

Summary
There has been exponential growth in the number of membrane protein structures determined. Nevertheless, these structures are usually resolved in the absence of their lipid environment. Coarse-grained molecular dynamics (CGMD) simulations enable insertion of membrane proteins into explicit models of lipid bilayers. We have automated the CGMD methodology, enabling membrane protein structures to be identified upon their release into the PDB and embedded into a membrane. The simulations are analyzed for protein-lipid interactions, identifying lipid binding sites, and revealing local bilayer deformations plus molecular access pathways within the membrane. The coarse-grained models of membrane protein/bilayer complexes are transformed to atomistic resolution for further analysis and simulation. Using this automated simulation pipeline, we have analyzed a number of recently determined membrane protein structures to predict their locations within a membrane, their lipid/protein interactions, and the functional implications of an enhanced understanding of the local membrane environment of each protein.
Graphical Abstract
Highlights
-
•
A simulation pipeline for predicting the location of a membrane protein in a bilayer
-
•
A protocol for identifying novel membrane protein structures in the PDB
-
•
Analysis of lipid binding sites and local bilayer deformation by membrane proteins
-
•
Functional implications from enhanced understanding of local membrane environments
Stansfeld et al. present MemProtMD, an automated pipeline for molecular simulations to insert membrane protein structures into explicit lipid bilayer membranes. An analysis of simulations of all known membrane protein structures reveals details of protein-induced membrane deformation and rules for residue distribution, and facilitates refinement of membrane proteins.
Introduction
Membrane proteins play a key role in the biology of the cell, accounting for ∼25% of genes. The structural biology of membrane proteins continues to progress, with the experimental determination of more than 2,000 structures. However, in the majority of cases determination of a membrane structure by X-ray diffraction, solution nuclear magnetic resonance (NMR), or, more recently, single-particle electron cryomicroscopy (cryo-EM) does not reveal the structure of protein in a membrane, but rather in a crystal lattice (albeit sometimes with lipids or detergents bound) or in a micelle or bicelle. Although structural data on the lipid interactions of membrane proteins are available (Killian and von Heijne, 2000; Lee, 2011; Palsdottir and Hunte, 2004), in the face of an exponential growth of the number of determined structures, there is a pressing need for computational methods to provide a general and accurate approach to modeling membrane protein/lipid bilayer interactions.
There are a number of methods, especially electron microscopy from two-dimensional crystals and solid-state NMR, which enable exploration of membrane proteins in their native membrane environment. It is also possible to use computational approaches to locate and orient a membrane protein relative to a simplified model of a lipid bilayer membrane environment, e.g., OPM (Lomize et al., 2012) and PDBTM (Kozma et al., 2013). However, these latter approaches do not generally include explicit lipid molecules, but rather model a bilayer as a hydrophobic slab of fixed dimensions. In the context of advances in our knowledge of membrane lipidomics (Coskun and Simons, 2011), it is important to develop accurate and reliable computational approaches that treat the lipid bilayer environment explicitly. This is of especial importance, as a number of experimental (Drachmann et al., 2014) and computational (Arnarez et al., 2013) studies have highlighted the structural and functional importance of the individual lipid molecules in the local environment of a membrane protein.
Molecular dynamics (MD) simulations allow the in silico reconstitution of membrane proteins into a bilayer environment. In particular, coarse-grained (CG) MD simulations (in which groups of ∼4 atoms are represented by a single particle or bead) allow one to self-assemble a lipid bilayer around a given membrane protein (Bond and Sansom, 2006; Marrink and Tieleman, 2013; Scott et al., 2008). This can be combined with conversion to atomistic (AT) resolution to enable more detailed MD simulations of, e.g., conformational dynamics in relation to function (Stansfeld and Sansom, 2011). It is therefore timely to develop a high-throughput simulation methodology to apply to all determined structures of membrane proteins.
Here, we describe a robust and accurate protocol for systematically identifying membrane protein structures in the PDB (Berman et al., 2000) and for embedding them in an explicit phosphatidylcholine (PC) bilayer. This enables determination of the dynamic interactions of membrane proteins with a lipid bilayer. All data produced by this automated pipeline are deposited online. A number of examples of the utility of simulated lipid-protein interactions are explored, providing insights into the relationship between membrane protein structure and function.
Results and Discussion
An Automated High-Throughput Pipeline for Membrane Proteins
MemProtMD provides a high-throughput computational pipeline for (re-)insertion of membrane protein structures into a detailed, atomic resolution model of their membrane environment. The whole process is automated, from the detection of membrane protein structures in the PDB upon its release, through preparation and running of CGMD simulations to their subsequent analysis, conversion to ATMD models, and deposition in the freely and publicly accessible MemProtMD database (http://sbcb.bioch.ox.ac.uk/memprotmd; Figure 1). The latter currently makes available both CG and AT models of the protein embedded in a phospholipid bilayer.
Figure 1.

The MemProtMD Pipeline for Inserting Membrane Proteins into Bilayers
The first step is to detect the protein structures from the PDB, here shown for the P2X4 receptor (PDB: 4DW0, trimeric biological assembly as annotated in the PDB). The second is to set up a lipid, water, and protein simulation system. CG simulations are then run (1 μs duration) to assemble and equilibrate a bilayer around each membrane protein structure. The CG simulation system is then converted to atomic resolution, allowing detailed analysis of lipid bilayer/protein interactions. See also Figure S1.
For the existing PDB, a keyword search for “membrane” identifies approximately 8,000 structures. However, many of these structures are of extra-membranous domains of integral membrane proteins, lacking the transmembrane (TM) portion, or are of peripheral proteins that have a degree of membrane association. Furthermore, it was apparent that this simple keyword search misses many well-known integral membrane protein structures. We therefore designed methods for the detection of α-helical and β-barrel membrane proteins instead of a keyword search.
For membrane proteins with α-helical TM domains, we use Octopus (Viklund and Elofsson, 2008) to predict TM helices based on the sequence of the protein, followed by a check to see if the TM elements in the structure are indeed α-helical and membrane accessible (Figure S1A). For possible β-barrel membrane proteins the length, accessibility to the membrane, and hydrophobicity (based on an OMPLA-derived hydrophobicity scale; Moon and Fleming, 2011) of the β strands is used to identify integral membrane proteins (Figure S1B). Combining these two methods captured a dataset of 2,294 integral membrane proteins, 320 of which were not present in the 8,000 proteins identified on the basis of the “membrane” keyword search alone (April 2015). To the best of our knowledge, this captures the vast majority of integral membrane protein structures, with false negatives forming less than 1% of the structures, as estimated by cross-referencing with the other online repositories (OPM, mpstruc, and PDBTM). A similar value is observed for false-positive structures, which are also eliminated during the quality control step (see below).
By using this approach, newly released membrane protein structures are identified upon their weekly release from the PDB. Simulation setup and an initial 0.1 μs of CGMD simulation, to embed the protein in a solvated bilayer, are ready within a few hours of release of the coordinates. This also acts as a quality control step, with systems that do not correctly assemble flagged for attention. This is observed as either a failure of the protein to insert or as irregular bilayer formation. This process can also identify false positives from the PDB screening procedure.
