

Abstract
In humans, atrial fibrillation is often triggered by ectopic pacemaking activity in the myocardium sleeves of the pulmonary vein (PV) and systemic venous return. The genetic programs that abnormally reinforce pacemaker properties at these sites and how this relates to normal sinoatrial node (SAN) development remain uncharacterized. It was noted previously that Nkx2-5, which is expressed in the PV myocardium and reinforces a chamber-like myocardial identity in the PV, is lacking in the SAN. Here we present evidence that in mice Shox2 antagonizes the transcriptional output of Nkx2-5 in the PV myocardium and in a functional Nkx2-5+ domain within the SAN to determine cell fate. Shox2 deletion in the Nkx2-5+ domain of the SAN caused sick sinus syndrome, associated with the loss of the pacemaker program. Explanted Shox2+ cells from the embryonic PV myocardium exhibited pacemaker characteristics including node-like electrophysiological properties and the capability to pace surrounding Shox2− cells. Shox2 deletion led to Hcn4 ablation in the developing PV myocardium. Nkx2-5 hypomorphism rescued the requirement for Shox2 for the expression of genes essential for SAN development in Shox2 mutants. Similarly, the pacemaker-like phenotype induced in the PV myocardium in Nkx2-5 hypomorphs reverted back to a working myocardial phenotype when Shox2 was simultaneously deleted. A similar mechanism is also adopted in differentiated embryoid bodies. We found that Shox2 interacts with Nkx2-5 directly, and discovered a substantial genome-wide co-occupancy of Shox2, Nkx2-5 and Tbx5, further supporting a pivotal role for Shox2 in the core myogenic program orchestrating venous pole and pacemaker development.
KEY WORDS: Shox2, Nkx2-5, Atrial fibrillation, Pulmonary vein, Sinoatrial node, Cell fate, Mouse, Human
Summary: Antagonism between Shox2 and Nkx2-5 in the cardiac venous pole of mouse embryos regulates cell fate, morphogenesis and the distinction between pacemaker cells and working myocardium.
INTRODUCTION
After the formation of a linear heart tube through fusion of the bilateral cardiogenic plates (the first heart field), additional progenitor cells are continually recruited from surrounding mesenchyme to both the inflow and outflow poles of the heart tube (Christoffels et al., 2006; Gittenberger-de Groot et al., 2007; Snarr et al., 2007; Xie et al., 2012). Anterior pole development has been studied extensively, but less information is available regarding posterior pole development. Many major issues, such as the functional identity of the pulmonary vein (PV) myocardium in relation to the myocardial sleeves of the systemic venous return, remain to be resolved (Douglas et al., 2011; Gittenberger-de Groot, 2011; Lescroart et al., 2012; Moorman and Anderson, 2011; van den Berg and Moorman, 2011). Ectopic triggers in the PV myocardium often account for focally induced atrial fibrillation (AF) (Douglas et al., 2011; Haïssaguerre et al., 1998). The PV myocardium was initially thought to be derived from atrial myocardium (Millino et al., 2000), but was recently reported to be differentiated from mesenchymal cells surrounding the developing venous pole (Mommersteeg et al., 2007a; Moorman et al., 2007; Peng et al., 2013). The PV myocardium is also positive for HNK-1 and CCS-lacZ, two putative cardiac conduction system (CCS) markers (Douglas et al., 2011; Jongbloed et al., 2004; Wenink et al., 2000). The association of mutations in NKX2-5 with AF patients (Huang et al., 2013; Xie et al., 2013), and the switch of the PV myocardium to an Hcn4+/Cx40− phenotype resembling that of the systemic venous myocardium in an Nkx2-5 hypomorphic mouse model (Martin, 2007; Mommersteeg et al., 2007a), suggest that Nkx2-5 acts as a repressor of the ‘default’ systemic venous genetic program in the PV myocardium, thus preventing this myocardium from pacemaker activity. Although melanocyte-like cells in the heart were also identified as non-myocardial triggers contributing to AF (Levin et al., 2009), factors that promote ectopic pacemaker fate in the PV myocardium remain to be identified.
The sinoatrial node (SAN), which is derived from the sinus venosus, acts as the primary cardiac pacemaker and can be morphologically identified in mice at embryonic day (E) 10.5 (Christoffels et al., 2006; Gittenberger-de Groot et al., 2007). Subsequently, the SAN is identified as a structure comprising an Nkx2-5− head region and an Nkx2-5+ sinoatrial (SA) junction or tail region (Liang et al., 2013; Liu et al., 2007; Wiese et al., 2009; Yamamoto et al., 2006), suggesting that the development of these two distinct SAN domains (Nkx2-5+ versus Nkx2-5−) is regulated by different mechanisms. The SAN is characterized by the expression of Hcn4, Tbx3, Isl1 and Shox2, but is negative for Cx40 (Gja5) and Nppa (Munshi, 2012).
The mouse and human Shox2 homeobox gene shares 99% identity at the amino acid level and encodes two alternatively spliced transcripts: Shox2a and Shox2b (Blaschke et al., 1998). Although SHOX2 has not been linked to any syndrome in humans, Shox2 inactivation in mice has revealed its essential role in the development of multiple organs, including the heart (Blaschke et al., 2007; Cobb et al., 2006; Espinoza-Lewis et al., 2009; Gu et al., 2008; Yu et al., 2005, 2007). Shox2 mutation results in a severely hypoplastic SAN, which is likely to be due to ectopic Nkx2-5 activation in the otherwise Nkx2-5− SAN head region (Blaschke et al., 2007; Espinoza-Lewis et al., 2009).
Despite the well-recognized role of Nkx2-5 as a core transcription factor in heart development, its function in venous pole development remains controversial. Nkx2-5 is expressed in the developing PV but is initially absent in the sinus venosus. Nkx2-5 was shown to be essential for maintaining the Cx40+/Hcn4− cell fate in the PV myocardium and for establishing a strict boundary of the SAN domain from the surrounding atrial working myocardium by inhibiting Hcn4 but activating Cx40 expression (Mommersteeg et al., 2007b). However, Nkx2-5 expression was also found in the SA junction region that is Hcn4+/Cx40− (Liang et al., 2013; Wiese et al., 2009), suggesting the existence of a mechanism that blocks the transcriptional output of Nkx2-5 (i.e. the transcription of Nkx2-5 target genes). Although Tbx3 blocks Cx40 activation in the SAN, Tbx3 is not required for Hcn4 expression (Frank et al., 2012; Wiese et al., 2009), implicating the involvement of other regulatory factors that are yet to be identified.
In this study, we provide evidence for a Shox2–Nkx2-5 antagonistic mechanism operating in the cardiac venous pole, particularly in the SAN and the PV myocardium, to regulate cell fate, morphogenesis and the distinction between pacemaker cells and working myocardium.
RESULTS
Expression of Shox2 in the developing venous pole
We and others have reported previously an essential role for Shox2 in SAN development (Blaschke et al., 2007; Espinoza-Lewis et al., 2009). To comprehensively document the Shox2 expression pattern in the developing heart, we created a knock-in allele (Shox2HA) that harbors a FLAG-HA-tagged Shox2a isoform coupled with IRES-DsRed sequences (Wang et al., 2014a). Using this allele, which allows for live imaging of Shox2 expression, we found a wide but specific Shox2 expression domain in the developing venous pole (Fig. 1A; supplementary material Fig. S1A). We confirmed this expression pattern by immunohistochemistry using anti-Shox2 antibodies (Fig. 1B). Given the essential role for Shox2 in SAN development, we also examined Hcn4 expression, a functional molecular marker for the CCS. Indeed, Hcn4 colocalized substantially with Shox2 in the venous pole, particularly in the sinus venosus and its derivatives including the coronary sinus, right sinus horn, SAN and venous valves (Fig. 1B). Intriguingly, Hcn4 also colocalized with Shox2 in the cTnT (Tnnt2)+ PV myocardium, although it was expressed at a relatively low level compared with the surrounding tissues (inset in Fig. 1B; supplementary material Fig. S1D,E). The PV myocardium was believed to be derived from a lineage, distinct from that of the systemic venous return that exhibits characteristics similar to pacemaker cells in the developing embryo (Ammirabile et al., 2012; Liang et al., 2013; Mommersteeg et al., 2007a; Vedantham et al., 2013), but the colocalization of Shox2 with Hcn4 in the PV myocardium suggests a similar genetic pathway and origin for pacemaker fate in these two structures. Notably, Shox2 expression was strong in the myocardial cells surrounding the forming PV from E11.5 onwards (supplementary material Fig. S1B,D).
Fig. 1.
