Clusters and subclusters described in the text are marked. Bootstrap values are given for the clusters described.
Although the 74 VIM producers were from 20 hospital laboratories, only six submitted isolates from more than three patients, and the set is therefore dominated by isolates from hospital laboratories London_17 (isolates from 15 patients, 4 screening isolates from unknown patient[s], and 5 environmental isolates), Wales_1 (isolates from 5 patients), London_9 (isolates from 6 patients and an environmental isolate), London_12 (isolates from 5 patients), London_26 (isolates from 5 patients and 5 environmental isolates), and London_28 (isolates from 4 patients). Some patients were known to have been transferred between hospitals; P37 was transferred between London_26 and London_28 (only the isolate from London_28 is included in the tree), P42 had a similar transfer history, and another patient was transferred between London_9 and London_11 (P51). Patient P8 from South East_10 attended London_17 ∼4 years later.
The VIM-producing isolates from London_9 and London_11 (cluster 1 in Fig. 2) clustered in a clade with very little diversity (maximum of 4 SNPs between any two isolates); a patient transfer (P51) had taken place between these hospitals, which are geographically close to each other. An isolate from London_6 was also present in the same clade with no obvious association based on patient records. A further cluster of geographically related isolates was from Wales_1 (cluster 2 with a maximum of 15 SNPs between isolates).
There was a further cluster of isolates from patient and environmental isolates from London_26 and some of those from London_28 (subcluster 3a); there were patient transfers (e.g., P37 and P42) between these two hospitals. Two of the patients were babies that had not been in other hospitals, and the epidemiological evidence strongly supports this cluster as did PFGE and VNTR analysis, which had indicated that these isolates were distinguishable from those from London_17. This was a subset within a much broader cluster (cluster 3 with samples differing by a maximum of 52 SNPs), mainly consisting of isolates from London_17, which experienced a long-running problem with this type and had implicated the sewers as a reservoir (23, 42), but also included isolates from London_1, London_6, London_12, London_14, South East_3, and South East_10. Notably, the isolates from London_12 were separated in time from one another (collected from 2010 to 2013) but had a putative common ancestor at the root of a subcluster that we have labeled 3b.
For some hospitals with multiple isolates, there is no clustering. For example P19 and P49 from London_7 were isolated >2 years apart and lie on separate long branches, which clearly indicates independent introduction. Isolates from North West_14, North West_16, Scotland_4, and West Midlands_5 lie on long branches separated from London clusters 1 to 4. There were also some isolates from London hospitals that are similarly distinct from the main London clusters, for example, those from London_24 and P39 and P58 from London_29.
There was no obvious clustering according to infection source, with most isolates clustering according to hospital/geographical region. For example, blood isolates were distributed in clusters 1 (from London_9) and 3 (from London_17, London_14, and London_28) and in outlying positions (P46_South East_6, 12.12, which clusters near the IMP producers, and P36, West Midlands_5, VIM-2, 06.12 on a long branch at the top of the tree). Although all isolates tested were resistant to most antibiotics, with the exception of colistin, there was variability in susceptibility to amikacin and gentamicin; all cluster 1 isolates tested were susceptible to gentamicin and resistant to amikacin, while all subcluster 3a isolates were resistant to the two aminoglycosides; the IMP set was resistant to gentamicin but had intermediate susceptibility to amikacin (MIC, 16 mg/liter). However, subcluster 3b isolates showed variable susceptibility to the two aminoglycosides. Gentamicin-susceptible isolates were distributed in multiple places across the tree, some appearing to be sporadic and some located in main clusters (e.g., P40, Scotland_4, VIM-2, 09.12 (edge of cluster 2); P17, North West_14, VIM, 03.10 (North West cluster); P33, London_28, VIM-2, 02.12 (between clusters 1 and 2); P36, West Midlands_5, VIM-2, 06.12 (sporadic); and P43_1, London_9, VIM-2, 11.12 (cluster 1).
