

Abstract
N6-methyladenosine (m6A), the most abundant internal RNA modification, functions in diverse biological processes, including regulation of embryonic stem cell self-renewal and differentiation. As yet, methods to detect m6A in the transcriptome rely on the availability and quality of an m6A antibody and are often associated with a high rate of false positives. Here, based on our observation that m6A interferes with A–T/U pairing, we report a microarray-based technology to map m6A sites in mouse embryonic stem cells. We identified 72 unbiased sites exhibiting high m6A levels from 66 PolyA RNAs. Bioinformatics analyses suggest identified sites are enriched on developmental regulators and may in some contexts modulate microRNA/mRNA interactions. Overall, we have developed microarray-based technology to capture highly enriched m6A sites in the mammalian transcriptome. This method provides an alternative means to identify m6A sites for certain applications.
Keywords: N6-methyladenosine, two-color microarray, RNA methylation, METTL3, METTL14, mouse embryonic stem cells
INTRODUCTION
Although PolyA RNA has long been known to bear m6A modification (Fu et al. 2014; Meyer and Jaffrey 2014), lack of knowledge on methylating enzymes and the absence of technologies useful to locate m6A in the transcriptome has hindered our understanding of its function. Recent advancement in both fronts has re-kindled interest in m6A function. In the past few years, both mammalian m6A methyltransferases (Liu et al. 2014; Ping et al. 2014; Schwartz et al. 2014; Wang et al. 2014b) and demethylases (Jia et al. 2011; Zheng et al. 2013) have been reported. Among them, m6A methyltransferase is a large protein complex, consisting in part of methyltransferase-like 3 (METTL3) and methyltransferase-like 14 (METTL14) catalytic subunits (Liu et al. 2014; Wang et al. 2014b). Both likely play important roles in mammalian development (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015). We have shown that in mouse ESCs (mESCs), m6A is enriched on and destabilizes lineage-specific genes to maintain cells in an undifferentiated state (Wang et al. 2014b).
In 2012, two studies independently reported a technology known as meRIP-seq (methylated RNA immunoprecipitation coupled with high-throughput sequencing) (Dominissini et al. 2012; Meyer et al. 2012) useful to locate m6A in the mammalian transcriptome. This method uses RNA fragmentation followed by RNA immunoprecipitation with an m6A antibody, library construction, and next-gen sequencing of IP'd RNA fragments. Identified sequences are then mapped to the mammalian transcriptome through bioinformatics analysis. Using meRIP-seq, authors of both studies revealed that m6A is enriched near stop codons at the 3′-UTR of thousands of mammalian mRNAs, suggesting that m6A regulates activity of numerous mammalian transcripts via 3′-UTR-related mechanisms. Since then, a handful of studies have reported improvements in meRIP-seq methodology to increase resolution to near the single-nucleotide level and decrease false-positive rates likely associated with immunoprecipitation (Schwartz et al. 2013; Chen et al. 2015a). Nonetheless, meRIP-seq, like any RIP method, still yields artifacts and requires at least 300 μg and up to milligrams of total RNA as starting material (Dominissini et al. 2012, 2013), minimizing its applicability to small samples. To overcome these issues, we have developed a microarray-based technology that represents the first nonantibody-based approach to map m6A on a large scale.
RESULTS
m6A interferes with A–U/T pairing
Enlightened by a previous study that detected novel yeast tRNA modifications by differential hybridization of modified versus unmodified RNA (Hiley et al. 2005), we asked whether m6A modification would alter hybridization of a transcript to an RNA or DNA probe. Methylation and A–U or A–T hydrogen bond formation occur on the same N6 position of adenine. Thus, we asked whether m6A interferes with A–U or A–T pairing by first carrying out complementation analysis using a Ribonuclease Protection Assay (RPA). To do so, we incubated equal amounts of in vitro-synthesized 140 nt RNA fragments of mouse β-actin mRNA, either modified or unmodified by m6A (Fig. 1A), with a complementary 32P-labeled 250-bp antisense actin RNA probe for 0.5, 2, or 4 h. Following annealing and digestion of unhybridized radiolabeled probe with RNAses, the reaction was loaded onto a PAGE gel to visualize and quantitate RNA duplex levels. We observed significant decreases in levels of radiolabeled RNA duplexes when we evaluated probes containing m6A after 4 h incubation (Fig. 1B,C), suggesting that m6A inhibits A–U or A–T pairing and indicating that microarray can be used to detect m6A.
FIGURE 1.
