

Abstract
Natural and artificial light-harvesting processes have recently gained new interest. Signatures of long-lasting coherence in spectroscopic signals of biological systems have been repeatedly observed, albeit their origin is a matter of ongoing debate, as it is unclear how the loss of coherence due to interaction with the noisy environments in such systems is averted. Here we report experimental and theoretical verification of coherent exciton–vibrational (vibronic) coupling as the origin of long-lasting coherence in an artificial light harvester, a molecular J-aggregate. In this macroscopically aligned tubular system, polarization-controlled 2D spectroscopy delivers an uncongested and specific optical response as an ideal foundation for an in-depth theoretical description. We derive analytical expressions that show under which general conditions vibronic coupling leads to prolonged excited-state coherence.
 Two-dimensional spectroscopy revealed oscillatory signals in photosynthesis' exciton dynamics, but crowded spectra impede the identification of what sustains the oscillations. Here the authors probe an J-aggregate, whose uncongested response shows that vibronic coupling is responsible for the sustained coherence.
Two-dimensional spectroscopy revealed oscillatory signals in photosynthesis' exciton dynamics, but crowded spectra impede the identification of what sustains the oscillations. Here the authors probe an J-aggregate, whose uncongested response shows that vibronic coupling is responsible for the sustained coherence.
The remarkably high efficiency in photosynthesis, where nine out of ten absorbed photons reach the reaction centre, is a fascinating field of modern research. In such photosynthetic complexes, structure, dynamics and function are inextricably linked. A conserved building block comprises strongly absorbing pigments arranged in close proximity to one another supported by the surrounding protein scaffold1,2. Typical inter-pigment distances are of order of 10 Å and photon absorption leads to the formation of delocalized excited electronic states (excitons) shared by two or more pigment molecules. Exciton creation, migration and trapping are central to the functionality of a photosynthetic apparatus. The controlled and adjustable arrangement of the pigments tunes the electronic network and the properties of its interaction with the vibrational environment that is associated with either the pigments or the protein. The detailed balance of these properties determines the efficiency of light-harvesting systems3,4.
Exciton dynamics can be efficiently probed by two-dimensional (2D) electronic spectroscopy5. This technique revealed oscillatory signals in the spectral response of a wide variety of photosynthetic aggregates6,7. Initially ascribed to excitonic beatings, oscillations have been found to persist up to several hundreds of femtoseconds at room temperature8,9,10. This timescale exceeds typical dephasing rates in the condensed phase and becomes comparable to exciton transfer times1, thus posing the question of the nature and functional relevance of these coherences4. Unfortunately, the complex structure of 2D signals makes the unambiguous identification of the underlying mechanisms that support such long-lived coherences a challenging task and several hypotheses to explain them have been formulated11,12,13,14,15,16,17,18,19,20,21. The different approaches can be classified into theories including coherent interaction of excitons with intra-pigment vibrations11,12,13,14,15 and theories focusing on incoherent exciton–protein interaction such as correlated fluctuations16,17,18. It is possible that some of these mechanisms may coexist on certain timescales and that one or another may become dominant depending on the system under consideration.
In this work, we show that the relatively simple excitonic structure of a molecular J-aggregate provides an ideal test case to identify the microscopic mechanism behind long-lived oscillations in electronic 2D signals. The investigated J-aggregate is tubular and aligns along the sample's flow direction when in solution. In addition, the J-aggregate exhibits excitonic bands with roughly orthogonal transition dipole moments. It is this combination of perpendicular excitonic transitions and macroscopic alignment that makes electronic 2D spectroscopy with polarization-controlled excitation pulses an ideal tool to study coherence effects between the excitonic bands. This approach significantly reduces the complexity of retrieved 2D signals, leading to only two peaks with oscillatory components in specific regions of the 2D maps, that is, one on the diagonal and one as a cross-peak for non-rephasing and rephasing signal components, respectively. Employing a vibronic model, we derive analytical expressions that show how system parameters such as electronic decoherence rates and exciton–vibrational resonance determine the amplitude and lifetime of oscillatory signals. Fitting the analytical expressions to measured data, the vibronic model achieves quantitative agreement with experimental observations. Concerning potential functional relevance of the observed oscillations, we show that the long-lived oscillatory signals in our system are dominated by excited-state coherence rather than ground-state coherence.
Results
The system
J-aggregates of cyanine dyes are promising candidates for artificial antenna systems22,23,24,25,26. They are chemically versatile and self-assemble into various extended supramolecular structures in aqueous solution27. Here a system that can be considered a macroscopically aligned synthetic light harvester was studied, namely a molecular J-aggregate of C8O3-monomers whose aggregation behaviour is well known28,29. As revealed by cryogenic transmission electron microscopy30, the aggregate structure is best described as a double-layered nanotube with outer diameter ∼11 nm and lamellar spacing of ∼2.2 nm between the chromophore layers. In addition, superhelical bundles of these tubes can also form though the addition of polyvinyl alcohol inhibits this process and thereby avoids single-layered tube formation24 and maintains a stable solution over several weeks31. A drawing of the J-aggregate under investigation, from here on referred to as C8O3, is shown in Fig. 1a. The bilayer configuration of C8O3 allows the effect of different decoherence rates to be studied as the outer solvent-exposed layer shows faster decoherence than the inner protected layer.