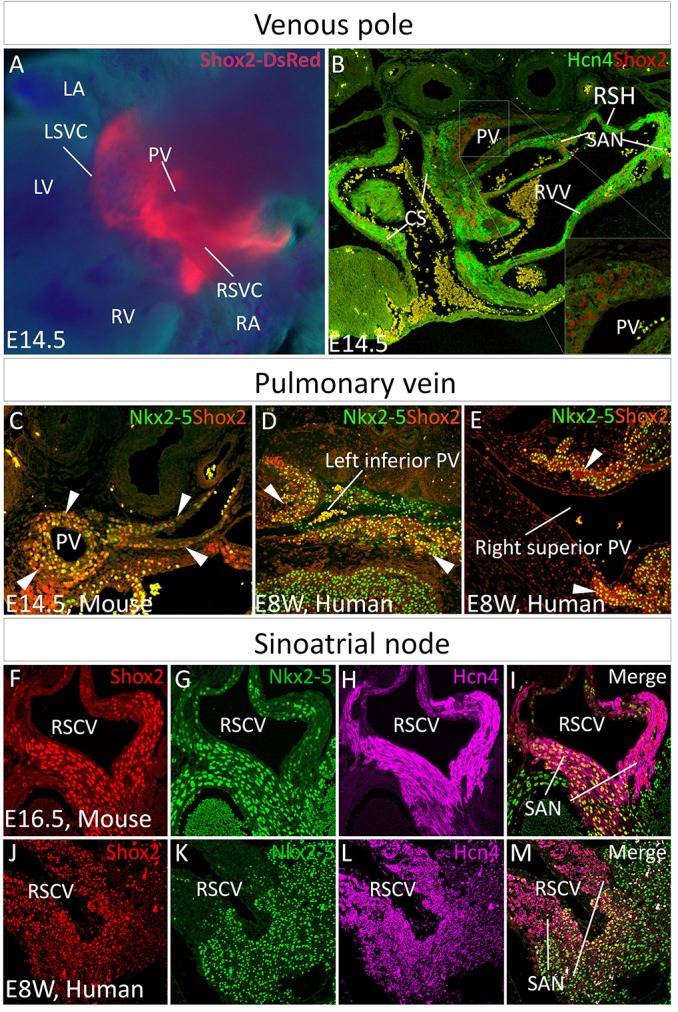
Shox2 expression in the developing venous pole. (A,B) Shox2 expression in the venous pole at E14.5, as revealed by whole-mount DsRed expression in a Shox2HA mouse embryo (A), and its colocalization with Hcn4, as revealed by immunohistochemistry (B). (C-E) Immunohistochemistry shows colocalization (arrowheads) of Shox2 and Nkx2-5 in the PV myocardium of mouse (C) and human (D,E) embryos. (F-M) Immunohistochemistry reveals colocalization of Shox2, Nkx2-5 and Hcn4 in the SAN of an E16.5 mouse embryo (F-I) and human embryo at 8 weeks gestation (J-M). CS, coronary sinus; LA, left atrium; LV, left ventricle; PV, pulmonary vein; RA, right atrium; RSH, right sinus horn; RV, right ventricle; RVV, right venus valve; SAN, sinoatrial node; LSVC, left superior vena cava; RSVC, right superior vena cava.
Since the PV myocardium expresses Nkx2-5, which distinguishes it from the adjacent sinus venosus-derived tissues (Mommersteeg et al., 2007a), we examined Shox2 and Nkx2-5 colocalization. Shox2 was co-expressed with Nkx2-5 in the developing PV in both mice and humans (Fig. 1C-E). It was reported previously that Shox2 regulates SAN development by preventing Nkx2-5 expression (Blaschke et al., 2007; Espinoza-Lewis et al., 2009). Such colocalization of Shox2 with Nkx2-5 in the PV myocardium prompted us to closely examine the expression patterns of Shox2, Nkx2-5 and Hcn4 in the developing SAN. Co-immunohistochemistry revealed a prominent domain in the SAN, primarily the SA junction/SAN tail, where Shox2, Nkx2-5 and Hcn4 colocalized in mouse and human embryos (Fig. 1F-M). This suggests an unidentified but conserved function of Shox2 and Nkx2-5 in regulating SAN development in mice and humans. The colocalization could be observed as early as E11.5 in mice (supplementary material Fig. S1C).
Shox2 sustains SAN cell fate in the Nkx2-5+ domain
To determine the specific role of Shox2 in the Nkx2-5+ domain in SAN development and function we compounded the Nkx2-5-IRESCre knock-in allele (Stanley et al., 2002), which exhibits Cre activity in the Nkx2-5+ domain of the SAN (supplementary material Fig. S2), with the floxed Shox2 allele (Cobb et al., 2006) to generate Nkx2-5IRESCre/+;Shox2F/F mice. Although mutant mice survived to adulthood with grossly normal size and appearance, the SAN function, as determined by surface ECG, was compromised. Nkx2-5IRESCre/+;Shox2F/F mice had severe bradycardia and highly irregular R-R intervals compared with age- and sex-matched controls at 2 months of age. The severely delayed P wave and short and variable P-R interval, as well as the lack of any obvious signs of atrial-ventrical (A-V) conduction block, indicated defective SAN function (Fig. 2A-D; supplementary material Fig. S3). A closer look at ECG lead II and lead III of the mutant mice revealed changes in P wave configuration, including abnormal diphasic P waves in lead II and upright P waves in lead III (Fig. 2B). The association of such changes in P wave configuration with the significantly variable P-P, R-R and P-R intervals compared with controls (Fig. 2D; supplementary material Fig. S3) indicated severe sick sinus syndrome and a potential shift of the dominant pacemaking site. However, the lead I P wave in Nkx2-5IRESCre/+;Shox2F/F mice remained upright in shape, indicating retention of the dominant pacemaking site in the right side of the heart. Associated with the sick sinus syndrome was the lack of identifiable SA junction structures in Nkx2-5IRESCre/+;Shox2F/F mice (Fig. 2E,F).
Fig. 2.

Sick sinus syndrome caused by Shox2 ablation in the Nkx2-5 expression domain. (A) Representative tracing of surface ECG lead I reveals bradycardia and irregular R-R intervals in Nkx2-5IRESCre/+;Shox2F/F (Shox2Nkx2-5Cre) mice as compared with controls at 8 weeks of age (n=12 each). (B) ECG tracing of lead I, II and III of Nkx2-5IRESCre/+;Shox2F/F mice shows various types of P wave disfiguration (red arrows), including diphasic P wave, no P wave and extremely shortened P-R interval. (C) Slower heartbeat rate of Nkx2-5IRESCre/+;Shox2F/F mice compared with controls at 8 weeks of age. **P<0.01. (D) Highly variable P-R intervals in the Nkx2-5IRESCre/+;Shox2F/F mice (n=12). (E,F) The absence of typical Nkx2-5+ SA junction structures in 8-week-old Nkx2-5IRESCre/+;Shox2F/F mice (F) compared with controls (E). Open arrowheads point to the SAN head; white arrowheads point to Nkx2-5+ cells; and arrows point to the SA junction. RA, right atrium; RSVC, right superior vena cava. BPM, beats per min.
Histological and molecular analyses were conducted to examine the developmental defects that contribute to the hypoplasia of the Nkx2-5+ SA junction and compromised SAN function in Nkx2-5IRESCre/+;Shox2F/F mice. At E12.5 the Nkx2-5+ domain in the SAN of Nkx2-5IRESCre/+;Shox2F/F mice had begun to exhibit discernible hypoplasia associated with dramatically reduced expression of Ki67 and the loss of SAN identity as assessed by the absence of Tbx3, Hcn4 and Isl1 expression but ectopic Cx40 activation (supplementary material Fig. S4). At E14.5, the altered expression patterns of these molecular markers persisted in the mutant SA junction, which became severely hypoplastic (Fig. 3A-H′). The elevated cTnT expression in the SA junction of Nkx2-5IRESCre/+;Shox2F/F mice further suggested its adoption of a working myocardium fate (Fig. 3H′). Interestingly, in the Nkx2-5− SAN head, where Shox2 expression was retained (Fig. 3B′,D′), the SAN identity remained unaltered, as evidenced by the persistent expression of SAN molecular markers (Fig. 3A′-H′). Three-dimensional reconstruction of the SAN in controls and mutants using Hcn4 expression as the key marker highlighting both the head and junction regions (Fig. 3I,I′), revealed a more than 64% reduction in the overall volume of the reconstructed SAN in Nkx2-5IRESCre/+;Shox2F/F mice (Fig. 3J).
Fig. 3.

Shox2 is required for maintaining the default SAN cell fate in the Nkx2-5+ domain. (A-H) Immunohistochemistry shows colocalization of Hcn4, Shox2, Tbx3 and Isl1, the absence of Cx40, as well as a relatively low level of cTnT in the Nkx2-5+ SA junction of the E14.5 wild-type embryo. (A′-H′) Immunohistochemistry reveals absence of Hcn4, Tbx3 and Isl1 expression but elevated Cx40 and cTnT levels in the Nkx2-5+ SA junction of E14.5 Nkx2-5IRESCre/+;Shox2F/F (Shox2Nkx2-5Cre) mice. The expression pattern of these genes remained unaltered in the Nkx2-5− SAN head region compared with controls. (I-J) 3D reconstruction and comparison of the SAN from E14.5 wild-type (I) and Nkx2-5IRESCre/+;Shox2F/F (I′) embryos based on the Hcn4+ domain revealed significantly reduced size of the SAN in the mutant (J) (n=3 each). White arrowheads point to the Nkx2-5+ SA junction and open arrowheads point to the Nkx2-5− SAN head. **P<0.01.