At the end point of a 1 μs simulation, a well-sampled model of the protein/bilayer/solvent interactions is returned from our local cluster for subsequent analysis and deposition. Of the identified structures, 2,219 are complete. This equates to more than 2 ms of total simulation time. At present, approximately 50 structures (∼2% of the total dataset) have not reached the end point of their 1 μs simulation.
Within the 2,294 identified structures there are 1,991 α-helical and 303 β-barrel proteins in MemProtMD, corresponding to 503 α-helical and 125 β-barrel unique membrane protein structures (Figure 2A; Figure S2). This updates the 533 unique structures, as annotated by mpstruc (http://blanco.biomol.uci.edu/mpstruc/) (White, 2009). Each entry in the dataset provides the coordinates of the first biological assembly in the PDB of the protein embedded in a PC lipid bilayer in both CG and AT representations (Figure 2B). The CG simulations associated with each entry (1 μs of CGMD in a bilayer) provide the basis for initial analysis of the protein/lipid interactions of a given membrane protein. Such interactions may also provide insights into membrane protein function, especially when a protein is known to interact with lipid-like ligands. Each entry also provides an initial setup for more detailed ATMD simulations to explore conformational dynamics of membrane proteins in relation to their mechanism of action. As a whole, the dataset provides the basis for more global analyses of membrane protein/bilayer interactions, including the propensities of each amino acid side-chain type to be located at a given position relative to the bilayer. In turn, local deviations from the global average distribution of side chains can provide pointers to key functional and structural aspects of the structure and function of the membrane protein. Finally, simulations based on individual entries may aid in evaluation and refinement of membrane protein structures.
Figure 2.

A Database of Membrane Proteins in Phospholipid Bilayers
(A) Snapshots of the final (i.e., 1 μs) configurations of 2,294 membrane proteins in a phospholipid (PC) bilayer.
(B) Zoomed-in representation of the 5-HT3 ligand-gated ion channel (PDB: 4PIR; pentameric asymmetric unit and biological assembly) in a PC bilayer. Lipid headgroups are shown as spheres representing the choline (blue), phosphate (red), and glycerol (yellow) particles of the CG model. See also Figure S2.
Identifying Lipid and Lipid-like Ligand Binding Sites
MemProtMD allows one to use simulation data to identify preferential lipid interaction sites on the surface of the protein. This can be done is by analyzing the relative frequency (i.e., an estimate of the probability) that a certain lipid CG-particle type occurs in a given region in the vicinity of the protein. To compare these predictions experimentally, we have selected three examples (Figure 3).
Figure 3.

Identifying Lipid Interaction Sites
Lipid binding sites for three membrane proteins, identified as regions with a high frequency of occurrence of the phosphate particles of PC molecules. In each case the corresponding crystal structure and MemProtMD simulation snapshot are shown on the left- and right-hand side, respectively. The relative dimensions and redness of the densities reflect the frequency of binding, with those sites that are repeatedly sampled larger in size and redder in color.
(A) Aquaporin Aqp0 (PDB: 3M9I) comparing the phosphorus atoms of the headgroups of the experimentally resolved PE lipids (orange spheres) with the phosphate particles from a CG simulation in a PC bilayer. While the phosphate binding sites agree well with the cryo-EM structures (as indicated by the arrows), the phosphates show preference for some sites over others, with a number of the more prominent sites residing at and either side of the subunit interfaces. Note that the biological assembly in the PDB is octameric, so the biological tetramer was regenerated using PISA. See also Figure S5.
(B) Cardiolipin molecules bound to the ADP/ATP carrier (AAC; PDB: 4C9G) compared with phosphates from a CG simulation. The level to which the phosphates bind reflects the composition of the binding site and suggests the most favorable interactions with pseudo-subunit X, then Y, then Z. Both yeast and bovine homologs of AAC show differences in the phosphate densities for each binding site.
(C) A Kir3.2 channel structure (PDB: 3SYA; tetrameric biological assembly from the PDB) with four bound molecules of a short-tail analog of PIP2, again compared with phosphates from a CG simulation.
The first example is the simplest in terms of lipids, and concerns the aquaporin Aqp0 for which a high-resolution electron diffraction structure is available, which resolves a full shell of annular phosphatidylethanolamine (PE) lipids (Hite et al., 2010). In a previous simulation study of aquaporins (Stansfeld et al., 2013), we have shown that simulations using the lipid species present in the crystal reproduce the experimentally observed annulus. Here, we test whether simulations embedding the Aqp0 tetramer (PDB: 3M9I) in a simple PC lipid bilayer as used in MemProtMD can also correctly identify the lipid headgroup interaction sites. The results (Figure 3A) show that, indeed, the Aqp0 annular shell can be correctly predicted. Furthermore, displaying the regions with highest probability of occurrence of the headgroup phosphate of an interacting PC molecule reveals a close correspondence between these and the locations of the phosphate groups of bound PE molecules in the crystal structure. Thus, CGMD simulations with MemProtMD using PC molecules are able to correctly predict the sites of bound PE molecules. One should note that the PC and PE headgroups differ in that the primary amine group of the latter is available as an H-bond donor to the protein structure.
A more complex example of a bound lipid is provided by cardiolipin (CL; 1,3-bis(sn-3′-phosphatidyl)-sn-glycerol), which has two phosphates in a headgroup shared between four acyl tails, and hence has a net charge of −2, and which binds to the mitochondrial ADP/ATP carrier (AAC). The structure of the AAC has six TM helices arranged in a 3-fold pseudo-symmetrical fashion, with three bound CL molecules. Simulations using MemProtMD with either the yeast (PDB: 4C9G; Figure 3B) (Ruprecht et al., 2014) or the bovine (PDB: 1OKC) (Pebay-Peyroula et al., 2003) AAC reveal a high probability for the (phosphates of) PC molecules at the three binding sites for CL. Closer inspection shows that two high-probability phosphate interaction sites are seen, corresponding to the two phosphates of the CL molecule. Thus the zwitterionic PC of the simulations can reveal the binding sites of the di-anionic CL molecules. It can also reveal the relative affinity of each site, with the binding site in the first pseudo-subunit for both organisms revealing the highest density. This appears to be controlled by the sequence composition of each site. For the bovine AAC there is one high and two medium-affinity sites, while the yeast AAC has high-, medium-, and low-affinity sites. The high-affinity site appears to be conferred by an arginine at the N-terminal end of the second TM helix. The medium-affinity site has either a lysine or glutamine at this site, while the low-affinity site in the yeast structures appears to be modulated by a neighboring aspartate (Asp266) residue at the N-terminal end of the amphipathic helix.
The third example concerns a more complex anionic lipid, namely PIP2 (phosphatidylinositol 4,5-bisphosphate), the headgroup of which has a charge of −5 (if one assumes all phosphates to be ionized). Four PIP2 molecules (with truncated acyl chains) have been observed bound to the crystal structure of the tetrameric potassium channel Kir3.2 (Whorton and MacKinnon, 2013). In previous studies we have shown that simulations in the presence of PIP2 molecules are able to identify these four binding sites (Schmidt et al., 2013; Stansfeld et al., 2009). Using MemProtMD we observe that the high-probability phosphate particle sites correspond almost exactly to the experimentally determined locations of the bound PIP2 molecules (Figure 3C). Thus, simulations using simple PC lipids are able to predict the likely binding sites of the considerably more complex PIP2 lipids.
