

Abstract
Peroxiredoxins (Prxs) are a ubiquitous family of cysteine-dependent peroxidase enzymes that play dominant roles in regulating peroxide levels within cells. These enzymes, often present at high levels and capable of rapidly clearing peroxides, display a remarkable array of variations in their oligomeric states and susceptibility to regulation by hyperoxidative inactivation and other post-translational modifications. Key conserved residues within the active site promote catalysis by stabilizing the transition state required for transferring the terminal oxygen of hydroperoxides to the active site (peroxidatic) cysteine residue. Extensive investigations continue to expand our understanding of the scope of their importance as well as the structures and forces at play within these critical defense and regulatory enzymes.
Keywords: antioxidant enzyme, peroxidase, redox signaling, antioxidant defense
Oxidative Stress Defenses and the Recently Recognized Importance of Peroxiredoxins
Peroxiredoxins (Prxs) are ubiquitous enzymes that have emerged as arguably the most important and widespread peroxide and peroxynitrite scavenging enzymes in all of biology [4, 5]. Discovered to be widely-distributed peroxidases in the mid-1990’s [6], the role of Prxs was long overshadowed by well-known oxidative stress defense enzymes such as catalase and glutathione peroxidase (Gpx). However, refined kinetics measurements now imply that Prxs reduce more than 90% of cellular peroxides [5, 7]. Helping awaken interest in Prxs were several developments in the early 2000’s. It was shown that as little as ~100 μM hydrogen peroxide caused rapid inactivation of human Prx I by hyperoxidation during catalytic turnover [8], and this sensitivity was shown to be due to conserved structural features within many eukaryotic Prxs [9]. The seemingly paradoxical finding that a peroxidase would be so easily inactivated by its own substrate led to the development of the ‘floodgate’ hypothesis [9], which posits that Prx inactivation enables peroxide-mediated signaling in eukaryotes, a phenomenon now known to regulate many normal cellular functions [10]. Prx hyperoxidation was also shown to be reversible in vivo [11] and the enzyme responsible for this “resurrection” was identified and named sulfiredoxin (Srx) [12]. Powerful addtional evidence that Prxs are crucial to proper cell regulation is that Prx I knockout mice develop severe hemolytic anemia as well as lymphomas, sarcomas and carcinomas by nine months of age [13].
Prxs have attracted the attention of cancer researchers not only for their apparent function as tumor suppressors (or in some circumstances promoters [14]), but also because they have elevated expression levels in various cancer tissues and immortalized cell lines. High Prx levels have been associated with the resistance of tumors and cancer-derived cell lines toward certain chemo- and radiotherapies [15–17]. Additional links of Prxs with disease are their abnormal nitration in early Alzheimer’s disease patients [18], and a role in promoting inflammation associated with ischemic brain injury [19]. Additionally, that pathogens rely on their Prxs to evade host immune systems makes them promising targets for the development of novel antibiotics [17, 20]. Such roles for Prxs in disease motivates continued exploration of their physiological roles as well as the molecular mechanisms at play in Prx enzymatic function and regulation. This review focuses on the state of our understanding of the biochemical and structural mechanisms involved in catalysis and hyperoxidation sensitivity across this widespread group of enzymes. Further, the potential mechanisms through which Prxs may regulate cell signaling are discussed, and a series of open questions in Prx biology and chemistry are posed.
The Catalytic Prowess of Prxs
Prxs are cysteine-based peroxidases that do not require any special cofactors for their activity. During their catalytic cycle, a peroxidatic Cys (CP) thiolate (CP-S−) contained within a universally-conserved PxxxTxxC motif (with T in some Prxs replaced by S) attacks a hydroperoxide substrate and is oxidized to a CP-sulfenic acid (CP-SOH), and then frequently to an inter- or intra-subunit disulfide, before being reduced (via a mixed disulfide with a reductant) to reform the thiolate (Fig. 1). As alluded to earlier, Prxs were long thought to be ~1000-times slower than the historically better-known catalase and glutathione peroxidases (Gpxs). However, by developing sensitive, spectral assays in which disulfide reduction was not rate limiting, it was shown (e.g., for the model bacterial Prx AhpC from Salmonella typhimurium and human PrxII) that the actual kcat/KM for H2O2 for some Prxs is as high as 107 to 108 M−1 s−1 [21–23].
Figure 1.

The catalytic and regulatory cycles of 2-Cys peroxiredoxins (Prxs). Shown in brown is the normal Prx cycle with the structure of the peroxidatic Cys (CP) residue shown for each redox state (carbons are colored gray, nitrogens blue, oxygens red, and sulfurs yellow; hydrogens are not shown for simplicity). The CP thiolate (RS−) in the fully folded (FF) active site is first oxidized by the peroxide to form the sulfenic acid (R-SOH) or sulfenate (R-SO−) (computational approaches suggest stabilization of the CP as the sulfenate, but the true protonation state is as yet uncertain). This sulfenate, which must undergo a conformational change to become locally unfolded (LU), then forms a disulfide bond with the resolving Cys (CR) in 2-Cys Prxs. Reductive recycling by thioredoxin (Trx) or a Trx-like protein or domain (e.g., tryparedoxin or the N-terminal domain of bacterial AhpF) then restores the thiolate in the FF active site for another catalytic cycle. Shown in blue is the redox-linked regulatory cycle of predominantly eukaryotic Prxs, wherein the CP sulfenate becomes further oxidized, in the presence of high peroxide levels, to the inactive sulfinate (R-SO2−). In some organisms and Prx isoforms the active enzyme is restored by the ATP-dependent activity of sulfiredoxin (Srx).
Also important in catalysis are conformational changes between a fully-folded (FF) conformation in which CP can react with peroxide, and a locally-unfolded (LU) conformation in which the CP is exposed and can form a disulfide with the so-called ‘resolving’ Cys (CR) present in many Prxs. In the CP-S− and CP-SOH forms, the FF and LU conformations rapidly equilibrate [24–28], but in disulfide forms Prxs become locked in the LU conformation (Fig. 1). As proposed in 2003 [9], stabilization of the FF conformation should effectively promote further oxidation of CP by peroxide, because of the greater opportunity to react with a second molecule of peroxide within the active site. Indeed, the CP-SOH form of those Prxs having a highly stabilized FF conformation does more readily form a Cys-sulfinate (CP-SO2−), rendering the Prx inactive (Fig. 1, redox regulation cycle) [28, 29]. In many eukaryotes, including humans, the enzyme Srx catalyzes repair of hyperoxidized Prxs in the Prx1 group (see next section), restoring their activity [17, 30]. Given that Prxs are abundant and highly reactive with cellular peroxides and peroxynitrite, and have activity that can be regulated, they are well suited to play roles not just in oxidant defense, but also in redox sensing and signaling and perhaps even in the recovery of oxidatively-damaged proteins [31–36]. The various physiological roles of Prxs can be better understood in the context of structural and functional features that are shared between certain members. Thus, we describe next how Prxs are classified into evolutionary subfamilies, and how these different subfamilies vary in phylogenetic distribution, cellular localization, oligomerization, conformation change, and susceptibility to hyperoxidative inactivation.