Figure 1. C8O3 and polarization-controlled 2D spectroscopy.

(a) Wire-guided window-free jet used for sample circulation, along with a schematic of the double-layered structure of the C8O3 aggregate. The aggregates align along the flow direction (white arrow). The transition dipole directions of bands 1–3 are displayed by arrows, which are mainly polarized along the tube axis (bands 1 and 2 shown in blue) or perpendicular to the axis (band 3 shown in orange). (b) Absorption spectra in arbitrary units, Abs. (a. u.), with light polarized parallel (blue) and perpendicular (perp.; orange) to the flow direction. (c) Non-resonant Raman spectra of the C8O3 monomer (black line) and aggregate (grey area). The vibrational frequencies ν1 and ν2 are close to the exciton energy splitting between bands 1 and 3 and bands 2 and 3, respectively. (d) Polarization-controlled 2D spectroscopy with three excitation pulses (k1 to k3) and a local oscillator (LO) for heterodyne detection of the signal field, depicted as an oscillating line. Polarization orientation (0° or 90°) is given with respect to the longitudinal axis of aligned C8O3.
The structural properties of the aggregate are remarkable: the 11-nm outer diameter is contrasted by a length of several micrometres. Circulating solvated C8O3 with a wire-guided jet (Fig. 1a) leads to a macroscopic orientation of the tubes because the longitudinal axis preferentially aligns along the flow direction. This creates anisotropy for linearly polarized light, as shown in Fig. 1b. Linear dichroism measurements31 and redox-chemistry studies32 assign bands 1 and 2 to longitudinal transitions localized on the inner and outer cylinders, respectively (Fig. 1a). Transitions to band 3 are preferentially polarized perpendicular to the long axis of C8O3 and are shared by both layers. A detailed description of sample-preparation methods and band assignments is given in the Supplementary Notes 1 and 2.
Fitting the well-defined absorption peaks of C8O3 with Lorentzian functions (see Supplementary Note 2) reveals an exciton energy difference between bands 1 and 3 of ΔΩ31≈690 cm−1 and ΔΩ32≈460 cm−1 for bands 2 and 3. Both exciton energy splittings are close to vibrational frequencies ν1≈668 cm−1 and ν2≈470 cm−1 observed in non-resonant Raman spectra33 (Fig. 1c). These vibrational frequencies are measured in both the monomer and aggregate Raman spectra, that is, they are not aggregation-induced Raman bands. Strongly enhanced modes at similar energies were observed in resonant Raman spectra of a related cyanine dye, and can be assigned to out-of-plane vibrations34. Such out-of-plane vibrations were shown to couple strongly to excitons35. The quasi-resonance between the vibrational frequencies ν1 and ν2 and exciton energy splittings ΔΩ31 and ΔΩ32 provides us with an interesting scenario of possible coherent interaction between bands (excitons) and vibrations11,13,14,21,36. Such exciton–vibrational coupling induces vibronic12 and vibrational coherences15, which can both lead to long-lived beating signals in 2D spectra. Here we emphasize that coherence in the electronic excited-state manifold is referred to as vibronic and in the ground-state manifold as vibrational. Identifying the dominant contribution is of fundamental importance because only vibronic coherence, which manifests in excited-state dynamics, can enhance exciton transport and thus support light-harvesting function37,38,39.
Experimental results
The absorption spectrum of a light-harvesting system may be heavily congested because of overlapping excitonic bands and the resulting 2D signal would exhibit significant overlap between diagonal and cross-peaks, thereby impeding further analysis. It has been suggested to employ laser pulses of different relative polarization to selectively address relevant excitation pathways to obtain a clearer 2D signal40. However, the advantage of polarization-controlled 2D spectroscopy has been limited by the isotropic nature of the investigated samples (an ensemble). In the experiment presented here, these problems are circumvented by the measurement of the macroscopically aligned C8O3. The transition dipole moments of bands 1 and 2 are preferentially parallel to the longitudinal axis while band 3 is orthogonal, thus allowing for optimal polarization selectivity. This combination reduces the obtained 2D maps to only two relevant peaks with negligible overlap and an up to 30 times stronger signal intensity as compared with the isotropic case41.
