

Abstract
Foxm1, a mammalian Forkhead Box M1 protein, is known as a typical proliferation-associated transcription factor. Here, we find that Foxm1 was essential for maintenance of hematopoietic stem cell (HSC) quiescence and self-renewal capacity in vivo in mice. Reducing expression of FOXM1 also decreased quiescence in human CD34+ HSCs and progenitor cells and its down-regulation was associated with a subset of myelodysplastic syndrome (MDS). Mechanistically, Foxm1 directly bound to the promoter region of Nurr1, inducing transcription while forced expression of Nurr1 reversed the loss of quiescence observed in Foxm1-deficient cells in vivo. Thus, our studies reveal a previously unrecognized role of Foxm1 as a critical regulator of HSC quiescence and self-renewal, mediated at least in part, by control of Nurr1 expression.
Hematopoietic stem cells (HSCs) have the ability to self-renew and differentiate into all blood cell lineages, and are critical for the maintenance of homeostasis of the hematopoietic system. HSCs predominantly exist in a quiescent state1, which is critical for preserving self-renewal capacity and enabling life-long hematopoiesis2. Elucidating the molecular regulation of HSC quiescence should increase our understanding of mechanisms important for tissue regeneration and perhaps indicate how these may become dysregulated in pathological conditions. The quiescent state of HSCs is tightly controlled by both intrinsic molecular mechanisms and extrinsic signals from the microenvironment. Several cell cycle regulators as well as the genes with functions in oxidative stress regulation, transcriptional regulation of hematopoiesis, or chromatin modification have been shown to regulate HSC quiescence by intrinsic mechanisms3,4.
Foxm1 belongs to a large family of Forkhead box (Fox) proteins. It is a key regulator of aspects of the cell cycle-G1/S-transition, S-phase progression, G2/M-transition and M-phase progression5, and is critical for DNA replication, mitosis6 and genomic stability7. Foxm1 has pleiotropic roles during embryonic development and tissue regeneration after injury5. Foxm1 is broadly expressed in embryonic tissues, while its expression in adult mice is restricted to the testes, thymus and intestinal crypts8–10. However, Foxm1 expression is re-activated after organ injury5,11. Studies demonstrate that Foxm1 plays a role in the proliferation of hepatocytes and pancreatic endocrine cells during liver and pancreatic regeneration12,13. Consistent with the critical role for Foxm1 in cell cycle progression, increased expression of FOXM1 has been found in numerous human tumors including lung cancer, breast cancer, liver cancer, glioblastoma and pancreatic cancer14. Collectively, Foxm1 was considered as a proliferation-specific transcription factor, required for cellular proliferation in various tissues. However, little is known of the function of Foxm1 during hematopoiesis. Deletion of Foxm1 during T cell lymphopoiesis reduces proliferation of early thymocytes and activates mature T cells but does not affect T cell differentiation15, while Foxm1 deletion within the myeloid lineage does not impact the proliferation or differentiation of myeloid cells16. Notably, the effects of loss of Foxm1 in HSCs or hematopoietic progenitor cells (HPCs) have not been examined.
Here we investigated the function of Foxm1 in HSCs and/or HPCs using conditional knockout mouse models. We found that Foxm1 loss reduced the frequency of quiescent HSCs, increased proliferation of both HSCs and HPCs, but did not affect the differentiation of HSCs and HPCs. As a consequence, Foxm1-deficient HSCs significantly reduced self-renewal capacity. Mechanistically, Foxm1 loss induced downregulation of cyclin-dependent kinase inhibitors, including p21 and p27, by directly suppressing the expression of Nurr1, encoding a critical regulator of HSC quiescence17. Notably, reducing expression of FOXM1 in human CD34+ primitive hematopoietic cells also decreased quiescence. and database analysis revealed that FOXM1 and NURR1 expression was both significantly down-regulated in CD34+ cells from a subset of patients with myelodysplastic syndrome (MDS). Together, our data provides the first evidence that Foxm1 is a critical regulator of HSC quiescence and self-renewal capacity through Nurr1-mediated pathways.
RESULTS
Perturbed hematopoiesis in Foxm1-deficient mice
A function for Foxm1 in hematopoietic stem/progenitor cells has not been previously reported. To determine its role, we characterized the expression of Foxm1 in subsets of primitive and mature bone marrow (BM) cells. Foxm1 was more highly expressed in primitive hematopoietic cells than in differentiated cells, including mature Mac-1+Gr-1+ myeloid cells, B220+ B cells, CD71+ Ter119+ erythroblasts, and CD4+ or CD8+ T cells (Fig. 1a). Notably, Foxm1 was expressed at relatively more in long-term HSCs (LT-HSC, Lin−Sca-1+c-Kit+CD48−CD150+) than in LSKs (Lin−Sca-1+c-Kit+) or HPCs (Lin−c-Kit+Sca-1−), suggesting that Foxm1 plays an important role in HSCs.
Figure 1. Foxm1 loss leads to abnormal hematopoiesis.
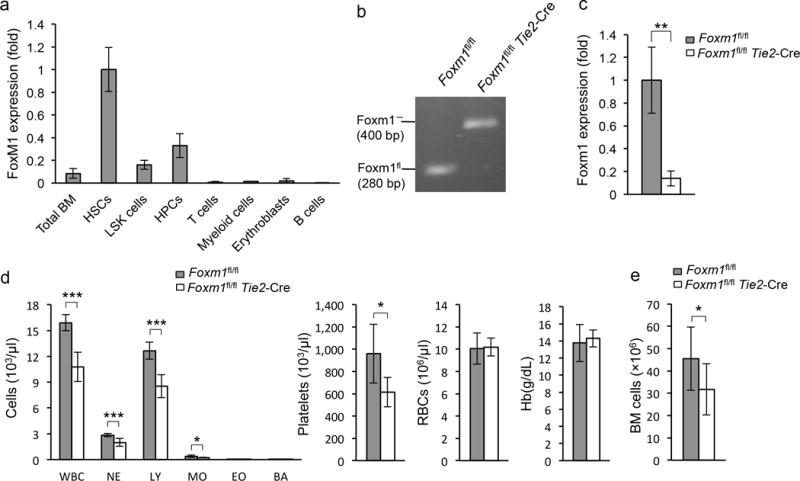
(a) Expression of Foxm1 in hematopoietic cells from bone marrow (BM) as determined by qRT-PCR. Gene expression was normalized initially to Actb expression. Values represent the fold changes in gene expression relative to that in HSCs.(b) Analysis of Foxm1 deletion as determined by semiquantitative PCR analysis of genomic DNA from BM LSK cells from Foxm1fl/flTie2-Cre or Foxm1fl/fl mice. (c) qRT-PCR analysis of mRNAs from BM LSK cells from both Foxm1fl/flTie2-Cre and Foxm1fl/fl mice. (d) Absolute number of white blood cells, neutrophils (NE), lymphocytes (LY), monocytes (MO), eosinophils (EO), basophils (BA), platelets (PLT), Red blood cells (RBC) and hemoglobin (HB) in peripheral bloodfrom Foxm1fl/flTie2-Cre and Foxm1fl/fl mice at 6 weeks of age (mean±SD, n=5). (e) Total number of BM cells in Foxm1fl/flTie2-Cre and Foxm1fl/fl mice. Data are from two independent experiments (a; mean±SD of n=3, per experiment), representative of three experiments (b), three independent experiments (c; mean±SD, n=3, per experiment), or two independent experiments (d,e, mean±SD, n=9, *, P<0.05; **, P<0.005; ***, P<0.0005).
To investigate the in vivo function of Foxm1 in normal hematopoiesis, we generated Foxm1 conditional knockout (CKO) mice by crossing Foxm1 floxed mice11 (Foxm1fl/fl) with Tie2-Cre transgenic mice, in which the Cre recombinase is expressed in endothelial cells and HSCs under the control of the Tie2 promoter18,19. High efficiency of Foxm1 deletion in BM cells was confirmed by semi-quantitative PCR analysis of genomic DNA isolated from BM cells (Supplementary Fig. 1a) or LSK cells (Fig. 1b) from both Foxm1fl/fl Tie2-Cre (referred to hereafter as Foxm1 CKO) and Foxm1fl/fl mice. Accordingly, qPCR analysis showed only trace amounts of Foxm1 mRNA in BM cells (Supplementary Fig. 1b) or LSK cells (Fig. 1c) from Foxm1 CKO mice. We analyzed the key hematological parameters in these mice at 6 weeks of age. Foxm1 CKO mice showed a markedly decreased number of White blood cells, Neutrophils, lymphocytes, monocytes and platelets (Fig. 1d). Total numbers of BM cells from Foxm1 CKO mice were reduced as compared to control littermates (Fig. 1e), with some Foxm1 CKO mice showing clearly hypocellular BM (Supplementary Fig. 1c). Likewise, flow cytometric analysis revealed that total numbers of mature myeloid cells, B cells, CD41+ megakaryocytes and F4/80+ macrophages were also decreased in BM from Foxm1 CKO mice compared to control littermates (Supplementary Fig. 2a). However, the frequencies of these mature blood cells in BM are comparable among Foxm1 CKO and control mice (Supplementary Fig. 2b–e), suggesting that loss of Foxm1 does not affect differentiation of myeloid or B cells under homeostatic conditions in young mice. Loss of Foxm1 did not interfere with erythroid lineage differentiation. Comparable frequencies of erythroid blasts in four stages of murine eythroid differentiation20, were detected in both the BM and spleen of Foxm1 CKO and control mice (Supplementary Fig. 2f,g,h,i). Thus, our data indicates that Foxm1 deletion leads to ineffective hematopoiesis, but does not impact mature blood cell differentiation.