Having shown that the MemProtMD methods can detect the phosphate interaction sites of more complex phospholipids binding to membrane proteins, we note that comparable analyses of the simulation dataset in terms of probability of occurrence of choline, glycerol, and lipid tail beads can also be used to predict novel potential lipid binding sites on a wide range of membrane protein structure for which lipid-occupied (or detergent-occupied) electron density is not resolved in available crystal structures. A more demanding test is whether MemProtMD simulations are able to reveal interactions in which the lipid molecules may be considered as lipids as proxies for lipid-like substrates and/or ligands important in the functional activity of membrane proteins. We illustrate this with three examples, two of enzymes and one of a lipid scramblase (Figure 4).
Figure 4.
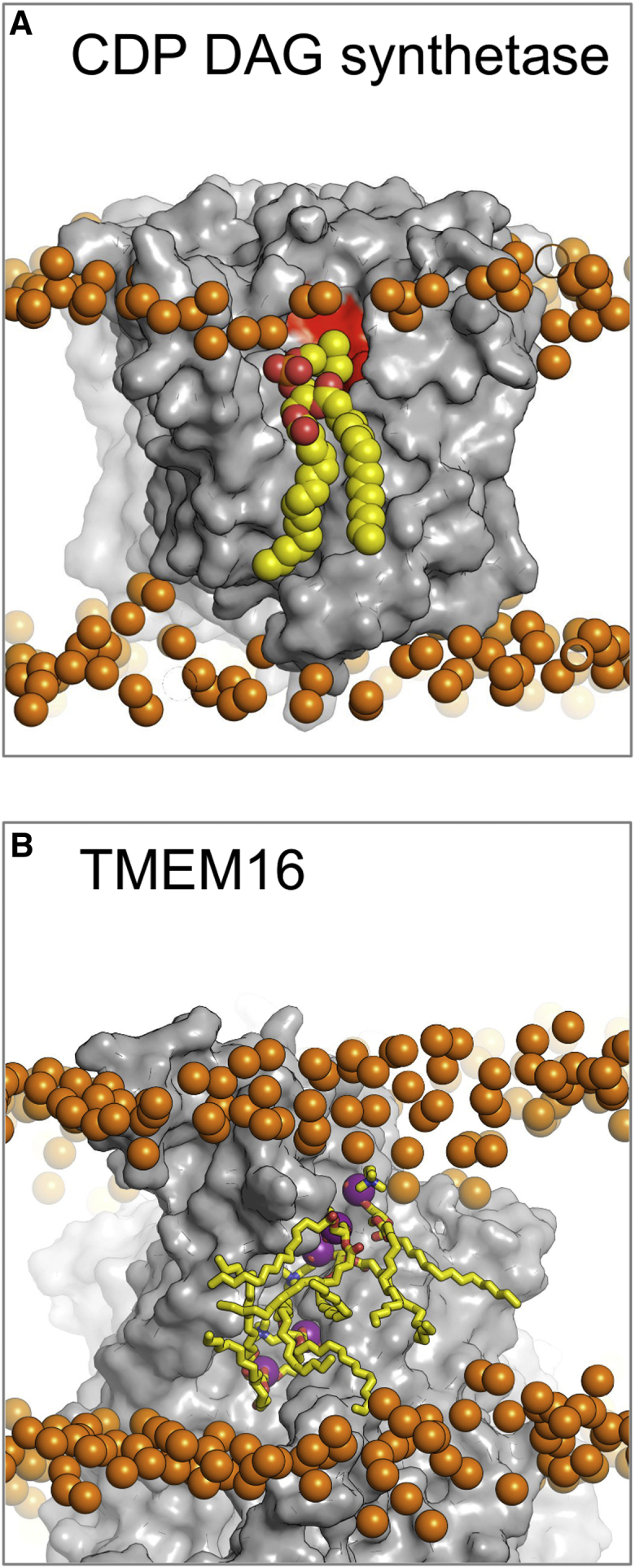
Interaction Sites for Lipid-like Ligands
(A) The CDP-DAG synthetase (PDB: 4Q2E; dimeric asymmetric unit and biological assembly in the PDB) is involved in a crucial step within the lipid biogenesis pathway. The crystal structures were resolved without bound lipid (gray surface). Here we propose the binding orientation of PC lipids (yellow spheres) to the active site (red surface). While this is not the native substrate or product for this enzyme, the binding reveals the overall orientation of lipid binding to the site.
(B) The fungal TMEM16 scramblase (PDB: 4WIS; dimeric asymmetric unit and biological assembly in the PDB) is responsible for permitting lipid flip-flop from one leaflet to the other. MemProtMD is able to predict lipid interaction sites within the permeation pathway, here illustrating five PC lipids (yellow sticks). In this case, not only do we observe lipid binding but also lipid migration, with approximately 15 lipids flipping from one leaflet to the other. Both structures illustrate a snapshot of the lipid binding after 1 μs. Phosphate atoms of the PC lipids are shown as orange spheres.
Closely interacting lipids can be identified either by taking the averaged lipid densities from the simulations or by inspecting the final 1-μs simulation snapshot. The first example is provided by the recently determined structure of CDP-DAG synthetase (PDB: 4Q2E), an enzyme central to the biogenesis of lipids. The binding site for phosphatidic acid at the active site this enzyme was suggested on the basis of its structure (Liu et al., 2014), but bound substrate or related ligands were not resolved in the crystal structure. From the MemProtMD simulations, one can identify close interactions of PC molecules with both proposed active sites of the dimeric enzyme. Both the headgroups and tails of the PC molecules fit tightly into the concave surface of the proposed binding site (Figure 4A).
The second example reveals a potentially more dynamic interaction between lipid and protein, suggesting the likely pathway for protein-induced lipid flip-flop across the membrane. This involves the interaction between PC lipids and the TMEM16A lipid scramblase (PDB: 4WIS and 4WIT). From the simulations, we are able to observe lipid headgroup interaction sites along the previously hypothesized TM surface of the protein (Figure 4B) (Brunner et al., 2014). Furthermore, the simulations reveal dynamic exchange of lipids between leaflets with approximately 15 lipids moving from the outer to the inner leaflet in 1 μs (PDB: 4WIS).
Another example of an integral membrane enzyme is provided by the membrane protease ZMPSTE24 (PDB: 4AW6) (Quigley et al., 2013). This catalyzes the cleavage of a peptide farnesylated at a cysteine residue, thus allowing the peptide to be partially inserted into the membrane. The structure of ZMPSTE24 consists of a seven TM helical barrel surrounding a water-filled intramembrane chamber, which is capped by a zinc metalloprotease domain such that the catalytic site faces into the aqueous chamber. Examination of the structure revealed fenestrations between TM helices M5 and M6, and between M1 and M2, which were suggested as likely entry and exit routes, respectively, for the farnesylated peptide. MemProtMD simulations of ZMPSTE24 reveal that the lipid tails intercalate between the helices that frame the proposed entry pathway, suggesting that the peptide may be targeted into the intramembrane chamber by the lipid-modified cysteine (Figure S3A).