Functional and structural subdivisions of Prxs
Prxs exist in six evolutionary subfamilies (Prx1, Prx5, Prx6, Tpx, PrxQ and AhpE), that vary in oligomeric states and interfaces, and in the locations of the resolving Cys [37]. In general, Prx1 subfamily enzymes –typically doughnut-shaped decamers – are the most highly expressed, making up 0.1–1% of the soluble protein in the cell [38]. These are the Prxs that have been commonly referred to as “typical 2-Cys” Prxs [39] as they were the original type of “2-Cys Prx” (having both a CP and CR) discovered [6]. In these proteins, the CR is near the C terminus of the second chain of a dimer, thus forming an intersubunit disulfide bond during the catalytic cycle. In mammals, Prx1 subfamily enzymes are in the cytosol and nucleus (PrxI and PrxII), the mitochondria (PrxIII) and the endoplasmic reticulum (PrxIV). Mammals have two other subfamilies: PrxV, which localizes to peroxisomes, mitochondria and the cytosol, and PrxVI, which is cytosolic [40]. The phylogenetic distribution of Prxs based on a bioinformatic analysis of >3500 Prxs from the 2008 GenBank database [37] demonstrated the widest biological distribution for Prx1 and Prx6 subfamilies; PrxQ members appear to be absent in animals, Prx5 members are apparently lacking in archaea, and Tpx and AhpE subfamily proteins are restricted almost exclusively to bacteria (Table 1) [37]. Bioinformatics approaches have also been used to track the prevalence and location of CR among members of each subfamily (Table 1), as described in more detail in Box 1.
Table I.
Summary of Prx subfamily phylogenetic distribution and structuresa
| Subfamily | Phylogenetic Distribution | Structural distinctions relative to Prx core fold | Oligomeric states and interfacesa | Typical Location and Conservation of CR (When Present) |
|---|---|---|---|---|
| Prx1 b | Archaea, Bacteria, Plants and Other Eukaryotes | Extended C-terminus | B-type dimers, (α2)5 decamers (and rare (α2)6 dodecamers) through A-interface | C-terminus of partner subunit (~99%) c |
| Prx6 d | Archaea, Bacteria, Plants and Other Eukaryotes | Long extended C-terminus | B-type dimers, some (α2)5 decamers through A-interface | No CR (≥41%) |
| AhpE e | Bacteria | Extended loop at N-terminus | A-type dimers |
|
| PrxQ f | Archaea, Bacteria, Plants and Fungi (not animals) | Extended helix α5 | Monomers and A-type dimers |
|
| Tpx g | Bacteria | N-terminal β-hairpin | A-type dimers | Helix α3 (>95%) |
| Prx5 h | Bacteria, Plants and Other Eukaryotes (not archaea) | π helix insertion in α2; ~20% fused with Grx domain | A-type dimers |
|
Structural information from Hall et al. [47] and bioinformatic analyses from Nelson et al. [33] updated using information from the October 2011 GenBank(nr) database, as available from http://www.csb.wfu.edu/prex/ [102].
Prx1 is also known as the “typical 2-Cys” Prx group and includes Salmonella typhimurium AhpC, Homo sapiens PrxI-PrxIV tryparedoxin peroxidases, Arabidopsis thaliana 2-Cys Prx, barley Bas1, and Saccharomyces cerevisiae TSA1 and TSA2.
The CR is near the C-terminus of the partner subunit within the homodimer; upon oxidation, intersubunit disulfide forms between the CP and the CR of the two chains.
The Prx6 subfamily (frequently referred to as the “1-Cys” group) includes H. sapiens PrxVI, Arenicola marina PRDX6, A. thaliana 1-Cys Prx and S. cerevisiae mitochondrial Prx1.
The canonical (and only well characterized) AhpE from Mycobacterium tuberculosis contains no CR and is dimeric. Distribution appears restricted to the order Actinomycetales.
The PrxQ group includes Escherichia coli PrxQ (previously referred to as BCP) and plant chloroplast PrxQ.
The Tpx subfamily includes bacterial proteins (e.g. from E. coli, Streptococcus pneumonia, Helicobacter pylori and Mycobacterium tuberculosis) named thiol peroxidase, p20, and scavengase.
The Prx5 subfamily includes H. sapiens PrxV, Populus trichocarpa PrxD, the plant type II Prxs [56], and a group of bacterial Prx5 proteins fused with a C-terminal glutaredoxin (Grx) domain.
The CR forms an intersubunit disulfide bond with CP, as exemplified in yeast Ahp1 [43].
BOX 1. Bioinformatic studies of Prxs.
When Prxs were first recognized to exist in distantly-related species [6], they were subdivided into “1-Cys” and “2-Cys” groups based on the presence or absence of the C-terminal CR now known to be largely characteristic of the Prx1 group (referred to as the “typical 2-Cys” group, Figure I). “1-Cys” and “2-Cys” are still useful as mechanistic designations (indicating the absence or presence of a CR, respectively), but bioinformatics approaches focused on structure/function/sequence relationships support more fine-grained subdivisions. Much like the initial classes proposed earlier using full sequence analyses [100], six subfamilies of Prxs were elucidated based on sequence comparisons among fragments plucked from a region within 10 Å of key active site residues in Prxs of known structure (29 structures at the time of this analysis) [37]. These sequences, arranged N- to C-terminus, comprised the functional-site signatures [101] extracted from each structure, subsequently aligned to create a profile. The six Prx functional-site profiles (one for each subfamily identified, named for a canonical member) were used to search the GenBank(nr) database (January, 2008 release) and a validated list of 3516 Prx sequences were unambiguously classified into one of the six subfamilies of Prxs, enabling a new, in-depth view of sequence conservation and species distribution within each of the subfamilies (Table 1) [37]. The Prx sequences thus identified, along with their subfamily assignments, accession numbers and associated genus and species information, were assembled into a web-based searchable database known as PREX (PeroxiRedoxin classification indEX) [102], with updated information from 2010 and 2011 GenBank(nr) searches also available (http://www.csb.wfu.edu/prex/). Summaries of the information extracted from these analyses are presented in Table 1 and Figure I. Panel B of Figure I illustrates the near homogeneity of members of the Prx1 and Tpx groups in the existence and location of CR, whereas members of the PrxQ and Prx5 subfamilies are more diverse in this regard. Moreover, the Prx6 group includes more bona fide “1-Cys” members than any other group, particularly true if most or all of the “uncertain” members of the Prx6 subfamily do in fact lack a CR. However, the designation “1-Cys Prx” should not be used interchangeably with “Prx6 subfamily” as they are not synonymous.
Figure I.

WebLogo plots for sequences proximal to CP and pie charts illustrating the prevalence of each CR location among all six subfamilies of Prxs. Subfamilies as defined by Nelson et al. [37] but updated from the October 2011 GenBank(nr) database were represented by the following number of members: Prx1 (1387), Prx6 (1102), AhpE (112), PrxQ (2060), Tpx (990), and Prx5 (1139). (A) The local sequences including the PxxxTxxC motif (conserved positions of the motif noted with asterisks) and 5 residues downstream of Cp were extracted from all members of each subfamily and used to create WebLogo plots to summarize sequence conservation around Cp. (B) Pie charts showing the frequency at which the CR is in a given location for each subfamily. Wedges are colored by CR position consistent with Fig. 2A: CR in the C-terminus (Ct, magenta), α2 (yellow), α3 (green), α5 (dark blue), or the N-terminus (Nt, orange). No CR (red) indicates that there is no other Cys in the protein, and uncertain (gray) indicates there are other Cys, but not in a recognized position of a CR. Hybrid proteins in the Prx5 subfamily with a C-terminally appended Grx domain, as exemplified by Haemophilus influenza Prx5-Grx, are represented in cyan; these proteins either have only one Cys (the Cp) in the Prx domain or have other Cys residues that do not align with recognized CR residues. The exact positions in canonical representatives (allowing a shift of up to 2 residues) are defined as follows: Ct aligns with Cys165 in S. typhimurium AhpC; α2 aligns with Cys50 in E. coli PrxQ (also known as BCP); α3 aligns with Cys95 in E. coli Tpx; α5 aligns with Cys151 in H. sapiens PrxV; and Nt aligns with Cys31 in S. cerevisiae Ahp1.