Cell-intrinsic control of HSC/HPC pools by Foxm1
During normal hematopoiesis, LT-HSCs have a capacity to self-renew with the potential for differentiation to common myeloid progenitors (CMPs) and more committed myeloid progenitor cells, including granulocyte-monocyte progenitors (GMP) and megakaryocyte-erythroid progenitors (MEP)21. We hypothesized that the abnormal hematopoiesis observed in Foxm1 CKO mice is likely a consequence of disturbance of HSCs and/or HPCs. We, thus, examined these compartments by flow cytometric analysis. We observed that both the frequency and total number of LSK cells, a stem-cell-enriched population, were significantly decreased by 5 to 6 fold in Foxm1 CKO mice compared to control wild-type mice (Fig. 2a,b). In addition, the total numbers of CD34− LSKs or LT-HSC were also significantly decreased (Fig. 2a,b). We also noted that both the frequency and total number of HPCs was decreased by 50% in Foxm1 CKO mice (Fig. 2a,d).
Figure 2. Marked decrease in HSC and HPC pools in Foxm1-deficient mice.
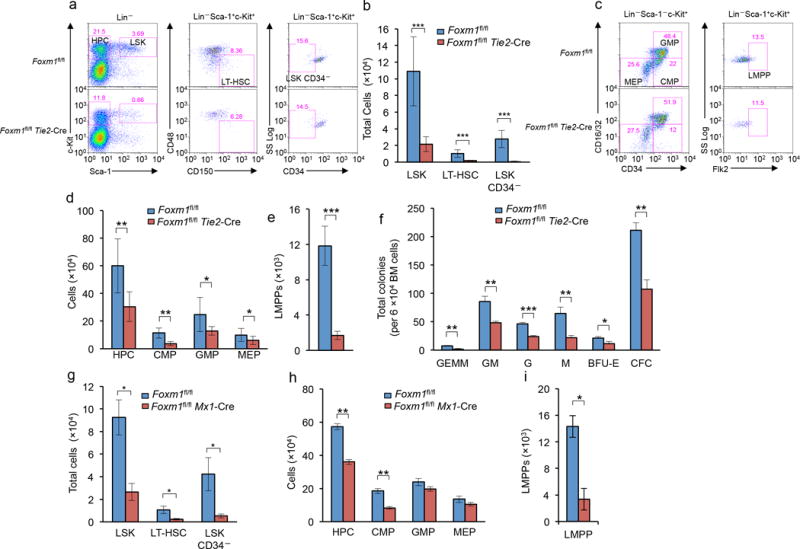
(a) Flow cytometric analysis of the frequency of LSK cells, LSKCD34− and LT-HSCs in representative Foxm1fl/flTie2-Cre and Foxm1fl/fl mice. (b) Total number of LSK cells, LSK CD34− and LT-HSCs in BM from Foxm1fl/flTie2-Cre and Foxm1fl/fl mice at 6–8 weeks of age. (c) Flow cytometric analysis of the frequency of HPCs and subsets of myeloid progenitors including CMPs, GMPs and MEPs and lymphoid-primed multipotent progenitor pool (LMPP) in representative Foxm1fl/flTie2-Cre and Foxm1fl/fl mice. (d,e) Total number of HPCs, CMPs, GMPs and MEPs (d), and LMPPs (e) in BM from Foxm1fl/fl Tie2-Cre and Foxm1fl/fl mice at 6–8 weeks of age. (f) In vitro colony assays: the number of BM CFU-GEMM, CFU-GM, CFU-G, CFU-M and BFU-E colonies was examined 10–12 days after plating. (g) Depiction of induction of Foxm1 deletion by pI-pC in Foxm1fl/flMx1-Cre mice. (h) Total number of LSK cells, LSKCD34− and LT-HSCs in BM from Foxm1fl/flMx1-Cre and Foxm1fl/fl mice at 6–8 weeks of age. Analysis was performed three weeks after three injections of pI-pC. *, P<0.05; LSK cells are defined as Lin− Sca-1+ c-Kit+ while LT-HSCs are defined as LSK, CD150+ CD48−. (i) Total number of HPCs, CMPs, GMPs and MEPs (left panel), and LMPPs (right panel) in BM from Foxm1fl/flMx1-Cre and Foxm1fl/fl mice at 6–8 weeks of age. Analysis was performed at three weeks after injection of pI-pC. HPCs are defined as Lin− Sca-1− c-Kit+ while CMPs, GMPs and MEPs are defined as CD34+/lo CD16/32int; CD34+CD16/32+, and CD34− CD16/32−, respectively. LMPPs are defined as Lin− Sca-1+ c-Kit+ Flk2+. *, P < 0.05; **, P < 0.005; ***, P < 0.0005, Data are representative of three experiments (a,c) or data are from three experiments (b,d,e; mean±SD, n=6–7 mice per genotype) or from two experiments (f,g,h,i, mean±SD, n=3).
We next characterized myeloid progenitors in Foxm1 CKO and wild-type mice. The frequency of CMPs was significantly lower in Foxm1 CKO mice than wild-type mice, while both Foxm1 CKO and control mice had comparable frequencies of GMPs and MEPs cells in vivo. However, the total number of all these subsets of myeloid progenitors was reduced in Foxm1 mice as compared to controls (Fig. 2c,d). The lymphoid-primed multipotent progenitor pool (LMPP, Lin− Sca-1+ c-Kit+ Flt3+) cells has been identified with the capacity to generate B cells, T cells and monocytes22. Foxm1 CKO mice had a significant reduction in the absolute number of LMPPs when compared to wild-type mice (Fig. 2e). We determined the frequency of myeloid progenitors in BM from Foxm1 CKO and control mice by in vitro colony-forming unit assays. Similar numbers of BM cells isolated from wild-type or Foxm1 CKO mice were plated in methylcellulose media containing interleukin 3 (IL-3), IL-6, stem cell factor (SCF) and erythropoietin (Epo) for 10–12 days. Foxm1-deficient BM cells gave rise to markedly lower numbers of total colony forming units (CFUs) as well as variety of myeloid precursors, including CFU-granulocyte, erythroid, macrophage, megakaryocyte (CFU-GEMM), CFU-granulocyte macrophage (CFU-GM), CFU-granulocyte (CFU-G), CFU-macrophage (CFU-M) and Burst-forming unit-erythroid (BFU-E) colonies, than did the BM cells from Foxm1 control mice (Fig. 2f).
Endothelial cells constitute a critical niche for HSCs23–25. To rule out the possibility that the endoethelial niche that HSCs are exposed to during development causes the observed hematopoietic defects in Foxm1fl/flTie2-Cre mice, we generated Foxm1fl/flMx1-Cre mice, in which deletion of Foxm1 in the hematopoietic compartments is efficiently induced in adult mice by intraperitoneal injection of the interferon-α inducer polyinosinic-polycytidylic acid (pI-pC) in adult mice (Supplementary Fig. 3a,b). Following induction of Foxm1 deletion, Foxm1fl/flMx1-Cre mice had reduced numbers of total BM cells (Supplementary Fig. 3c) and develop a similar defect in hematopoietic stem cell compartment as that observed in Foxm1 CKO mice (Fig. 2g,h). Foxm1fl/flMx1-Cre mice, with abnormal hematopoiesis (Supplementary Table 1) characterized by significantly decreased numbers of total LSK cells, CD34− LSKs and LT-HSCs (Fig. 2g), and concomitant reductions in the number of HPCs, CMPs and LLMPs (Fig. 2h,i). Together, these data suggest that loss of Foxm1 significantly interferes with the maintenance of adult HSCs and early HPCs during normal hematopoiesis, and the defect appears to be independent of the endothelial niche.