Shox2 potentiates a pacemaker-like phenotype in the PV myocardium
As a positive regulator of SAN development, Shox2 is co-expressed with Hcn4 in the proximal PV and the adjacent coronary sinus (Fig. 1B), another site that triggers and sustains AF in humans (Eckardt, 2002; Habib et al., 2009; Lemola et al., 2005). This suggests that the Shox2+ cells in the PV and coronary sinus possess pacemaking activity. To test this hypothesis, we first compared the contraction rate between Shox2+ cells and the surrounding Shox2− atrial myocardial cells in cultured cell clumps. We isolated the proximal PV tissue from E14.5 Shox2HA reporter mice and separated Shox2+ from Shox2− cells by FACS based on DsRed expression (Fig. 4A). FACS-sorted cells were re-aggregated to form cell clumps that exhibited synchronized contraction, and beating rate was counted (Fig. 4B). The results showed significantly higher beating rates in Shox2+ cell aggregates than in those made of only Shox2− cells, which comprised over 90% cTnT+ cardiomyocytes (data not shown) (Fig. 4D).
Fig. 4.

Shox2 potentiates a pacemaker-like phenotype in the PV myocardium. (A) Separation of Shox2+ cells from the surrounding Shox2− cells of the PV myocardium of E14.5 Shox2HA embryo by FACS. (B) A representative beating clump composed of Shox2+ cells. (C) An isolated DsRed+ cell from E14.5 Shox2HA PV myocardium used for whole-cell patch recording. (D) Comparison of beating rate of cell clumps consisting of Shox2+, or Shox2−, or mixed cells isolated from the PV of E14.5 Shox2HA mice (n=5). **P<0.01. (E) Two representative configurations (PV-1 and PV-2; n=3 each) in Shox2+ cells from the E14.5 PV myocardium, as compared with that seen in SAN cells isolated from an embryo of the same stage. (F-I) Immunohistochemistry shows reduced Hcn4 and enhanced Cx40 levels (arrowheads) in the PV myocardium of E14.5 Nkx2-5IRESCre/+;Shox2F/F (Shox2Nkx2-5Cre) mice (G,I), as compared with controls (F,H). (J-M) Immunohistochemistry shows complementary patterns of Hcn4 and Nkx2-5, despite the lack of Shox2, in the coronary sinus (arrowheads) of E14.5 Shox2Nkx2-5Cre mice (K,M), as compared with controls (J,L; note that panel J is the same as Fig. 1B). (N,O) Hcn4 is ablated in the coronary sinus myocardial cells (arrowheads) at E16.5 when Nkx2-5 protein accumulation becomes prominent in Shox2Nkx2-5Cre mice (O) compared with controls (N). Open arrowheads point to cells that escape Shox2 deletion and express Hcn4. (P,Q) Hcn4 expression is sustained in Nkx2-5− SAN head (open arrowheads) but ablated from the Nkx2-5+ SAN junction (arrowheads) of E11.5 Shox2−/− mice (Q), as compared with the control (P). CS, coronary sinus; PV, pulmonary vein.
We next tested whether, in mixed cell clumps, the rapidly contracting Shox2+ cells could pace Shox2− cells. Indeed, such cell clumps exhibited synchronized contraction and at a rate almost identical to that seen in the Shox2+ aggregates (Fig. 4D), suggesting that these Shox2+ cells from the PV are able to pace Shox2− myocardial cells in the absence of the SAN. Whole-cell patch clamp on individual DsRed+ cells (Fig. 4C) isolated from the PV provided electrophysiological evidence for pacemaking activity of the Shox2+ cells. As shown in Fig. 4E, two subtypes of configuration of action potentials (APs) were identified in the Shox2+ cells, which resembled APs seen in SAN cells isolated from an embryo of the same age, including spontaneous APs and a marked diastolic depolarization phase. Interestingly, subtype PV-1 markedly resembles that of SAN cells and of SAN cells transduced from cardiac fibroblasts (Nam et al., 2014), while the PV-2 subtype is similar to that reported in chicken sinus venosus cells (Lieberman and Paes de Carvalho, 1965). The existence of two subtypes of AP configuration suggests that the embryonic PV myocardial cells exhibit a primitive pacemaker-like phenotype. To further characterize the AP configurations of Shox2+ PV cells and compare them with that from other myocytes, we quantified parameters such as the amplitude, diastolic slopes and APD 50/90 from Shox2+ PV cells as well as SAN cells and Shox2− atrial cardiomyocytes. The AP configurations in PV cells appeared more similar to that in SAN cells rather than atrial cells, suggesting that Shox2+ PV cells and SAN cells have similar electrophysiological properties at this stage (supplementary material Fig. S5).
Since Shox2 expression also overlaps with Nkx2-5 in the PV and coronary sinus myocardium (Fig. 1), we investigated whether Shox2 is also required for Hcn4 expression and is involved in cell fate regulation. Similar to that observed in the Nkx2-5+ domain of the SAN, Hcn4 expression was ablated whereas Cx40 expression was greatly enhanced in the PV myocardium of E14.5 Nkx2-5IRESCre/+;Shox2F/F embryos (Fig. 4F-I), indicating a cell fate change. Reconstruction of the venous pole in control and mutant embryos based on cTnT expression and a comparison of the relative volume of Hcn4+ domains revealed that Shox2 inactivation did not lead to a discernible morphological defect but caused significant reduction in the Hcn4 expression domain in the PV (supplementary material Fig. S6). Surprisingly, Shox2 expression was ablated in the mutant coronary sinus myocardium, whereas Hcn4 expression was unchanged (Fig. 4J,K). Further examination of Nkx2-5 by immunohistochemistry revealed a barely detectable level of Nkx2-5 protein in the coronary sinus myocardium as compared with that in the PV myocardium at this stage (Fig. 4L,M). However, in Nkx2-5IRESCre/+;Shox2F/F mice at E16.5, Nkx2-5 protein had accumulated, and Hcn4 expression became abolished in the coronary sinus wall (Fig. 4N,O). Interestingly, a few cells that escaped Shox2 ablation expressed Hcn4 (Fig. 4O). Such selective inhibition of Hcn4 in Nkx2-5+ cells but not in Nkx2-5− cells was also found in the SAN of Shox2 null mice in which Hcn4 expression was abolished in the Nkx2-5high SA junction but retained in the Nkx2-5low/negative SAN head (Fig. 4P,Q). These observations indicate that Shox2 is essential for Hcn4 expression in Nkx2-5+ cells in the SAN and PV myocardium, but does not activate Hcn4 directly. We hypothesize that Shox2 acts to antagonize the repressive activity of Nkx2-5 on the expression of the pacemaker program, including Hcn4. Based on this model, Shox2 is needed for normal Hcn4 expression when Nkx2-5 is expressed.
Shox2 antagonizes Nkx2-5 repression of the pacemaker program
To test if Shox2 functionally antagonizes the repressive effect of Nkx2-5 on the pacemaker program, we examined whether a reduction in Nkx2-5 dose would rescue the genetic defects in the SAN of Shox2 mutants. We took advantage of an established Nkx2-5 hypomorphic model that harbors the Nkx2-5-IRESCre allele and an Nkx2-5 null allele (the Nkx2-5-Cre knock-in allele was used in this study) and shows ∼75% reduction in Nkx2-5 protein level (Mommersteeg et al., 2007a; Prall et al., 2007). Compounding the Nkx2-5 hypomorphic alleles (referred as Nkx2-5Cre/IRESCre) with the floxed Shox2 allele generated mice carrying a Shox2 deletion in the Nkx2-5+ domain with Nkx2-5 hypomorphism (Nkx2-5Cre/IRESCre;Shox2F/F). For ease of visual comparison, we focused on the transition domain of the Nkx2-5− SAN head and Nkx2-5+ SA junction. Immunohistochemistry revealed that, compared with the SAN of the Nkx2-5IRESCre/+;Shox2F/F embryo, in which Hcn4 expression was eliminated whereas Cx40 became expressed in the Nkx2-5+ cells (Fig. 5B,B′), hypomorphism for Nkx2-5 in the context of Nkx2-5Cre/IRESCre;Shox2F/F re-established the Hcn4+/Cx40− fate in the SAN, similar to controls (Fig. 5A,A′,C,C′). Furthermore, expression of Isl1 and Tbx3 was also re-established in the SA junction of Nkx2-5Cre/IRESCre;Shox2F/F mice, although the hypoplastic defect was not rescued (Fig. 5D-I). These results demonstrate an antagonistic action of Shox2 on the repressive function of Nkx2-5 on the pacemaker program. When Nkx2-5 is absent or present at low levels, Shox2 is dispensable in maintaining the pacemaker cell fate.
Fig. 5.