As noted above and in Box 1, the resolving Cys, when present, can be located in at least 5 different positions on the thioredoxin (Trx)-like fold of Prxs (Fig. 2A), varying both across and within subfamilies. For instance, the PrxQ subfamily has members with CR in the α2 helix, CR in α3, or lacking CR altogether (Box 1 and Table 1). Such variation in CR locations brings up two important points. First, to form the CP-CR disulfide, a set of very different conformational changes (i.e. local unfolding) must occur (Fig. 2B). Second, each of these unique conformational changes provides an opportunity for a “kinetic pause” in the catalytic cycle that can be controlled structurally and dynamically to tune the sensitivity to hyperoxidation [9, 28].. Additionally, several lines of evidence suggest that the CR residue of Prx1 proteins [41, 42] and the yeast Prx5 protein (also known as Ahp1p, cTpxIII, or YLR109W) [43] is the point-of-attack for Trx or Trx-like reductases, highlighting another way in which local unfolding and disulfide bond formation can promote the catalytic cycle.
Figure 2.

Variable locations of the resolving Cys (CR) and oxidation-driven conformational changes across Prxs with distinct CR locations. (A) Shown are the various positions of the peroxiredoxin CR (colored sidechains) in relation to the active site peroxidatic Cys (CP, circled and in red). Intramolecular CP-CR disulfides are formed for the α2 (yellow), α3 (green), and α5 (blue) types, and intermolecular disulfides are formed for the N-terminal (Nt, orange CR in the gold chain) and C-terminal (Ct, magenta CR in the black chain) types. (CR residues are mapped onto a composite structure based on S. typhimurium AhpC (StAhpC), Protein Databank Identifier 4MA9). (B) Shown are the transitions from fully folded to locally unfolded conformations for representative Prxs: Nt, Saccharomyces cerevisiae Ahp1; α2, Aeropyrum pernix PrxQ; α3, E. coli Tpx; α5, Homo sapiens PrxV; Ct, StAhpC. The fully folded conformation is shown in white and the locally unfolded conformation is shown in black, with structural changes that occur in the transition highlighted in yellow. CP is highlighted in red, and CR is highlighted in blue. For clarity, residues 169–186 of StAhpC are not shown. Note substantial movement of CR when in the C-terminus (making an intersubunit disulfide bond), and of both CP and CR when CR is in α2 [25].
The Specialized Active Site of Prxs: Chemical and Kinetic Properties
Most protein sulfhydryl groups in biological systems are in their protonated (thiol, R-SH) form as the reference pKa for Cys, at ~8.5 [44], is well above neutral pH. However, the pKa of any Cys residue can be substantially influenced by its protein microenvironment; for instance, at the active sites of some Cys-dependent enzymes a positively-charged environment lowers the pKa by stabilizing the anionic thiolate form (R-S−) [45, 46]. In Prxs, the CP pKa values determined for nearly a dozen representatives are clustered between 5.1 and 6.3 [47, 48]. Using the Henderson-Hasselbalch equation, this equates to over 83% of the CP residues being present in the thiolate form at pH 7, [46]. Because thiolate nucleophilicity decreases linearly as its pKa decreases [49], the observed values represent a compromise that creates a thiolate form that is highly prevalent at neutral pH, and still highly reactive. But as small molecule thiolates only react with peroxides at about 20 M−1 s−1 [50], additional factors must contribute to the exquisitely selective high reactivity of Prxs with hydroperoxides (including peroxynitrite), but not other, more electrophilic compounds [5, 22, 48, 51].
Recent structural and computational work has provided a plausible proposal for the special features of the Prx active site that support its unique reactivity. Describing the Prx reaction with peroxide (HOA-OBR) as a nucleophilic displacement (SN2) reaction, the CP thiolate sulfur attacks the terminal (i.e. CP proximal) oxygen (OA) of the hydroperoxide substrate, breaking the OA–OB bond (with OB associated with the alkoxide or hydroxide leaving group, Fig. 3A) [52–54]. Supporting this view of the reaction, a 2010 survey of Prx crystal structures documented a “track” of oxygen binding sites in the active site [52] that defined a path along which OA and OB could move, toward the thiolate sulfur and away from each other, during the course of the reaction. Surrounding the H2O2 (or ROOH) substrate in the Prx active site are the conserved Thr and Arg residues and two backbone amides that hydrogen bond (HB) with the peroxide substrate so as to orient and polarize it for attack (Fig. 3A). It appears that these interactions would be optimized when the OB-OA bond is partially broken and the CP-Sγ…OA bond is partially formed (Fig. 3A), leading to preferential stabilization of the transition state [52]. Fully supporting this proposed oxygen track and mechanism is a remarkable authentic Michaelis complex structure (Fig. 3B) with H2O2 bound in the active site [55].
Figure 3.

The universally-conserved Prx active site. (A) Cartoon representation of the putative active-site transition state conformation. The stabilizing interactions between key atoms from the backbone and the four conserved residues, and with the ROOH substrate, are indicated. In the transition state, a bond is forming between the S atom of the CP and the OA of ROOH, and a bond is breaking between the OA and OB atoms of ROOH. The geometry of the active site appears nicely set up to stabilize the larger distance between the OA and OB atoms as the bond is broken. Based on a figure from Hall et al., 2010 [52]. (B) Shown is the Michaelis complex of a hydrogen peroxide-bound Prx (ApTpx, Protein Databank Identifier code 3A2V) with the four conserved residues of the active site motif highlighted in pale yellow and key active site hydrogen bonds noted with dashed lines. The interaction between Cp and the OA of H2O2 where the bond will form is depicted with dots.
Site-directed mutagenesis and hybrid quantum mechanics-molecular mechanics (QM-MM) studies support this model and further provide evidence that the stabilizing interactions with the CP-thiolate change upon substrate binding to transiently raise the thiolate pKa and promote its nucleophilicity [53, 54]. Based on an Arrhenius analysis of the catalytic rates of Mycobacterium tuberculosis AhpE over a range of temperatures, Zeida et al. also report a large entropic penalty (TΔS) that may reflect an increase in the order of side chains in the transition state; this penalty appears to be more than offset by a large enthalpic change (ΔH), leading to an overall decrease in free energy of activation [54]. Many details are still under discussion, such as whether the conserved Thr hydroxyl donates a strong HB to the CP thiolate in the Michaelis complex, as represented in the QM-MM simulations described [53, 54], or, as inferred from crystal structures [52], donates a HB to a nearby backbone carbonyl oxygen (as shown in Fig. 3). And regarding the conserved Arg, agreed are that it stabilizes the CP thiolate and the developing negative charge on the peroxide, but the specific HB interactions it makes are uncertain [52–54, 56, 57]. Interestingly, in both QM-MM studies of the Prx reaction with H2O2 [53, 54], the proton originally on the peroxide OA-atom is transferred to the OB hydroxyl leaving group to form water, providing a plausible explanation for the noted lack of a potential active site acid that could protonate the ROB− leaving group.
Complementing the structural and simulation work, the knowledge of steady state and presteady state kinetics of Prxs has increased greatly since a 2007 review on this topic [58]. The Prx1 group has been by far the most widely studied by full bisubstrate kinetic analyses, and specificities range from broad to narrow (both for peroxides and reductants), with kcat/KM values for reduction of the preferred peroxide substrates on the order of 1 × 105 – 7 × 107 M−1 s−1 at pH 7–7.4 and 20–25 °C [58–60]. Escherichia coli and Mycobacterium tuberculosis Tpx proteins have kcat/KMperoxide values similar to those of the Prx1 proteins [58], while these values for PrxQ members from bacteria and plants have been ~104 M−1 s−1 [58, 61, 62]. All Prxs show double displacement (i.e., ping pong) kinetics with two independent half reactions: first, reduction of the peroxide by the Prx (resulting in oxidized enzyme), and second, reestablishment of reduced Prx by electron transfer from a reductant [58, 63]. Kinetic complexities can occur however, particularly since oligomeric properties can change with concentration and redox state of some Prxs (see Box 2 for details).