BEAST analysis (Fig. 3) using a relaxed molecular clock indicated that all the isolates were linked by a common ancestor from approximately 50 years ago (the 1960s), with confidence intervals that stretch back to 90 years, with the IMP producers and most of the MBL-negative isolates diverging at that time from the rest. The remaining isolates appeared to have a common ancestor dating back about 20 to 30 years. Interestingly, one of the VIM-producing isolates from London_7 (P49, London_7, VIM, 01.13) appeared to have diverged from other VIM producers at around this time. The isolates from the North West hospitals clustered in a branch with the West Midlands isolate from a common ancestor from approximately the early 2000s. This analysis also supported the close linkage of isolates from London_12, with all five isolates branching from a much more recent common ancestor (from around 5 years earlier [2009]). Overall, the clusters corresponded to those identified on the maximum-likelihood SNP tree, with the common ancestor for the broad cluster 3 apparently dating back to approximately 15 years earlier (late 1990s). Reassuringly, the members of each pair from the six patients from whom two isolates were included were adjacent (P8, P42, P52, and P54) or close (P43 and P51) to each other in each case, sharing a recent common branch. According to the BEAST analysis, the isolate from London_1 dating from 2007 shared a common ancestor with those in subcluster 3a from approximately 11 years previously.
FIG 3.

Bayesian evolutionary analysis sampling trees (BEAST) analysis of representatives of ST-111 from United Kingdom hospitals collected between 2004 and 2014 calculated using a relaxed molecular clock. Isolates and labeling are as in Fig. 2. The horizontal position of isolates on the tree is according to the date that they were isolated; calculated times for putative common ancestors (at the nodes) can be read from the scale (in years). Time zero is that of the most recent isolate (April 2014). Horizontal bars indicate degrees of confidence.
Accessory gene profiling (Fig. 4 and 5) shows clear differences in accessory gene content among these isolates with clear blocks of genes found exclusively in some clusters. Note the large block of genes associated with cluster 3 (which includes subclusters 3a and 3b) and not found in clusters 1 and 2. Note also the clear distinction of the isolate from P19 (from London_7, at the bottom of Fig. 4) from the rest of the set with a large set of genes present in this isolate that are not found in most of the other isolates. These blocks of genes contain many encoding hypothetical proteins and genes commonly associated with phages. The full list of genes present or absent in each isolate is available in Table S3 in the supplemental material.
FIG 4.
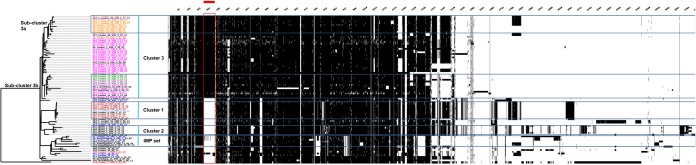
Presence/absence of accessory genes in representatives of ST-111 from United Kingdom hospitals collected between 2005 and 2014 obtained from an iterative-BLAST approach. Vertical black bars represent presence of accessory genes displayed against a phylogenetic tree. A cluster of genes distinguishing the cluster 3 isolates is marked with the red bar and box. A full list of the genes presented is available (see Table S3 in the supplemental material).
FIG 5.

Snapshot of accessory gene comparison for some genes of a subset of 24 isolates from various clusters from CD-HIT analysis. Dark squares indicate isolates with particular sets of genes, the number of genes in the set being indicated in the bottom row, e.g., the starred set indicates 44 genes exclusive to clusters 1 and 2 isolates among the set.
Analysis of genes exclusive to cluster 1 showed a high number coding for hypothetical proteins, but many associated with bacteriophage, often closely matching (≥95% similarity) genes from bacteriophage F10 (http://www.ncbi.nlm.nih.gov/nuccore/DQ163912) (e.g., nucleotides (nt) 27178-27570, 27690-27493, and 29414-29749) described by Kwan et al. (43). They also included ATP binding genes (e.g., P797_09120 of GenBank accession number CP008739.2), a prepilin-like leader sequence (T222_04300 of GenBank accession number CP006982.1), and genes for transposase and integrase hypothetical proteins (e.g., T226_04070 of GenBank accession number CP006985.1).