Shox2 antagonizes Nkx2-5 transcriptional output on the pacemaker program. (A-C′) A comparison of Hcn4 and Cx40 expression in the transition region of the Nkx2-5− SAN head and the Nkx2-5+ SA junction reveals a clear boundary (dotted lines) between the SAN cells and atrial cells in controls (A,A′), the conversion of Hcn4+/Cx40− cells to Hcn4−/Cx40+ cells in the Nkx2-5IRESCre/+;Shox2F/F (Shox2Nkx2-5Cre) SA junction (B,B′), and the rescue of this phenotype in Nkx2.5Cre/IRESCre;Shox2F/F (Nkx2-5hypo;Shox2F/F) mice (C,C′). (D-I) Expression of SAN markers, including Hcn4, Isl1 and Tbx3, is restored in the SA junction of Nkx2-5hypo;Shox2F/F mice. Dotted lines delineate the border between the SAN and atrial tissue that can be otherwise clearly observed in controls. (J-L′) Comparison of Hcn4 and Cx40 expression in the PV myocardium of control (J,J′), Nkx2-5hypo (K,K′) and Nkx2-5hypo;Shox2F/F (L,L′) mice. Arrowheads point to PV myocardium.
It was reported previously that the genetic program of the PV myocardium was converted from Cx40+/Hcn4− to Cx40−/Hcn4+ in Nkx2-5 hypomorphic mice (Mommersteeg et al., 2007a), suggesting the existence of a similar repressive effect of Nkx2-5 on the pacemaker program in the PV myocardium. We examined whether simultaneous deletion of Shox2 on the Nkx2-5 hypomorphic background could reverse the Cx40−/Hcn4+ cell fate back to the Cx40+/Hcn4− working myocardial fate in the PV. Our analyses of Nkx2-5Cre/IRESCre embryos concurred primarily with the previous report that Hcn4 expression is significantly elevated and the Cx40 level dramatically reduced, compared with controls (Fig. 5J-K′). We also observed, albeit at low level, Hcn4 expression in the wild-type PV myocardium (Fig. 1B, Fig. 5J′,K′; supplementary material Fig. S7A,B,D,E,G,H). Nevertheless, Shox2 inactivation by the Nkx2-5Cre/IRESCre alleles resumed the working myocardial fate (Cx40+/Hcn4−) in the PV (Fig. 5L,L′; supplementary material Fig. S7C,F,I), further supporting a pro-pacemaker role for Shox2.
Thus, a Shox2–Nkx2-5 antagonistic mechanism operates in the venous pole, particularly in the SAN and the PV myocardium, to regulate cell fate. Disturbing the balance of Shox2 and Nkx2-5 interaction shifts cell fate between pacemaker and working myocardium.
Shox2+/Nkx2-5+/Hcn4+ pacemaker-like cells are present in differentiated embryoid bodies
It is well established that in vitro differentiated embryoid bodies (EBs) from embryonic stem cells (ESCs) are able to form cardiac cells, including atrial-like, ventricular-like and pacemaker-like cells (Boheler et al., 2002). We sought to determine whether Shox2 is also co-expressed with Nkx2-5 and Hcn4 in EBs. Using the ESC line that carries the Shox2HA allele and displays DsRed activity when Shox2 is activated (Fig. 1A), we identified Shox2+ cells within the spontaneously contracting region in each EB (Fig. 6A). Immunostaining revealed cTnT expression in almost every Shox2+ cell, an indicator of cardiac character (Fig. 6B,D). Supporting the idea that Shox2 is a pro-pacemaker factor, the majority of Shox2+ cells (94.4%) also expressed Hcn4 (Fig. 6C,D). Consistent with the findings described above, a large percentage (80%) of Shox2+ cells also expressed Nkx2.5 (Fig. 6B-D). We next tested if the Shox2–Nkx2-5 antagonistic machinery also functions in EBs to regulate Hcn4 expression. Indeed, silencing of Nkx2-5 by siRNA in EBs promoted Hcn4 expression (Fig. 6E), suggesting the adoption of the Shox2–Nkx2.5 antagonistic mechanism in regulating the differentiation of potential pacemaker-like cells in EBs.
Fig. 6.

Co-expression of Shox2 with cTnT, Hcn4 and Nkx2-5 in differentiated pacemaker-like cells in EBs. (A) Shox2 expression, as revealed by DsRed (arrowheads), in an EB differentiated from stem cells bearing the Shox2HA allele. (B,C) Immunocytochemistry shows colocalization of Shox2 with cTnT, Nkx2-5 and Hcn4. (B) White arrowheads point to Shox2+/cTnT+/Nkx2.5+ cells, and open arrowheads point to Shox2+/cTnT+ cells. (C) White arrowhead points to a Shox2+/Nkx2.5+/Hcn4+ cell, and open arrowhead points to a Shox2+/Hcn4+ cell. (D) Percentage of cells that co-express Shox2 and other molecular markers. (E) qPCR reveals elevated Hcn4 expression in differentiated EBs with Nkx2-5 silencing by siRNA. **P<0.01.
Genome-wide co-occupancy by Shox2 and Nkx2-5
To understand the functional mechanism of Shox2 in venous pole development, we performed ChIP-Seq on E12.5 hearts from Shox2HA/+ mice, in which HA-tagged Shox2 is expressed at a physiological level and can be immunoprecipitated by anti-HA antibody after crosslinking (supplementary material Fig. S8A). Immunoprecipitated chromatins from two independent preparations were subjected to deep sequencing. Peaks were called by MACS2 from ∼65 million uniquely mapped reads using ∼60 million input reads as controls. Gene ontology (GO) analysis of annotated genes associated with Shox2 binding peaks indicated that Shox2 regulates genes that are associated with human phenotypes involving nervous system, bone and those that are linked to arrhythmia (Fig. 7A), consistent with the reported expression and function of Shox2 in the development of these organs (Abdo et al., 2011; Blaschke et al., 2007; Dougherty et al., 2013; Rosin et al., 2013; Vickerman et al., 2011; Yu et al., 2007). Interestingly, the GO analysis also revealed a direct association of Shox2 with cellular machineries responsible for active proliferation and transcription (supplementary material Fig. S8B).
Fig. 7.

Genome-wide co-occupancy by Shox2 and Nkx2-5. (A) GO analysis of phenotypes of annotated genes associated with Shox2 binding peaks. (B,C) Genome-wide co-occurrence of Shox2, Nkx2-5 and Tbx5 binding sites. (D) Plot of Shox2, Nkx2-5 and Tbx5 co-bound peaks around TSSs. (E) Co-occurrence of Shox2, Nkx2-5 and Tbx5 binding sites on representative genes (read numbers above 0.15/million reads are shown). (F) Co-immunoprecipitation shows physical interaction between Shox2 and Nkx2-5. (G) Direct co-regulation of target genes by Shox2, Nkx2-5 and Tbx5 underlies the Shox2–Nkx2-5 antagonistic mechanism.
Based on the evident antagonistic action of Shox2 on the transcriptional targets of Nkx2-5 and the concept that cardiac transcription factors may co-occupy the same regulatory elements (He et al., 2011; van den Boogaard et al., 2012), we further conducted Nkx2-5 ChIP-Seq on E12.5 hearts using anti-Nkx2-5 antibodies. Motif discovery, GO analysis and position relative to the transcription start site (TSS) are summarized in supplementary material Fig. S9. We aligned Shox2 with Nkx2-5 binding peaks and looked for genome-wide co-occurrence of Shox2 and Nkx2-5 binding sites. Nkx2-5 peaks co-occurred in 79% of Shox2 binding sites (Fig. 7B).
To further clarify the relevance of such co-occurrence of Shox2 and Nkx2-5 binding sites in venous pole development, we intersected the Shox2 binding sites and Shox2–Nkx2-5 co-occupied sites with the published binding peaks of Tbx5 (He et al., 2011), a transcription factor that is crucial for CCS development and may act upstream of Shox2 (Bruneau et al., 1999; Moskowitz et al., 2004; Puskaric et al., 2010). We found that 73% of Shox2 binding sites co-occur with Tbx5 binding peaks and, most significantly, Tbx5 binding peaks account for 67% of the Shox2–Nkx2-5 co-occupied sites (53% of the total Shox2 binding peaks) (Fig. 7B). The location of the Shox2, Nkx2-5 and Tbx5 bound sites relative to Shox2 binding peaks (Fig. 7C) indicates that the Shox2–Nkx2-5 co-occupied sites are highly relevant in the context of venous pole development.