BOX 2. Dynamic oligomeric states of Prxs.
Most Prxs exist in solution as oligomers, with only some members of the PrxQ subfamily known to be stable and active as monomers (Table 1). The formation of stable dimers is therefore a common theme across all Prx subfamilies, although there are two distinct interfaces through which dimers can be formed depending upon the Prx (Figure I). The A interface (denoting the presumed ancestral interface) supports dimerization in an end-to-end fashion (if the central beta-sheet is likened to the palm of a hand, the interaction is through the fingertips). This is observed for Tpx, Prx5 and the dimeric PrxQ subfamily members. Dimeric Prx1, Prx6 and AhpE proteins are built by bringing together the beta-sheets of two monomers in an antiparallel manner to generate an extended, 14-strand sheet using the B (or β-sheet) interface (bringing the “palms” together side-by-side, but in a head-to-tail manner). For the Prx1 and Prx6 proteins, extended (relative to the other subfamilies) C-terminal helices stabilize these “tail swapped” B-type dimers. In nearly all of the Prx1 family members (>96%), the resolving Cys (CR) which partners with the oxidized peroxidatic Cys (CP) to form an intersubunit disulfide during catalysis is located in this C-terminal extended region. Moreover, the B-type dimers can interact to form toroidal complexes, coming together through interactions at their A interfaces to form (α2)5 decamers [and rare (α2)6 dodecamers] (Figure I). Higher order complexes can also be built up under some conditions to form even larger oligomers in either spherical aggregates or open-ended linear polymers that have been proposed to be related to hyperoxidation state and chaperone function [30, 59, 103].
A remarkable feature of many decameric Prx1 proteins is the modulation of the strength of the A-interface interactions by the redox state of the CP, which is close enough to the decamer-building A interface to exert such an influence [51, 59]. In analytical ultracentrifugation analyses of bacterial AhpC, the reduced enzyme was decameric at all concentrations studied, while the disulfide-bonded (i.e. oxidized) protein ranged from dimeric (at low concentration) to decameric (at high concentration) [104]. Similar redox-dependent oligomerization properties have been observed in other Prx1 family members, as well, although the stability of oligomers varies with the Prx origin and isoform; for example, oxidation of human PrxIII, but not PrxIV, leads to decamer dissociation [27].
Figure I.
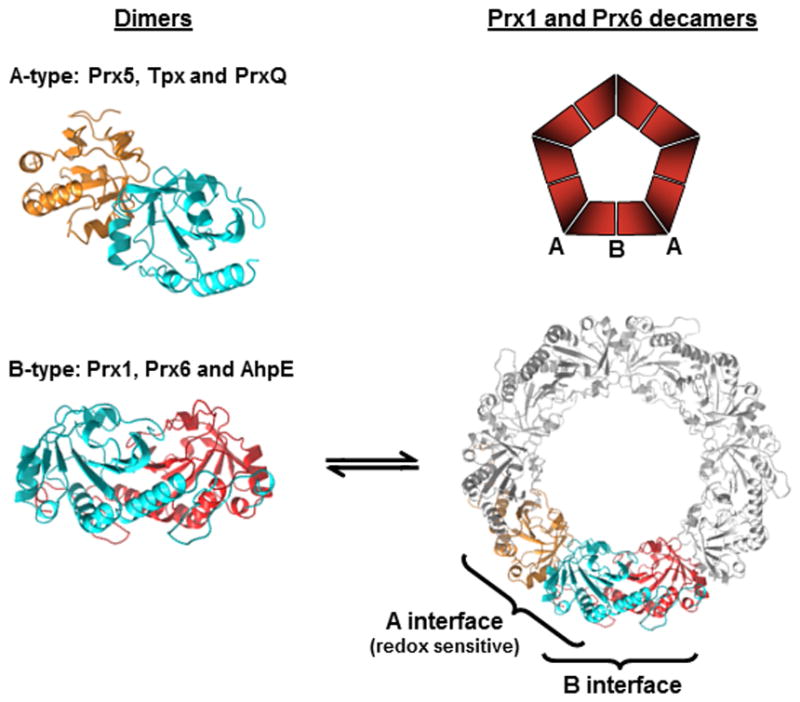
Quaternary structures of Prxs. Dimeric α2 complexes are formed using either an A-type interface, where the monomers interact near helix α3, or B-type dimers where the interaction is through the β-strands, generating an extended 10–14 strand β-sheet. Further interactions at the A-interfaces of some Prx1 and Prx6 members generate (α2)5 decamers [or in rare cases (α2)6 dodecamers]. The blue subunit is displayed at approximately the same position in each of the structures to illustrate these interaction interfaces that together build the decamer. For a number of Prx1 members, the structural change upon disulfide bond formation destabilizes the A-type dimer interface, and the decamer dissociates to B-type dimers. The structures depicted are: Aeropyrum pernix PrxQ (A-type dimer, Protein Data Bank Identifier 4GQF), and wild type S. typhimurium AhpC (B-type dimer and decamer, Protein Data Bank Identifier 4MA9).
Importantly, peroxide reactivity can be assessed even when the reaction is very rapid and/or the physiological reductant is unknown or unavailable by monitoring changes in Trp fluorescence or assessing Prxs competing with horseradish peroxidase (HRP) for H2O2. Since the first assessment of Prx1 family members from budding yeast using the HRP competition assay [64], similar second order rate constants (107 – 108 M−1 s−1) have been measured for many additional Prx1 family members [22, 23, 65–67], as well as Arenicola marina Prx6 [68], human PrxV [69], and PrxQ from Xylella fastidiosa [61]. For AhpE [70, 71] and other PrxQ members [62], the values are only 104 – 105 M−1 s−1, but it is not fully understood why these enzymes are slower. These approaches have also been used to assess the peroxynitrite reductase activity of Prxs, first seen for bacterial Prx1 group members [72], and now confirmed for additional Prx1 members [23, 58], and members of the Prx5 and Tpx [58], AhpE [70, 71], and PrxQ [61] subfamilies. As the only enzymes known to catalyze the reduction of peroxynitrite to nitrite, it has been argued that tightly controlling this highly reactive species may be one of the most important roles of Prxs [58].
Studies using rapid reaction and competitive kinetic approaches to measure Prx disulfide bond formation rates have also yielded a better understanding of some of the kinetic distinctions that exist between different Prxs [29, 67, 69, 71]. Interestingly, human PrxII, which is sensitive toward inactivation during turnover with H2O2, undergoes disulfide bond formation at a rate (1.7 s−1) that is over 10-fold slower than the less sensitive PrxIII (at 22 s−1) [29]. In comparison, the rates for human PrxV [69] and S. typhimurium AhpC, a highly robust Prx [67], were 15 s−1 and 75–80 s−1, respectively. Also, the pre-steady state data of S. typhimurium AhpC show large and rapid fluorescence changes after addition of peroxides, implying that active site rearrangements are associated with peroxide binding. Moreover, interpreting this phase as an initial binding step leads to an inferred Kd for H2O2 of ~400 nM, remarkably tight given the small size and polarity of this small molecule. This finding implies that there may be more to this initial interaction than simple binding [67]. In addition, these studies showed that higher KM organoperoxide substrates are poorer by virtue of slower binding rather than enhanced dissociation as is typical for enzymes [67]. These results with respect to peroxide binding are all suggestive of substrate-induced, active site conformational changes that enhance reactivity in the Prx active site.