The ideal pulse sequence to isolate beating signals between states with orthogonal transition dipole moments, that is, bands 1 and 3 in the present case, is depicted in Fig. 1d, where the phase-matched direction for measuring rephasing spectra is displayed: non-rephasing spectra can be measured along the same phase-matched signal direction by changing the order of the first two pulses (see Methods). After subtraction of the non-oscillatory background, we performed a Fourier transformation along waiting time t2 for all points on the 2D (ω1, ω3) map. The resulting ω2 plots allow the lineshape of beating signal with frequency ω2 to be visualized as a function of position in (ω1, ω3) space. The slice at the exciton energy splitting between bands 1 and 3 (ω2=705±20 cm−1 with the experimental resolution of ±20 cm−1) reveals a non-rephasing diagonal peak N11 and a rephasing cross-peak R31 as shown in Fig. 2a,b, respectively. N11 is centred at (ω1, ω3)=(Ω1, Ω1) with exciton energy Ω1≈16,405 cm−1 of band 1 and a symmetric linewidth 2Γg1≈130 cm−1 along both ω1 and ω3 axes (Fig. 2a). The centre of R31 is located at (ω1, ω3)=(Ω3, Ω1) with exciton energy Ω3≈17,125 cm−1 of band 3 and asymmetric linewidths 2Γg3≈300 cm−1 and 2Γg1≈130 cm−1 along ω1 and ω3 axes, respectively (Fig. 2b). In peak amplitude, R31 is ∼30% of N11. Turning to the ω2 slice corresponding to the energy splitting between bands 2 and 3 (ω2=462±20 cm−1), Fig. 2e,f reveal a diagonal non-rephasing peak N22, which is centred at (ω1, ω3)=(Ω2, Ω2) with the exciton energy Ω2≈16,672 cm−1 of band 2 and a symmetric linewidth 2Γg2≈225 cm−1 along ω1 and ω3 axes. The amplitude of N22 is only 5% of N11.
Figure 2. Experimental and theoretical 2D spectra.

(a,b) The Fourier-transform (FT) amplitude maps of non-rephasing (non-reph.) and rephasing (reph.) spectra at ω2=705±20 cm−1, which reveal the presence of a non-rephasing diagonal peak N11 and a rephasing cross-peak R31. These peaks stem from the coherent interaction of bands 1 and 3 with the quasi-resonant vibrational mode with frequency ν1≈668 cm−1. The amplitude of N11 is about three times larger than R31. The lineshape of N11 is symmetric along both ω1 and ω3 axes, while that of R31 is elongated along ω1 axis. (c,d) The simulated spectra at ω2=705 cm−1 with N11 and R31. (e) The FT amplitude map at ω2=462±20 cm−1 reveals coherent interaction of bands 2 and 3 with the quasi-resonant vibrational mode with frequency ν2≈470 cm−1. However, as depicted in (f) the associated non-rephasing peak N22 at ω1,3≈16,670 cm−1 is weak and only amounts to 5% of N11 at ω2=705±20 cm−1 (see a). The diagonal peak at ω1,3≈16,400 cm−1 in e stems from N11, with a peak centred at ω2=705±20 cm−1, but broad enough to appear at ω2=462±20 cm−1. All measurements were carried out at room temperature.
Theoretical model
To describe the long-lived oscillations in N11 and R31, a vibronic model is employed that describes the coupling of bands 1 and 3 to a quasi-resonant vibrational mode with frequency ν1. Consider a system with electronic ground state |gk〉 and excited states for bands 1 and 3, denoted by |1k〉 and |3k〉, respectively, where k=0 and 1 denote the vibrational ground and excited states, respectively (Fig. 3a). The vibronic coupling between the quasi-resonant states |30〉 and |11〉 leads to unnormalized vibronic eigenstates 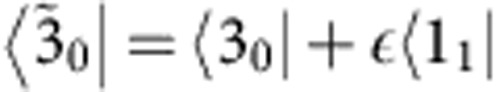 and
and 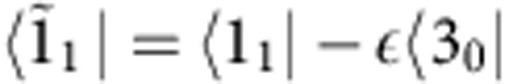 . Here
. Here 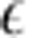 represents the degree of vibronic mixing defined by
represents the degree of vibronic mixing defined by
Figure 3. Vibronic model.

(a) We consider a vibronic model for bands 1 and 3 coupled to a vibrational mode with frequency ν1≈668 cm−1 (see Supplementary Note 2). The vibronic states |k0〉 and |k1〉 denote the vibrational ground and first excited states of an electronic state |k〉, respectively, with the single index states |g〉, |1〉 and |3〉 denoting the electronic ground state and bands 1 and 3, respectively. The exciton energy splitting ΔΩ31=Ω3−Ω1 between bands 1 and 3 is quasi-resonant with the vibrational frequency ν1, where the detuning is denoted by Δν1=ΔΩ31−ν1. The exciton–vibrational coupling between uncoupled states |30〉 and |11〉 leads to vibronic eigenstates 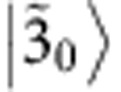 and
and 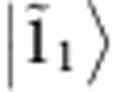 , each of which is a superposition of |30〉 and |11〉, leading to an energy-level shifting by δω. (b) The time trace of N11 in normalized intensity (Norm. Int.) against waiting time t2, where the experimental results are shown as light red circles and the theoretical simulation is shown as a full red line. (c) The time trace of R31 where the experimental results are shown as light blue circles and the simulated data are depicted as a full blue line. The root mean squared deviation between the experimental results and theoretical simulation in b and c is 0.92 and 0.59, respectively.