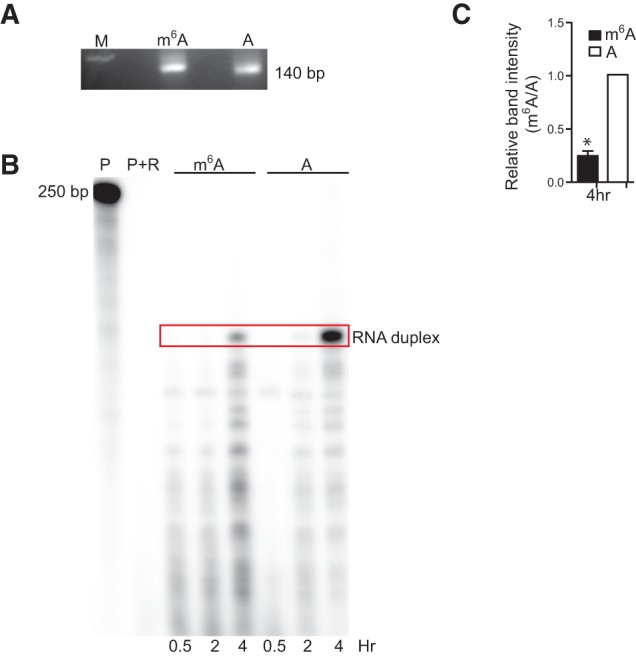
m6A interferes with A–U or A–T pairing. (A) Agarose gel showing equal loading of 1 μg sense actB RNA containing either m6A or A after PAGE purification. (M) marker, (m6A) sense actB RNA containing only m6A, (A) sense actB RNA containing only A. (B) RNA protection assay (RPA) showing that m6A-containing RNA does not anneal to a complementary RNA as efficiently as does an A-containing probe. (P) radiolabeled probe, (P + R) radiolabeled probe digested with RNAses A and T1 for 0.5, 2, or 4 h, and (Red box) sense/antisense RNA duplex. (C) Quantification of RPA results at 4 h. (*) Student's t-test, P ≤ 0.0018, n = 2.
Two-color tiling microarray can detect m6A
We next undertook a pilot microarray experiment using in vitro-transcribed RNA containing A or m6A as input. Eight randomly chosen DNA regions served as templates for the reaction (Supplemental Table S1), resulting in two RNA pools containing either A or m6A residues. As the percentage of m6A at specific site varies, we estimated array sensitivity by mixing A and m6A-RNA to obtain samples containing different proportions of m6A (0%, 2%, 8%, 16%, 32%, and 100%) (Supplemental Fig. S1). We chose the Agilent two-color array as the platform as it allows concurrent hybridization of Cy3-labeled RNA containing unmodified A (A-RNA) and Cy5-labeled m6A-RNA to the same array to compare signals within rather than between arrays, a condition that reduces experimental variation. To increase array sensitivity, 25-bp short oligonucleotide probes in reverse complementary to cDNA sequences were designed. Probes were also tiled at 5-nt intervals to minimize the possibility that a probe cannot pick up the m6A signal because of the presence of m6A in a corresponding region of the probe. We also tested different formamide concentrations to optimize hybridization conditions (Fig. 2B; Supplemental Table S1, “samples” sheet). Importantly, unlike conventional microarrays in which RNA is first transcribed into cDNA for hybridization, Cy3- and Cy5-labeled RNAs were mixed and directly hybridized to the array to preserve m6A signals. Following hybridization, we calculated overall Cy5/Cy3 ratios. As the Cy3 A-RNA sample is the same across all arrays, a lower Cy5/Cy3 ratio would suggest less hybridization of m6A- versus A-RNA to DNA probes. Figure 2B shows raw Cy5/Cy3 signal ratios in log scale from six different input samples with various m6A proportions and using different formamide concentrations. At 0% m6A, Cy5- and Cy3- channels contained the same amount of A-RNA, and we detected a log2 [Cy5/Cy3] of ∼1, as expected, as Cy5 is a stronger dye than Cy3 when the signal is preserved under ozone-free conditions, as we used here. Importantly, increasing m6A levels gradually decreased the Cy5/Cy3 ratio, particularly when m6A-RNA proportions exceeded 32% (Fig. 2B), demonstrating that abundant m6A indeed perturbs RNA hybridization. As use of 10% formamide resulted in highest detection sensitivity, and 20% formamide led to significant signal variation (Fig. 2B), we used 10% formamide in experiments reported below.
FIGURE 2.