However, the Mx1-Cre transgene also induces gene deletion in non-hematopoietic cells including the BM stromal compartment and liver26. To rule out the possibility that the effects of Foxm1 deletion on HSCs and HPCs are dependent on BM microenvironment, we transplanted BM cells from Foxm1fl/flMx1-Cre and Foxm1fl/fl mice (CD45.2) into lethally-irradiated, wild-type syngeneic recipient (CD45.1) mice to generate Foxm1fl/fl Mx1-Cre and Foxm1fl/fl chimeric mice. Foxm1fl/flMx1-Cre and control Foxm1fl/fl BM cells resulted in comparable engraftment efficiency with more than 95% of BM cells in recipient mice replaced with BM cells from donor mice (Supplementary Fig. 4a). Deletion of Foxm1 in the chimeric Foxm1fl/flMx1-Cre mice was induced by pI-pC injection six weeks post-transplantation, and, two months later, the HSC and HPC compartments were examined by flow cytometry. The numbers of total LSK cells, CD34− LSKs and LT-HSCs were significantly lower in Foxm1fl/flMx1-Cre chimeric mice than control Foxm1fl/fl chimeric mice (Supplementary Fig. 4b). In addition, the number of HPCs and CMPs were also significantly decreased in the Foxm1fl/flMx1-Cre chimeric mice (Supplementary Fig. 4c). In summary, our results indicate that Foxm1 is an intrinsic regulator of HSCs and HPCs.
Loss of Foxm1 impairs HSC long-term self-renewal
To further investigate the effects of Foxm1 deletion on hematopoietic reconstitution in situ in primary mice, Foxm1 CKO and Foxm1fl/fl mice were injected weekly with the cell-cycle-dependent myelotoxic agent 5-fluorouracil (5-FU)27, which kills proliferating cells including hematopoietic progenitors, thus stimulating HSCs to proliferate and replenish the hematopoietic system. We monitored the treated mice for up to three weeks. 80% of Foxm1-deficient mice succumbed after the second 5-FU injection and all remaining mice died after a third injection of 5-FU. In contrast, more than 90% of control mice were still alive after third injection of 5-FU (Fig. 3a). Analysis of BM cells and HPCsof another cohort of 5-FU-treated mutant and control mice indicated that Foxm1-deficient HSCs failed to efficiently replenish BM cells and HPCs (Fig. 3b,c). To further evaluate the long-term self-renewal capacity of Foxm1-deficient HSCs, we performed serial bone marrow transplantation assays. BM cells (5 × 106) derived from 2–3 Foxm1 CKO or Foxm1fl/fl mice were serially transplanted into lethally irradiated wild-type recipients. While wild-type HSCs ceased to reconstitute the lethally irradiated mice after the fourth transplantation cycle, Foxm1 CKO HSCs failed to reconstitute hematopoiesis upon secondary transplantation (Fig. 3d), suggesting Foxm1-deficient HSCs have a severe defect in self-renewal capacity.
Figure 3. Foxm1 depletion decreases the self-renewal capacity of HSCs.
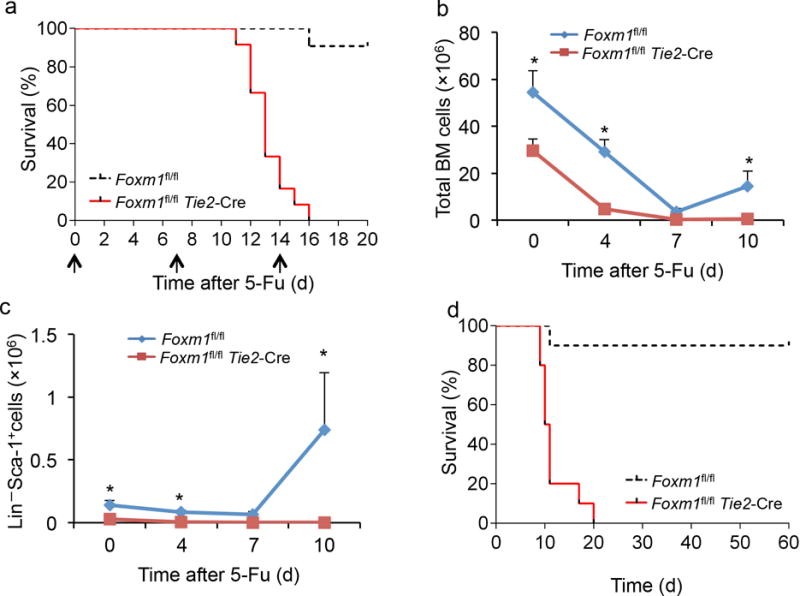
(a) Kaplan-Meier survival curve of Foxm1fl/flTie2-Cre and Foxm1fl/fl mice receiving multiple injections of 5-FU (indicated by arrows, 150mg/kg) (P < 0.0001, n=12). (b,c) The total number of BM (b) and Lin− Sca-1+ (c) cells present in Foxm1fl/flTie2-Cre and Foxm1fl/fl mice before (Day 0) and after 5-FU injection (200 mg/kg) (Days 4, 7 and 10). (d) Kaplan-Meier survival curve of recipient mice transplanted with BM cells derived from primary Foxm1fl/flTie2-Cre and Foxm1fl/fl transplanted mice (P=0.0001, n=10). Data are from one experiment (a,d) or three experiments (b,c; mean±SD, n=3).
Next, we examined the reconstituting capacity of Foxm1-deficient HSCs in a competitive situation by performing competitive serial transplantation assays. Equal numbers of BM cells or sorted LT-HSCs from Foxm1 CKO or corresponding littermate control mice (CD45.2) were transplanted into lethally irradiated Ly5.1(CD45.1) recipients together with competitor BM cells from Ly5.1/B6 mice (CD45.1/CD45.2). Four months after transplantation, equal numbers of BM cells derived from a cohort of primary transplantation mice were transplanted into a second set of lethally irradiated mice. Donor-derived peripheral blood cells (CD45.2) and competitor-derived peripheral blood cells were assessed by flow cytometric analysis every month post transplantation. Of note, unfractionated Foxm1-deficient BM cells showed a progressive repopulation defect with 80% less repopulation than that of wild-type cells at one month and 90% less repopulation at four months after primary transplantation (Fig. 4a, left). Purified Foxm1-deficient LT-HSCs also showed a progressive decrease in repopulation capacity with 25% less repopulation compared to wild-type LT-HSCs after one month and 90% less at four months after transplantation. Peripheral blood cells derived from Foxm1-deficient BM cells and LT-HSCs were barely detected at four months after the second transplantation (Fig. 4a). We also performed competitive repopulation assays using BM cells from Foxm1fl/flMx1-Cre and Foxm1fl/fl mice. Without pI-pC induction of Foxm1 deletion, Foxm1fl/flMx1-Cre and Foxm1fl/fl BM cells had a comparable repopulation capacity (Fig. 4b). Delayed deletion of Foxm1 induced at six weeks post-transplantation consistently resulted in a progressive decrease in HSC repopulation capacity (Fig. 4b). Flow cytometric analysis of total mature cells (Fig. 4c) and myeloid, B cell and T cell lineages in peripheral blood revealed that the repopulation of all three lineages was impaired (Supplementary Fig. 5a–c). Additionally, immature BM cells derived from Foxm1-deficient donor cells were barely detected at four months after the second transplantation (Fig. 4d, Supplementary Fig. 5d,e), indicative of a HSC defect. Consistent with the data in Foxm1 CKO mice, Foxm1 loss did not affect the lineage differentiation during repopulation of hematopoietic cells in recipient mice (Supplementary Fig. 5f). In summary, these studies provide strong evidence that loss of Foxm1 exerts a negative effect on HSC long-term regenerative capacity.
Figure 4. Foxm1-deficient HSCs have a significant decrease in repopulation capacity.
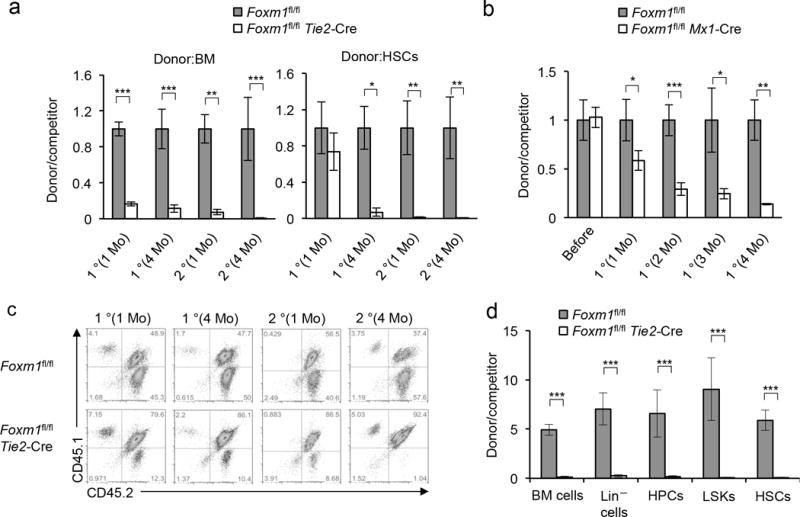
(a) The histogram shows the relative ratio of donor-derived CD45.2+CD45.1− versus competitor-cell-derived CD45.1+ CD45.2+ peripheral blood cells in chimeric mice reconstituted with BM cells (left panel) or HSCs (right panel) from Foxm1fl/flTie2-Cre or Foxm1fl/fl mice examined at 1 month or 4 months after the first transplantation and at 1 month or 4 months after secondary transplantation. (b) The histogram shows the relative ratio of CD45.2+ CD45.1− versus CD45.1+ CD45.2+ peripheral blood cells in chimeric mice reconstituted with BM cells from Foxm1fl/flMx1-Cre or Foxm1fl/fl examined at 1, 2, 3, or 4 months after the first transplantation (*, P<0.05; **, P<0.005; ***, P<0.0005, n=4–5). (c) Flow cytometric analysis of donor-derived CD45.2+ CD45.1− and competitor-cell-derived CD45.1+ CD45.2+ peripheral blood cells from representative chimeric mice at 1 month or 4 months after the first transplantation, and at 1 month or 4 months after secondary transplantation. The numbers indicate the percentage of cells in each population. (d) The ratio of donor-derived and competitor-cell-derived cells in total BM cells, Lin− cells, HPCs (lin− Sca-1− c-Kit+ cells), LSKs and HSCs from the recipients 4 months after the secondary transplantation is shown (*, P < 0.05; **, P < 0.005; ***, P < 0.0005, n=5). Data are from one experiment (a,b,c; mean±SD, n=4–5) or two experiments (d; mean±SD, n=5).