, each of which is a superposition of |30〉 and |11〉, leading to an energy-level shifting by δω. (b) The time trace of N11 in normalized intensity (Norm. Int.) against waiting time t2, where the experimental results are shown as light red circles and the theoretical simulation is shown as a full red line. (c) The time trace of R31 where the experimental results are shown as light blue circles and the simulated data are depicted as a full blue line. The root mean squared deviation between the experimental results and theoretical simulation in b and c is 0.92 and 0.59, respectively.
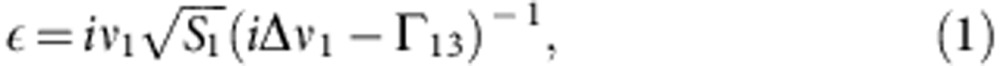 |
where Δν1=(Ω3−Ω1)−ν1 denotes the detuning between |30〉 and |11〉, that is, between the exciton energy splitting and vibrational frequency, and S1 denotes the Huang–Rhys factor of the vibrational mode, which in turn quantifies the strength of the vibronic coupling (see Supplementary Note 2 for details of the derivation). The electronic decoherence rate Γgk describes the exponential decay rate of the coherence between electronic ground state and band k, while Γ13 represents the overall exponential decay rate of the inter-exciton coherence between bands 1 and 3. In our model, we do not consider inhomogeneous broadening, which is justified by the observation that the experimentally measured absorption spectrum is well matched to a sum of Lorentzian functions with the linewidths 2Γgk (see Supplementary Note 2). This is valid when homogeneous broadening dominates the linewidths and the Huang–Rhys factors are sufficiently small, as is the case here. In addition, the lineshape of N11 (Fig. 2a) is not elongated along the diagonal ω1=ω3, implying our 2D signal is dominated by homogeneous broadening. The same conclusion is reached from analysing 2D correlation spectra33.
In nonlinear spectroscopy, the molecular response to laser excitation is described by response functions42. According to the vibronic model described above, the response function for the oscillatory signals in N11 reads
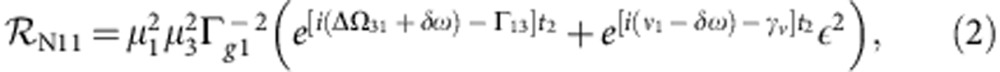 |
with μ1 and μ3 denoting the transition dipole moment of bands 1 and 3, respectively. The prefactor Γg1−2 stems from the lineshape of N11, γv denotes the dissipation rate of the vibrations and δω stands for the frequency shift of the vibronic eigenstates 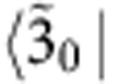 and
and 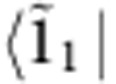 relative to the uncoupled states 〈30| and 〈11| due to the vibronic coupling (see Fig. 3a and Supplementary Note 2 for further details). The coupling was found to be sufficiently strong to induce non-negligible vibronic mixing
relative to the uncoupled states 〈30| and 〈11| due to the vibronic coupling (see Fig. 3a and Supplementary Note 2 for further details). The coupling was found to be sufficiently strong to induce non-negligible vibronic mixing 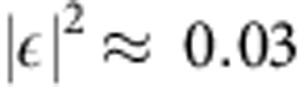 , which leads to a long-lived beating signal in N11 up to t2≈800 fs, as shown in Fig. 3b. These results imply that the initial excitonic part of
, which leads to a long-lived beating signal in N11 up to t2≈800 fs, as shown in Fig. 3b. These results imply that the initial excitonic part of 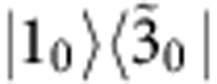 decays rapidly with 1/e decay time of Γ13−1≈66 fs, while the vibronic coherence
decays rapidly with 1/e decay time of Γ13−1≈66 fs, while the vibronic coherence 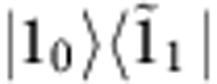 explains a long-lived oscillatory signal in N11: here
explains a long-lived oscillatory signal in N11: here 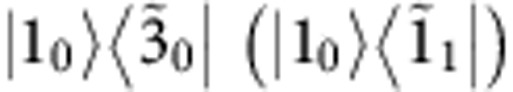 represents coherence between two vibronic states |10〉 and
represents coherence between two vibronic states |10〉 and 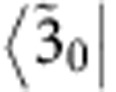 (|10〉 and
(|10〉 and 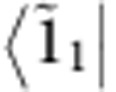 ), respectively.
), respectively.