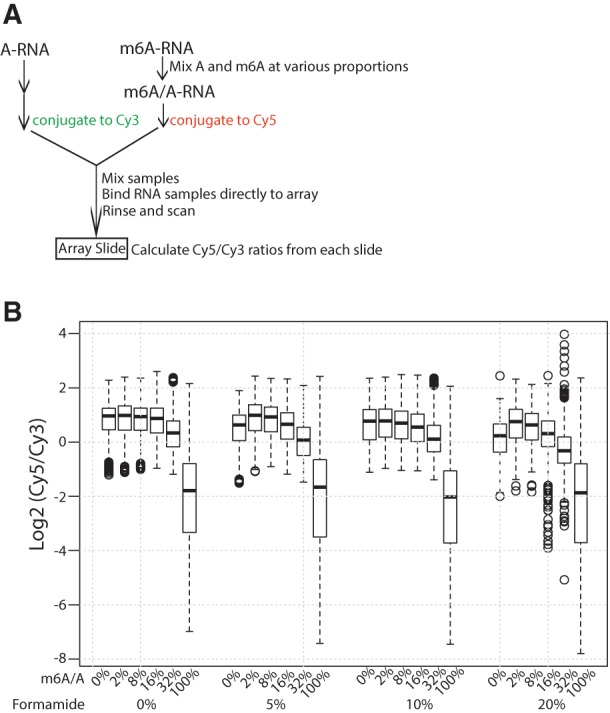
m6A is detected by a two-color microarray. (A) Schematic drawing of pilot array experiment. In vitro-synthesized A-RNA or different mixed m6A/A-RNA samples were labeled with Cy3 or Cy5, respectively. Cy3 and Cy5 samples were mixed and directly hybridized to an Agilent two-color tiling array, and Cy5/Cy3 signal ratios were calculated. (B) Overall Cy5/Cy3 signal ratios of A versus m6A/A mixed RNAs. Signal intensities in the Cy5 channel correspond to a RNA population that lacks m6A (i.e., background).
Detection of highly enriched m6A in the mESC transcriptome
Encouraged by pilot experiments, we used the same platform and experimental conditions to evaluate biological samples. Previously, using meRIP-seq, we identified 13,324 meRIP-seq peaks in mESCs (Wang et al. 2014b). As m6A sites are more likely to be located within these peaks than in the rest of transcriptome, we designed 1 × 1 million (1 M) microarrays with 947,952 probes with 6 nt tiling intervals covering these meRIP-seq peaks to examine m6A sites. All RNAs were DNAse-treated, PolyA-enriched and rRNA-depleted, and we used three sample pairs as starting materials. Sample 1 consisted of a population of Cy3-labeled RNAs from mESCs expressing scramble shRNA (Scr mESC) plus Cy5-labeled RNAs from METTL14 KD mESCs; Sample 2 was the same population of Cy3-labeled RNAs from shRNA control mESCs plus Cy5-labeled RNAs from cells treated with the global methylation inhibitor 3-deaza-adenosine (3-DZA) (Fig. 3A). Both METTL14 KD and DZA treatment decrease cellular m6A levels (Fustin et al. 2013; Wang et al. 2014b). In principle, an increase in the Cy5/Cy3 signal for a given probe reflects increased hybridization of an RNA molecule to the array following METTL14 KD or DZA treatment, relative to treatment with the scramble control (Fig. 3B). Sample #3 functioned as a negative control in which both Cy3 and Cy5 channels consist of RNAs extracted from METTL14 KD mESCs. Analysis was done in triplicate, requiring nine arrays. Upon array normalization, we searched for significant probes (SPs) that potentially hybridize to RNA regions containing m6A based on two independent analyses, the peak call and the sample test methods. Sample test method takes into consideration the biological variability in a given sample. We performed t-tests on each probe using means estimated from all probes within the same meRIP-seq peak and variance from the three biological replicates. A candidate SP was required have a value of P < 0.05 in both test conditions (METTL14 KD versus Scr and DZA versus Scr) but show no significant difference with the negative control (METTL14 KD versus METTL14 KD). Furthermore, based on the assumption that m6A likely alters hybridization of several tiled probes, the Cy5/Cy3 signal from the probe immediately upstream of a candidate SP also has to exhibit statistical significance relative to test samples but not the negative control. A different analysis, the peak call method, requires that a candidate SP must exhibit increased hybridization to RNA compared with surrounding probes, defining a SP within a local RNA context. Specifically, the Cy5/Cy3 ratio of a candidate SP from test versus negative control samples had to be significantly greater (False Discovery Rate or FDR <0.1) than the average Cy5/Cy3 ratios of nine tiling probes surrounding and including the candidate SP (that is, four probes immediately upstream of candidate SP, the candidate SP, and four downstream probes). Nine probes were used to model local signal variation for each anchor probe based on our array design in which any pair of adjacent probes overlaps by 19 nt; thus, each probe overlaps with four probes upstream and four probes downstream. As microarray methods are often associated with high signal variation, we defined a SP as a probe showing statistical significance in the both sample test and peak call methods.
FIGURE 3.