To further quantify the degree of genome-wide co-occupancy between Shox2, Nkx2-5 and Tbx5, we plotted co-occupancy using ChIP signal (in bedGraph format) by multiple wiggle correlation (Liu et al., 2011) (supplementary material Fig. S10). Similar to the previously described multiple transcription factor binding loci that were bound simultaneously by several transcription factors (He et al., 2011), most sites co-occupied by Shox2, Nkx2-5 and Tbx5 were close to a TSS (Fig. 7D). As shown in representative Genome Browser views (Fig. 7E), Shox2 binding peaks on several representative genes, as confirmed by ChIP assay on E12.5 hearts and HL-1 cells (supplementary material Fig. S8E and Fig. S11), overlap with those of Nkx2-5 and Tbx5. Among these genes, Baf250a (Arid1a) was shown to be essential for maintaining Hcn4 expression and SAN function (Wu et al., 2014), and Cdk6 and Anapc10 are known downstream targets of Tbx5 (Xie et al., 2012), suggesting a co-regulation of these target genes by Shox2, Nkx2-5 and Tbx5 directly. The reported physical interaction between Nkx2-5 and Tbx5 (He et al., 2011; Hiroi et al., 2001) and the evident interaction between Shox2 and Nkx2-5 (Fig. 7F), as well as the interaction between Shox2 and Tbx5 (our unpublished results), further support the co-occupancy of these factors on the same regulator elements (Fig. 7G), and suggest a mechanism for the antagonistic action between Shox2 and Nkx2-5 at the transcription level.
DISCUSSION
Developmental origin of the PV myocardium
The lineage composition of the PV myocardium is still enigmatic, and discussions have surrounded whether the PV myocardium possesses properties similar to primary heart field derivatives or the sinus venosus, or even to the derivatives of the dorsal mesenchyme protrusion that contributes to atrial septation (Gittenberger-de Groot, 2011; Mommersteeg et al., 2007a; Moorman and Anderson, 2011). Whereas genetic lineage-tracing studies using Tbx18-Cre suggest an independent lineage origin of the PV myocardium (Moorman and Anderson, 2011), retrospective lineage-tracing methods that do not rely on the provenance of assumed lineage markers indicate that the PV myocardium and coronary sinus/left superior vena cava myocardium are clonally related (Lescroart et al., 2012). In the current study, we provide evidence that the PV myocardium and sinus venosus derivatives share Shox2 expression during venous pole development (Fig. 1). Furthermore, the majority of the PV myocardium appears to be derived from Shox2+ cells, as revealed by fate mapping using the Shox2-Cre allele (supplementary material Fig. S12A and Fig. S13B) (Sun et al., 2013), suggesting a pro-pacemaker origin of the PV myocardium that shares pacemaker characteristics with that of the systemic venous return. By contrast, dorsal mesenchyme protrusion derivatives, as labeled by the Mef2c-AHF-Cre allele (Verzi et al., 2005), only contributed to a small subset of the PV cells (supplementary material Fig. S12B). The Shox2-Cre-labeled PV myocardial cells at the opening of the PV into the left atrium further support that the lineage origin of PV myocardial cells is distinct from that of the atrial cells (supplementary material Fig. S1D and Fig. S13B1).
Shox2–Nkx2-5 antagonism in cell fate decisions in the SAN and PV myocardium
Extensive research has been conducted on the genetic regulation of SAN development as a whole unit. However, the existence of genetically distinguishable domains, i.e. the Nkx2-5+ SA junction and Nkx2-5− SAN head, within the developing SAN indicates the involvement of different regulatory mechanisms for these two domains. Shox2 was originally thought to regulate SAN development by preventing ectopic Nkx2-5 activation in the SAN (Blaschke et al., 2007; Espinoza-Lewis et al., 2009). However, as shown in the present study, the situation is more complicated. We provide unambiguous evidence that Shox2 is co-expressed with Nkx2-5 in the SA junction during SAN development, and loss of Shox2 in the Nkx2-5+ domain leads to severely hypoplastic and eventually unidentifiable SA junction structures. The compromised SAN function in the mutant mice, which is manifested as severe bradycardia, irregular R-R intervals and variable P-R intervals, demonstrates for the first time the requirement for Shox2 in the SA junction for the functional integrity of the SAN. The current study was conducted on anesthetized mice by surface ECG, and further characterization of this model by ambulatory ECG telemetry and intracardiac ECG will provide more detailed information on the physiological function of this subdomain of the SAN and whether this subdomain functions by contributing to SAN-atrium (S-A) conduction and if S-A exit block underlies the sick sinus syndrome observed in Shox2Nkx2-5Cre mice.
In a developmental context, we also identified a novel function for Shox2 in maintaining the pacemaker program, including Hcn4, Tbx3 and Isl1 in the Nkx2-5+ domain of the SAN, by antagonizing Nkx2-5 transcriptional output. Importantly, the co-expression of Shox2, Nkx2-5 and Hnc4 in the developing PV myocardium suggests a role for Shox2 in PV development as well. Our genetic evidence suggests that Shox2 plays a similar pro-pacemaker function in the cell fate decision in the PV myocardium by employing similar machinery to that utilized in the Nkx2-5+ domain of the SAN. Thus, the presence of a regulatory circuit similar to that found in the SAN appears to prime the PV myocardium as a latent pacemaker that is held in check by Nkx2-5 expression, providing an explanation for why Shox2+ cells from the embryonic PV myocardium exhibit node-like APs and the capacity to pace adjacent Shox2− cells ex vivo. These observations also potentially explain why the PV myocardium is prone to develop ectopic pacemaker activity. The potential pacemaking function of the PV myocardium appears to be subservient to the SAN under normal conditions, but could become active when SAN function is compromised. The absence of Tbx3 and Isl1 expression, which are also positive regulators of SAN development, in the PV myocardium (Christoffels et al., 2010; our unpublished data) supports a unique role for Shox2 in controlling a pacemaker program independently of Tbx3 and Isl1. However, the presence of Nkx2-5 prevents the adoption of pacemaker function in the PV myocardium, providing an explanation for the association of AF with NKX2-5 mutations in humans (Huang et al., 2013; Wang et al., 2014b; Xie et al., 2013). We therefore propose a seesaw model for the function of Shox2–Nkx2-5 antagonism in the regulation of cell fate during venous pole development (Fig. 8). In this model, the ‘weight shift’ between Shox2 and Nkx2-5 transcriptional output determines either pacemaker fate or working myocardial fate, with higher Nkx2-5 activity promoting working myocardial fate, and higher Shox2 promoting pacemaker fate. Abnormal weight shift would alter cell fate and lead to sick sinus syndrome or AF. In support of this notion, it was reported that Pitx2, a left-sided transcriptional factor in the developmental left/right asymmetry pathway, is expressed in the PV myocardium and negatively regulates Shox2 expression, with Pitx2 haploinsufficiency leading to risk of AF (Wang et al., 2010).
Fig. 8.
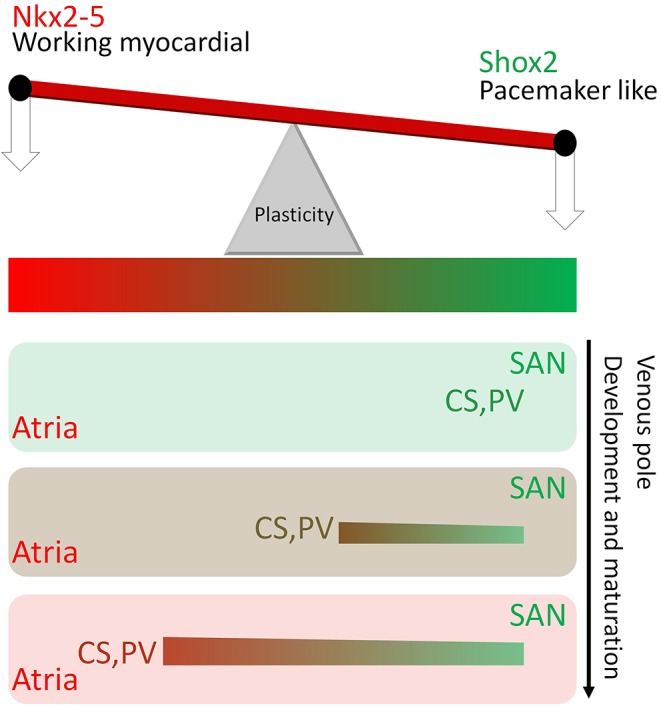
A Shox2–Nkx2-5 antagonism seesaw model links the pacemaker program in the PV myocardium to the SAN. A seesaw model explains the genetic ‘weight shift’ between Shox2 and Nkx2-5 to determine cell fate in the venous pole derivatives. In this model it is proposed that, at the early developmental stage, Shox2 is expressed ‘heavily’ in the venous pole derivatives including the SAN, PV and CS that exhibit pacemaker charateristics. As the venous pole develops and becomes mature, the ‘weight’ of Nkx2-5 activity increases, enabling the venous pole derivatives, except the SAN, to adopt an atrial-like working myocardial fate gradually.