Thus, much insight into Prx peroxidase activity has been provided by experimental structures, computational analyses and simulations, and improved kinetic assays. However, it remains intriguing that despite a shared catalytic mechanism Prxs exhibit vastly different susceptibilities to inactivation by hyperoxidation. The biological ramifications of this are discussed next, focusing on how Prxs in eukaryotes contribute to peroxide-mediated signaling that influences a host of cellular processes.
Localization of H2O2-Mediated Signaling Processes
Over the past 15 years, recognition of the importance of Prxs in biology has expanded tremendously. They are no longer seen as just oxidative stress defense enzymes, but as regulators of peroxide levels in vivo that are involved both in stress response signaling (e.g., in bacteria and yeast) and in non-stress related signaling. It is now widely recognized that Prxs are intimately linked to the regulation of signaling processes because, as initially shown for platelet-derived growth factor (PDGF) signaling [73], H2O2 is a critical small molecule second messenger in many signaling processes, including growth factor signaling, angiogenesis, toll-like receptor and cytokine signaling [10, 74]. Activated NADPH oxidase (Nox) complexes are the predominant source of the signaling-related H2O2, produced either directly (e.g., by Nox4 and DUOXs) or via the dismutation of initially-produced superoxide [75, 76]. In growth factor signaling, this Nox-generated H2O2 apparently crucially augments the steady state levels of the active phosphotyrosine forms of the receptors or other signaling proteins, at least in part by inhibiting tyrosine phosphatases. Thus one key target of Nox-derived H2O2 is the active site Cys in protein tyrosine phosphatases (PTPs) and “phosphatase and tensin homologue deleted on chromosome 10” (PTEN), which are sensitive to oxidative inactivation [77, 78]. Importantly, this Cys oxidation in the phosphatase active site is reversible, allowing the reduced active Cys thiols to reform and act to “brake” kinase-mediated phosphorylation cascades [77].
Because phosphatases are only moderately reactive with H2O2, the H2O2 levels must rise substantially for direct inactivation to occur. Yet, if this were to happen throughout the cell, many proteins and lipids would be nonspecifically oxidized. Two non-exclusive explanations for this exist: (1) that the H2O2 buildup is kept highly localized within the cell [34, 38], and/or (2) that Prxs, Gpxs or other highly reactive redox proteins act as mediators (i.e., redox relays) that enhance the oxidation of specific targets [5, 32, 35, 79]. As an example of such a redox relay, human Prx II has recently been reported to directly transmit its oxidizing equivalents to the signaling mediator STAT3 in the context of cytokine signaling in HEK293T cells [80]. The role of Prxs as redox relays of H2O2 signals is appealing given their abundance, high reactivity, and potential for specificity through protein-protein interactions. However, multiple lines of evidence also support the importance of localized peroxide buildup in signaling. First, Nox complexes are themselves localized, for example to specialized lipid raft regions of membranes (Nox1 and Nox2) or to endoplasmic reticulum (ER) membranes (Nox4) [76, 81]. These expected “hot spots” of H2O2 generation also localize with signaling complexes including receptor tyrosine kinases and Src family kinases [82]. An elegant study of epidermal growth factor (EGF) stimulation of ER-localized Nox4 showed that the native ER-localized PTP1B became oxidatively inactivated and that the ER targeting of PTB1B was required for its inhibition and for effective EGF-stimulated proliferation [81]. Also, recent imaging of H2O2 generation [83, 84] and protein oxidation [85, 86] show that localized buildup of H2O2 occurs proximally to growth factor receptors and oxidatively-modified targets. In endosome-mediated growth factor and cytokine signaling, the H2O2 generation localized proximal to these so-called “redoxosomes” is required for effective downstream signaling [74, 85]. Thus, models for Prx involvement in regulating H2O2-mediated cell signaling processes must be compatible with the necessity for H2O2 to act locally during signal transduction.
Potential Roles for Prxs in Stress and Non-Stress Related Cell Signaling
In 2003, it was proposed that the sensitivity to hyperoxidative inactivation by H2O2 that is inherent to some eukaryotic Prxs is an evolutionary adaptation that would facilitate the local buildup and signaling activity of H2O2 [9]. In this “floodgate” model, Prxs would be inactivated proximally to activated Nox complexes, enhancing a gradient of H2O2 concentration while simultaneously removing active Prxs from the signaling zone, where they would otherwise compete for reaction with H2O2. Since the original study, much has been learned about how sequence features that differentially stabilize the FF conformation affect the rate of Prx disulfide formation and how disfavoring disulfide bond formation augments the sensitivity to hyperoxidation [17, 87, 88]. Prxs are now considered as centrally important regulators of non-stress related peroxide signaling but not yet known is the extent to which they act indirectly as floodgates that modulate peroxide levels or directly as mediators that enhance the oxidation of downstream target proteins [89]. Of course, both may be important mechanisms in the wide array of signaling processes.
In terms of non-stress-related signaling, there is to date only one well-documented example of a floodgate-enabled buildup of H2O2 [90]. This study showed that, as part of the circadian synthesis of corticosteroids in the adrenal cortex mitochondria, a cytochrome P450 produces localized H2O2. In concert with PrxIII hyperoxidation, this causes a H2O2 buildup that leads to p38 activation and a negative feedback suppression of steroidogenesis. Another intriguing example of apparent floodgate-like regulation of signaling occurs in the Src kinase-driven phosphorylation of Tyr194 in Prx I by growth factor or immune receptors. This phosphorylation causes the inactivation of Prx I, and the phosphorylated status appears to be enhanced by a transient H2O2 buildup around the receptor complexes that inactivates PTPs and potentially other oxidation-sensitive signaling components [82]. This second example emphasizes that post-translational modifications of Prxs besides hyperoxidation also provide ways for Prxs to be reversibly inactivated for modulating peroxide levels (i.e. for the floodgate to be opened). That such control mechanisms may be widespread is indicated by the variety of in vivo modifications of Prxs that modulate its properties. These include Tyr nitration [91], Ser and Thr phosphorylation, acetylation at the N terminus or near the C terminus, proteolytic truncation, and S-nitrosylation or glutathionylation of catalytic or non-catalytic Cys residues (reviewed in [40, 92]).
More examples are known of stress-related regulatory activities of Prxs. Chief among these are examples of redox relay activity. For instance, fission yeast Tpx1 promotes oxidation of the transcriptional regulator Pap1 [93], and Prxs and Gpx homologues are necessary mediators of transcriptional responses to H2O2 in budding yeast [94]. Also stress-related is the Prx2-STAT3 interaction mentioned earlier that also occurred upon treatment of cells with 100 μM H2O2 [80]. In addition, three studies have shown how hyperoxidation of Prxs can modulate stress-related responses [33]. First, Day et al. [31] showed that in fission yeast exposed to very high (>1 mM) peroxide, hyperoxidative inactivation of Tpx1 functions as a triage event [33] conserving cellular reducing equivalents to repair proteins damaged by the H2O2 assault. Second, hyperoxidized Prxs produced during peroxide stress or heat shock in budding yeast aggregate to ‘high molecular weight forms’ which act as chaperones to help restore proper folding and function of other proteins [95]. Such a chaperone function may be relevant to higher organisms, as well [16, 34, 92]. And third, in mouse C10 lung epithelial cells the accumulation of hyperoxidized Prx serves as a “dosimeter” of peroxide exposure that links to cell cycle arrest [96].