Other antibiotic resistance genes.
The WGS identified the intrinsic OXA-50-like gene (variant blaOXA-50d), blaPAO, aph(3)-lb, and catB7 in most, but not all, isolates tested (see Table S1 in the supplemental material). The other antibiotic resistance genes found were consistent with the VIM or IMP containing integrons found by conventional sequencing (L. Wright, unpublished data). All but one (P49, London_7) of the VIM isolates carried blaVIM-2 in In59-like structures consisting of two aminoglycoside resistance gene cassettes (aacA29a and aacA29b) and blaVIM-2; the WGS identified the aacA29 in most, but not all, cases. The IMP-only isolates clustered according to the IMP variant (IMP-1 or IMP-13) by WGS; this fitted with the distinct integron structures found by conventional sequencing. Interestingly, most of the MBL-negative isolates (from patients P39, P41, P46, P58, and P64 and environmental isolate E11) carried PSE-1 genes, together with aacA4 (aac6′Ib) and aadA2, which were linked in the same contig in assemblies made of the sequences and are likely all to be on the same integron, as described by Peña et al. (44), as did all those that carried IMP-13, the only exception being the isolate from P28 from London_17.
DISCUSSION
In this study, three complementary methods of analysis were used, each providing information that the others did not. The SNP analysis generated the phylogenetic tree, which showed that P. aeruginosa ST-111 in the United Kingdom consists of a number of distinct clusters, albeit with evidence for some spread within and between hospitals; this was consistent with the epidemiological information, where it was available. It is encouraging that such resolution is possible among these closely related isolates belonging to an international lineage and strongly indicates that WGS should play an increasing role in cross-infection investigations of such clones in real time. While BEAST analysis is computationally demanding, it can give enhanced information for isolates spanning a number of years and, crucially, can indicate timelines for putative common ancestors that link the more recent isolates. This indicated a common ST-111 ancestor dating back ∼50 years, with the main VIM-2 group having a common ancestor from 20 to 30 years ago. Correspondingly, the first description of VIM-2 in P. aeruginosa was from a blood culture taken in 1996 (45). Finally, the accessory gene profiling supported the SNP and BEAST analyses in showing blocks of genes associated with particular clusters, supporting that they are distinct from other clusters and that the isolates within each cluster are linked to one another. Accessory gene profiling identified genes that were specific to individual clusters (e.g., the bacteriophage genes associated with cluster 1), and these may be useful as PCR targets for rapidly identifying isolates within a cluster or for assessing cross-infection based on the presence or absence of a subset of these genes. We have previously found such an approach helpful in providing discrimination among ST-27 isolates (22).
Most isolates with no MBLs or with MBLs other than VIM-2 clustered apart from those with VIM; the only exceptions was P28, London_17, 02.11, which differed from other London_17 isolates in lacking VIM-2 but, nevertheless, was similarly resistant to carbapenems, and P49, London_7, VIM-2, 01.13, which also carried IMP-18. The fact that there were representatives with various MBLs or none at all is typical of an international clone. The WGS analysis clearly showed that one can consider isolates with different MBLs as only distantly related to one another.
Among the VIM set, cluster 1 was clearly separated from the rest of the isolates (as was cluster 2). The tight clustering of the isolates in this cluster suggests that a single introduction resulted in a short outbreak that was effectively managed by the hospitals concerned, at least during the main outbreak, which occurred between November 2012 and February 2013. The involvement of two hospitals (London_9 and London_11) during this time is explained by the fact that there were shared patients between them. However, later isolates from London_6 (P60, London_6, VIM-2, 11.13) and London_9 (P62, London_9, 01.14) that fall within this cluster as well as one from London_25 (P63, London_25, VIM-2, 03.14) that falls on the edge of it during late 2013/early 2014 indicate that this has extended to further hospitals and may have reemerged in one of the original hospitals.