Shox2 as a transcription factor in venous pole development
We show that Shox2 and Nkx2-5 are co-expressed in multiple regions of the venous pole, where Shox2 acts to antagonize Nkx2-5 transcriptional output that promotes working myocardial fate while inhibiting pacemaker fate. However, Shox2 does not activate the expression of pacemaker genes directly, but shields their expression from inhibition by Nkx2-5. Isl1 was reported previously as a direct transcriptional target of Shox2 (Hoffmann et al., 2013), but our results demonstrate that, instead of activating Isl1 directly, Shox2 allows Isl1 expression by antagonizing the inhibitory effect of Nkx2-5. The evidence that Shox2 interacts with Nkx2-5 directly and their substantial genome-wide co-occupancy provide a potential mechanistic explanation for the antagonistic function of Shox2. However, although the inhibitory effect of Nkx2-5 over Isl1, Tbx3 and Hcn4 has been documented (Cambier et al., 2014; Mommersteeg et al., 2007b; Prall et al., 2007), the cis-regulatory elements associated with these genes that mediate such effects remain to be characterized. It is unclear whether Nkx2-5 inhibits the expression of these pacemaker genes directly, although it inhibits cardiac progenitor genes directly (Watanabe et al., 2012). In our current studies, we could not identify co-occupancy of Shox2 and Nkx2-5 on the regulatory elements of any of these genes. Whether Shox2 antagonizes Nkx2-5 function by competing with it for binding sites in these genes warrants future investigation.
Shox2 might antagonize Nkx2-5 transcriptional output at a generic level instead of on pacemaker-specific genes. In line with this notion, we found a significant co-occurrence of Shox2 and Nkx2-5 binding sites in the promoters of genes essential for gene expression regulation, such as those involved in chromatin and chromosome organization, RNA processing, translation and post-transcriptional regulation (supplementary material Fig. S8B). This is best exemplified by the co-occupancy of Shox2 and Nkx2-5 on the regulatory elements of Baf250a (Fig. 7E), a key regulatory component of the ATP-dependent chromatin remodeling complex SWI/SNF and a factor proven to be essential for maintaining Hcn4 expression and SAN function (Wu et al., 2014).
Most importantly, the genome-wide binding sites of Shox2, and the coincident binding peaks of Shox2 and Nkx2-5, overlap extensively with those of Tbx5. Such co-occupancy pinpoints the functional importance of Shox2 in how the key regulators of venous pole development exert their function. Furthermore, consistent with the positive role of Shox2 in cell proliferation, we frequently found Shox2 binding sites in the cis-regulatory elements of genes involved in cell proliferation.
In summary, Shox2 functions as a key pro-pacemaker factor, together with Nkx2-5 and possibly Tbx5 as well as other factors, and regulates cell fate decisions and morphogenesis during venous pole development.
MATERIALS AND METHODS
Mouse models
Generation of Nkx2-5Cre/+, Shox2+/−, Shox2F/F, Nkx2-5IRESCre/+, Shox2HA and Shox2Cre mice has been reported previously (Cobb et al., 2006; Moses et al., 2001; Stanley et al., 2002; Sun et al., 2013; Wang et al., 2014a; Yu et al., 2005). The Tulane University Institutional Animal Care and Use Committee approved the animal experiments in this study.
Collection of embryos, histology, immunohistochemistry and section plans
Embryos or hearts from timed pregnant females were fixed in ice-cold 4% paraformaldehyde (PFA), embedded in paraffin, and sectioned at 8 µm for standard Hematoxylin and Eosin (H&E) staining and immunohistochemical analyses. Human embryos obtained by legally terminated gestation were provided by the Hospital for Women and Children of Fujian Province, China, with the permission of the Ethics Committee of Fujian Normal University. Immunofluorescence was performed with citrate-based antigen retrieval solution (Vector Labs) and mounted samples were visualized and photographed under a Nikon Eclipse Ti confocal microscope or a Nikon Eclipse E600 fluorescence microscope. Information on the antibodies used is provided in the supplementary Materials. Consecutive sections through the SAN or PV are shown in supplementary material Fig. S13 to detail the section plane and orientation of representative sections presented in the current studies.
FACS, electrophysiology and cell-clump cultures
The DsRed+ proximal PV domain was dissected out from E14.5 Shox2HA embryos under a fluorescence dissecting scope, and subjected to digestion by a cocktail of collagenase I, II, IV, followed by a brief trypsin treatment. Suspended cells were subjected to FACS. DsRed+ cells were allowed to attach onto fibronectin-gelatin-coated coverslips and recover overnight before whole-cell patch clamp recordings were performed, as detailed in the supplementary Materials. To generate beating cell clumps, about 200 DsRed+, or DsRed−, or a mixture of 100 DsRed+ and 100 DsRed− cells were aggregated in 96-well round-bottom ultra-low attachment plates for 24 h. Subsequently, cell clumps were transferred to gelatin-coated plates, and the contraction rate in each clump was counted after 24 h in culture.
Differentiation of embryoid bodies
G4 ESCs carrying the Shox2HA allele were cultured in RESGRO medium (Millipore) and differentiated into EBs in differentiation medium as reported previously (Behrens et al., 2013). Detailed procedures, including siRNA treatment (supplementary material Fig. S14), are provided in the supplementary Materials.
Surface electrocardiography (ECG)
Isoflurane-anesthetized 2-month-old mice, maintained under 1.5% isoflurane supplemented with O2 at 1.5 l/min during recording, were placed in prone position on a Mouse Monitor S (Indus Instruments) and recorded according to the manufacturer's recommended settings for 5 min under the default filter set.
Co-IP, ChIP and ChIP-Seq
Co-immunoprecipitation (Co-IP) and western blotting were performed as described previously (Yang et al., 2014). The procedures for ChIP and ChIP-Seq and information concerning ChIP-Seq data are described in the supplementary Materials. ChIP-Seq data reported in this study have been deposited at GEO with accession number GSE70332.
Statistical analysis
All experiments were repeated at least three times. Quantification results are presented as mean±s.d., and statistical analysis was conducted using Student's t-test. For qPCR, reactions for each sample were also performed in triplicate. P<0.05 was considered significant.
Supplementary Material
Acknowledgements
We thank Dr Min Zhang of Baylor College of Medicine for help with ChIP-Seq data processing and analysis.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
We acknowledge financial support by grants from the National Institutes of Health [R01DE017792 to Y.C.; R01HL118761 to J.F.M.], an American Heart Association (AHA) Predoctoral Fellowship [13PRE1375003 to W.Y.], AHA Scientist Development Grant [14SDG19840000 to J.W.] and a grant [WKJ-FJ-24] from the National Health and Family Planning Commission of China. The bioinformatics analysis described in this study was also assisted by the Tulane Cancer Crusaders Next Generation Sequence Analysis Core Facility funded by the NIH COBRA Project [P20GM103518]. Deposited in PMC for release after 12 months.