Detection of highly enriched m6A sites in mESCs. (A) Samples used for microarray analysis. For each, three independent arrays were used in triplicate. (B) Schematic showing expected outcome. Tiling probes were used to cover a given RNA region. If m6A is present on Cy3-labeled RNA extracted from the Scramble (Scr) control but not from METTL14 KD or DZA treated samples (Cy5), we predicted increased hybridization in treated versus Scr samples, hence a high Cy5/Cy3 ratio (increased red color) of tiling probes hybridized to the m6A-containing region versus nearby probes. (C) The Dtx4 transcript, one of 66 m6A-RNA targets we identified, illustrates how we identified significant probes (SPs). Columns 1–9 display log ratios of Cy5/Cy3 for Sample 1 (METTL14 KD versus Scr; red bar; columns 1–3), sample 2 (DZA treatment versus Scr; yellow bar; columns 4–6), and sample 3 (METTL14 KD versus METTL14 KD; blue bar; columns 7–9). Each row represents a probe. Adjacent probes overlap by 19 nt. Red and blue colors indicate high and low Cy5/Cy3 ratios, respectively. Columns 10 and 11 demonstrate the peak call method. A green square represents a probe whose Cy5/Cy3 ratio from Sample 1 and/or 2 relative to 3 is significantly greater (FDR < 0.1) than the average Cy5/Cy3 ratios from surrounding probes. Panels 12–14 demonstrate the sample test method. A green square represents a probe whose average Cy5/Cy3 ratio from the biological triplicates is significantly greater (P < 0.05) than average Cy5/Cy3 ratios of all probes within the same meRIP-seq peak in Samples 1 and 2 but not 3. A significant probe (SP, marked by asterisks [**]) is defined as a probe displaying statistical significance in both methods and whose upstream probe shows statistical significance in the sample test method. (D). Cumulative density curve demonstrating that meRIP-seq peaks containing the 206 SPs exhibit significantly higher enrichment scores than those from remaining probes (P < 8.06 × 10−7, one-sided Wilcoxon rank-sum test) using previously generated MeRIP-seq data.
RNA encoding Dtx4 serves as an example of how we defined a SP (Fig. 3C). Using the same stringent conditions as those used in that example, we identified 206 SPs against 64 annotated RNAs (all protein-coding) and two intergenic regions (Supplemental Tables S2, S3). As expected, some SPs overlapped and 206 SPs accounted for 72 distinct regions on 66 RNAs. Based on our pilot experiment, we expected that the array method would detect sites showing a high percentage of m6A (Fig. 2B). Indeed, based on analysis of our published meRIP-seq data (Wang et al. 2014b), peaks containing SPs were much more enriched in m6A than were remaining m6A peaks (Fig. 3D, P < 8.06 × 10−7).
m6A modification may modulate microRNA/mRNA interactions
We next carried out various bioinformatic analyses to characterize detected RNA regions. Location analysis revealed that many SPs are located in coding sequences (CDS), and also at 3′UTRs and in some introns or 5′-UTRs (Fig. 4A). Relevant to CDSs, it is noteworthy that many SPs were more enriched on long exons, suggesting that m6A modification functions in their retention (Fig. 4B). We then evaluated potential mechanisms underlying high enrichment of m6A sites. Using published mESC m6A meRIP-seq data, we found that SP-containing peaks were much more enriched in the methylation motif R(A/G)RACH(A/C/T) (Dominissini et al. 2012; Meyer et al. 2012) than were SP-absent m6A peaks (Fig. 4C, P < 6.11 × 10−5). MEME analysis detected a motif containing RRACH from SP probes (Supplemental Fig. S2, P < 3.5 × 10−6). To assess potential functions of these sites, we carried out GO analysis on mRNAs containing an SP. As shown in Figure 4D, those RNAs functioned primarily in development, consistent with differentiation defects detected in methyltransferase-deficient ESCs or mice (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015).
FIGURE 4.
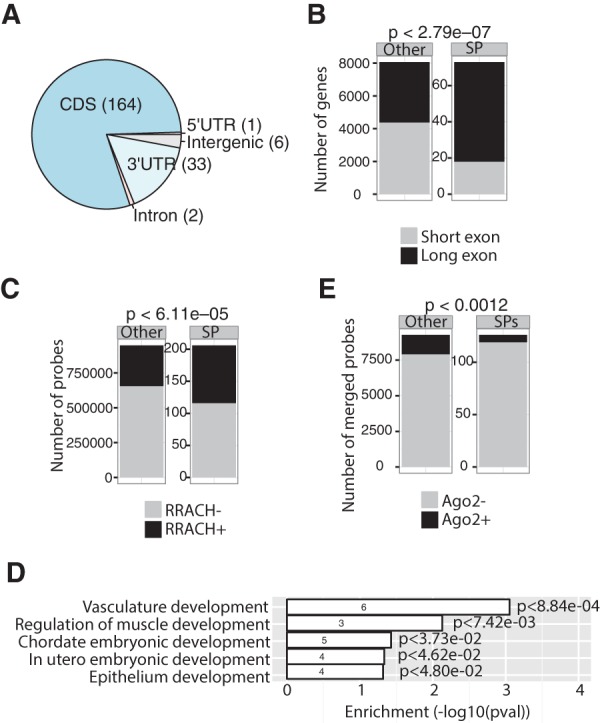
m6A interferes with A–U or A–T pairing. (A) Pie chart indicating genic location of significant probes (SPs). (B) Bar plots show specific enrichment of SPs on long (>300 nt) versus short (≤300 nt) exons (P < 2.79 × 10−7; Fisher's exact test). (C) Bar plots showing enrichment of the RRACH motif within 206 SPs and remaining probes (P < 6.11 × 10−5; Fisher's exact test). (D) GO analysis of 64 protein-coding genes containing SPs (P < 0.05). The number of gene hits in a GO category is indicated within the bars. (E). Bar plots showing that meRIP-seq peaks containing SPs exhibit significantly decreased binding to AGO2 than do those from remaining probes. Fisher's exact test, P < 0.0012.