In contrast, the extended isolation of ST-111 experienced by London_17, which dates from 2005 and later (although our set only includes those from 2009 and later) and the broad clustering of isolates from this hospital and others in cluster 3, suggests a wide reservoir of the organism, likely the sewer system, giving rise to a long-running outbreak, which was extremely difficult to control (23). Intervention measures to contain this kind of scenario are likely to differ considerably from those associated with a single introduction outbreak. We therefore intend to carry out WGS on future representatives of ST-111 as we receive them so that we can see where they cluster on the tree, which may help to shed light not only on transmission patterns but also on the measures necessary to contain them.
Our set included six patient pairs, which generally showed relatively little variation between the first and second isolate in each case. A study among multiple isolates from patients with cystic fibrosis focusing on hypermutators (which have a mutation rate approximately 40-fold higher than those in normal populations) found a mutation rate of approximately 100 SNPs per year (35) and extensive within-patient diversification of populations, which were composed of a number of different sublineages. Without diversification, we would therefore have expected approximately 10 SNPs between isolates from the same patient separated by 4 years. The study by Synder et al. (4) on representatives of the Nottingham strain estimates an accumulation of SNPs at a rate of 1 every 4 to 5 months. Based on this estimate, we would have expected to see 12 SNPs between isolates separated by 4 years. For the isolates from patient P8 separated by this time period, we found 18 SNPs, which is within the expected range, especially when one considers that there almost certainly were variants within the population. However, the SNPs we identified may be an underestimate of the total since some SNP positions may not have been included in our analysis due to technical reasons such as low coverage for some isolates.
There were marked differences in the accessory genome among the set of isolates, which is perhaps surprising given the relatively short time period over which they were isolated and given that the BEAST analysis indicates that they all share an ancestor dating back only ∼25 to 90 years. However, this is an international lineage, and there are many opportunities for introductions into United Kingdom hospitals from other countries, which may well have increased the diversity seen. Certainly, the isolate from P19 (from London_7) has a very different accessory gene content from the rest of the isolates, suggesting a different origin. In contrast, the VIM-2 gene seems to be extremely stable in the main VIM clade, with only one representative lacking it. The diversity of accessory genes found among representatives of this high-risk clone may reflect loss of the clustered, regularly interspaced short palindromic repeats system (CRISPR-Cas), which acts as a bacterial defense system against foreign DNA, such as phage DNA and plasmids, as a result of phage-derived anti-CRISPR proteins, encoded by elements that may also be found in mobile genetic elements in P. aeruginosa (46). Multidrug-resistant isolates of Enterococcus faecalis and P. aeruginosa are associated with compromised CRISPR-cas genome defense systems, making them better able to acquire elements carrying antibiotic resistance genes (47, 48); this would also explain the marked accessory genome diversity among the set. We searched the sequences of isolates in this study for subtype I-F (cas1, cas3,csy1, csy2, csy3, and csy4) and subtype I-E (cas2, cas1, cas6e, cas5, cas7, cse2, and cas3) genes and did not find them in any of the isolates (L. Wright and M. Doumith, unpublished data).
This investigation was carried out amid concerns that spread had occurred, particularly among London hospitals, of a sequence type associated with MBLs that had caused major, long-running problems in at least one London hospital. The WGS analysis revealed a mixed picture with clearly separated outbreaks in some cases but evidence of spread within and between hospitals in others. There were cases of single introductions (e.g., P49) where no onward transmission was observed, at least among the isolates in our set. There was a case of a short-lived outbreak probably deriving from a single introduction (cluster 1). There was also an example of a large group of related samples from one hospital (London_17), which exhibited considerable diversity (maximum of 35 SNPs between any pair of London_17 samples within the cluster), that was potentially the result of an environmental source seeding human infection multiple times. The route by which this potential environmental contamination, presumably originating from patients, is reaching further patients may involve periodic blockage of hospital waste systems. Consequent backflow of contaminated materials into the clinical environment then exposes patients to antimicrobial-resistant organisms, such as Pseudomonas. Effective intervention requires the implementation of measures to prevent hospital waste system blockage (23, 42).