Local Distortions of the Lipid Bilayer Membrane
Using an explicit lipid (and hence flexible) bilayer in the MemProtMD simulations allows one to characterize possible local membrane deformations induced by an inserted integral membrane protein. This is not readily achievable with a continuum model. The thickness of the (unperturbed) PC bilayer used in our CG simulations, measured from phosphate particle to phosphate particle, is 39.9 ± 0.6 Å. For about 75% of the membrane protein structures only minor (<2 Å) local deformations, predominantly thinning, of the simple PC bilayer are observed. However, for a number of proteins local deformation of the bilayer is observed and is likely to be of structural and/or functional importance.
In the case of the bacterial OMPs there is generally a degree of bilayer/protein mismatch, as the PC bilayer used in MemProtMD is thicker than that of a typical bacterial outer membrane (Piggot et al., 2011; Pogozheva et al., 2013). Thus, the PC bilayer usually thins to 34.6 ± 1.4 Å in the annular shell of an OMP to compensate for the lipid/protein mismatch. A clear example of this is seen for the outer membrane porin AlgE (PDB: 4AZL) (Tan et al., 2014) (Figure S3B). From this it is evident that the bilayer can readily deform to accommodate the relatively rigid OMP structure.
An example of a functionally important bilayer deformation is provided by the enzyme diacylglycerol kinase (DgkA; PDB: 3ZE5). For DgkA it is important that the active site of the enzyme is within the lipid headgroup region, so that the substrate diacylglycerol may be phosphorylated to yield phosphatidic acid (Li et al., 2013). This location is maintained by amphipathic α helices that anchor the protein to the cytoplasmic leaflet of the bilayer. In turn, this may lead to a partial (and asymmetric) mismatch between the protein and the bilayer at the opposite (periplasmic) surface of the membrane (Figure 5A), such that overall the membrane deforms by 3–4 Å. As this enzyme is stimulated by osmotic stress (Badola and Sanders, 1997), it is conceivable that the enzyme may be modulated in response to changes in membrane thickness.
Figure 5.

Local Distortions of the Lipid Bilayer
Local distortions of the bilayer induced by membrane proteins may be analyzed by evaluating the positions of the phosphate headgroups of the lipids over the course of the simulations. The extent to which the bilayer deforms can then be compared against the standard DPPC bilayer thickness of 40 Å. The deformations are shown as a mesh on a red-white-blue scale, with points greater than 20 Å from the midplane of the bilayer being colored on a blue gradient and those that are less than 20 Å colored on a red scale.
(A) For the enzyme DgkA (PDB: 3ZE5; trimeric asymmetric unit and biological assembly in the PDB), amphipathic helices anchor the protein to one (cytoplasmic) leaflet of the bilayer, such that local distortions are limited to the opposite leaflet.
(B) The ABC transporter ABCB10 (PDB: 4AYX; dimeric biological assembly in the PDB) locally perturbs the bilayer such that lipids enter the central core of the protein, suggestive of a proposed pathway for lipid flip-flop catalyzed by related ABC transporters. See also Figure S3.
In other cases, local bilayer distortions may indicate a key mechanistic involvement. For the human ABC transporter ABCB10 (Shintre et al., 2013) in an inward-facing conformation (PDB: 4AYX), the analysis of the lipid bilayer shows protein-induced deformation revealing a pathway adopted by lipids as they leave the bilayer and partition into the cavity of the ABC transporter (Figure 5B). This appears to be determined by the lipid headgroup, which interacts with the hydrophilic central cavity. Similar partitioning events occur for other ABC transporters, including those in the outward-facing state, e.g., Sav1866 (Dawson and Locher, 2006). It is likely that the observed pathways are linked to the proposed lipid flippase or drug-efflux activity of such transporters (van Meer et al., 2006). Lipid partitioning into the central cavity is also observed for the P-glycoprotein (PgP) structures (Jin et al., 2012; Li et al., 2014; Ward et al., 2013). However, with the Caenorhabditis elegans PgP structure (PDB: 4F4C), an N-terminal helical hairpin blocks one of the lateral openings and, therefore, a lipid pathway is only apparent from one side of the transporter. Furthermore, the partitioning into mouse PgP structures appears to be determined by the lipid tails, which interact with the hydrophobic cavity; therefore, for these structures the lipids do not flip within the cavity.
Rules for Membrane Proteins
The MemProtMD dataset can also be analyzed to provide a more global description of protein/bilayer interactions, yielding the probabilities of occurrence of amino acid side chains with respect to the bilayer (Figure 6; Figure S4). This extends previous analyses (Baeza-Delgado et al., 2013; Yarov-Yarovoy et al., 2006) to a significantly larger unique dataset. As expected the hydrophobic residues, Phe, Ile, Val, and Leu, are all found within the core of the membrane. Charged and larger polar residues (Arg, Lys, Asp, Asn, Glu, Gln) are found at the membrane/water interface, with Arg and Lys found with a higher frequency at the cytosolic interface than the outer leaflet, conforming with the positive-inside rule (Heijne, 1986) for α-helical membrane proteins. Small polar side chains (Ser, Thr) are found at both the interfaces and within the bilayer, the latter presumably reflecting their ability to form intra- and inter-helix H bonds within the hydrophobic core (Eilers et al., 2000). Tyr, His, and, most notably, Trp residues are observed at the boundary between the hydrophobic core and headgroup region of the bilayer, reflecting their role in anchoring membrane proteins within membranes (Landolt-Marticorena et al., 1993). Glycine residues are found throughout the bilayer, reflecting their roles in mediating TM helix packing (Russ and Engelman, 2000). Prolines also occur with high frequency within the bilayer, consistent with their formation of TM helix kinks of structural and functional importance. These rules for residue localization are also relevant to TM helix recognition and membrane insertion by the translocon (Cymer et al., 2015).
Figure 6.

Amino Acid Distributions
Frequencies of occurrence of membrane-interacting amino acid side chains within the TM domains of membrane proteins. The data are shown with respect to the bilayer for all unique α-helical membrane proteins, with the midplane of the bilayer marked as zero and the headgroup region marked by gray lines. In all instances the cytoplasmic side of the membrane (In) relates to the negative distance from the bilayer center of mass. See also Figure S4.
These distributions provide global averages for residue occurrence within a bilayer, and so can aid efforts in membrane protein modeling (Kelm et al., 2010; Nugent and Jones, 2012). They also can be used to flag, e.g., polar side chains with an unusual location relative to the bilayer (Figure 7), which are of possible functional and/or structural importance. For example, one may identify polar and charged residues within the hydrophobic core of the membrane. In some cases (Figures 7A and 7B) these correspond to Arg and Lys residues, the terminal charged groups of which snorkel toward the phosphate headgroups of the lipids, aided by local deformation of the membrane around such sites. However, there are also charged/polar residues that are located in the core of the bilayer. For ionizable residues this may indicate either an altered pKa and/or protein-protein interactions within the membrane.
Figure 7.

Unexpected Residue Locations
(A–D) The observed frequencies of occurrence of residues relative to a bilayer (see Figure 6) may be used as a guideline to identify residues in unusual locations within the membrane. In many instances where residues appear misplaced, bilayer deformations and/or snorkeling of side chains occurs to accommodate charged amino acids within the bilayer (shown as yellow spheres). In some other cases the residue (shown as red spheres) is buried deeply within the bilayer and so is unlikely to be accommodated by such mechanisms. Examples are shown for (A) Lys288 in LeuT (PDB: 4MM8; monomeric biological assembly-1 in the PDB), while Lys196 and Arg453 are able to snorkel; (B) Asn58 in MelB (PDB: 4M64; monomeric biological assembly in the PDB), while Arg52 can escape the hydrophobic core; (C) Lys 972 in SERCA (PDB: 4BEW; monomeric biological assembly in the PDB); and (D) Glu 73 in VDAC1 (PDB: 4C69; monomeric biological assembly in the PDB). In each case the considered protein is monomeric.