The response function for the oscillatory contributions to R31 is given by
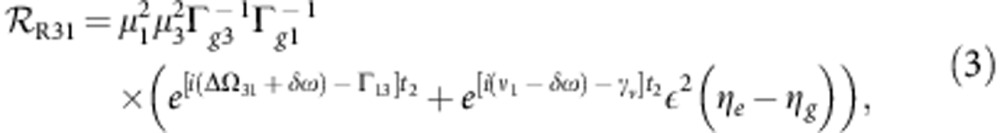 |
where 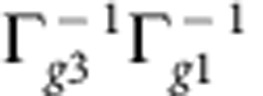 derives from the asymmetric lineshape of R31 (see Fig. 2b,d). Here ηe and ηg represent the contribution of excited-state vibronic coherence
derives from the asymmetric lineshape of R31 (see Fig. 2b,d). Here ηe and ηg represent the contribution of excited-state vibronic coherence 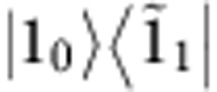 and ground-state vibrational coherence |g0〉〈g1|, respectively, to the long-lived beating signal in R31 (see Supplementary Note 2). The vibrational coherence in the electronic ground-state manifold does not play a role in exciton transfer dynamics, but nonetheless modulates the 2D spectra. A fit of model parameters to experimental results (Fig. 3c) shows that |ηe|≈2.5|ηg|. This means the long-lived beating signal in R31 is dominated by the excited-state coherence
and ground-state vibrational coherence |g0〉〈g1|, respectively, to the long-lived beating signal in R31 (see Supplementary Note 2). The vibrational coherence in the electronic ground-state manifold does not play a role in exciton transfer dynamics, but nonetheless modulates the 2D spectra. A fit of model parameters to experimental results (Fig. 3c) shows that |ηe|≈2.5|ηg|. This means the long-lived beating signal in R31 is dominated by the excited-state coherence 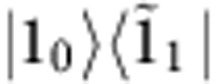 . The short-lived beating signal in R31 is induced by
. The short-lived beating signal in R31 is induced by 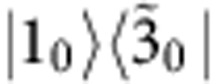 , as is the case for N11. We note that the signal at N11, with approximately three times the amplitude of R31, is exclusively determined by excited-state contributions. Details of this vibronic model and the corresponding Feynman diagrams for the spectral components N11 and R31 are discussed in the Supplementary Note 2.
, as is the case for N11. We note that the signal at N11, with approximately three times the amplitude of R31, is exclusively determined by excited-state contributions. Details of this vibronic model and the corresponding Feynman diagrams for the spectral components N11 and R31 are discussed in the Supplementary Note 2.
These results demonstrate how an excitonic system within a noisy environment can exhibit long-lasting coherent features: the observed long-lived oscillations are the result of coherent interaction of excitonic bands with an underdamped, quasi-resonant vibration. This vibronic mechanism requires the vibrational dissipation rate γv to be much slower than the electronic decoherence rate Γ13, which is the case for C8O3, where 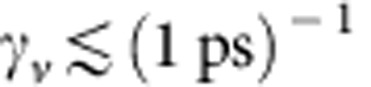 and Γ13≈(66 fs)−1. The difference in electronic and vibrational decoherence rates can be rationalized from the fact that excitons and vibrations are related to the motion of electrons and nuclei, respectively. The lower mass of electrons as compared with nuclei makes excitons more mobile and therefore more sensitive to environmental fluctuations, such as local electric fields, than vibrations. We note that the vibronic mixing leading to long-lived beating signals in 2D electronic spectra is described by a vibronic coupling that induces coherent energy exchange between excitons and quasi-resonant vibrations (see Supplementary Note 2 for further details):
and Γ13≈(66 fs)−1. The difference in electronic and vibrational decoherence rates can be rationalized from the fact that excitons and vibrations are related to the motion of electrons and nuclei, respectively. The lower mass of electrons as compared with nuclei makes excitons more mobile and therefore more sensitive to environmental fluctuations, such as local electric fields, than vibrations. We note that the vibronic mixing leading to long-lived beating signals in 2D electronic spectra is described by a vibronic coupling that induces coherent energy exchange between excitons and quasi-resonant vibrations (see Supplementary Note 2 for further details):
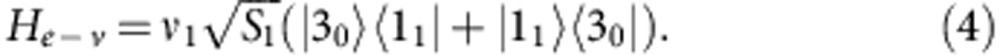 |
This implies that the vibronic coupling not only induces long-lasting electronic excited-state coherences but also can mediate population transfer between excitonic bands. In a combination with thermal relaxation of exciton populations, the vibronic coupling may further enhance exciton population transfer and as a result could, in principle, have functional relevance in exciton transport14,38,43,44,45.