Two final intriguing observations also may relate to the roles of Prxs in cellular regulation. First, circadian cycles of Prx hyperoxidation seen in a variety of organisms were suggested to be a fundamental, transcription-independent mechanism underlying circadian clocks [97, 98]. However, there is no evidence yet that this is related to signaling, and rather than being a driver of rhythms, it may be a consequence of other rhythmic cellular activities. In fact, the circadian rhythm involving Prx II in human red blood cells was recently shown to be caused by peroxide produced by the autoxidation of hemoglobin; the hyperoxidzed Prx was not repaired by sulfiredoxin, but was instead degraded by the 20S proteasome [99]. Second, and again of unknown relevance, specific interactions of Prxs with a number of proteins have been reported over the years, including interactions with the protein and lipid phosphatase PTEN and the signaling kinase Mst1, some of which are dependent on Prx redox status [92]. Unfortunately, the molecular details of these interactions are lacking [34, 92] so that it is not yet possible to assess their meaning.
Concluding Remarks
With the wide array of modifications and peroxidase-dependent and independent secondary functions, as well as the delicate balance regulating the oligomeric state and functional attributes of Prxs, excitement is building as we continue to learn more about the pivotal roles Prxs play in organismal growth and survival across the biome. It is remarkable that despite being separated by billions of years of evolutionary optimization, Prxs in organisms as distantly related as humans and deep sea archaea possess the same active site components. The universal conservation of the Thr and Arg active site residues speaks to their essential role in enhancing the peroxidatic reactivity of the active site Cys, and although details remain to be fleshed out, with ~120 experimentally-determined structures of Prxs, and advances in kinetic assays, a reasonable understanding of the mechanism appears to be in hand.
The larger open questions regard the nature of and mechanisms of regulatory functions served by Prxs, especially as a part of normal growth and development. A complex interplay exists between peroxide levels and peroxidases, as most eukaryotes possess multiple Prx isoforms, as well as other peroxidases such as catalase and Gpxs. It remains to be determined how this interplay and regulatory behavior varies at different developmental stages, between tissue types, in disease states and across species. Investigating such questions and unraveling these complex relationships will be crucial for understanding the roles Prxs play in cellular homeostasis, as well as in cancer development and drug resistance, Alzheimer’s disease, and ischemic brain injury. A list of open questions in the field is provided in Box 3.
BOX 3. Outstanding questions related to Prx function.
Activity and Oligomerization
Why are some Prxs inherently slower reacting?
What is the in vivo relevance of the dimer↔decamer transitions for Prx1 enzymes?
To what extent do Prxs react in vivo with other reactive compounds such HOCl and chloramines?
What is the physiological significance of the reported chaperone activities of Prxs?
Signaling and Regulation
What aspects of normal cell growth and development are influenced by Prx inactivation?
What dictates whether Prx activity will be modulated by hyperoxidation or another posttranslational modification?
Why do knockouts and knockdowns of individual Prx isoforms cause such dramatic phenotypes, even when there are other isoforms in the same cellular compartments (e.g. for PrxI and PrxII in mice)?
Which organisms utilize non-stress-related peroxide signaling?
To what extent does peroxide locally build up in cells as part of normal signaling?
To what extent do Prxs colocalize with other proteins involved in peroxide-mediated signaling?
To what extent are Prxs direct transducers promoting disulfide formation in specific downstream target proteins?
How does the interplay of sulfiredoxin and peroxiredoxins influence aging?
Why do some organisms without sulfiredoxin seem to have Prx isoforms sensitive to hyperoxidation?
Why do some cyanobacteria have sulfiredoxin, but not other bacteria?
Are Prxs key players in the regulation of circadian rhythms?
Highlights.
Peroxiredoxins are ubiquitous defense enzymes and in some cases signaling regulators
ROOH orientation and polarization in the active site promotes peroxidase activity
The activated, catalytic cysteine can be susceptible to inactivation by hyperoxidation
Sulfiredoxin-mediated repair of hyperoxidation restores activity in some organisms
Regulated peroxiredoxins link peroxide metabolism and signaling
Acknowledgments
The authors thank many collaborators and colleagues who have contributed to the field and this review in various ways, and regret the omission of important contributions due to space limitations. Support from the National Institutes of Health (R01 GM050389 to L.B.P. and P.A.K., and R33 CA177461 to L.B.P. and C. M. Furdui) is gratefully acknowledged.
GLOSSARY
- Arrhenius analysis
determination of the activation energy (Ea) for a chemical reaction by measuring the reaction rate at multiple temperatures and fitting the experimental data to the Arrhenius equation for how a rate constant (k) depends on absolute temperature (T): k = Ae−Ea/RT, where R is the universal gas constant, and A is a factor accounting for the fraction of substrate molecules that have the kinetic energy to react. The Eyring equation gives the temperature dependence of the entropy (ΔS‡) and the enthalpy (ΔH‡) of formation. Eyring equation: , where N is Avagadro’s number and h is Plank’s constant.
- Disumtation of superoxide
a chemical process whereby one molecule of superoxide donates one electron to another molecule of superoxide, generating one molecule each of oxygen and hydrogen peroxide. This process can be accelerated by the enzyme superoxide dismutase, but is also quite rapid in the absence of enzyme.
- Double displacement/ping pong kinetics
a mechanism in which the enzyme first reacts with one substrate to release a product and give a modified, intermediate form of the enzyme, then reacts with the second substrate to return to its original state and release a second product. A double displacement reaction gives a characteristic family of parallel lines when the concentrations of both substrates are varied and the initial rates are plotted on a double reciprocal, Lineweaver-Burk plot [1].
- Henderson-Hasselbalch equation
- an equation relating the negative log of the hydrogen ion (H+) concentration (pH) to the ratio of the concentrations of deprotonated (A−, the conjugate base) to protonated (HA, the conjugate acid) forms of a chemical species for which the acid dissociation constant (Ka), and therefore the negative log of the Ka (pKa), is known [1]:
Used here, HA represents the thiol group (protonated Cys) and A− represents the thiolate group (deprotonated Cys). - Michaelis complex
the enzyme-substrate complex formed prior to any chemical change.
- Oxidizing equivalent
a species that has the propensity to accept one or two electrons depending on the reaction in question.
- Quantum mechanics-molecular mechanics (QM-MM) studies
a computational method for simulating enzyme reactions. A small part of the system directly participating in the chemical reaction (typically some atoms of the substrate and enzyme) is simulated using quantum mechanics (QM), which analyzes the electronic changes involved in the making and breaking of bonds. The remainder of the system is simulated using a simpler and less computationally expensive molecular mechanics (MM) forcefield that treats atoms as fixed quantities [2]
- Redox relay
the transfer of redox state from one molecule to another, typically in the context of this review by the transfer of two electrons at a time between thiols, disulfides or sulfenic acid centers. These transfers often occur via a transient mixed-disulfide intermediate linking the two centers during thiol-disulfide interchange [3].