Carrying out real-time analysis on future isolates and adding them to the tree generated here should inform future epidemiological investigations of this international lineage and may shed light on the interventions required to contain them. Analysis of international lineages such as ST-111 requires the enhanced resolution provided by whole-genome based analyses coupled with a consideration of the epidemiological information that may, or may not, link the isolates concerned.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to colleagues in United Kingdom hospital laboratories for submitting these isolates to the reference laboratory and to staff in AMRHAI for carrying out typing and resistance determinations on them. We are indebted to consultant medical microbiologists in the hospitals concerned who have discussed the results with us and provided epidemiological details. We thank Jack Turton for carrying out the CD-HIT analysis of the accessory genome and for generating Fig. 5.
PHE's AMRHAI Reference Unit has received financial support for conference attendance, lectures, research projects, or contracted evaluations from numerous sources, including Achaogen Inc., Allecra Antiinfectives GmbH, Amplex, AstraZeneca UK Ltd., Becton Dickinson Diagnostics, The British Society for Antimicrobial Chemotherapy (BSAC), Cepheid, Check-Points B.V., Cubist Pharmaceuticals, Department of Health, Food Standards Agency, GlaxoSmithKline Services Ltd., Henry Stewart Talks, IHMA Ltd., Merck Sharp & Dohme Corp., Meiji Seika Kaisha Ltd., Momentum Biosciences Ltd., Nordic Pharma Ltd., Norgine Pharmaceuticals, Rempex Pharmaceuticals Ltd., Rokitan GmbH, Smith & Nephew UK Ltd., Trius Therapeutics, VenatoRx, and Wockhardt Ltd.
This study was funded by Public Health England.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.00505-15.
REFERENCES
- 1.Woodford N, Turton JF, Livermore DM. 2011. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35:736–755. doi: 10.1111/j.1574-6976.2011.00268.x. [DOI] [PubMed] [Google Scholar]
- 2.Mulet X, Cabot G, Ocampo-Sosa AA, Domínguez MA, Zamorano L, Juan C, Tubau F, Rodríguez C, Moyà B, Peña C, Martínez-Martínez L, Oliver A; Spanish Network for Research in Infectious Diseases (REIPI). 2013. Biological markers of Pseudomonas aeruginosa epidemic high-risk clones. Antimicrob Agents Chemother 57:5527–5535. doi: 10.1128/AAC.01481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snitkin ES, Zelazny AM, Thomas PJ, Stock F; NISC Comparative Sequencing Program Group, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med 4:148ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder LA, Loman NJ, Faraj LA, Levi K, Weinstock G, Boswell TC, Pallen MJ, Ala'Aldeen DA. 2013. Epidemiological investigation of Pseudomonas aeruginosa isolates from a six-year-long hospital outbreak using high-throughput whole genome sequencing. Euro Surveill 18(42): pii=20611 http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20611. [DOI] [PubMed] [Google Scholar]
- 5.Le VT, Diep BA. 2013. Selected insights from application of whole-genome sequencing for outbreak investigations. Curr Opin Crit Care 19:432–439. doi: 10.1097/MCC.0b013e3283636b8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castanheira M, Deshpande LM, Costello A, Davies TA, Jones RN. 2014. Epidemiology and carbapenem resistance mechanisms of carbapenem-non-susceptible Pseudomonas aeruginosa collected during 2009-11 in 14 European and Mediterranean countries. J Antimicrob Chemother 69:1804–1814. doi: 10.1093/jac/dku048. [DOI] [PubMed] [Google Scholar]