Interestingly, the different decoherence rates Γg3≈2Γg1 of bands 1 and 3 lead to different amplitudes of the short-lived beating signals in N11 and R31 (Fig. 3b,c), which are determined by the prefactors 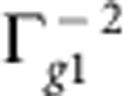 and
and 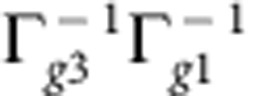 , respectively. The lower decoherence rate of band 1 can be explained by band 1 being localized on the inner layer, while band 3 is delocalized over both the inner and outer layers46. As shown by the response functions for N11 and R31, the overall strength of the beating signals is proportional to the inverse of the electronic decoherence rates. It is therefore expected that the beating signal amplitude would diminish with an increase of the decoherence rate. This is the case for N22, where the physical situation in terms of exciton–vibrational resonance (ΔΩ32≈ν2≈470 cm−1) is equivalent to N11 (ΔΩ31≈ν1≈668 cm−1). The crucial difference is that band 2 has a higher decoherence rate than band 1, as band 2 is localized on the outer layer exposed to solvent46. This explains the broader linewidth of band 2 in absorption and 2D spectra. Using an estimated value of Γg2≈(47 fs)−1, the presented theory predicts the strength of N22 to be 5% of N11 (see Supplementary Note 2), which is in line with the experimental observations (Fig. 2f). These results indicate that the experimentally observed long-lived beating signals, induced by vibronic mixing, require adequately low electronic decoherence rates, highlighting that resonance between exciton energy splitting and vibrational frequency alone is not sufficient47.
, respectively. The lower decoherence rate of band 1 can be explained by band 1 being localized on the inner layer, while band 3 is delocalized over both the inner and outer layers46. As shown by the response functions for N11 and R31, the overall strength of the beating signals is proportional to the inverse of the electronic decoherence rates. It is therefore expected that the beating signal amplitude would diminish with an increase of the decoherence rate. This is the case for N22, where the physical situation in terms of exciton–vibrational resonance (ΔΩ32≈ν2≈470 cm−1) is equivalent to N11 (ΔΩ31≈ν1≈668 cm−1). The crucial difference is that band 2 has a higher decoherence rate than band 1, as band 2 is localized on the outer layer exposed to solvent46. This explains the broader linewidth of band 2 in absorption and 2D spectra. Using an estimated value of Γg2≈(47 fs)−1, the presented theory predicts the strength of N22 to be 5% of N11 (see Supplementary Note 2), which is in line with the experimental observations (Fig. 2f). These results indicate that the experimentally observed long-lived beating signals, induced by vibronic mixing, require adequately low electronic decoherence rates, highlighting that resonance between exciton energy splitting and vibrational frequency alone is not sufficient47.
The presented vibronic model achieves quantitative agreement with the experimental observations. Crucially, the constraints imposed by the observed asymmetric decoherence rates Γg3≈2Γg1 and fast relaxation of exciton population in C8O3 on sub-picosecond timescales33 rule out incoherent models, where long-lived oscillations are sustained by Markovian correlated fluctuations (see Supplementary Note 3 for a detailed analysis). This further supports our conclusion that the observed experimental data provide evidence for vibronic mixing being the mechanism at play in our system.
We note that our results do not imply that correlated fluctuations can be universally ruled out, as this mechanism could be in place in certain pigment–protein complexes. The notion of correlated fluctuations has been developed for photosynthetic complexes where pigments are embedded in a protein scaffold. The protein has been considered as the potential source of correlated fluctuations in natural light harvesters16,17. For C8O3, a structural frame such as a protein scaffold is absent and therefore correlated fluctuations are unlikely to induce long-lived oscillatory 2D signals, which is in line with our observations.
Discussion
We have verified, theoretically and experimentally, that coherent vibronic coupling in the electronic excited-state manifold is responsible for the long-lived beating signals observed in 2D spectra of an artificial light harvester. The relatively simple electronic and vibrational structure of the investigated molecular aggregate along with its macroscopic alignment allowed us to rule out the presence of correlated fluctuations. The specific geometry of our system allowed us to gain further insights by illustrating the conditions under which intra-pigment vibrations can prolong electronic coherent effects. The moderately low decoherence rate of band 1, localized on the inner layer and protected from solvent, is the basis for exciton–vibrational coupling as the source of long-lived beating signals. The outer band 2, even though resonantly coupled to a vibration, exhibits a higher decoherence rate and therefore fails to produce observable oscillations. We conclude that the mere resonance between excitons and vibrations does not suffice to explain long-lived beating signals. An adequately low electronic decoherence rate, determined by the interaction between system and bath, is an equally important prerequisite.