Foxm1 deficiency increases cycling of HSC/HPC
Previous findings indicating that Foxm1 plays a role in G1/S cell cycle transition28–30, and also contributes to the regulation of the G2/M transition in mammalian cells7,29. As Foxm1 is a critical cell cycle regulator, we examined whether Foxm1 loss leads to cell cycle defects in hematopoietic stem and progenitor cells. We observed a greater accumulation of cells in the S, and G2/M phase in the Foxm1-deficient HPCs, LSKs and LT-HSCs compared to wild-type controls (Fig. 5a–c). To determine whether the accumulation of cells in the S, and G2/M phase in Foxm1-deficient LSKs/LT-HSCs was due to the block of cell cycle in S, and G2/M transition or an overall increase in cycling cells, we performed in vivo BrdU incorporation assay to assess the kinetics of cell cycle in both Foxm1-deficient and wild-type control LSKs/LT-HSCs. While ~13% of Foxm1fl/fl LSK cells (or ~5% of Foxm1fl/fl LT-HSCs) incorporated BrdU, ~50% of the Foxm1 CKO LSK cells (or ~35% of Foxm1 CKO LT-HSCs) were BrdU positive (Fig. 5d,e), indicating that absence of Foxm1, increases proliferation of LT-HSCs and LSK cells. In addition, loss of Foxm1 also leads to accumulation of cells in G2/M transition in LT-HSCs and LSKs (Fig. 5d,e). Collectively, our study suggests that Foxm1 regulates the cell cycle in a context-dependent manner. As quiescence is crucial for the function of HSCs, we next determined the fraction of the stem cell-enriched population (LSK cells) in the G0 phase by assessing RNA and DNA content using pyronin Y/HOECHST 33342 staining as previously described31,32. The proportion of quiescent cells was 3-fold less among Foxm1-deficient LSK cells than in control LSK cells, suggesting that Foxm1 deficiency regulates G0 phases in HSCs (Fig. 5f). Similarly, Mx1-Cre-induced Foxm1 deletion in both primary and chimeric mice (Supplementary Figs. 6) resulted in a loss of G0 maintenance in LSK cells and defects in cell cycle progression in HSCs, LSK cells and HPCs. Thus, these data suggest that Foxm 1 is critical for maintenance of HSC quiescence, and in its absence, HSCs more easily enter the cell cycle.
Figure 5. Foxm1 deletion leads to an accumulation of cells in the S and G2/M phases in the cell cycle and loss of quiescence in hematopoietic stem/progenitor cells.
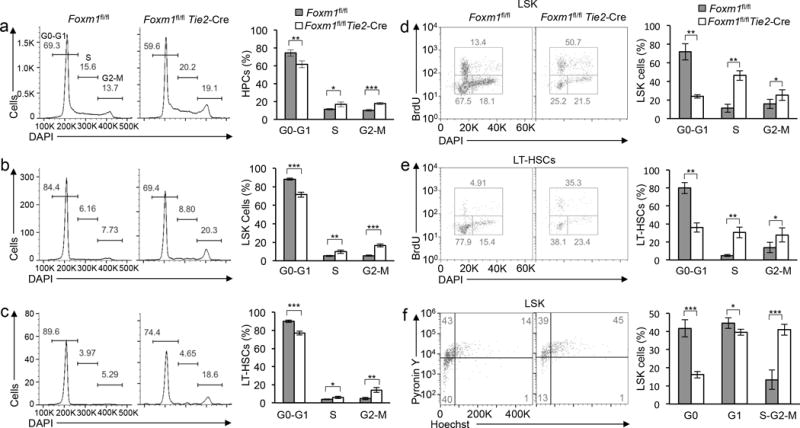
(a–e) Representative histograms (left panel) of flow cytometric analysis of the cell cycle in HPCs (a), LSK cells (b), and LT-HSCs (c), stained with DAPI, and in LSK cells (d) and LT-HSCs (e), labeled with BrdU. The histogram (right panel) depicts the cell cycle profile of HPCs (a), LSK cells (b), and LT-HSCs (c), stained with DAPI and in LSK cells(d) and LT-HSCs (e), labeled with BrdU. The cell cycle was analyzed in Foxm1fl/flTie2-Cre and Foxm1fl/fl mice (mean±SD, n=4). (f) Representative histograms (left panel) of flow cytometric analyses of G0-G1 cell cycle in LSKs. The histogram (right panel) depicts cell cycle status of LSK cells in Foxm1fl/flTie2-Cre and Foxm1fl/fl mice (mean±SD, n=7). *, P < 0.05; ***, P < 0.0005. Data are from two experiments or representative of two experiments (a–f; mean±SD of n=5 mice per genotype in a-c; 4 mice per genotype in d,e; 7 mice per genotype in f).
Foxm1 loss reduces HSC/HPC survival under stress
As cell survival contributes to the maintenance of HSCs and HPCs, we examined Foxm1-deficient and control HSCs and HPCs for evidence of apoptosis using Annexin V staining. We found that Foxm1 loss did not affect the frequency of apoptosis in LSK cells and HPCs in Foxm1 CKO mice (Supplementary Fig. 7a). Interestingly, we found that the frequency of apoptosis was significantly increased in LSK cells and HPCs from Foxm1 CKO and Foxm1fl/flMx1-Cre chimeric mice (Supplementary Fig. 7b,c). As HSC transplantation exposes HSCs and HPCs to various stresses33, our studies suggest that loss of Foxm1 reduced the survival of HSCs and HPCs under stress.
Foxm1 deficiency affects multiple pathways in HSCs
To identify Foxm1-dependent genes and the molecular pathways involved in the regulation of HSC function, we performed global gene expression profiling of LT-HSCs isolated from Foxm1 CKO or control Foxm1fl/fl mice at 2 months of age. We found that 70 genes were differentially expressed greater than two-fold in Foxm1-deficient versus wild-type HSCs (Fig. 6a, Supplementary Table 2). The differentially expressed genes are classified into seven functional groups including genes with nucleic acid binding transcription factor, binding, catalytic and structural activity (Fig. 6b). Accumulating evidence suggests that the ribosome has a regulatory function in gene expression34. Deregulation of protein synthesis by altering ribosome function impairs HSC functions35. Notably, several ribosomal proteins including Rpl38, Utp14a, Rpl29, Mrpl24 and Rpl21 were significantly increased in HSCs with absence of Foxm1 compared to control HSCs (Supplementary Table 2). Next we employed Gene Set Enrichment Analysis (GSEA)36 to determine whether an a priori-defined set of genes shows statistically significant differences in HSCs with presence or absence of Foxm1 using a data set with 20,000 transcripts. GSEA analysis revealed that genes downregulated in Foxm1-deficient HSCs were enriched for gene set associated with cytokine signaling in immune system (Fig. 6c, left). Notably, the genes targeted by AML1 (Runx1), a critical regulator of HSC self-renewal, were also down-regulated in Foxm1-deficient HSCs (Fig. 6c, right). Consistent with the observed increase in cycling HSCs in Foxm1-deficient mice, genes upregulated in Foxm1-deficient HSCs were enriched for a gene set associated with S phase progression (Fig. 6d, left). E2F1 is a positive regulator of G1/S cell cycle37, and E2F1-targeted genes were also enriched in the upregulated genes in Foxm1-deficient HSCs (Fig. 6d, right), providing additional evidence for enhanced cell cycling in Foxm1-deficient HSCs. Thus, Foxm1 deficiency-mediated transcriptional changes implicate that absence of Foxm1 perturbs multiple stem cell-maintenance mechanisms.
Figure 6. Genes and molecular pathways dysregulated in Foxm1-deficient HSCs.