- 7.Cornaglia G, Giamarellou H, Rossolini GM. 2011. Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis 11:381–393. doi: 10.1016/S1473-3099(11)70056-1. [DOI] [PubMed] [Google Scholar]
- 8.Poirel L, Bonnin RA, Nordmann P. 2012. Genetic support and diversity of acquired extended-spectrum β-lactamases in Gram-negative rods. Infect Genet Evol 12:883–893. doi: 10.1016/j.meegid.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Pasteran F, Faccone D, Gomez S, De Bunder S, Spinelli F, Rapoport M, Petroni A, Galas M, Corso A. 2012. Detection of an international multiresistant clone belonging to sequence type 654 involved in the dissemination of KPC-producing Pseudomonas aeruginosa in Argentina. J Antimicrob Chemother 67:1291–1293. doi: 10.1093/jac/dks032. [DOI] [PubMed] [Google Scholar]
- 10.Wright LL, Turton JF, Livermore DM, Hopkins KL, Woodford N. 2015. Dominance of international “high-risk clones” among metallo-β-lactamase-producing Pseudomonas aeruginosa in the United Kingdom. J Antimicrob Chemother 70:103–110. doi: 10.1093/jac/dku339. [DOI] [PubMed] [Google Scholar]
- 11.Pirnay JP, Bilocq F, Pot B, Cornelis P, Zizi M, Van Eldere J, Deschaght P, Vaneechoutte M, Jennes S, Pitt T, De Vos D. 2009. Pseudomonas aeruginosa population structure revisited. PLoS One 4:e7740. doi: 10.1371/journal.pone.0007740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edalucci E, Spinelli R, Dolzani L, Riccio ML, Dubois V, Tonin EA, Rossolini GM, Lagatolla C. 2008. Acquisition of different carbapenem resistance mechanisms by an epidemic clonal lineage of Pseudomonas aeruginosa. Clin Microbiol Infect 14:88–90. doi: 10.1111/j.1469-0691.2007.01874.x. [DOI] [PubMed] [Google Scholar]
- 13.Liakopoulos A, Mavroidi A, Katsifas EA, Theodosiou A, Karagouni AD, Miriagou V, Petinaki E. 2013. Carbapenemase-producing Pseudomonas aeruginosa from central Greece: molecular epidemiology and genetic analysis of class I integrons. BMC Infect Dis 13:505. doi: 10.1186/1471-2334-13-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koutsogiannou M, Drougka E, Liakopoulos A, Jelastopulu E, Petinaki E, Anastassiou ED, Spiliopoulou I, Christofidou M. 2013. Spread of multidrug-resistant Pseudomonas aeruginosa clones in a university hospital. J Clin Microbiol 51:665–668. doi: 10.1128/JCM.03071-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van der Bij AK, Van der Zwan D, Peirano G, Severin JA, Pitout JD, Van Westreenen M, Goessens WH; MBL-PA Surveillance Study Group. 2012. Metallo-β-lactamase-producing Pseudomonas aeruginosa in the Netherlands: the nationwide emergence of a single sequence type. Clin Microbiol Infect 18:E369–E372. doi: 10.1111/j.1469-0691.2012.03969.x. [DOI] [PubMed] [Google Scholar]
- 16.Gilarranz R, Juan C, Castillo-Vera J, Chamizo FJ, Artiles F, Alamo I, Oliver A. 2013. First detection in Europe of the metallo-β-lactamase IMP-15 in clinical strains of Pseudomonas putida and Pseudomonas aeruginosa. Clin Microbiol Infect 19:E424–E427. doi: 10.1111/1469-0691.12248. [DOI] [PubMed] [Google Scholar]