The influence of vibronic coupling on energy transport in molecular aggregates has been extensively studied in the past, as recently reviewed44. The vibronic coupling has recently gained new interest (see ref. 48 for a recent tutorial overview), as it was suggested as a feasible mechanism to explain long-lived oscillations in the 2D spectra of several natural light-harvesting complexes and a photosynthetic reaction centre9,10. The requirement of exciton–vibrational resonance is readily satisfied in such systems, given their numerous excitonic bands and rich vibrational structures. Incoherent models based on correlated fluctuations were not ruled out though. Our work provides a quantum mechanical foundation for enhanced energy transfer based on vibronic coupling. As recently demonstrated, this mechanism is not limited to natural light harvesting, vibronic coupling is also of key importance in photovoltaic devices49.
Methods
Polarization-controlled 2D electronic spectroscopy
In 2D electronic spectroscopy, three ultrashort laser pulses generate an optical response of a molecular ensemble, which is spectrally resolved along both absorption (ω1) and detection (ω3) frequencies within the laser pulse spectrum. The absorption frequency ω1 is obtained by precise scanning of the time delay between the first two pulses and subsequent Fourier transformation (t1→ω1). In detection, the signal is spectrally dispersed, leading directly to the detection frequency ω3. Varying time delay t2 between pulses 2 and 3 provides information about evolution of the system on a femtosecond timescale50,51,52. To retrieve the purely absorptive part, the signal induced by pulses 1–3 is detected in a heterodyned fashion by interfering it with a phase-stable local oscillator pulse. Polarization control is achieved by the combination of λ/4 wave plates and wire grid polarizers for each of the laser beams to select the desired polarization with high accuracy. Polarization-resolved 2D experiments change the relative contributions of distinct pathways depending on the polarization of the laser pulses, orientation of the transition dipole moments and isotropy of the sample40. Rephasing spectra were acquired with a polarization sequence of (90°, 0°, 90°, 0°) for pulses (1, 2, 3, local oscillator), in contrast to non-rephasing spectra, where the time ordering of the first two pulses is reversed, leading to a polarization sequence of (0°, 90°, 90°, 0°). The polarization scheme used for rephasing spectra (Fig. 1d) shows 0° was defined to be parallel to the sample flow direction, depicted as a white arrow in Fig. 1a. For a macroscopically aligned sample, this particular polarization sequence selects pathways stemming from interband coherences and vibronic mixing12,15, discussed throughout the paper, while pathways with all-parallel transition dipole moments such as ground-state bleach, stimulated emission, excited-state absorption and also vibrational wave packet excitation are suppressed. For the details regarding the experimental methods, see Supplementary Note 1. To subtract the non-oscillatory signals from 2D spectra, we employed a decay-associated spectra analysis33, where the population decays were fitted by a sum of three 2D spectra with individual decay constants. The ω2 maps in Fig. 2 were obtained using Fourier transformation (t2→ω2) with zero-padding up to 27 data points. All measurements were carried out at room temperature.
Additional information
How to cite this article: Lim, J. et al. Vibronic origin of long-lived coherence in an artificial molecular light harvester. Nat. Commun. 6:7755 doi: 10.1038/ncomms8755 (2015).
Supplementary Material
Supplementary Figures 1-7, Supplementary Notes 1-3 and Supplementary References
Acknowledgments
We thank Valentyn I. Prokhorenko for help in 2D-DAS analysis. C.N.L. and J.H. acknowledge funding by the Austrian Science Fund (FWF): START project Y 631-N27 and by COST Action CM1202—PERSPECT-H2O. J.L., F.C.-S., S.F.H. and M.B.P. acknowledge funding by the EU STREP PAPETS and QUCHIP, the ERC Synergy Grant BioQ, the Deutsche Forschungsgemeinschaft (DFG) within the SFB/TRR21 and an Alexander von Humboldt Professorship. J.P. acknowledges funding by the Spanish Ministerio de Economía y Competitividad under Project No. FIS2012-30625. D.P. and D.Z. acknowledge funding by the Swedish Research Council and Knut and Alice Wallenberg Foundation.
Footnotes
Author contributions D.P., D.Z. and J.H. designed and conducted experiments; H.v.B. was responsible for sample preparation, structural characterization and Raman measurements; J.L., F.C.-S., C.N.L., D.P., J.P. and J.H. analysed the data; J.L., F.C.-S., S.F.H., J.H. and M.B.P. developed theory; all authors discussed the results and wrote the manuscript.