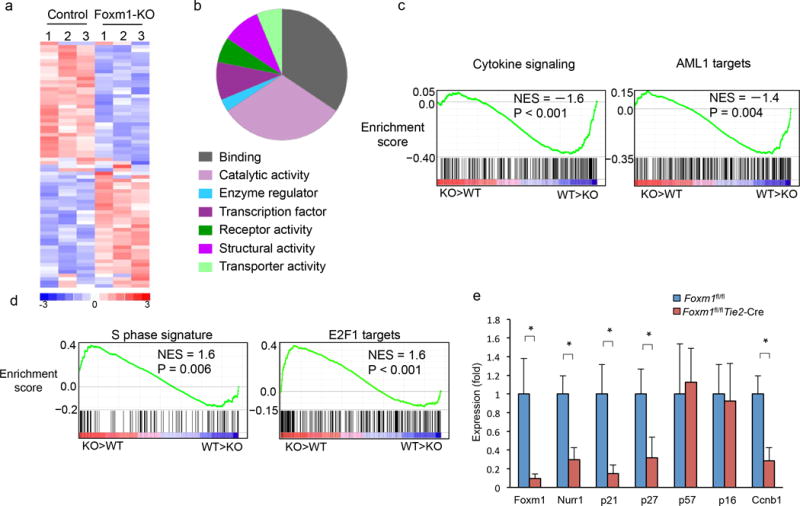
(a) Heat map shows the expression of the 70 Foxm1-regulated genes in LT-HSCs from Foxm1-deficient or wild-type mice (≥ 2 fold, P≤0.05). The experiments were performed in triplicate. Upregulated and down-regulated genes are represented as red and blue, respectively. (b)Pie charts show the distribution of 70 genes which are differentially expressed in Foxm1-deficent and control LT-HSCs cells into functional groups. (c–d) Enrichment plots of selected gene sets from GSEA analysis. Expression data with 20,000 transcripts was used for analysis. (e) qPCR analysis of the expression of selected genes in LT-HSCs. Gene expression was initially normalized to actb expression. Values represent the fold changes in gene expression relative to that in control HSCs. *, P < 0.05.
Nurr1 mediates Foxm1 function in regulating HSC quiescence
Of the genes that are significantly down-regulated in Foxm1-deficient HSCs (Fig. 6a), the orphan nuclear receptor Nurr1 (known as Nr4a2) stands out due to its important role in the maintenance of HSC quiescence. Loss of a single allele of Nurr1 is sufficient to induce HSCs to enter into the cell cycle and proliferate17. This effect is associated with downregulation of cyclin-dependent kinase inhibitors (CKIs) including p21 and p27 in Nurr1-null HSCs17. Notably, our expression profiling data (confirmed by qRT-PCR, Fig. 6e) revealed that the absence of Foxm1 in HSCs correlates with downregulation of Nurr1, p21 and p27. In contrast, p57 and p16 expression are comparable in both Foxm1-deficient and wild-type HSCs. Together, the data suggests that Foxm1 deletion promotes cell cycling of HSCs at molecular level. Ccnb1, which is associated with G2/M transition, is a down-stream target of Foxm1 in other cell types7. Our gene expression data (confirmed by qRT-PCR, Fig. 6e) indicates that Ccnb1 was down-regulated in Foxm1-deficient HSCs, which is consistent with our finding that G2/M transition is delayed in these cells.
To determine whether Foxm1 modulates Nurr1 expression through direct transcriptional activation, we searched for the consensus Foxm1 binding site [T(G/A)TTT(G/A)TT]38 in the proximal promoter region of Nurr1. Two putative Foxm1 binding sites were identified upstream of the Nurr1 transcription start site (TSS) (Fig. 7a). We next performed dual luciferase reporter assays using cells expressing either the wild-type Nurr1 promoter or a mutant promoter with mutations of the predicted Foxm1 binding sites. The luciferase activity of a construct containing binding site 1 but not mutated binding site 1 or site 2 alone, was activated by Foxm1 expression (Fig. 7b,c), suggesting that integrity of the consensus site 1 in the Nurr1 promoter is required for Foxm1-mediated activation of Nurr1 expression. The chromatin immunoprecipitation (ChIP) assay using Lin− BM cells revealed that Foxm1 directly bound to the site 1 but not site 2 of Nurr1 promoter (Fig. 7d).
Figure 7. Forced expression of Nurr1 rescues Foxm1-deletion-induced loss of quiescence in vivo.
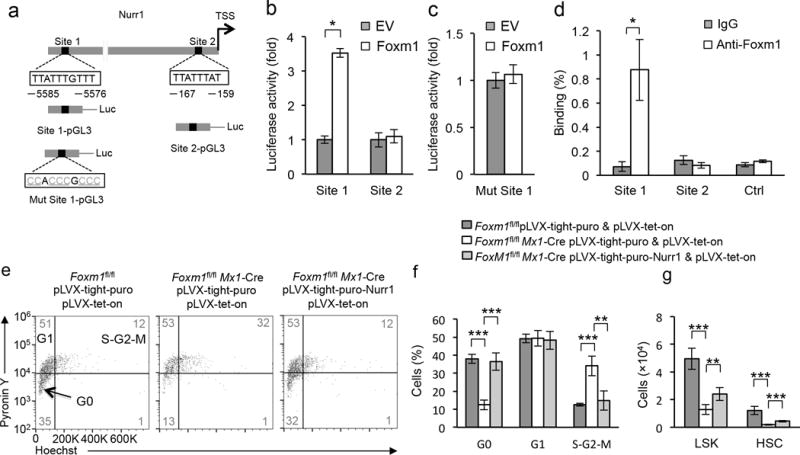
(a) Schematic diagram of the mouse Nurr1 upstream promoter region, the wild-type and the mutated Nurr1 promoter luciferase constructs. Predicted Foxm1 binding regions are shown: site 1 and site 2. Arrows indicate the transcription start site. The core motif TTATTTGTTT was mutated into CCACCCGCCC in the mutant Nurr1 promoter luciferase construct. (b,c) Luciferase reporter assays. 293T cells were transfected with wild-type (b), or mutated (c) Nurr1 promoter luciferase constructs, and either pMSCV-PIG-Foxm1 (Foxm1), or empty vector (EV). (d) Endogenous binding of Foxm1 to the upstream region of Nurr1 as determined by ChIP assay in Lin− BM cells. IgG was used as a negative control. (e,f) Induced expression of Nurr1 increased the proportion of Foxm1-deficient LSK cells in G0 phase. Representative histograms (e) of flow cytometric analyses of G0-G1 cell cycle in LSK cells from Foxm1fl/fl control chimeric mice with expression of control vector, Foxm1fl/flMx1-Cre chimeric mice with expression of Nurr1 or control vector. The histogram (f) depicts cell cycle status of LSK cells these mice (mean±SD, n=5). (g) The histogram depicts the total number of LSK cells and HSCs from Foxm1fl/fl control chimeric mice, Foxm1fl/flMx1-Cre chimeric mice with expression of Nurr1 or control vector (mean±SD, n=5). **,<0.005; ***, P<0.0005. Data are from three experiments (b,c,d; mean±SD, n=3) or two experiments (f,g; mean±SD of n=5 mice per genotype) or are representative of two experiments (e).
To determine whether Nurr1 downregulation is critical for the loss of quiescence seen in Foxm1-deficient LSK cells, we examined the effect of enforced expression of Nurr1 on the quiescence of Foxm1fl/flMx1-Cre LSK cells in vivo. Nurr1 was expressed under control of PTight, a modified Tet-responsive promoter39 in a lentiviral vector. The Foxm1fl/fl or Foxm1fl/flMx1Mx1-Cre BM cells infected with pLVX-Tet-on and Nurr1- or vector- lentivirus were transplanted into the letheally-irradiated recipient mice to generate chimeric mice. The induction of Foxm1 depletion and expression of Nurr1 in these mice was induced by pI-pC and Doxycycline (Dox) respectively 6 weeks post-transplantation. The induction of Nurr1 expression in BM cells from chimeric mice was confirmed by immunoblot (Supplementary Fig. 8a). Notably, the frequency of quiescent cells in Nurr1-transduced Foxm1fl/flMx1-Cre LSKs was significantly increased to the level similar to that in vector-transduced control Foxm1fl/fl LSK cells in chimeric mice (Fig. 7e,f). The frequency of LSK cells and LT-HSCs was increased in BM cells from Nurr1-transduced Foxm1fl/flMx1-Cre chimeric mice as compared to control vector- transduced Foxm1fl/flMx1-Cre chimeric mice even after induction of Nurr1 expression for two weeks (Fig. 7g), indicating that Nurr1 expression prevents LT-HSCs and LSK cells depletion induced by absence of Foxm1. In addition, both p21 and p27 were upregulated by Nurr1 expression, reversing the downregulation of p27 and p21 expression in Foxm1-deficient LSK cells (supplementary Fig. 8b). Together these data suggest that Nurr1 insufficiency mediates Foxm1-depletion-induced quiescence loss of LSK cells.