Finally, to understand how m6A might regulate these mRNAs, we asked whether m6A perturbs microRNA targeting, based on our finding that m6A interferes with A–U/T pairing. Using published Argonaute 2 (Ago2) CLIP-seq (photo-cross-linking immunoprecipitation followed by deep sequencing) data in mESCs (Leung et al. 2011), we found that compared with other m6A peaks detected by m6A meRIP-seq, SP-containing peaks showed significantly decreased binding to Ago2 (Fig. 4E), suggesting that m6A blocks mRNA degradation by microRNA. These analyses support the idea that m6A regulates developmental genes in mESCs potentially through the microRNA pathway.
DISCUSSION
Our understanding of how RNA modifications impact RNA activity, particularly that of mRNA, is still at its infancy, partly because of technical challenges encountered in locating these modifications. In this study, we developed a microarray-based method to map the most abundant internal mRNA modification, m6A, on a large scale. Our method has several technical advantages over current m6A detection methods.
First, we consider m6A sites detected by microarray to be high confidence sites: Although meRIP-seq is associated with a high rate of false-positives, microarray analysis is in fact more typically associated with false-negative signals, as not all probes covering modified nucleotides are more efficiently bound by unmodified versus modified RNAs. Therefore, sites detected using this approach are more likely to be valid. To further validate our findings, we also required that m6A sites detected become unmodified following both DZA treatment and METTL14 KD and that an m6A site had to be detected by more than one consecutive tiling probe. Because of these stringent experimental and analytical conditions, we identified 72 m6A-containing RNA regions. These sites are of high abundance (Figs. 2B, 3D). Unlike some tRNA or rRNA modifications in which almost 100% of the RNA molecules are modified (Hori 2014), the extent of m6A enrichment varies widely on different mammalian RNAs. For example, using a method known as SCARLET (for, site-specific cleavage and radioactive-labeling followed by ligation-associated extraction and thin-layer chromatography) to detect m6A on a specific RNA, Liu et al. (2013) reported that the m6A fraction at different sites varies from 6% to 80%. Highly enriched m6A sites detected in our study likely play a functional role in regulating RNA activities.
Second, our method is relatively high resolution. As yet, no genome-wide method can capture the m6A methylome at single-base resolution. Although bioinformatics methods have been developed to predict precise m6A sites (Schwartz et al. 2013, 2014; Chen et al. 2015a), the resolution of IP-based methods depends experimentally on the size of RNA fragment, which typically exceeds 100 bp. By microarray analysis, we can now narrow down an m6A site to within the span of a DNA probe, making it possible to mutate some or all A residues within a 25-bp region for further analysis.
In addition, as m6A meRIP requires a fair amount of starting material, m6A analysis has typically been carried out using mouse tissues or cultured cell lines (Dominissini et al. 2012; Meyer et al. 2012; Schwartz et al. 2013, 2014; Wang et al. 2014a,b; Chen et al. 2015a). In contrast, Agilent two-color array uses minimally 1.65 μg initial RNA for 1 × 1 million array and 300 ng for 8X60K arrays (Manual: Two-Color Microarray-Based Gene Expression Analysis, Agilent Technologies, http://www.chem.agilent.com/library/usermanuals/Public/G4140-90050_GeneExpression_TwoColor_6.8.pdf); thus, this method is particularly useful to detect m6A on a subset of RNAs from small tissue blocks or clinical samples.
Our method does, however, have some disadvantages: Specifically, it is far less sensitive than meRIP-seq and cannot detect low abundance m6A sites. As m6A detection by microarray is a novel concept, we anticipate that our current study will provide an impetus for investigators to devise novel ways to increase its sensitivity in the future.