Author contributions
W.Y. and Y.C. conceived the project. W.Y. performed most experiments, collected and analyzed data, prepared figures and wrote the manuscript. J.W. and J.F.M. provided reagents and helped in ChIP-Seq studies. Y.S., C.S. and C.L. helped in histology and immunohistochemistry experiments. D.Y., F.C. and L.S. conducted whole-cell patch recordings and analysis. Y.Z., R.P.H. and F.W. provided necessary reagents. R.P.H. and J.F.M. provided insights on the project and helped in manuscript editing. Y.C. conducted final revision and editing of the manuscript.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.120220/-/DC1
References
- Abdo H., Li L., Lallemend F., Bachy I., Xu X.-J., Rice F. L. and Ernfors P. (2011). Dependence on the transcription factor Shox2 for specification of sensory neurons conveying discriminative touch. Eur. J. Neurosci. 34, 1529-1541. 10.1111/j.1460-9568.2011.07883.x [DOI] [PubMed] [Google Scholar]
- Ammirabile G., Tessari A., Pignataro V., Szumska D., Sutera Sardo F., Benes J. Jr, Balistreri M., Bhattacharya S., Sedmera D. and Campione M. (2012). Pitx2 confers left morphological, molecular, and functional identity to the sinus venosus myocardium. Cardiovasc. Res. 93, 291-301. 10.1093/cvr/cvr314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A. N., Iacovino M., Lohr J. L., Ren Y., Zierold C., Harvey R. P., Kyba M., Garry D. J. and Martin C. M. (2013). Nkx2-5 mediates differential cardiac differentiation through interaction with Hoxa10. Stem Cells Dev. 22, 2211-2220. 10.1089/scd.2012.0611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke R. J., Monaghan A. P., Schiller S., Schechinger B., Rao E., Padilla-Nash H., Ried T. and Rappold G. A. (1998). SHOT, a SHOX-related homeobox gene, is implicated in craniofacial, brain, heart, and limb development. Proc. Natl. Acad. Sci. USA 95, 2406-2411. 10.1073/pnas.95.5.2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke R. J., Hahurij N. D., Kuijper S., Just S., Wisse L. J., Deissler K., Maxelon T., Anastassiadis K., Spitzer J., Hardt S. E. et al. (2007). Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation 115, 1830-1838. 10.1161/CIRCULATIONAHA.106.637819 [DOI] [PubMed] [Google Scholar]
- Boheler K. R., Czyz J., Tweedie D., Yang H.-T., Anisimov S. V. and Wobus A. M. (2002). Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ. Res. 91, 189-201. 10.1161/01.RES.0000027865.61704.32 [DOI] [PubMed] [Google Scholar]
- Bruneau B. G., Logan M., Davis N., Levi T., Tabin C. J., Seidman J. G. and Seidman C. E. (1999). Chamber-specific cardiac expression of Tbx5 and heart defects in Holt–Oram syndrome. Dev. Biol. 211, 100-108. 10.1006/dbio.1999.9298 [DOI] [PubMed] [Google Scholar]
- Cambier L., Plate M., Sucov H. M. and Pashmforoush M. (2014). Nkx2-5 regulates cardiac growth through modulation of Wnt signaling by R-spondin3. Development 141, 2959-2971. 10.1242/dev.103416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels V. M., Mommersteeg M. T. M., Trowe M.-O., Prall O. W. J., de Gier-de Vries C., Soufan A. T., Bussen M., Schuster-Gossler K., Harvey R. P., Moorman A. F. M. et al. (2006). Formation of the venous pole of the heart from an Nkx2-5-negative precursor population requires Tbx18. Circ. Res. 98, 1555-1563. 10.1161/01.RES.0000227571.84189.65 [DOI] [PubMed] [Google Scholar]
- Christoffels V. M., Smits G. J., Kispert A. and Moorman A. F. M. (2010). Development of the pacemaker tissues of the heart. Circ. Res. 106, 240-254. 10.1161/CIRCRESAHA.109.205419 [DOI] [PubMed] [Google Scholar]
- Cobb J., Dierich A., Huss-Garcia Y. and Duboule D. (2006). A mouse model for human short-stature syndromes identifies Shox2 as an upstream regulator of Runx2 during long-bone development. Proc. Natl. Acad. Sci. USA 103, 4511-4515. 10.1073/pnas.0510544103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty K. J., Zagoraiou L., Satoh D., Rozani I., Doobar S., Arber S., Jessell T. M. and Kiehn O. (2013). Locomotor rhythm generation linked to the output of spinal shox2 excitatory interneurons. Neuron 80, 920-933. 10.1016/j.neuron.2013.08.015 [DOI] [PubMed] [Google Scholar]
- Douglas Y. L., Jongbloed M. R. M., DeRuiter M. C. and Gittenberger-de Groot A. C. (2011). Normal and abnormal development of pulmonary veins: state of the art and correlation with clinical entities. Int. J. Cardiol. 147, 13-24. 10.1016/j.ijcard.2010.07.004 [DOI] [PubMed] [Google Scholar]
- Eckardt L. (2002). Automaticity in the coronary sinus. J. Cardiovasc. Electrophysiol. 13, 288-289. 10.1046/j.1540-8167.2002.00288.x [DOI] [PubMed] [Google Scholar]
- Espinoza-Lewis R. A., Yu L., He F., Liu H., Tang R., Shi J., Sun X., Martin J. F., Wang D., Yang J. et al. (2009). Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev. Biol. 327, 376-385. 10.1016/j.ydbio.2008.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D. U., Carter K. L., Thomas K. R., Burr R. M., Bakker M. L., Coetzee W. A., Tristani-Firouzi M., Bamshad M. J., Christoffels V. M. and Moon A. M. (2012). Lethal arrhythmias in Tbx3-deficient mice reveal extreme dosage sensitivity of cardiac conduction system function and homeostasis. Proc. Natl. Acad. Sci. USA 109, E154-E163. 10.1073/pnas.1115165109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittenberger-de Groot A. C. (2011). The development of the pulmonary vein revisited. Int. J. Cardiol. 147, 463-464. 10.1016/j.ijcard.2011.01.008 [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot A. C., Mahtab E. A. F., Hahurij N. D., Wisse L. J., Deruiter M. C., Wijffels M. C. E. F. and Poelmann R. E. (2007). Nkx2.5-negative myocardium of the posterior heart field and its correlation with podoplanin expression in cells from the developing cardiac pacemaking and conduction system. Anat. Rec. 290, 115-122. 10.1002/ar.20406 [DOI] [PubMed] [Google Scholar]
- Gu S., Wei N., Yu L., Fei J. and Chen Y. (2008). Shox2-deficiency leads to dysplasia and ankylosis of the temporomandibular joint in mice. Mech. Dev. 125, 729-742. 10.1016/j.mod.2008.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib A., Lachman N., Christensen K. N. and Asirvatham S. J. (2009). The anatomy of the coronary sinus venous system for the cardiac electrophysiologist. Europace 11 Suppl. 5, v15-v21. 10.1093/europace/eup270 [DOI] [PubMed] [Google Scholar]
- Haïssaguerre M., Jaïs P., Shah D. C., Takahashi A., Hocini M., Quiniou G., Garrigue S., Le Mouroux A., Le Métayer P. and Clémenty J. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659-666. 10.1056/NEJM199809033391003 [DOI] [PubMed] [Google Scholar]
- He A., Kong S. W., Ma Q. and Pu W. T. (2011). Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. USA 108, 5632-5637. 10.1073/pnas.1016959108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi Y., Kudoh S., Monzen K., Ikeda Y., Yazaki Y., Nagai R. and Komuro I. (2001). Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat. Genet. 28, 276-280. 10.1038/90123 [DOI] [PubMed] [Google Scholar]
- Hoffmann S., Berger I. M., Glaser A., Bacon C., Li L., Gretz N., Steinbeisser H., Rottbauer W., Just S. and Rappold G. (2013). Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res. Cardiol. 108, 339 10.1007/s00395-013-0339-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. T., Xue S., Xu Y. J., Zhou M. and Yang Y. Q. (2013). A novel NKX2.5 loss-of-function mutation responsible for familial atrial fibrillation. Int. J. Mol. Med. 31, 1119-1126. 10.3892/ijmm.2013.1316 [DOI] [PubMed] [Google Scholar]
- Jongbloed M. R. M., Schalij M. J., Poelmann R. E., Blom N. A., Fekkes M. L., Wang Z., Fishman G. I. and Gittenberger-De Groot A. C. (2004). Embryonic conduction tissue: a spatial correlation with adult arrhythmogenic areas. J. Cardiovasc. Electrophysiol. 15, 349-355. 10.1046/j.1540-8167.2004.03487.x [DOI] [PubMed] [Google Scholar]
- Lemola K., Mueller G., Desjardins B., Sneider M., Case I., Good E., Han J., Tamirisa K., Tschopp D., Reich S. et al. (2005). Topographic analysis of the coronary sinus and major cardiac veins by computed tomography. Heart Rhythm 2, 694-699. 10.1016/j.hrthm.2005.04.016 [DOI] [PubMed] [Google Scholar]
- Lescroart F., Mohun T., Meilhac S. M., Bennett M. and Buckingham M. (2012). Lineage tree for the venous pole of the heart: clonal analysis clarifies controversial genealogy based on genetic tracing. Circ. Res. 111, 1313-1322. 10.1161/CIRCRESAHA.112.271064 [DOI] [PubMed] [Google Scholar]
- Levin M. D., Lu M. M., Petrenko N. B., Hawkins B. J., Gupta T. H., Lang D., Buckley P. T., Jochems J., Liu F., Spurney C. F. et al. (2009). Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J. Clin. Invest. 119, 3420-3436. 10.1172/jci39109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X., Wang G., Lin L., Lowe J., Zhang Q., Bu L., Chen Y., Chen J., Sun Y. and Evans S. M. (2013). HCN4 dynamically marks the first heart field and conduction system precursors. Circ. Res. 113, 399-407. 10.1161/CIRCRESAHA.113.301588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman M. and Paes de Carvalho A. (1965). The electrophysiological organization of the embryonic chick heart. J. Gen. Physiol. 49, 351-363. 10.1085/jgp.49.2.351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Dobrzynski H., Yanni J., Boyett M. R. and Lei M. (2007). Organisation of the mouse sinoatrial node: structure and expression of HCN channels. Cardiovasc. Res. 73, 729-738. 10.1016/j.cardiores.2006.11.016 [DOI] [PubMed] [Google Scholar]