Down-regulation of FOXM1 in a subset of MDS patients
To determine the function of FOXM1 in cell cycle regulation of human hematopoietic stem/progenitor cells, we investigated whether downregulation of FOXM1 expression in human cold blood CD34+ hematopoietic cells could lead to the same perturbations in cell cycle that we observed in Foxm1 null mice. We down-regulated FOXM1 expression in human cord blood CD34+ cells by shRNAs. Consistent with previously published results40, a significant number of CD34+ hematopoietic stem cells were quiescent after several days of in vitro culture (Fig. 8a,b). We observed increased proliferation (percentage in S phase) and decreased numbers of quiescent cells (percentage in G0) when FOXM1 expression was reduced by shRNAs (Fig. 8a,b). Thus, decreasing FOXM1 expression in these human cells reduces quiescence. Decreased HSC function contributes to the development of myelodysplastic syndrome (MDS), which is a clonal HSC disorder characterized by infective hematopoiesis41. By analyzing the published microarray dataset of CD34+ HSPCs from 183 MDS patients with different cytogenetic abnormalities and 17 healthy controls42, we found that Foxm1 and Nurr1 were all down-regulated in CD34+ cells from MDS patients with deletion of chromosome 5q [5q(del)] as compared to the cells from healthy individuals. Notably, reducing FOXM1 expression by shRNAs in human primary CD34+ cells led to down-regulation of NURR1 (Fig. 8d). Our findings raise the possibility that FOXM1-mediated pathway plays a critical role in the pathogenesis of 5q(del) MDS through deregulating HSC function in patients.
Figure 8.
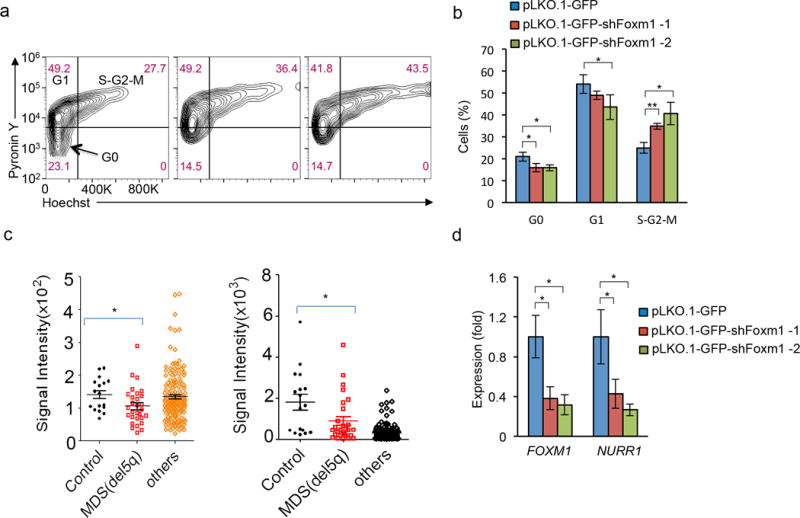
Foxm1 downregulation is associated with a subset of 5q(del) MDS and reduced expression of Foxm1 in human primitive hematopoietic cells decreases quiescence. (a) Representatitive histograms of flow cytometric analysis of quiescence of CD34+ cells expressing PLKO-GFP vector and PLKO-GFP-shRNAs against Foxm1. (b) The histogram depicts cell cycle status of CD34+ cells. The experiments were repeated three times. (c) Microarray analysis of FOXM1 and NURR1 expression in CD34+ cells from 41 MDS patients with del(5q) or 142 other genetic abnormalities. The CD34+ cells from 17 healthy individuals were used as controls. (d). qRT-PCR analysis of FOXM1 and NURR1 expression in human CD34+ cells expressing control vector or shRNAs against FOXM1. Gene expression was initially normalized to ACTB expression. Values represent the fold changes in gene expression relative to that in CD34+ cells expressing control vector. *, P < 0.05. Data are representative of two experiments (a), or from two experiments (b,d; mean±SD, n=3).
DISCUSSION
In this study, we have identified a previously unrecognized role for Foxm1 in the maintenance of HSCs and HPCs. Using conditional knockout murine models, we have shown that Foxm1 is essential for maintaining HSC quiescence and self-renewal in vivo, and that it intrinsically regulates HSC and HPC proliferation, but not HSC and HPC differentiation. We also have identified Nurr1 as a novel downstream effector of Foxm1, which can be directly activated by Foxm1 and can mediate the function of Foxm1 in regulating HSC quiescence.
Foxm1 has been consistently implicated as a ‘pro-proliferation’ factor in proliferating cells through promoting cell cycle progression5. However, some studies have suggested a role for Foxm1 in self-renewal of stem cells. Foxm1 is involved in maintenance of ES (embryonic stem) cell pluripotency43. Interestingly, deletion of Foxm1 in the neural cortical stem/progenitor cells from Day 14.5 embryonic cortical tissue reduced their formation of neurosphere in vitro, implicating a possible role of Foxm1 in self-renewal of neural stem/progenitor cells44. Foxm1 also regulates glioma-initiating cell (GIC) self-renewal in vitro by promoting β-catenin nuclear localization (and thus activation of its target genes)45. However, the in vivo role of Foxm1 in adult stem cells has not been previously characterized. Our findings provide the first in vivo evidence that Foxm1 appears to have divergent functions in HSCs. We observed that either embryonic or adult induction of Foxm1 loss in HSCs (by either Tie2-Cre or Mx1-Cre respectively) had almost identical effects on HSC and HPC functions, suggesting that Foxm1 intrinsically regulates adult HSC self-renewal and quiescence. Foxm1 is required for injury-induced regeneration of adult liver, lung and pancreas5. Our findings raise the possibility that the role of Foxm1 in tissue regeneration may be, in part, mediated by the impact of Foxm1 on relevant tissue-specific stem cells.
Previous studies showed that Foxm1 stimulated proliferation by promoting cell cycle entry into S phase and M phase5. In contrast, we observe here that Foxm1 increases proliferation of HSCs and HPCs. In line with our observation that Foxm1 loss promotes proliferation of HSCs and HPCs, our expression profiling data showed that E2F1 target genes and other genes promoting S phase transition were significantly upregulated when Foxm1 was absent in HSCs, providing molecular evidence for the observed promotion of S phase progression in HSCs. Moreover, Foxm1 loss led to downregulation of Nurr1 and CKIs including p21 and p27. Of interest, previous studies have shown that loss of even a single allele of Nurr1 markedly impairs HSC quiescence and their capacity for self-renewal, and is associated with downregulation of p21 and p27 in HSCs17. We found that ectopic expression of Nurr1 reversed down-regulation of p21 and p27 induced by Foxm1 deletion, suggesting that these effects are likely mediated by Nurr1. Finally, our studies indicate that Foxm1 activates Nurr1 expression by directly binding its promoter and that forced expression of Nurr1 reverses the loss of quiescence in Foxm1-deficient stem cell-enriched populations. Together, these results suggest that Foxm1 regulates HSC quiescence through control of Nurr1-mediated downstream pathways. Nonetheless, analysis of global gene expression in Foxm1-deficient and control LT-HSCs suggests that Foxm1 regulates the function of HSCs through multiple molecular pathways. Additional studies are required to determine whether the other downstream pathways are important for mediating Foxm1 function in HSCs.
There are several known downstream targets of Foxm1 including sox2 and Bmi1, which appear to be important for self-renewal of neural stem cells44. However, we did not observe changes in sox2 or Bmi1 expression in Foxm1-deficient HSCs compared with control HSCs. In addition, we did not detect significant changes in Foxm1-deficient HSCs in the expression of β-catenin target genes (regulated by Foxm1 in GICs45). Thus, we conclude that Foxm1 regulates gene expression in a tissue specific manner, perhaps to confer the divergent functions of Foxm1 in various tissues.
The role of Foxm1 in the regulation of apoptosis is controversial46. We found that Foxm1 depletion did not affect apoptosis of HSCs and HPCs at steady state. However, absence of Foxm1 induced apoptosis of HSCs and HPCs upon transplantation into chimeric mice, a condition that exposes HSCs and HPCs to various stresses33. These data indicate a role for Foxm1 in maintaining HSC or HPC survival under conditions of cellular stress.
The role of FOXM1 in MDS is unknown. By analyzing the public database, we find that FOXM1 and its down-stream target NURR1 are both significantly down-regulated in a subset of MDS patients. More importantly, reduced expression of FOXM1 promotes loss of quiescence and down-regulates NURR1 expression in human primary CD34+ hematopoietic stem/progenitor cells. Thus, our data implicate a critical role of FOXM1 in maintaining human stem/progenitor cells and that deregulation of FOXM1 expression may contribute to human hematological malignant diseases such as MDS by affecting the function of HSCs/HPCs. In summary, we provide functional and molecular evidence that Foxm1 acts a critical regulator of self-renewal of adult HSCs through control of HSC quiescence and provide evidence in support of a tissue-specific multi-faceted role of Foxm1 in cell cycle regulation.
ONLINE METHODS
Mice and blood cell counts
For conditional deletion of Foxm1 in vivo, Foxm1fl/fl mice were mated to Tie2-Cre or Mx1-Cre transgenic mice to generate Foxm1fl/flTie2-Cre or Foxm1fl/flMx1-Cre mice. Mx1-Cre expression was induced by 3 intraperitoneal (i.p.) injections of 6–10 μg polyI–polyC (pI-pC) (GE Healthcare) per gram of body weight every second day for a total of three injections. Times after pI-pC injection are counted from the third pI-pC injection. Peripheral blood samples were collected by tail bleeding into tubes containing EDTA. Complete blood counts and differentials were obtained using a Hemavet 950FS (Drew Scientific). All animal research was approved by the University of Illinois at Chicago Institutional Animal Care and Use Committee.