Finally, we note that complementary base-pairing underlies many RNA activities, such as establishment of secondary structure, microRNA targeting of mRNA, anti-codon recognition by tRNA, and RNA/DNA interactions. Therefore, our finding that mono-methylation at the N6 position of adenosine hinders formation of a Watson–Crick base pair that includes m6A has wide biochemical implications. Indeed, two studies report that at specific positions and concentrations, the presence of m6A alters stability of an RNA duplex (Micura et al. 2001; Roost et al. 2015), supporting our observation. Based on our finding that m6A interferes with A–U pairing, one potential function of highly enriched m6A sites detected here could be to hinder microRNA targeting to mRNA. Interestingly, a genome-wide inverse correlation between m6A peaks and predicted microRNA binding sites has been reported (Meyer et al. 2012). If m6A were located within a microRNA target site on mRNA, particularly in the seed region, it could antagonize microRNA/mRNA binding to prevent mRNA slicing by Ago2. Our bioinformatics analysis showed significantly decreased Ago2 binding to RNA regions containing SPs (Fig. 4E), supporting this mechanism. Recent studies report that microRNA facilitates m6A formation on its targets (Chen et al. 2015b) and that m6A tags primary microRNA for processing (Alarcon et al. 2015). Together, these studies suggest that m6A and microRNA interact at different stages, likely from biogenesis to targeting.
In summary, we have developed a new technology to capture the m6A-RNA methylome. Our method detects high abundance m6A modifications and is advantageous in part due to its requirement for small amounts of tissue for certain applications.
MATERIALS AND METHODS
Ribonuclease protection assay (RPA)
This assay was conducted using Ribonuclease Protection Assay (RPA) III kit (Ambion). A 250 nt of mouse β-actin antisense RNA (actin-as-250) was radiolabeled by transcribing pTRI-Actin-Mouse vector provided by RPA kit using T7 Maxiscript Kit (Ambion) with the presence of α-32P-UTP. A 140-bp sense fragment complementary to the probe was amplified by PCR reaction using pTRI-Actin-Mouse vector as template and forward primer 5′-CGGggtaccgacggccaggtcat cactat-3′ and reverse primer 5′-ATAAGAATGCGGCCGCcggatgtca acgtcacactt-3′ containing a T7 promoter; 140 nt mouse β-actin sense RNA (actin-s-140) was in vitro-transcribed using T7 Maxiscript Kit (Ambion) with the presence of either ATP or N6-methyl-ATP (Trilink) and was purified by polyacrylamide gel electrophoresis (PAGE). Actin-as-250 was mixed with actin-s-140-m6A or actin-s-140-A at a molecular ratio 5:1 and coprecipitated. RNA pellets were resuspended in hybridization buffer, aliquoted, and incubated for 0.5, 2 and 4 h for sense–antisense annealing and duplex formation. After incubation, unhybridized RNA were digested by RNases A and T1. RNA samples were precipitated and then separated on a 5% acrylamide denaturing gel (National diagnostics). The gel was exposed to a storage phosphor screen and scanned by FujiFilm FLA-5100 imager.
In vitro RNA synthesis containing A or m6A
RNA was in vitro synthesized by AmpliScribe T7-Flash Transcription Kit (Epicenter) per manufacturer's instruction. For RNA oligo containing m6A, ATP is substituted with N6-Methyl-ATP (Trilink) for the in vitro synthesis reaction.
Dot blot analysis
Synthesized RNAs with or without m6A were spotted on Sure Blot nylon membrane (Millipore). After UV cross-linking in XL-1000 UV Crosslinker (Spectroline), the membrane was washed by TBST, blocked with 5% of nonfat milk in TBST, then incubated with anti-m6A antibody (1:5000, Synaptic Systems) at 4°C overnight. After incubating with HRP-conjugated anti-rabbit IgG secondary antibody, the membrane was visualized by ECL (Millipore). To check RNA loading, the same membrane was stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2).
Experimental procedure for microarray analysis
Purified DNAse-free polyA+/Ribo- RNAs were labeled using the ULS Fluorescent Labeling Kit for Agilent arrays with Cy3 and Cy5 (Kreatech) according to manufacturer's instruction. A measure of 2.5 μg fragmented and labeled mRNA with Cy3 or Cy5 were mixed in the presence of 10% formamide and microarray was performed by the Gene Expression Hybridization Kit (Agilent Technologies) following Two-Color Microarray-Based Gene Expression Analysis version 6.6 (Agilent Technologies).
Pilot microarray analysis
The pilot microarray contains 15,260 probes with Cy3 measuring the intensities of unmethylated RNAs and Cy5 dedicated for the signal intensities of RNAs with the methylated proportion equal to 0%, 2%, 8%, 16%, 32%, or 100%. Thus, lower Cy5/Cy3 signals indicated weaker hybridization attributable to the RNA methylation. Additionally, for each methylation concentration, we also examine the effects of varying formamide concentration at 0%, 5%, 10%, and 20%. The raw intensities were read in by read.maimages from limma package (Smyth et al. 2005). After background subtraction, distributions of log2 Cy5/Cy3 over various conditions were visualized by boxplot as shown in Figure 2B.