- Liu T., Ortiz J. A., Taing L., Meyer C. A., Lee B., Zhang Y., Shin H., Wong S. S., Ma J., Lei Y. et al. (2011). Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 12, R83 10.1186/gb-2011-12-8-r83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J. F. (2007). Left right asymmetry, the pulmonary vein, and a-fib. Circ. Res. 101, 853-855. 10.1161/CIRCRESAHA.107.164079 [DOI] [PubMed] [Google Scholar]
- Millino C., Sarinella F., Tiveron C., Villa A., Sartore S. and Ausoni S. (2000). Cardiac and smooth muscle cell contribution to the formation of the murine pulmonary veins. Dev. Dyn. 218, 414-425. [DOI] [PubMed] [Google Scholar]
- Mommersteeg M. T. M., Brown N. A., Prall O. W. J., de Gier-de Vries C., Harvey R. P., Moorman A. F. M. and Christoffels V. M. (2007a). Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ. Res. 101, 902-909. 10.1161/CIRCRESAHA.107.161182 [DOI] [PubMed] [Google Scholar]
- Mommersteeg M. T. M., Hoogaars W. M. H., Prall O. W. J., de Gier-de Vries C., Wiese C., Clout D. E. W., Papaioannou V. E., Brown N. A., Harvey R. P., Moorman A. F. M. et al. (2007b). Molecular pathway for the localized formation of the sinoatrial node. Circ. Res. 100, 354-362. 10.1161/01.RES.0000258019.74591.b3 [DOI] [PubMed] [Google Scholar]
- Moorman A. F. M. and Anderson R. H. (2011). Development of the pulmonary vein. Int. J. Cardiol. 147, 182 10.1016/j.ijcard.2010.12.034 [DOI] [PubMed] [Google Scholar]
- Moorman A. F. M., Christoffels V. M., Anderson R. H. and van den Hoff M. J. B. (2007). The heart-forming fields: one or multiple? Philos. Trans. R. Soc. Lond. B Biol. Sci. 362, 1257-1265. 10.1098/rstb.2007.2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses K. A., DeMayo F., Braun R. M., Reecy J. L. and Schwartz R. J. (2001). Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 31, 176-180. 10.1002/gene.10022 [DOI] [PubMed] [Google Scholar]
- Moskowitz I. P. G., Pizard A., Patel V. V., Bruneau B. G., Kim J. B., Kupershmidt S., Roden D., Berul C. I., Seidman C. E. and Seidman J. G. (2004). The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development 131, 4107-4116. 10.1242/dev.01265 [DOI] [PubMed] [Google Scholar]
- Munshi N. V. (2012). Gene regulatory networks in cardiac conduction system development. Circ. Res. 110, 1525-1537. 10.1161/CIRCRESAHA.111.260026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y.-J., Lubczyk C., Bhakta M., Zang T., Fernandez-Perez A., McAnally J., Bassel-Duby R., Olson E. N. and Munshi N. V. (2014). Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development 141, 4267-4278. 10.1242/dev.114025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T., Tian Y., Boogerd C. J., Lu M. M., Kadzik R. S., Stewart K. M., Evans S. M. and Morrisey E. E. (2013). Coordination of heart and lung co-development by a multipotent cardiopulmonary progenitor. Nature 500, 589-592. 10.1038/nature12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prall O. W. J., Menon M. K., Solloway M. J., Watanabe Y., Zaffran S., Bajolle F., Biben C., McBride J. J., Robertson B. R., Chaulet H. et al. (2007). An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 128, 947-959. 10.1016/j.cell.2007.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskaric S., Schmitteckert S., Mori A. D., Glaser A., Schneider K. U., Bruneau B. G., Blaschke R. J., Steinbeisser H. and Rappold G. (2010). Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart. Hum. Mol. Genet. 19, 4625-4633. 10.1093/hmg/ddq393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin J. M., Abassah-Oppong S. and Cobb J. (2013). Comparative transgenic analysis of enhancers from the human SHOX and mouse Shox2 genomic regions. Hum. Mol. Genet. 22, 3063-3076. 10.1093/hmg/ddt163 [DOI] [PubMed] [Google Scholar]
- Snarr B. S., Wirrig E. E., Phelps A. L., Trusk T. C. and Wessels A. (2007). A spatiotemporal evaluation of the contribution of the dorsal mesenchymal protrusion to cardiac development. Dev. Dyn. 236, 1287-1294. 10.1002/dvdy.21074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley E. G., Biben C., Elefanty A., Barnett L., Koentgen F., Robb L. and Harvey R. P. (2002). Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3′UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int. J. Dev. Biol. 46, 431-439. [PubMed] [Google Scholar]
- Sun C., Zhang T., Liu C., Gu S. and Chen Y. (2013). Generation of Shox2-Cre allele for tissue specific manipulation of genes in the developing heart, palate, and limb. Genesis 51, 515-522. 10.1002/dvg.22397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg G. and Moorman A. F. M. (2011). Development of the pulmonary vein and the systemic venous sinus: an interactive 3D overview. PLoS ONE 6, e22055 10.1371/journal.pone.0022055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boogaard M., Wong L. Y. E., Tessadori F., Bakker M. L., Dreizehnter L. K., Wakker V., Bezzina C. R., ‘t Hoen P. A. C., Bakkers J., Barnett P. et al. (2012). Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Invest. 122, 2519-2530. 10.1172/JCI62613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantham V., Evangelista M., Huang Y. and Srivastava D. (2013). Spatiotemporal regulation of an Hcn4 enhancer defines a role for Mef2c and HDACs in cardiac electrical patterning. Dev. Biol. 373, 149-162. 10.1016/j.ydbio.2012.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzi M. P., McCulley D. J., De Val S., Dodou E. and Black B. L. (2005). The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 287, 134-145. 10.1016/j.ydbio.2005.08.041 [DOI] [PubMed] [Google Scholar]
- Vickerman L., Neufeld S. and Cobb J. (2011). Shox2 function couples neural, muscular and skeletal development in the proximal forelimb. Dev. Biol. 350, 323-336. 10.1016/j.ydbio.2010.11.031 [DOI] [PubMed] [Google Scholar]
- Wang J., Klysik E., Sood S., Johnson R. L., Wehrens X. H. T. and Martin J. F. (2010). Pitx2 prevents susceptibility to atrial arrhythmias by inhibiting left-sided pacemaker specification. Proc. Natl. Acad. Sci. USA 107, 9753-9758. 10.1073/pnas.0912585107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Bai Y., Li N., Ye W., Zhang M., Greene S. B., Tao Y., Chen Y., Wehrens X. H. T. and Martin J. F. (2014a). Pitx2-microRNA pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proc. Natl. Acad. Sci. USA 111, 9181-9186. 10.1073/pnas.1405411111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang D.-F., Sun Y.-M. and Yang Y.-Q. (2014b). A novel PITX2c loss-of-function mutation associated with familial atrial fibrillation. Eur. J. Med. Genet. 57, 25-31. 10.1016/j.ejmg.2013.11.004 [DOI] [PubMed] [Google Scholar]
- Watanabe Y., Zaffran S., Kuroiwa A., Higuchi H., Ogura T., Harvey R. P., Kelly R. G. and Buckingham M. (2012). Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2-5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc. Natl. Acad. Sci. USA 109, 18273-18280. 10.1073/pnas.1215360109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenink A. C. G., Symersky P., Ikeda T., DeRuiter M. C., Poelmann R. E. and Gittenberger-de Groot A. C. (2000). HNK-1 expression patterns in the embryonic rat heart distinguish between sinuatrial tissues and atrial myocardium. Anat. Embryol. 201, 39-50. 10.1007/PL00008227 [DOI] [PubMed] [Google Scholar]
- Wiese C., Grieskamp T., Airik R., Mommersteeg M. T. M., Gardiwal A., de Gier-de Vries C., Schuster-Gossler K., Moorman A. F. M., Kispert A. and Christoffels V. M. (2009). Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ. Res. 104, 388-397. 10.1161/CIRCRESAHA.108.187062 [DOI] [PubMed] [Google Scholar]
- Wu M., Peng S., Yang J., Tu Z., Cai X., Cai C.-L., Wang Z. and Zhao Y. (2014). Baf250a orchestrates an epigenetic pathway to repress the Nkx2.5-directed contractile cardiomyocyte program in the sinoatrial node. Cell Res. 24, 1201-1213. 10.1038/cr.2014.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L., Hoffmann A. D., Burnicka-Turek O., Friedland-Little J. M., Zhang K. and Moskowitz I. P. (2012). Tbx5-hedgehog molecular networks are essential in the second heart field for atrial septation. Dev. Cell 23, 280-291. 10.1016/j.devcel.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W. H., Chang C., Xu Y. J., Li R. G., Qu X. K., Fang W. Y., Liu X. and Yang Y. Q. (2013). Prevalence and spectrum of Nkx2.5 mutations associated with idiopathic atrial fibrillation. Clinics 68, 777-784. 10.6061/clinics/2013(06)09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M., Dobrzynski H., Tellez J., Niwa R., Billeter R., Honjo H., Kodama I. and Boyett M. R. (2006). Extended atrial conduction system characterised by the expression of the HCN4 channel and connexin45. Cardiovasc. Res. 72, 271-281. 10.1016/j.cardiores.2006.07.026 [DOI] [PubMed] [Google Scholar]
- Yang G., Yuan G., Ye W., Cho K. W. and Chen Y. (2014). An atypical canonical BMP signaling pathway regulates msh homeobox 1 (Msx1) expression during odontogenesis. J. Biol. Chem. 289, 31492-31502. 10.1074/jbc.M114.600064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Gu S., Alappat S., Song Y., Yan M., Zhang X., Zhang G., Jiang Y., Zhang Z., Zhang Y. et al. (2005). Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development 132, 4397-4406. 10.1242/dev.02013 [DOI] [PubMed] [Google Scholar]
- Yu L., Liu H., Yan M., Yang J., Long F., Muneoka K. and Chen Y. (2007). Shox2 is required for chondrocyte proliferation and maturation in proximal limb skeleton. Dev. Biol. 306, 549-559. 10.1016/j.ydbio.2007.03.518 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.