Flow cytometry
Single-cell suspensions were prepared from bone marrow (femurs and tibiae), spleen, thymus and peripheral blood. Red cells were lysed with ammonium-chloride-potassium (ACK) buffer. Cells were incubated with antibodies for 20 minutes on ice. The following fluorochrome- or biotin-conjugated antibodies to mouse molecules were used: Gr-1, Ter119, B220, CD19, IgM, IL-7R, CD3 for lineage markers, Streptavidin-PE-Cy5, PE-Sca-1, APC-Cy7-c-Kit, PE-Cy7-CD48, and APC-CD150 for HSC, LSK, and HPC analysis; Streptavidin-APC-Cy7, PE-Sca-1, APC-c-Kit, PE-Cy5-Flt3 for LMPPs analysis; Streaptavidin-APC-Cy7, PE-Sca-1, APC-c-Kit, PE-Cy7-CD16/32, eFluor450-CD34 for CMPs, GMPs, MEPs analysis47,48; PE-B220 and APC-IgM for B cells; PE-Gr-1 and APC-Mac-1 for myeloid cells; APC-Ter119 and PE-CD71 for erythroid cells; CD4 and CD8 for mature T cells; PE-Cy7-CD41and APC-c-Kit for megakaryocyte; PE-F4/80 and APC-MAC for macrophage; APC-Cy7-CD45.2 or FITC-CD45.2 and PE-CD45.1 were used to identify the donor cells in recipient mice. All of the antibodies are purchased from eBioscience, except APC-CD150 (from Biolegend) (Supplementary Table 3). For cell cycle analysis, 5×106 BM cells were stained for HSCs, then fixed with 1% formaldehyde in PBS, treated with 0.1% tritionX-100, stained with 5 μg/ml DAPI, and analyzed on Gallios™ Flow Cytometer. For the cell cycle analysis using Hoechst 33342 and Pyronin Y, BM Cells were incubated with 5 μg/ml Hoechst dye for 45 min at 37 °C, and then further incubated with Hoechst dye and 1 μg/ml Pyronin Y at 37 °C for 45 min. Cells were stained with lineage cocktail, and then the cells were stained with Streptavidin-APC-Cy7, FITC-Sca-1and APC-c-Kit. For the detection of apoptosis, BM cells were stained with antibody conjugates, Annexin V, and DAPI.
Colony-forming assays
6×104 Bone marrow cells from Foxm1fl/fl or Foxm1fl/flTie2-Cre mice were plated in triplicate in 35-mm tissue culture dishes containing Mouse Methylcellulose Complete Media (HSC007, R&D systems), after 10 days of incubation at 37 °C in 5% CO2, CFU-GEMM, CFU-GM, CFU-G, CFU-M, BFU-E were scored under an inverted microscope.
Bone marrow transplantation assays
For competitive reconstitution assays, total BM cells (2.5 × 106) or sorted CD150+CD48−LSK cells (2 × 102) from Foxm1fl/flTie2-Cre, Foxm1fl/flMx1-Cre or corresponding littermate control mice (CD45.2) were transplanted into lethally irradiated Ly5.1 (CD45.1) recipients together with competitor BM cells (2.5 × 106) for BM cell analysis or (0.4 × 106) for HSC analysis from Ly5.1/B6 mice (CD45.1+ CD45.2+). For second transplantation, BM cells (2 × 106) were transferred to a second set of lethally irradiated mice. For bone marrow transplantation assays, BM cells (5 × 106) from Foxm1fl/fl, Foxm1fl/flMx1-Cre were transplanted into lethally irradiated Ly5.1 mice. For serial transplantation analysis, BM cells (1 × 106) were obtained from recipient at 4 months after first transplantation and were transferred to a second set of lethally irradiated mice.
5-FU treatment
5-FU was administrated to mice intraperitoneally (i.p.) at a dose of 150 mg/kg once per week for 3 weeks, and the survival of individual mice was monitored daily.
BrdU incorporation assay
Mice were injected intraperitoneally with 100 μl of 10 mg/ml BrdU (Sigma) 24 h before the mice were sacrificed for analysis. BM cells were stained with fluorochrome-conjugated antibodies, followed by fixation and permeabilization with Cytofix/Cytoperm (BD Biosciences), treatment with DNaseI (Sigma), and staining with a BrdU-specific antibody (eBioscience) according to the BrdU flow kit instruction manual (BD Pharmingen). The cells were analyzed by flow cytometry using a CyAn ADP flow cytometer.
Microarray and bioinformatics analyses
Total RNA was extracted from sorted LT-HSC cells (CD150+CD48−LSK) from 2-month old Foxm1fl/fl and Foxm1fl/flTie2-Cre mice with QIAGEN RNeasy Micro Kit (Qiagen) and amplified with Ovation® Pico WTA System V2 (NuGEN Technologies, Inc.). Labeled cDNA was hybridized to the Mouse Gene 1.0 ST Array (Affymetrix). Chip quality was analyzed and determined using Affymetrix GCOS software. All RNA samples and arrays used in this study passed the established quality criteria. The raw data were normalized using R/Bioconductor package “affy”49. Gene set enrichment analysis was performed with GSEA v2.0 software available from the broad institute (http://www.broad.mit.edu/gsea).
RNA extraction and quantitative RT-PCR analysis
Total RNA was isolated using an QIAGEN RNeasy Mini Kit, or QIAGEN Rneasy Micro Kit. cDNAs were reverse-transcribed from total RNA using SuperScript III First-Strand Synthesis System (Life technologies). Then cDNAs were subjected to real-time PCR using SYBR Green Supermix (BIO-RAD) in realtime PCR System I-cycler (BIO-RAD). All of the samples were run in triplicate. Amplification of Actb was used for sample normalization. Primers are provided in Supplementary Table 4.
Chip assay
Lin− BM cells from wild-type mice were fixed with 1% of formaldehyde at 37 °C for 10 min, the reaction was stopped by adding glycine to a final concentration of 125 mM at 25°C for 5 min. Cells were washed 2 times with cold PBS, and lysed in Szak RIPA buffer (150 mM Nacl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris pH8.0, 5 mM EDTA) with protease inhibitors cocktail, After cell lysis, cross-linked chromatin was sheared using a sonicator. The 5 μg of anti-Foxm1 (K-19; Santa Cruz Biotechnology) or control IgG antibodies were used for immunoprecipitation. After eluting DNA from precipitated immune complexes, quantitative real-time PCR analysis was performed using specific primers to amplify Site1and Site2 in Nurr1 promoter region, and a control site reside in 8kb downstream of the last exon of ccnb1 gene. The sequences of the primers are provided in Supplementary Table 4.
Luciferase reporter assay
Two putative Foxm1 binding sites were identified by searching the consensus Foxm1 binding site [T(G/A)TTT(G/A)TT] upstream of the Nurr1 transcription start site. The corresponding genomic region which included these putative binding sites were amplified by PCR from WT C57BL/6 mouse genomic DNA, subsequently subcloned into a pGL3-Basic luciferase reporter vector, and named Site 1-pGL3, Site 2-pGL3. Mut-Site 1-pGL3 was generated by site-directed mutagenesis of Foxm1 binding site in Site 1-pGL3. Primer sequences are provided in the Supplementary Table 4. Site 1-pGL3, Site 2-pGL3 or Mut-Site 1-pGL3 were co-transfected into 293T cells using lipid-based 293T TransIT reagent (Mirus Bio) with either MigR1 empty vector or MigR1-Foxm1, as well as phRL-SV40 vector as an internal control. Cells were collected 36 h after transfection and both luciferase activities were determined using Dual-Luciferase reporter assay system (Promega) following the manufacturer’s instructions.
Purification of human CD34+ cells from cord blood
Human cord blood cells, purchased from New York Blood Center (New York, NY), were diluted with an equal volume of PBS containing 20% fetal bovine serum (Gibco) and 0.5 mM EDTA (Gibco). The diluted cells were then separated on a Ficoll density gradient (GE health care) at 400g for 30 min at 25°C. The mononuclear cells at the interface were collected and washed with PBS containing 2% fetal bovine serum once at 300g for 10 min and twice at 200g for 10 min. CD34+ cells were isolated from the mononuclear population by positive selection using the CD34 MicroBead Kit (Miltenyi Biotec) according to the manufacturer’s instructions. To obtain high purity, the cells were first passed through the LS column and then through the MS column (Miltenyi Biotec). The purity of CD34+ cells was above 90% as determined by flow cytometry analysis.