- Second order rate constant
a rate constant which, when multiplied by the concentrations of two reactants in a bimolecular chemical reaction, yields the rate of the reaction. Typical units for a second order rate constant are M−1 s−1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Segel IH. Biochemical Calculations. John Wiley & Sons, Inc; 1976. [Google Scholar]
- 2.van der Kamp MW, Mulholland AJ. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry. 2013;52:2708–2728. doi: 10.1021/bi400215w. [DOI] [PubMed] [Google Scholar]
- 3.Winther JR, Thorpe C. Quantification of thiols and disulfides. Biochim Biophys Acta. 2014;1840:838–846. doi: 10.1016/j.bbagen.2013.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karplus PA. A primer on peroxiredoxin biochemistry. Free Radic Biol Med. 2014 doi: 10.1016/j.freeradbiomed.2014.10.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 6.Chae HZ, et al. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci USA. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adimora NJ, et al. A model of redox kinetics implicates the thiol proteome in cellular hydrogen peroxide responses. Antioxid Redox Signal. 2010;13:731–743. doi: 10.1089/ars.2009.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang KS, et al. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J Biol Chem. 2002;277:38029–38036. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]
- 9.Wood ZA, et al. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 10.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woo HA, et al. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 12.Biteau B, et al. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 13.Neumann CA, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 14.Gong F, et al. Peroxiredoxin 1 promotes tumorigenesis through regulating the activity of mTOR/p70S6K pathway in esophageal squamous cell carcinoma. Medical oncology (Northwood, London, England) 2015;32:455. doi: 10.1007/s12032-014-0455-0. [DOI] [PubMed] [Google Scholar]
- 15.Ishii T, et al. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. Journal of clinical biochemistry and nutrition. 2012;50:91–105. doi: 10.3164/jcbn.11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nystrom T, et al. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Genes Dev. 2012;26:2001–2008. doi: 10.1101/gad.200006.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perkins A, et al. Tuning of peroxiredoxin catalysis for various physiological roles. Biochemistry. 2014;53:7693–7705. doi: 10.1021/bi5013222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed TT, et al. Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J Cell Mol Med. 2009;13:2019–2029. doi: 10.1111/j.1582-4934.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shichita T, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- 20.Gretes MC, et al. Peroxiredoxins in parasites. Antioxid Redox Signal. 2012;17:608–633. doi: 10.1089/ars.2011.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parsonage D, et al. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peskin AV, et al. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 23.Manta B, et al. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch Biochem Biophys. 2009;484:146–154. doi: 10.1016/j.abb.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 24.Aden J, et al. Extraordinary mus-ms backbone dynamics in Arabidopsis thaliana peroxiredoxin Q. Biochim Biophys Acta. 2011;1814:1880–1890. doi: 10.1016/j.bbapap.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 25.Perkins A, et al. Mapping the active site helix-to-strand conversion of CxxxxC Peroxiredoxin Q enzymes. Biochemistry. 2012;51:7638–7650. doi: 10.1021/bi301017s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiorillo A, et al. The crystal structures of the tryparedoxin-tryparedoxin peroxidase couple unveil the structural determinants of Leishmania detoxification pathway. PLoS neglected tropical diseases. 2012;6:e1781. doi: 10.1371/journal.pntd.0001781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao Z, et al. Crystal structure of reduced and of oxidized peroxiredoxin IV enzyme reveals a stable oxidized decamer and a non-disulfide-bonded intermediate in the catalytic cycle. J Biol Chem. 2011;286:42257–42266. doi: 10.1074/jbc.M111.298810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perkins A, et al. The sensitive balance between the fully folded and locally unfolded conformations of a model peroxiredoxin. Biochemistry. 2013;52:8708–8721. doi: 10.1021/bi4011573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peskin AV, et al. Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem. 2013;288:14170–14177. doi: 10.1074/jbc.M113.460881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowther WT, Haynes AC. Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxid Redox Signal. 2011;15:99–109. doi: 10.1089/ars.2010.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Day AM, et al. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Mol Cell. 2012;45:398–408. doi: 10.1016/j.molcel.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 32.Flohé L. Changing paradigms in thiology from antioxidant defense toward redox regulation. Methods Enzymol. 2010;473:1–39. doi: 10.1016/S0076-6879(10)73001-9. [DOI] [PubMed] [Google Scholar]
- 33.Karplus PA, Poole LB. Peroxiredoxins as molecular triage agents, sacrificing themselves to enhance cell survival during a peroxide attack. Mol Cell. 2012;45:275–278. doi: 10.1016/j.molcel.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 35.Randall LM, et al. Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods Enzymol. 2013;527:41–63. doi: 10.1016/B978-0-12-405882-8.00003-9. [DOI] [PubMed] [Google Scholar]
- 36.Teixeira F, et al. Mitochondrial peroxiredoxin functions as crucial chaperone reservoir in Leishmania infantum. Proc Natl Acad Sci U S A. 2015;112:E616–624. doi: 10.1073/pnas.1419682112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson KJ, et al. Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins. 2011;79:947–964. doi: 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall A, et al. Typical 2-Cys peroxiredoxins - structures, mechanisms and functions. Febs J. 2009;276:2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood ZA, et al. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 40.Rhee SG, et al. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J Biol Chem. 2012;287:4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Budde H, et al. Verification of the interaction of a tryparedoxin peroxidase with tryparedoxin by ESI-MS/MS. Biol Chem. 2003;384:1305–1309. doi: 10.1515/BC.2003.146. [DOI] [PubMed] [Google Scholar]
- 42.Jönsson TJ, et al. Cysteine reactivity and thiol-disulfide interchange pathways in AhpF and AhpC of the bacterial alkyl hydroperoxide reductase system. Biochemistry. 2007;46:5709–5721. doi: 10.1021/bi7001218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lian FM, et al. Structural snapshots of yeast alkyl hydroperoxide reductase Ahp1 peroxiredoxin reveal a novel two-cysteine mechanism of electron transfer to eliminate reactive oxygen species. J Biol Chem. 2012;287:17077–17087. doi: 10.1074/jbc.M112.357368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kyte J. Structure in Protein Chemistry. Garland Publishing, Inc; 1995. [Google Scholar]
- 45.Marino SM, Gladyshev VN. Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J Mol Biol. 2010;404:902–916. doi: 10.1016/j.jmb.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med. 2015;80:148–157. doi: 10.1016/j.freeradbiomed.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poole LB, et al. Overview of peroxiredoxins in oxidant defense and redox regulation. In: Maines Mahin D, et al., editors. Current protocols in toxicology. Chapter 7. 2011. pp. 7.9.1–7.9.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrer-Sueta G, et al. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol. 2011;24:434–450. doi: 10.1021/tx100413v. [DOI] [PubMed] [Google Scholar]
- 49.Whitesides GM, et al. Rates of thiol-disulfide interchange reactions between mono- and dithiols and Ellman’s reagent. J Org Chem. 1977;42:332–338. [Google Scholar]
- 50.Winterbourn CC, Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic Biol Med. 1999;27:322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 51.Hall A, et al. Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid Redox Signal. 2011;15:795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hall A, et al. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J Mol Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Portillo-Ledesma S, et al. Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry. 2014 doi: 10.1021/bi500389m. [DOI] [PubMed] [Google Scholar]
- 54.Zeida A, et al. The extraordinary catalytic ability of peroxiredoxins: a combined experimental and QM/MM study on the fast thiol oxidation step. Chemical communications (Cambridge, England) 2014;50:10070–10073. doi: 10.1039/c4cc02899f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakamura T, et al. Crystal structure of peroxiredoxin from Aeropyrum pernix K1 complexed with its substrate, hydrogen peroxide. Journal of biochemistry. 2010;147:109–115. doi: 10.1093/jb/mvp154. [DOI] [PubMed] [Google Scholar]