For example, Lys288 in TM7 of LeuT (PDB: 4MM8) is found in the central hydrophobic region of the hydrophobic bilayer, exposed to the lipid tails and at a point too distant from the interface to initiate snorkeling (Figure 7A). Functional studies indicate that a K288A mutant has enhanced transport activity (Piscitelli et al., 2010), while other computational studies highlight membrane deformation, water permeation, and unfavorable contacts induced by the wild-type Lys288 (Mondal et al., 2013). Two other basic residues, Lys196 and Arg453, have their amino acid backbone trapped within the hydrophobic core, but their side chains are able to escape by snorkeling to the membrane interface, which in turn exhibits a degree of local thinning. The melibiose transporter (MelB) (PDB: 4M64) also has a polar side chain exposed to the hydrophobic membrane core (Asn58) (Figure 7B). A previous study of this residue has revealed that it is important for Na+-stimulated galactoside transport (Franco and Wilson, 1996). However, a further membrane-exposed residue on the same helix, Arg52, is able to escape the hydrophobic core by snorkeling to the interface and inducing local membrane deformation. Initial examination of the calcium ATPase (SERCA) structure suggested that the surrounding membrane could be as thin as 21 Å (Lee, 2002). This was principally due to the position of Lys972 in TM10 (PDB: 4BEW). However, MemProtMD simulations suggest that this residue is not located near the water/bilayer interface. While there are some local membrane deformations, none is substantial enough to allow the Lys972 side chain to avoid exposure to the hydrophobic core of the bilayer. This leads us to speculate it may have a key functional role, possibly via interaction with other (protein) components of the native membrane.
Such out-of-place residues are not only found in α-helical TM proteins. The voltage-dependent anion channel (VDAC) porin, found in the outer membrane of mitochondria, has a glutamate residue (Glu73) on the surface of its β-barrel TM domain that points directly into the hydrophobic core of the surrounding bilayer (PDB: 4C69) (Choudhary et al., 2014). Experimental studies reveal that this residue is important for hexokinase-I mediated gating and apoptosis inhibition (Zaid et al., 2005), and has a functional role in the dynamics of the protein (Bayrhuber et al., 2008; Villinger et al., 2010). Computational studies using pKa calculations indicate that this residue is likely to be protonated when embedded in a membrane (Zachariae et al., 2012).
Refining Membrane Protein Structures within Membranes
Not all membrane protein structures are determined at high resolution, and despite advances in the use of novel methods, e.g., lipidic cubic phases to obtain better diffracting crystals (Caffrey, 2015), this may continue to be the case, especially as more membrane protein structures are determined by single-particle cryo-EM (Liao et al., 2014). One possible problem in interpretation of medium-resolution structural data for membrane proteins is that of inaccuracies in the register of TM helices (Cross et al., 2013). The simulation approach used in MemProtMD can be used to identify, and in some cases remedy, such issues, thus providing a role for simulations in aiding the refinement of membrane protein structures within a bilayer.
An example is provided by two structures (4OH3 at 3.25 Å resolution, and 4CL4 at 3.7 Å) of the same membrane protein, the plant nitrate transporter NRT1.1 (Parker and Newstead, 2014; Sun et al., 2014). The individual subunits of the structures were nearly identical except for TM12, the register of which differs between the two structures by one helix turn (Figure 8). Both structures were simulated and analyzed by MemProtMD followed by subsequent 100-ns AT simulations. Helix TM12 of 4OH3 remained stable (Figure 8A; average TM12 helix Cα root-mean-square deviation [RMSD] 2.7 Å) throughout the simulation, whereas for 4CL4 the position of TM12 within the membrane altered rapidly (within the first 10 ns), suggestive of conformational instability (Cα RMSD 5.8 Å). More detailed examination (Figure 8B) revealed an outward helix shift relative to the bilayer during the simulation of 4CL4. Evaluation of the membrane interactions of TM12 in the 4CL4 simulation reveals that Asn555 forms a number of contacts with the lipid tails, while this residue is more shielded from the membrane in 4OH3. We also considered the exposure of the six aromatic (Trp and Tyr) residues, which flank either end of the TM12 helix. In 4OH3, four of the six residues are exposed to the membrane, with the other two interacting with the core of the protein. In contrast, in 4CL4 only two of these residues make direct lipid interactions, therefore reducing the extent by which the helix is anchored in the membrane. Refinement of TM12 in the 4CL4 structure in light of this simulation-derived information leads to a more stable TM12 helix in 100 ns of MD simulation (Cα RMSD 2.8 Å). The refined models of PDB: 4CL4, 4CL5 have been updated in the PDB, with new accession numbers PDB: 5A2N, 5A2O, respectively.
Figure 8.

Refining Membrane Protein Structures within Membranes
Two structures and associated simulations (see main text for details) are shown for the MFS transporter NRT1.
(A) A higher-resolution structure (PDB: 4OH3), showing little change in the conformation of TM12 (in blue) on simulation. Both a tyrosine (∗1) and a tryptophan (∗2) interact with the headgroups of the lipid bilayer, potentially anchoring the helix.
(B) A lower-resolution structure (PDB: 4CL4), showing a (partial) change in register of TM12 (with the red arrows indicating the positions of the helix before and after in the simulation). Only a tyrosine (∗1) interacts with the membrane in this helix register.
Conclusions and Future Directions
MemProtMD provides a pipeline for re-embedding membrane protein structures in lipid bilayers, allowing their lipid/protein interactions and related properties to be examined in detail, both for individual proteins and more globally. Comparisons of, e.g., predicted phospholipid interactions of selected proteins agree well with available crystal structures. Initial uptake of MemProtMD by the structural biology community has been very promising, via a number of collaborative studies of membrane proteins: ZMPSTE24 (Quigley et al., 2013); the TatA and TatC proteins of the twin-arginine translocase (Rodriguez et al., 2013; Rollauer et al., 2012); ABCB10 (Shintre et al., 2013); an outer membrane protein, LptDE (Dong et al., 2014); membrane enzyme DgkA (Li et al., 2013); an MFS transporter (CHL1/NRT1) (Parker and Newstead, 2014); and an outer membrane porin (AlgE) (Tan et al., 2014). In all cases the simulations provided structural and/or mechanistic insights, suggesting that this approach can be a useful addition to experimental studies of membrane proteins.
In addition to the simulation and protein/lipid interaction analysis functionalities of MemProtMD described above, a range of other analyses is available online. These include details of predicted lipid binding sites, local bilayer deformations, protein-lipid interactions, and residue distributions relative to the membrane. In addition, coordinates from simulation snapshots and bilayer deformations are available for download, along with parameters for starting both CG and AT simulations.