Lentiviral constructs and packaging
To generate the Dox-inducible Flag-Nurr1 expression vector, the Nurr1 cDNA were obtained by PCR, and cloned into pLVX-Tight-Puro (Clontech, PT3996-5). The cloned Nurr1 gene was confirmed by DNA sequencing. pLVX-Tight-Puro is a lentiviral expression vector that allows tightly regulated, doxycycline-controlled expression of a gene of interest. To generate the Foxm1 shRNA vector, we first constructed pLKO.1maxGFP2aPuro vector by cloning maxGFP2aPuro fragment into BamHI and NsiI sites of pLKO.1 vector. Then, we cloned Foxm1 shRNA 1#, 2# into the pLKO.1maxGFP2aPuro vector. The sequences of the primers are provided in Supplementary Table 4.
For lentivirus production, 293T cells were transfected with pCMV△R8.91 helper plasmid and pMD.G using Lipofectamine 3000. Medium was replaced with fresh medium 12 h after transfection. The culture supernatants were collected at 36 h after transfection, and concentrated by ultracentrifugation in a Beckman Optima L-90K ultracentrifuge using a SW32 Ti rotor at 100,000 × g for 2.5 h, 4 °C. Virus pellet was resuspended in fresh medium, and stored at −80 °C until use.
Lentiviral infection of cells
Human CD34+ cells were cultured in stemspam serum-free expansion medium (Stemcell technologies) with 10 ng/ml human SCF, 20 ng/ml human TPO, 10 ng/ml FGF-1, 500 ng/ml ANGPTC and 10 ng/ml heparin as previously described50. For lentiviral infection of human CD34+ cells, lentiviral stock was added to CD34+ cells, and then transferred onto RetroNectin (Takara Mirus) – coated 96-well plates. After incubation at 37 °C overnight, supernatants were removed, washed with PBS, and cells were resuspended in fresh medium. Similar methods were used for infection of mouse Lin− BM cells. BM cells were collected from Foxm1fl/fl and Foxm1fl/flMx1-Cre mice, and Lin+ cells were depleted with Dynabeads (Life Technologies).
Statistical analysis
Statistical significance was calculated using the two tailed Student’s t test.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health grant RO1 CA140979 (to Z.Q.). We thank M.A. Goodell, Baylor College of Medicine (Houston, TX), for providing the plasmid MSCV-FlagNurr1.
Footnotes
Accession codes
The gene expression data for the HSCs in Foxm1fl/fl and Foxm1fl/flTie2-Cre has been deposited in the Gene Expression Omnibus (GEO) under accession number: (GSE62360).
AUTHOR CONTRIBUTIONS
Y.H.(Yu Hou), W.L., Y.S., Z.Z., L.L and Y.H (Yong Huang). performed research; Z.Q. and Y.H.(Yu Hou) designed research and performed data analysis; W.W., J.G.Q., D.P. T.Z. and Y.Z. provided advice and new reagents/analytic tools; Z.Q., Y.H.(Yu Hou), D.P. and J.Q. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Wilson A, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 2.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 3.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nature reviews Molecular cell biology. 2013;14:329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pietras EM, Warr MR, Passegue E. Cell cycle regulation in hematopoietic stem cells. J Cell Biol. 2011;195:709–720. doi: 10.1083/jcb.201102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalin TV, Ustiyan V, Kalinichenko VV. Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle. 2011;10:396–405. doi: 10.4161/cc.10.3.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa RH. FoxM1 dances with mitosis. Nat Cell Biol. 2005;7:108–110. doi: 10.1038/ncb0205-108. [DOI] [PubMed] [Google Scholar]
- 7.Laoukili J, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 8.Ye H, et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. 1997;17:1626–1641. doi: 10.1128/mcb.17.3.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalin TV, et al. Forkhead Box m1 transcription factor is required for perinatal lung function. Proc Natl Acad Sci U S A. 2008;105:19330–19335. doi: 10.1073/pnas.0806748105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ustiyan V, et al. Forkhead box M1 transcriptional factor is required for smooth muscle cells during embryonic development of blood vessels and esophagus. Developmental biology. 2009;336:266–279. doi: 10.1016/j.ydbio.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci U S A. 2002;99:16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. 2008;57:3069–3077. doi: 10.2337/db08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye H, Holterman AX, Yoo KW, Franks RR, Costa RH. Premature expression of the winged helix transcription factor HFH-11B in regenerating mouse liver accelerates hepatocyte entry into S phase. Mol Cell Biol. 1999;19:8570–8580. doi: 10.1128/mcb.19.12.8570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koo CY, Muir KW, Lam EW. FOXM1: From cancer initiation to progression and treatment. Biochim Biophys Acta. 2012;1819:28–37. doi: 10.1016/j.bbagrm.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Xue L, Chiang L, He B, Zhao YY, Winoto A. FoxM1, a forkhead transcription factor is a master cell cycle regulator for mouse mature T cells but not double positive thymocytes. PLoS One. 2010;5:e9229. doi: 10.1371/journal.pone.0009229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren X, et al. Forkhead box M1 transcription factor is required for macrophage recruitment during liver repair. Mol Cell Biol. 2010;30:5381–5393. doi: 10.1128/MCB.00876-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sirin O, Lukov GL, Mao R, Conneely OM, Goodell MA. The orphan nuclear receptor Nurr1 restricts the proliferation of haematopoietic stem cells. Nat Cell Biol. 2010;12:1213–1219. doi: 10.1038/ncb2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kisanuki YY, et al. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Developmental biology. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 19.Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell. 2008;2:484–496. doi: 10.1016/j.stem.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Socolovsky M, et al. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 21.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 22.Adolfsson J, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler JM, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kobayashi H, et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2010;12:1046–1056. doi: 10.1038/ncb2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 27.Cheng T, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 28.Ma RY, et al. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J Cell Sci. 2005;118:795–806. doi: 10.1242/jcs.01657. [DOI] [PubMed] [Google Scholar]
- 29.Wang IC, et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25:10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang IC, et al. FoxM1 regulates transcription of JNK1 to promote the G1/S transition and tumor cell invasiveness. J Biol Chem. 2008;283:20770–20778. doi: 10.1074/jbc.M709892200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yilmaz OH, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 32.Qian Z, Chen L, Fernald AA, Williams BO, Le Beau MM. A critical role for Apc in hematopoietic stem and progenitor cell survival. J Exp Med. 2008;205:2163–2175. doi: 10.1084/jem.20080578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison DE, Stone M, Astle CM. Effects of transplantation on the primitive immunohematopoietic stem cell. J Exp Med. 1990;172:431–437. doi: 10.1084/jem.172.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xue S, et al. RNA regulons in Hox 5′ UTRs confer ribosome specificity to gene regulation. Nature. 2015;517:33–38. doi: 10.1038/nature14010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509:49–54. doi: 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Helin K. Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev. 1998;8:28–35. doi: 10.1016/s0959-437x(98)80058-0. [DOI] [PubMed] [Google Scholar]
- 38.Wierstra I, Alves J. Despite its strong transactivation domain, transcription factor FOXM1c is kept almost inactive by two different inhibitory domains. Biological chemistry. 2006;387:963–976. doi: 10.1515/BC.2006.120. [DOI] [PubMed] [Google Scholar]
- 39.Gossen M, et al. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 40.Lacorazza HD, et al. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell. 2006;9:175–187. doi: 10.1016/j.ccr.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 41.Nimer SD. MDS: a stem cell disorder–but what exactly is wrong with the primitive hematopoietic cells in this disease? Hematology Am Soc Hematol Educ Program. 2008:43–51. doi: 10.1182/asheducation-2008.1.43. [DOI] [PubMed] [Google Scholar]
- 42.Pellagatti A, et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia. 2010;24:756–764. doi: 10.1038/leu.2010.31. [DOI] [PubMed] [Google Scholar]
- 43.Xie Z, et al. Foxm1 transcription factor is required for maintenance of pluripotency of P19 embryonal carcinoma cells. Nucleic Acids Res. 2010;38:8027–8038. doi: 10.1093/nar/gkq715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, et al. FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res. 2011;71:4292–4302. doi: 10.1158/0008-5472.CAN-10-4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang N, et al. FoxM1 promotes beta-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wierstra I. The transcription factor FOXM1 (Forkhead box M1): proliferation-specific expression, transcription factor function, target genes, mouse models, and normal biological roles. Adv Cancer Res. 2013;118:97–398. doi: 10.1016/B978-0-12-407173-5.00004-2. [DOI] [PubMed] [Google Scholar]
- 47.Qian Z, Chen L, Fernald AA, Williams BO, Le Beau MM. A critical role for Apc in hematopoietic stem and progenitor cell survival. J Exp Med. 2008;205:2163–2175. doi: 10.1084/jem.20080578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou Y, et al. FHL2 regulates hematopoietic stem cell functions under stress conditions. Leukemia. 2014 doi: 10.1038/leu.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 50.Zhang CC, Kaba M, Iizuka S, Huynh H, Lodish HF. Angiopoietin-like 5 and IGFBP2 stimulate ex vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation. Blood. 2008;111:3415–3423. doi: 10.1182/blood-2007-11-122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.