- 56.Nagy P, et al. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J Biol Chem. 2011;286:18048–18055. doi: 10.1074/jbc.M111.232355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tairum CA, Jr, et al. Disulfide biochemistry in 2-cys peroxiredoxin: requirement of Glu50 and Arg146 for the reduction of yeast Tsa1 by thioredoxin. J Mol Biol. 2012;424:28–41. doi: 10.1016/j.jmb.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 58.Trujillo M, et al. Kinetics of peroxiredoxins and their role in the decomposition of peroxynitrite. Sub-cellular biochemistry. 2007;44:83–113. doi: 10.1007/978-1-4020-6051-9_5. [DOI] [PubMed] [Google Scholar]
- 59.Dietz KJ. Peroxiredoxins in plants and cyanobacteria. Antioxid Redox Signal. 2011;15:1129–1159. doi: 10.1089/ars.2010.3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bang YJ, et al. Distinct characteristics of two 2-Cys peroxiredoxins of Vibrio vulnificus suggesting differential roles in detoxifying oxidative stress. J Biol Chem. 2012;287:42516–42524. doi: 10.1074/jbc.M112.421214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horta BB, et al. Structural and biochemical characterization of peroxiredoxin Qbeta from Xylella fastidiosa: catalytic mechanism and high reactivity. J Biol Chem. 2010;285:16051–16065. doi: 10.1074/jbc.M109.094839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reeves SA, et al. Kinetic and thermodynamic features reveal that Escherichia coli BCP is an unusually versatile peroxiredoxin. Biochemistry. 2011;50:8970–8981. doi: 10.1021/bi200935d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nogoceke E, et al. A unique cascade of oxidoreductases catalyses trypanothione-mediated peroxide metabolism in Crithidia fasciculata. Biol Chem. 1997;378:827–836. doi: 10.1515/bchm.1997.378.8.827. [DOI] [PubMed] [Google Scholar]
- 64.Ogusucu R, et al. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radic Biol Med. 2007;42:326–334. doi: 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 65.Cox AG, et al. Redox potential and peroxide reactivity of human peroxiredoxin 3. Biochemistry. 2009;48:6495–6501. doi: 10.1021/bi900558g. [DOI] [PubMed] [Google Scholar]
- 66.Nelson KJ, et al. Cysteine pKa values for the bacterial peroxiredoxin AhpC. Biochemistry. 2008;47:12860–12868. doi: 10.1021/bi801718d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parsonage D, et al. Dissecting Peroxiredoxin Catalysis: Separating Binding, Peroxidation, and Resolution for a Bacterial AhpC. Biochemistry. 2015 doi: 10.1021/bi501515w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Loumaye E, et al. Kinetic studies of peroxiredoxin 6 from Arenicola marina: rapid oxidation by hydrogen peroxide and peroxynitrite but lack of reduction by hydrogen sulfide. Arch Biochem Biophys. 2011;514:1–7. doi: 10.1016/j.abb.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 69.Trujillo M, et al. Pre-steady state kinetic characterization of human peroxiredoxin 5: taking advantage of Trp84 fluorescence increase upon oxidation. Arch Biochem Biophys. 2007;467:95–106. doi: 10.1016/j.abb.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 70.Hugo M, et al. Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: kinetics, acidity constants, and conformational dynamics. Biochemistry. 2009;48:9416–9426. doi: 10.1021/bi901221s. [DOI] [PubMed] [Google Scholar]
- 71.Reyes AM, et al. Oxidizing substrate specificity of Mycobacterium tuberculosis alkyl hydroperoxide reductase E: kinetics and mechanisms of oxidation and overoxidation. Free Radic Biol Med. 2011;51:464–473. doi: 10.1016/j.freeradbiomed.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 72.Bryk R, et al. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 73.Sundaresan M, et al. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 74.Oakley FD, et al. Signaling Components of Redox Active Endosomes: The Redoxosomes. Antioxid Redox Signal. 2009;11:1313–1333. doi: 10.1089/ars.2008.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nisimoto Y, et al. Nox4: a hydrogen peroxide-generating oxygen sensor. Biochemistry. 2014;53:5111–5120. doi: 10.1021/bi500331y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rhee SG, et al. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 78.Haque A, et al. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell. 2011;147:185–198. doi: 10.1016/j.cell.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Forman HJ, et al. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sobotta MC, et al. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol. 2015;11:64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 81.Chen CH, et al. Reactive oxygen species generation is involved in epidermal growth factor receptor transactivation through the transient oxidization of Src homology 2-containing tyrosine phosphatase in endothelin-1 signaling pathway in rat cardiac fibroblasts. Mol Pharmacol. 2006;69:1347–1355. doi: 10.1124/mol.105.017558. [DOI] [PubMed] [Google Scholar]
- 82.Woo HA, et al. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 83.Mishina NM, et al. Does cellular hydrogen peroxide diffuse or act locally? Antioxid Redox Signal. 2011;14:1–7. doi: 10.1089/ars.2010.3539. [DOI] [PubMed] [Google Scholar]
- 84.Malinouski M, et al. Hydrogen peroxide probes directed to different cellular compartments. PloS one. 2011;6:e14564. doi: 10.1371/journal.pone.0014564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klomsiri C, et al. Endosomal H2O2 production leads to localized cysteine sulfenic acid formation on proteins during lysophosphatidic acid-mediated cell signaling. Free Radic Biol Med. 2014;71C:49–60. doi: 10.1016/j.freeradbiomed.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paulsen CE, et al. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2011;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haynes AC, et al. Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J Biol Chem. 2013;288:29714–29723. doi: 10.1074/jbc.M113.473470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nelson KJ, et al. Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Methods Enzymol. 2013;527:21–40. doi: 10.1016/B978-0-12-405882-8.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Winterbourn CC, Hampton MB. Redox biology: Signaling via a peroxiredoxin sensor. Nat Chem Biol. 2015;11:5–6. doi: 10.1038/nchembio.1722. [DOI] [PubMed] [Google Scholar]
- 90.Kil IS, et al. Feedback control of adrenal steroidogenesis via H(2)O(2)-dependent, reversible inactivation of Peroxiredoxin III in mitochondria. Mol Cell. 2012;46:584–594. doi: 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 91.Randall LM, et al. Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J Biol Chem. 2014;289:15536–15543. doi: 10.1074/jbc.M113.539213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chae HZ, et al. Protein glutathionylation in the regulation of peroxiredoxins: a family of thiol-specific peroxidases that function as antioxidants, molecular chaperones, and signal modulators. Antioxid Redox Signal. 2012;16:506–523. doi: 10.1089/ars.2011.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Calvo IA, et al. Dissection of a redox relay: H2O2-dependent activation of the transcription factor Pap1 through the peroxidatic Tpx1-thioredoxin cycle. Cell reports. 2013;5:1413–1424. doi: 10.1016/j.celrep.2013.11.027. [DOI] [PubMed] [Google Scholar]
- 94.Fomenko DE, et al. Thiol peroxidases mediate specific genome-wide regulation of gene expression in response to hydrogen peroxide. Proc Natl Acad Sci U S A. 2011;108:2729–2734. doi: 10.1073/pnas.1010721108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jang HH, et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 96.Phalen TJ, et al. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Robinson I, Reddy AB. Molecular mechanisms of the circadian clockwork in mammals. FEBS Lett. 2014;588:2477–2483. doi: 10.1016/j.febslet.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 98.O’Neill JS, Feeney KA. Circadian redox and metabolic oscillations in mammalian systems. Antioxid Redox Signal. 2014;20:2966–2981. doi: 10.1089/ars.2013.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cho CS, et al. Circadian rhythm of hyperoxidized peroxiredoxin II is determined by hemoglobin autoxidation and the 20S proteasome in red blood cells. Proc Natl Acad Sci U S A. 2014;111:12043–12048. doi: 10.1073/pnas.1401100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Copley SD, et al. Divergence of function in the thioredoxin fold suprafamily: evidence for evolution of peroxiredoxins from a thioredoxin-like ancestor. Biochemistry. 2004;43:13981–13995. doi: 10.1021/bi048947r. [DOI] [PubMed] [Google Scholar]
- 101.Cammer SA, et al. Structure-based active site profiles for genome analysis and functional family subclassification. J Mol Biol. 2003;334:387–401. doi: 10.1016/j.jmb.2003.09.062. [DOI] [PubMed] [Google Scholar]
- 102.Soito L, et al. PREX: PeroxiRedoxin classification indEX, a database of subfamily assignments across the diverse peroxiredoxin family. Nucleic Acids Res. 2011;39:D332–337. doi: 10.1093/nar/gkq1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saccoccia F, et al. Moonlighting by different stressors: crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure. 2012;20:429–439. doi: 10.1016/j.str.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wood ZA, et al. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]