- 17.Libisch B, Watine J, Balogh B, Gacs M, Muzslay M, Szabó G, Füzi M. 2008. Molecular typing indicates an important role for two international clonal complexes in dissemination of VIM-producing Pseudomonas aeruginosa clinical isolates in Hungary. Res Microbiol 159:162–168. doi: 10.1016/j.resmic.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Samuelsen O, Toleman MA, Sundsfjord A, Rydberg J, Leegaard TM, Walder M, Lia A, Ranheim TE, Rajendra Y, Hermansen NO, Walsh TR, Giske CG. 2010. Molecular epidemiology of metallo-beta-lactamase-producing Pseudomonas aeruginosa isolates from Norway and Sweden shows import of international clones and local clonal expansion. Antimicrob Agents Chemother 54:346–352. doi: 10.1128/AAC.00824-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sardelic S, Bedenic B, Colinon-Dupuich C, Orhanovic S, Bosnjak Z, Plecko V, Cournoyer B, Rossolini GM. 2012. Infrequent finding of metallo-β-lactamase VIM-2 in carbapenem-resistant Pseudomonas aeruginosa strains from Croatia. Antimicrob Agents Chemother 56:2746–2749. doi: 10.1128/AAC.05212-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duljasz W, Gniadkowski M, Sitter S, Wojna A, Jebelean C. 2009. First organisms with acquired metallo-beta-lactamases (IMP-13, IMP-22, and VIM-2) reported in Austria. Antimicrob Agents Chemother 53:2221–2222. doi: 10.1128/AAC.01573-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hentschke M, Goritzka V, Campos CB, Merkel P, Ilchmann C, Lellek H, Scherpe S, Aepfelbacher M, Rohde H. 2011. Emergence of carbapenemases in Gram-negative bacteria in Hamburg, Germany. Diagn Microbiol Infect Dis 71:312–315. doi: 10.1016/j.diagmicrobio.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 22.Martin K, Baddal B, Mustafa N, Perry C, Underwood A, Constantidou C, Loman N, Kenna DT, Turton JF. 2013. Clusters of genetically similar isolates of Pseudomonas aeruginosa from multiple hospitals in the UK. J Med Microbiol 62:988–1000. doi: 10.1099/jmm.0.054841-0. [DOI] [PubMed] [Google Scholar]
- 23.Witney AA, Gould KA, Pope CF, Bolt F, Stoker NG, Cubbon MD, Bradley CR, Fraise A, Breathnach AS, Butcher PD, Planche TD, Hinds J. 2014. Genome sequencing and characterization of an extensively drug-resistant sequence type 111 serotype O12 hospital outbreak strain of Pseudomonas aeruginosa. Clin Microbiol Infect 20:O609–O618. doi: 10.1111/1469-0691.12528. [DOI] [PubMed] [Google Scholar]
- 24.Cholley P, Thouverez M, Hocquet D, van der Mee-Marquet N, Talon D, Bertrand X. 2011. Most multidrug-resistant Pseudomonas aeruginosa isolates from hospitals in eastern France belong to a few clonal types. J Clin Microbiol 49:2578–2583. doi: 10.1128/JCM.00102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vatcheva-Dobrevska R, Mulet X, Ivanov I, Zamorano L, Dobreva E, Velinov T, Kantardjiev T, Oliver A. 2013. Molecular epidemiology and multidrug resistance mechanisms of Pseudomonas aeruginosa isolates from Bulgarian hospitals. Microb Drug Resist 19:355–361. doi: 10.1089/mdr.2013.0004. [DOI] [PubMed] [Google Scholar]
- 26.Correa A, Montealegre MC, Mojica MF, Maya JJ, Rojas LJ, De La Cadena EP, Ruiz SJ, Recalde M, Rosso F, Quinn JP, Villegas MV. 2012. First report of a Pseudomonas aeruginosa isolate coharboring KPC and VIM carbapenemases. Antimicrob Agents Chemother 56:5422–5423. doi: 10.1128/AAC.00695-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turton JF, Turton SE, Yearwood L, Yarde S, Kaufmann ME, Pitt TL. 2010. Evaluation of a nine-locus variable-number tandem-repeat scheme for typing of Pseudomonas aeruginosa. Clin Microbiol Infect 16:1111–1116. doi: 10.1111/j.1469-0691.2009.03049.x. [DOI] [PubMed] [Google Scholar]