A remaining challenge for MemProtMD is that simulations are automatically performed on the deposited biological assembly present in the PDB. If this is not supplied, the monomeric asymmetric unit is prepared for simulation. In the great majority of cases this does not present any problems. However, there remain some instances whereby the oligomeric state of the PDB entry is ambiguously or incorrectly annotated, e.g., Aqp0 (PDB: 3M9I) (Figure 3A). It is possible to use tools such as PISA (http://www.ebi.ac.uk/pdbe/pisa/pistart.html) to help assess the likely biological significance of macromolecular interfaces observed in a crystal structure (Krissinel and Henrick, 2007). However, this remains complex for membrane proteins. In the context of MemProtMD, we can consider three examples.
The first is that of a membrane protein monomer, which is favorably inserted in the membrane by MemProtMD, as by the 3FB6 structure for the potassium channel KcsA (see Figure S5A). For this PDB entry, the biological assembly is a monomer rather than a tetramer. MemProtMD is able to flag residues that correspond to the oligomerization interface, due to their unusual exposure to the hydrophobic core of the membrane, e.g., Thr74 of the selectivity filter.
The second example is that of a non-biological oligomer, which may be identified using MemProtMD by two criteria: (1) individual monomers of the oligomer adopt radically different orientations relative to the bilayer; and (2) as a consequence the bilayer is distorted. Thus Aqp0, 2B6O, for which “biological assembly 1” in the PDB is a non-biological octamer (whereas biological assembly 2 is the tetramer), is identifiable in this manner (see Figure S5B).
The third example is that of a non-biological oligomer, which we might highlight by its behavior in MemProtMD but for which we would need additional (biochemical) information to decide on the correct oligomerization state. Thus for the 1WPG structure of SERCA, the biological assembly and asymmetric unit in the PDB is an anti-parallel tetramer (see Figure S5C). This is identified by the previously mentioned test of “different monomers of the oligomer adopt radically different orientations relative to the bilayer.” However, the monomers in this structure are anti-parallel with respect to one another, which, though unlikely, is not unknown. Therefore, curation requires biochemical insight, i.e., SERCA does not adopt an anti-parallel dimeric orientation in cell membranes.
More broadly, this is an area where simulations may be able to provide a useful tool in the future. Simulation studies of the free energy of membrane protein oligomerization within a bilayer environment (Periole et al., 2012) suggest such approaches might be productively linked with MemProtMD to help us to decide between alternative possible oligomeric states of a protein in a membrane.
One may predict the likely future need for MemProtMD. By extrapolation it is expected that by 2020 there will be about 2,000 unique membrane protein structures and close to 4,000 structures in total. Alongside a possible acceleration in determination of membrane protein structures there will clearly be a requirement for methods such as MemProtMD, which allow protein structures to be linked to more complex models of cell membrane systems (Ingolfsson et al., 2014; Koldso et al., 2014).
Experimental Procedures
Membrane Protein Identification
The amino acid sequence of each protein structure in the PDB is parsed through a TM helix prediction algorithm, Octopus (Viklund and Elofsson, 2008) (http://octopus.cbr.su.se/). If at least one TM segment is identified within the sequence, DSSP (Kabsch and Sander, 1983) is used to assess both the secondary structure and accessibility of the predicted region in the protein structure. A region is considered a true TM helix if there are ≥15 α-helical residues, with a helix end-to-end distance of ≥20 Å. The predicted TM helices are also defined on the basis that at least one is accessible to the membrane, rather than hydrophobic helices buried in the core of a soluble protein (Figure S1A).
Outer membrane proteins and related β-barrel membrane proteins are detected as containing β strands that are ≥8 residues in length. A hydrophobic-accessible score was defined by combining an OMPLA hydrophobicity scale (Moon and Fleming, 2011) and a DSSP accessibility score for each residue in the strand. A β-barrel membrane protein must contain ≥5 such hydrophobic β strands in close proximity to one another (Figure S1B). This filters the number of structures extracted from the PDB from ∼100,000 to ∼2,000.
Simulation System Setup
Once identified and downloaded, a structure is checked for any non-native subunits or insertions, e.g., antibody fragments, nanobodies, or chimeric constructs (Rosenbaum et al., 2007). If present, these units are removed before the PDB-deposited biological unit is parsed through PDB2PQR to add any missing atoms (Dolinsky et al., 2007). If more than 10% of the total number of atoms is absent from the structure, Modeller is used to rebuild the missing side chains (Sali and Blundell, 1993). If the deposited structure only contains a Cα trace, or greater than 50% of the total atoms are absent, Pulchra is used to reconstruct the protein structure (Rotkiewicz and Skolnick, 2008). In the latter two instances, PDB2PQR is subsequently used to remove any steric clashes.
The output structure is oriented using MEMEMBED (Nugent and Jones, 2013) such that the bilayer should form in the xy plane, with the TM helices or strands lying approximately parallel to the z axis. This step also filters out any proteins that do not have a TM region and have evaded our initial screen, described above. The topology from the Octopus TM helix prediction is used to orient α-helical membrane proteins so that the cytosolic regions of the protein correspond to negative z coordinates relative to the bilayer. For bacterial outer membrane proteins, the first N-terminal residue is always assumed to be on the periplasmic side of the membrane.
The protein structure is placed within a periodic box of varying dimensions, at a minimum distance of 30 Å from the edge of the box in the x and y directions. The initial size of the box in the z direction is 80 Å. The protein is then converted to CG before dipalmitoylphosphatidylcholine (DPPC) lipid molecules are systematically added to the periodic box in a random orientation. The length of the z axis is extended so that the minimum distance between the protein and all six faces of the box is 30 Å. This allows the formation of the bilayer in the xy plane. The system is then flooded with water particles. CG Na+ and Cl− ions are then added to a final concentration of ∼0.15 M to neutralize the system.
Molecular Dynamics Simulations
CGMD simulations are run for 100 ns, using the MARTINI 2.1 force field, at 323 K to permit the initial assembly of the bilayer around the protein (Monticelli et al., 2008). In these simulations an elastic network, between all Cα atoms within 10 Å, is used to harmonically restrain all Cα particles within the protein, with a force constant of 1,000 kJ/mol. The system is then assessed to make sure that the bilayer has suitably formed, with any lipids not in the bilayer removed. If more than ten lipids are removed the simulation is restarted with different initial velocities, as this suggests malformation of the lipid bilayer. Where the bilayer has formed, the simulations are extended for a further 900 ns.
The final snapshot at 1 μs of CGMD simulation is converted back to atomic detail by using the CG2AT-align method, described previously (Stansfeld and Sansom, 2011).
Analysis and Output
Analyses used GROMACS tools (Hess et al., 2008), MDANALYSIS (Michaud-Agrawal et al., 2011), and locally written code. All are performed on the final 800 ns of the CGMD simulations, when the bilayer is expected to have reached equilibrium. Images of structures are generated using PyMOL (http://www.pymol.org). The website is written in PHP and Javascript and is connected to a MySQL database.
Author Contributions
P.J.S. performed the research. P.J.S., J.E.G., M.C., E.P.C., J.L.P., S.N., and M.S.P.S. designed the research and wrote the manuscript. M.S.P.S. supervised the project.
Acknowledgments
We thank members of the M.S.P.S. laboratory for helpful discussions. This work was supported by Biotechnology and Biological Sciences Research Council grants BB/L002531/1 and BB/I019855/1, the Wellcome Trust, the Science Foundation Ireland (12/IA/1255), and the Irish Centre for High End Computing.