Cell culture and RNA preparation
J1 mESC lines expressing scramble shRNA or shRNA against mettl14 and the culture conditions have been described (Wang et al. 2014b). J1-scr was treated by 3-deaza-adenosine (3-DZA, Cayman Chemical Co.) for 24 h. Total RNA was extracted using TRIzol reagent (Invitrogen). RNA was treated with RNAse-free DNAse I (Roche) to eliminate DNA contamination. PolyA RNA was purified using GenElute mRNA Miniprep Kit (Sigma-Aldrich) and residual ribosomal RNA was depleted (Ambion) per manufacturer's instruction.
Microarray analysis
Each custom two-channel Agilent tiling array harbors 947,952 probes covering the 13,324 m6A peaks we previously identified from MeRIP-seq data (Wang et al. 2014b). Each probe is 25 nt, and any two adjacent probes in the genomic coordinate overlap each other by 19 nt. The Cy5 or red channel corresponds to Mettl14 knock down (M14) or DZA mutant mESC cell line, and Cy3 or green channel is associated with wild-type cell line treated with scramble hairpin (SCR). Thus, in principle a higher Cy5/Cy3 signal for each probe reflects an increased hybridization to the oligonucleotide because of lack of methylation of a particular RNA molecule in M14 or DZA condition relative to the SCR control. Moreover, we used additional arrays with both channels dedicated for M14 as an external control for technical difference between the Cy5 and Cy3 dye (details below). For each comparison, we have three biological replicates, and therefore there are nine tiling arrays in total (i.e., three arrays for M14 versus SCR, three arrays for DZA versus SCR, and three arrays for M14 versus M14). After image processing by Agilent software, the raw data were read into R environment by function read.maimages from limma package (Smyth et al. 2005). The raw intensities were subject to background correction, log2 transformation of Cy5/Cy3 ratios, loess normalization within each array (Yang et al. 2002), and quantile normalization between arrays to ultimately enable inter-array comparison.
As an overview of detecting m6A probes from the normalized data, we devised two independent methods, namely “peak call” and “sample test.” We then took as confidence m6A probes as implicated in both detection algorithms. In the peak call approach, we first averaged the signals across three replicates for M14/SCR, DZA/SCR, and M14/M14. We then grouped probes belonging to the same putative methylation region or peak identified from our previous MeRIP-seq experiment, and ordered them based on their genomic coordinates. To detect methylated probe, we took a model-based approach as follows. For probe within a peak, we assessed the statistical significance of its Cy5/Cy3 ratio in the treatment condition (i.e., M14/SCR or DZA/SCR) based on its “local Gaussian distribution” with mean equal to max (0, mu_c, mu_t), where mu_c and mu_t are the average signals of four upstream and four downstream probes including probe itself from M14/M14 background and treatment, respectively; the standard deviation was also estimated from same probes from M14/M14. As a result, a significance P-value is associated with each probe indicating how much its signal deviate from the cognate local Gaussian distribution. The P-value was then subject to multiple testing corrections with Benjamini–Hochberg method to produce the false discovery rate (FDR). We used FDR<0.1 to select significantly methylated probes.
For the sample test approach, we performed one-sided t-test on each probe for M14/SCR, DZA/SCR, and M14/M14 separately, using mean estimated from the probes belonging to the same peak and variance from the three biological replicates (for the same probe). For a probe to be considered as a candidate, we required that it must have P-value <0.05 in both M14/SCR and DZA/SCR but not in M14/M14. Because adjacent probes overlap each other by 19 nt, we reasoned that a bona fide methylation site should lead to multiple significant probes in vicinity. According to this rationale, we performed a cumulative scoring approach on the ordered probes belonging to the same peak. Specifically, we kept a running score with initial value equal to 0. At a particular probe, we incremented the running score by 1 if that probe satisfied the above criteria; otherwise, we deducted the score by 1 if the score was positive.
Finally, we integrated the predictions from the above two methods and identified a confidence set of methylated probes as having FDR<0.1 from the peak call method in both M14/SCR and DZA/SCR AND running score ≥2 from the sample test method.
Comparison of enrichment scores from MeRIP-seq data
Recall that each probe was originally derived from the 13324 m6A peaks that we previously detected using peak calling method MACS (Zhang et al. 2008) on our MeRIP-seq data by comparing the MeRIP library with RNA input libraries in mESC. Thus, we expected that the 206 significant probes we identified by the microarray analysis were originated from peak regions with stronger MeRIP-enrichment score than the remaining peaks. To this end, we divided the peaks into two groups by whether they encompass the 206 positive probes. We then compared between the two groups by their enrichment score calculated by MACS as the −log10 (P-value) based on local Poisson background distribution (Fig. 3D).