- 28.Ellington MJ, Kistler J, Livermore DM, Woodford N. 2007. Multiplex PCR for rapid detection of genes encoding acquired metallo-β-lactamases. J Antimicrob Chemother 59:321–322. [DOI] [PubMed] [Google Scholar]
- 29.Mushtaq S, Irfan S, Sarma JB, Doumith M, Pike R, Pitout J, Livermore DM, Woodford N. 2011. Phylogenetic diversity of Escherichia coli strains producing NDM-type carbapenemases. J Antimicrob Chemother 66:2002–2005. doi: 10.1093/jac/dkr226. [DOI] [PubMed] [Google Scholar]
- 30.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Cornell University Library. arXiv:1303.3997. [Google Scholar]
- 32.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 34.Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees, p 1–8. In Gateway Computing Environments Workshop (GCE). IEEE, New York, NY. [Google Scholar]
- 35.Feliziani S, Marvig RL, Luján AM, Moyano AJ, Di Rienzo JA, Krogh Johansen H, Molin S, Smania AM. 2014. Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet 10:e1004651. doi: 10.1371/journal.pgen.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 39.Witney AA, Marsden GL, Holden MT, Stabler RA, Husain SE, Vass JK, Butcher PD, Hinds J, Lindsay JA. 2005. Design, validation, and application of a seven-strain Staphylococcus aureus PCR product microarray for comparative genomics. Appl Environ Microbiol 71:7504–7514. doi: 10.1128/AEM.71.11.7504-7514.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thuong M, Arvaniti K, Ruimy R, de la Salmonière P, Scanvic-Hameg A, Lucet JC, Régnier B. 2003. Epidemiology of Pseudomonas aeruginosa and risk factors for carriage acquisition in an intensive care unit. J Hosp Infect 53:274–282. doi: 10.1053/jhin.2002.1370. [DOI] [PubMed] [Google Scholar]
- 42.Breathnach AS, Cubbon MD, Karunaharan RN, Pope CF, Planche TD. 2012. Multidrug-resistant Pseudomonas aeruginosa outbreaks in two hospitals: association with contaminated hospital waste-water systems. J Hosp Infect 82:19–24. doi: 10.1016/j.jhin.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Kwan T, Liu J, Dubow M, Gros P, Pelletier J. 2006. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J Bacteriol 188:1184–1187. doi: 10.1128/JB.188.3.1184-1187.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peña C, Suarez C, Tubau F, Juan C, Moya B, Dominguez MA, Oliver A, Pujol M, Ariza J. 2009. Nosocomial outbreak of a non-cefepime-susceptible ceftazidime-susceptible Pseudomonas aeruginosa strain overexpressing MexXY-OprM and producing an integron-borne PSE-1 betta-lactamase. J Clin Microbiol 47:2381–2387. doi: 10.1128/JCM.00094-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poirel L, Naas T, Nicolas D, Collet L, Bellais S, Cavallo JD, Nordmann P. 2000. Characterization of VIM-2, a carbapenem-hydrolyzing metallo-β-lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob Agents Chemother 44:891–897. doi: 10.1128/AAC.44.4.891-897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pawluk A, Bondy-Denomy J, Cheung VH, Maxwell KL, Davidson AR. 2014. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-cas system of Pseudomonas aeruginosa. mBio 5(2):e00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palmer KL, Gilmore MS. 2010. Multidrug-resistant enterococci lack CRISPR-cas. mBio 1(4):e00227–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kos VN, Déraspe M, McLaughlin RE, Whiteaker JD, Roy PH, Alm RA, Corbeil J, Gardner H. 2015. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother 59:427–436. doi: 10.1128/AAC.03954-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.