Published: June 11, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.str.2015.05.006.
Supplemental Information
References
- Arnarez C., Mazat J.P., Elezgaray J., Marrink S.J., Periole X. Evidence for cardiolipin binding sites on the membrane-exposed surface of the cytochrome bc1. J. Am. Chem. Soc. 2013;135:3112–3120. doi: 10.1021/ja310577u. [DOI] [PubMed] [Google Scholar]
- Badola P., Sanders C.R., 2nd Escherichia coli diacylglycerol kinase is an evolutionarily optimized membrane enzyme and catalyzes direct phosphoryl transfer. J. Biol. Chem. 1997;272:24176–24182. doi: 10.1074/jbc.272.39.24176. [DOI] [PubMed] [Google Scholar]
- Baeza-Delgado C., Marti-Renom M.A., Mingarro I. Structure-based statistical analysis of transmembrane helices. Eur. Biophys. J. 2013;42:199–207. doi: 10.1007/s00249-012-0813-9. [DOI] [PubMed] [Google Scholar]
- Bayrhuber M., Meins T., Habeck M., Becker S., Giller K., Villinger S., Vonrhein C., Griesinger C., Zweckstetter M., Zeth K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA. 2008;105:15370–15375. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond P.J., Sansom M.S.P. Insertion and assembly of membrane proteins via simulation. J. Am. Chem. Soc. 2006;128:2697–2704. doi: 10.1021/ja0569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner J.D., Lim N.K., Schenck S., Duerst A., Dutzler R. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. 2014;516:207–212. doi: 10.1038/nature13984. [DOI] [PubMed] [Google Scholar]
- Caffrey M. A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr. F Struct. Biol. Commun. 2015;71:3–18. doi: 10.1107/S2053230X14026843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary O.P., Paz A., Adelman J.L., Colletier J.P., Abramson J., Grabe M. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat. Struct. Mol. Biol. 2014;21:626–632. doi: 10.1038/nsmb.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun U., Simons K. Cell membranes: the lipid perspective. Structure. 2011;19:1543–1548. doi: 10.1016/j.str.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Cross T.A., Murray D.T., Watts A. Helical membrane protein conformations and their environment. Eur. Biophys. J. 2013;42:731–755. doi: 10.1007/s00249-013-0925-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cymer F., von Heijne G., White S.H. Mechanisms of integral membrane protein insertion and folding. J. Mol. Biol. 2015;427:999–1022. doi: 10.1016/j.jmb.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R.J., Locher K.P. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G., Baker N.A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H., Xiang Q., Gu Y., Wang Z., Paterson N.G., Stansfeld P.J., He C., Zhang Y., Wang W., Dong C. Structural basis for outer membrane lipopolysaccharide insertion. Nature. 2014;511:52–56. doi: 10.1038/nature13464. [DOI] [PubMed] [Google Scholar]
- Drachmann N.D., Olesen C., Moller J.V., Guo Z., Nissen P., Bublitz M. Comparing crystal structures of Ca(2+)-ATPase in the presence of different lipids. FEBS J. 2014;281:4249–4262. doi: 10.1111/febs.12957. [DOI] [PubMed] [Google Scholar]
- Eilers M., Shekar S.C., Shieh T., Smith S.O., Fleming P.J. Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. USA. 2000;97:5796–5801. doi: 10.1073/pnas.97.11.5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco P.J., Wilson T.H. Alteration of Na(+)-coupled transport in site-directed mutants of the melibiose carrier of Escherichia coli. Biochim. Biophys. Acta. 1996;1282:240–248. doi: 10.1016/0005-2736(96)00062-4. [DOI] [PubMed] [Google Scholar]
- Heijne G. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J. 1986;5:3021–3027. doi: 10.1002/j.1460-2075.1986.tb04601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B., Kutzner C., van der Spoel D., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theor. Comp. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Hite R.K., Li Z.L., Walz T. Principles of membrane protein interactions with annular lipids deduced from aquaporin-0 2D crystals. EMBO J. 2010;29:1652–1658. doi: 10.1038/emboj.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolfsson H.I., Melo M.N., van Eerden F.J., Arnarez C., Lopez C.A., Wassenaar T.A., Periole X., de Vries A.H., Tieleman D.P., Marrink S.J. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014;136:14554–14559. doi: 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Jin M.S., Oldham M.L., Zhang Q., Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012;490:566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W., Sander C. Dictionary of protein secondary structure: pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kelm S., Shi J., Deane C.M. MEDELLER: homology-based coordinate generation for membrane proteins. Bioinformatics. 2010;26:2833–2840. doi: 10.1093/bioinformatics/btq554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killian J.A., von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 2000;25:429–434. doi: 10.1016/s0968-0004(00)01626-1. [DOI] [PubMed] [Google Scholar]
- Koldso H., Shorthouse D., Helie J., Sansom M.S. Lipid clustering correlates with membrane curvature as revealed by molecular simulations of complex lipid bilayers. PLoS Comput. Biol. 2014;10:e1003911. doi: 10.1371/journal.pcbi.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma D., Simon I., Tusnady G.E. PDBTM: Protein Data Bank of transmembrane proteins after 8 years. Nucleic Acids Res. 2013;41:D524–D529. doi: 10.1093/nar/gks1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Landolt-Marticorena C., Williams K.A., Deber C.M., Reithmeier R.A.F. Nonrandom distribution of amino-acids in the transmembrane segments of human type-I single span membrane-proteins. J. Mol. Biol. 1993;229:602–608. doi: 10.1006/jmbi.1993.1066. [DOI] [PubMed] [Google Scholar]
- Lee A.G. Ca2+-ATPase structure in the E1 and E2 conformations: mechanism, helix-helix and helix-lipid interactions. Biochim. Biophys. Acta. 2002;1565:246–266. doi: 10.1016/s0005-2736(02)00573-4. [DOI] [PubMed] [Google Scholar]
- Lee A.G. Biological membranes: the importance of molecular detail. Trends Biochem. Sci. 2011;36:493–500. doi: 10.1016/j.tibs.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Li D., Lyons J.A., Pye V.E., Vogeley L., Aragao D., Kenyon C.P., Shah S.T., Doherty C., Aherne M., Caffrey M. Crystal structure of the integral membrane diacylglycerol kinase. Nature. 2013;497:521–524. doi: 10.1038/nature12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Jaimes K.F., Aller S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M., Cao E., Julius D., Cheng Y. Single particle electron cryo-microscopy of a mammalian ion channel. Curr. Opin. Struct. Biol. 2014;27:1–7. doi: 10.1016/j.sbi.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Yin Y., Wu J., Liu Z. Structure and mechanism of an intramembrane liponucleotide synthetase central for phospholipid biosynthesis. Nat. Commun. 2014;5:4244. doi: 10.1038/ncomms5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S.J., Tieleman D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013;42:6801–6822. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- Michaud-Agrawal N., Denning E.J., Woolf T.B., Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S., Khelashvili G., Shi L., Weinstein H. The cost of living in the membrane: a case study of hydrophobic mismatch for the multi-segment protein LeuT. Chem. Phys. Lipids. 2013;169:27–38. doi: 10.1016/j.chemphyslip.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L., Kandasamy S.K., Periole X., Larson R.G., Tieleman D.P., Marrink S.J. The MARTINI coarse grained force field: extension to proteins. J. Chem. Theor. Comp. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- Moon C.P., Fleming K.G. Side-chain hydrophobicity scale derived from transmembrane protein folding into lipid bilayers. Proc. Natl. Acad. Sci. USA. 2011;108:10174–10177. doi: 10.1073/pnas.1103979108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent T., Jones D.T. Accurate de novo structure prediction of large transmembrane protein domains using fragment-assembly and correlated mutation analysis. Proc. Natl. Acad. Sci. USA. 2012;109:E1540–E1547. doi: 10.1073/pnas.1120036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent T., Jones D.T. Membrane protein orientation and refinement using a knowledge-based statistical potential. BMC Bioinformatics. 2013;14:276. doi: 10.1186/1471-2105-14-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsdottir H., Hunte C. Lipids in membrane protein structures. Biochim. Biophys. Acta. 2004;1666:2–18. doi: 10.1016/j.bbamem.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Parker J.L., Newstead S. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature. 2014;507:68–72. doi: 10.1038/nature13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebay-Peyroula E., Dahout-Gonzalez C., Kahn R., Trezeguet V., Lauquin G.J., Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44. doi: 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Periole X., Knepp A.M., Sakmar T.P., Marrink S.J., Huber T. Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J. Am. Chem. Soc. 2012;134:10959–10965. doi: 10.1021/ja303286e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piggot T.J., Holdbrook D.A., Khalid S. Electroporation of the E. coli and S. aureus membranes: molecular dynamics simulations of complex bacterial membranes. J. Phys. Chem. B. 2011;115:13381–13388. doi: 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- Piscitelli C.L., Krishnamurthy H., Gouaux E. Neurotransmitter/sodium symporter orthologue LeuT has a single high-affinity substrate site. Nature. 2010;468:1129–1132. doi: 10.1038/nature09581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogozheva I.D., Tristram-Nagle S., Mosberg H.I., Lomize A.L. Structural adaptations of proteins to different biological membranes. Biochim. Biophys. Acta. 2013;1828:2592–2608. doi: 10.1016/j.bbamem.2013.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley A., Dong Y.Y., Pike A.C., Dong L., Shrestha L., Berridge G., Stansfeld P.J., Sansom M.S., Edwards A.M., Bountra C. The structural basis of ZMPSTE24-dependent laminopathies. Science. 2013;339:1604–1607. doi: 10.1126/science.1231513. [DOI] [PubMed] [Google Scholar]
- Rodriguez F., Rouse S.L., Tait C.E., Harmer J., De Riso A., Timmel C.R., Sansom M.S., Berks B.C., Schnell J.R. Structural model for the protein-translocating element of the twin-arginine transport system. Proc. Natl. Acad. Sci. USA. 2013;110:E1092–E1101. doi: 10.1073/pnas.1219486110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollauer S.E., Tarry M.J., Graham J.E., Jaaskelainen M., Jager F., Johnson S., Krehenbrink M., Liu S.M., Lukey M.J., Marcoux J. Structure of the TatC core of the twin-arginine protein transport system. Nature. 2012;492:210–214. doi: 10.1038/nature11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D.M., Cherezov V., Hanson M.A., Rasmussen S.G., Thian F.S., Kobilka T.S., Choi H.J., Yao X.J., Weis W.I., Stevens R.C. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Rotkiewicz P., Skolnick J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 2008;29:1460–1465. doi: 10.1002/jcc.20906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht J.J., Hellawell A.M., Harding M., Crichton P.G., McCoy A.J., Kunji E.R. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. USA. 2014;111:E426–E434. doi: 10.1073/pnas.1320692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ W.P., Engelman D.M. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schmidt M.R., Stansfeld P.J., Tucker S.J., Sansom M.S. Simulation-based prediction of phosphatidylinositol 4,5-bisphosphate binding to an ion channel. Biochemistry. 2013;52:279–281. doi: 10.1021/bi301350s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott K.A., Bond P.J., Ivetac A., Chetwynd A.P., Khalid S., Sansom M.S.P. Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure. 2008;16:621–630. doi: 10.1016/j.str.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Shintre C.A., Pike A.C., Li Q., Kim J.I., Barr A.J., Goubin S., Shrestha L., Yang J., Berridge G., Ross J. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. USA. 2013;110:9710–9715. doi: 10.1073/pnas.1217042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P.J., Sansom M.S.P. From coarse-grained to atomistic: a serial multi-scale approach to membrane protein simulations. J. Chem. Theor. Comp. 2011;7:1157–1166. doi: 10.1021/ct100569y. [DOI] [PubMed] [Google Scholar]
- Stansfeld P.J., Hopkinson R.J., Ashcroft F.M., Sansom M.S.P. The PIP2 binding site in Kir channels: definition by multi-scale biomolecular simulations. Biochemistry. 2009;48:10926–10933. doi: 10.1021/bi9013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P.J., Jefferys E.E., Sansom M.S. Multiscale simulations reveal conserved patterns of lipid interactions with aquaporins. Structure. 2013;21:810–819. doi: 10.1016/j.str.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J., Bankston J.R., Payandeh J., Hinds T.R., Zagotta W.N., Zheng N. Crystal structure of the plant dual-affinity nitrate transporter NRT1.1. Nature. 2014;507:73–77. doi: 10.1038/nature13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Rouse S.L., Li D., Pye V.E., Vogeley L., Brinth A.R., El Arnaout T., Whitney J.C., Howell P.L., Sansom M.S. A conformational landscape for alginate secretion across the outer membrane of Pseudomonas aeruginosa. Acta Crystallogr. D Biol. Crystallogr. 2014;70:2054–2068. doi: 10.1107/S1399004714001850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G., Halter D., Sprong H., Somerharju P., Egmond M.R. ABC lipid transporters: extruders, flippases, or flopless activators? FEBS Lett. 2006;580:1171–1177. doi: 10.1016/j.febslet.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Viklund H., Elofsson A. OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics. 2008;24:1662–1668. doi: 10.1093/bioinformatics/btn221. [DOI] [PubMed] [Google Scholar]
- Villinger S., Briones R., Giller K., Zachariae U., Lange A., de Groot B.L., Griesinger C., Becker S., Zweckstetter M. Functional dynamics in the voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA. 2010;107:22546–22551. doi: 10.1073/pnas.1012310108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A.B., Szewczyk P., Grimard V., Lee C.W., Martinez L., Doshi R., Caya A., Villaluz M., Pardon E., Cregger C. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA. 2013;110:13386–13391. doi: 10.1073/pnas.1309275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S.H. Biophysical dissection of membrane proteins. Nature. 2009;459:344–346. doi: 10.1038/nature08142. [DOI] [PubMed] [Google Scholar]
- Whorton M.R., MacKinnon R. X-ray structure of the mammalian GIRK2-betagamma G-protein complex. Nature. 2013;498:190–197. doi: 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V., Schonbrun J., Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62:1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae U., Schneider R., Briones R., Gattin Z., Demers J.P., Giller K., Maier E., Zweckstetter M., Griesinger C., Becker S. Beta-barrel mobility underlies closure of the voltage-dependent anion channel. Structure. 2012;20:1540–1549. doi: 10.1016/j.str.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaid H., Abu-Hamad S., Israelson A., Nathan I., Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–760. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.