GO enrichment analysis
We examined the functional enrichment of the 64 protein-coding genes covering the 200 of 206 significant probes (six other probes came from intergenic regions) using DAVID online tool (Huang da et al. 2009). Because our gene set is rather small, we used a rather lenient cutoff of ordinary P-value <0.05 to identity significantly enriched GO terms or pathways associated with developmental biology. The number of gene hits and the enrichment as −log (P-value) were plotted in Figure 4D.
Motif analysis
We merged the 206 significant probes into 125 larger nonoverlapping genomic regions, which were subject to motif enrichment analysis by MEME (Bailey and Elkan 1994). Additionally, we assessed by Fisher's exact test whether the 206 probes were significantly enriched for the canonical motif of RRACH (IUPAC Code, where R for A or G, H for A or C or T) relative to the 947,672 remaining probes (Fig. 4C).
AGO2 binding sites
The published AGO2 binding data in mESC (Leung et al. 2011) were downloaded from doRiNA database (Anders et al. 2012). The chromosomal coordinates from the mm9 build were converted to mm10 by LiftOver using R package rtracklayer (Lawrence et al. 2009). We then merged the 206 significant probes or the remaining probes into nonoverlapping regions, overlapped these regions with AGO2 binding sites, and assessed the significant association between methylated regions and AGO2 localization using Fisher's exact test (Fig. 4E).
DATA DEPOSITION
Raw and processed microarray data derived from Agilent arrays were deposited to GEO under access code GSE62530.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Weiss from Agilent for performing custom two-color microarrays at the Agilent facility. This work is supported by a CIRM Training Grant (TG2-01162) to Y.W.; a CIRM Bridges to Stem Cell Research grant (TB1-01193) to A.V.Z.; an AACR-Aflac Inc. Career Development Award for Pediatric Cancer Research (12-20-10-ZHAO), NIH P30 CA023168, and R01 GM110090 grants to J.C.Z.; a CIHR operating grant (No. 115194) and a Natural Sciences and Engineering Research Council (NSERC) Discovery Grant (327612-2009) to Z.Z.; and an NSERC Canada Graduate Scholarship to Y.L.
Author contributions: J.C.Z. conceived the project; Y.W., Y.L., and J.C.Z. designed experiments; Y.L. and Z.Z. performed bioinformatics analysis; Y.W. performed most experiments assisted by A.V.Z.; and J.C.Z., Y.L., and Y.W. wrote the paper with suggestions from Z.Z.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.051474.115.
REFERENCES
- Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. 2015. N6-methyladenosine marks primary microRNAs for processing. Nature 519: 482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders G, Mackowiak SD, Jens M, Maaskola J, Kuntzagk A, Rajewsky N, Landthaler M, Dieterich C. 2012. doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res 40: D180–D186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. 2014. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Lu Z, Wang X, Fu Y, Luo GZ, Liu N, Han D, Dominissini D, Dai Q, Pan T, et al. 2015a. High-resolution N(6) -methyladenosine (m(6) A) map using photo-crosslinking-assisted m(6) a sequencing. Angew Chem Int Ed Engl 54: 1587–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, Wu Y, Lv Y, Hao J, Wang L, et al. 2015b. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16: 289–301. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. 2013. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc 8: 176–189. [DOI] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet 15: 293–306. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. 2013. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155: 793–806. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. 2015. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347: 1002–1006. [DOI] [PubMed] [Google Scholar]
- Hiley SL, Jackman J, Babak T, Trochesset M, Morris QD, Phizicky E, Hughes TR. 2005. Detection and discovery of RNA modifications using microarrays. Nucleic Acids Res 33: e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H. 2014. Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet 5: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, Carey V. 2009. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25: 1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, Sharp PA. 2011. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol 18: 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. 2013. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19: 1848–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. 2014. A METTL3-METTL14 complex mediates mammalian nuclear RNA N(6)-adenosine methylation. Nat Chem Biol 10: 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. 2014. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol 15: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micura R, Pils W, Hobartner C, Grubmayr K, Ebert MO, Jaun B. 2001. Methylation of the nucleobases in RNA oligonucleotides mediates duplex-hairpin conversion. Nucleic Acids Res 29: 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. 2014. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roost C, Lynch SR, Batista PJ, Qu K, Chang HY, Kool ET. 2015. Structure and thermodynamics of N-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc 137: 2107–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. 2013. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155: 1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. 2014. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK, Michaud J, Scott HS. 2005. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21: 2067–2075. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. 2014a. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. 2014b. N(6)-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP. 2002. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.