References
- van Amerongen H., Valkunas L. & van Grondelle R. Photosynthetic Excitons World Scientific (2000). [Google Scholar]
- Blankenship R. E. Molecular Mechanisms of Photosynthesis Blackwell Science (2002). [Google Scholar]
- Renger T., May V. & Kühn O. Ultrafast excitation energy transfer dynamics in photosynthetic pigment-protein complexes. Phys. Rep. 343, 137–254 (2001). [Google Scholar]
- Huelga S. F. & Plenio M. B. Vibrations, quanta and biology. Contemp. Phys. 54, 181–207 (2013). [Google Scholar]
- Jonas D. M. Two-dimensional femtosecond spectroscopy. Annu. Rev. Phys. Chem. 54, 425–463 (2003). [DOI] [PubMed] [Google Scholar]
- Engel G. S. et al. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 446, 782–786 (2007). [DOI] [PubMed] [Google Scholar]
- Dostál J., Mančal T., Vácha F., Pšenčík J. & Zigmantas D. Unraveling the nature of coherent beatings in chlorosomes. J. Chem. Phys. 140, 115103 (2014). [DOI] [PubMed] [Google Scholar]
- Collini E. et al. Coherently wired light-harvesting in photosynthetic marine algae at ambient temperature. Nature 463, 644–647 (2010). [DOI] [PubMed] [Google Scholar]
- Romero E. et al. Quantum coherence in photosynthesis for efficient solar-energy conversion. Nat. Phys. 10, 676–682 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller F. D. et al. Vibronic coherence in oxygenic photosynthesis. Nat. Chem. 6, 706–711 (2014). [DOI] [PubMed] [Google Scholar]
- Chin A. W. et al. The role of non-equilibrium vibrational structures in electronic coherence and recoherence in pigment-protein complexes. Nat. Phys. 9, 113–118 (2013). [Google Scholar]
- Plenio M. B., Almeida J. & Huelga S. F. Origin of long-lived oscillations in 2D-spectra of a quantum vibronic model: electronic versus vibrational coherence. J. Chem. Phys. 139, 235102 (2013). [DOI] [PubMed] [Google Scholar]
- Chin A. W., Huelga S. F. & Plenio M. B. Coherence and decoherence in biological system: principles of noise assisted transport and the origin of long-lived coherences. Phil. Trans. R. Soc. A 370, 3638–3657 (2012). [DOI] [PubMed] [Google Scholar]
- Kolli A., O'Reilly E. J., Scholes G. D. & Olaya-Castro A. The fundamental role of quantized vibrations in coherent light harvesting by cryptophyte algae. J. Chem. Phys. 137, 174109 (2012). [DOI] [PubMed] [Google Scholar]
- Tiwari V., Peters W. K. & Jonas D. M. Electronic resonance with anticorrelated pigment vibrations drives photosynthetic energy transfer outside the adiabatic framework. Proc. Natl Acad. Sci. USA 110, 1203–1208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Cheng Y.-C. & Fleming G. R. Coherence dynamics in photosynthesis: protein protection of excitonic coherence. Science 316, 1462–1465 (2007). [DOI] [PubMed] [Google Scholar]
- Ishizaki A., Calhoun T. R., Schlau-Cohen G. S. & Fleming G. R. Quantum coherence and its interplay with protein environments in photosynthetic electronic energy transfer. Phys. Chem. Chem. Phys. 12, 7319–7337 (2010). [DOI] [PubMed] [Google Scholar]
- Hayes D., Griffin G. B. & Engel G. S. Engineering coherence among excited states in synthetic heterodimer systems. Science 340, 1431–1434 (2013). [DOI] [PubMed] [Google Scholar]
- Christensson N. et al. High frequency vibrational modulations in two-dimensional electronic spectra and their resemblance to electronic coherence signatures. J. Phys. Chem. B 115, 5383–5391 (2011). [DOI] [PubMed] [Google Scholar]
- Caycedo-Soler F., Chin A. W., Almeida J., Huelga S. F. & Plenio M. B. The nature of the low energy band of the Fenna-Matthews-Olson complex: vibronic signatures. J. Chem. Phys. 136, 155102 (2012). [DOI] [PubMed] [Google Scholar]
- Christensson N., Kauffmann H. F., Pullerits T. & Mančal T. Origin of long-lived coherences in light-harvesting complexes. J. Phys. Chem. B 116, 7449–7454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijs D.-J., Dijkstra A. G. & Knoester J. Ultrafast pump-probe spectroscopy of linear molecular aggregates: effects of exciton coherence and thermal dephasing. Chem. Phys. 341, 230–239 (2007). [Google Scholar]
- Würthner F., Kaiser T. E. & Saha-Möller C. R. J-aggregates: from serendipitous discovery to supramolecular engineering of functional dye materials. Angew. Chem. Int. Ed. 50, 3376–3410 (2011). [DOI] [PubMed] [Google Scholar]
- Eisele D. M. et al. Robust excitons inhabit soft supramolecular nanotubes. Proc. Natl Acad. Sci. USA 111, E3367–E3375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen-Zhou J. et al. Coherent exciton dynamics in supramolecular light-harvesting nanotubes revealed by ultrafast quantum process tomography. ACS Nano 8, 5527–5534 (2014). [DOI] [PubMed] [Google Scholar]
- Qiao Y. et al. Nanotubular J-aggregates and quantum dots coupled for efficient resonance excitation energy transfer. ACS Nano 9, 1552–1560 (2015). [DOI] [PubMed] [Google Scholar]
- von Berlepsch H. & Böttcher C. in J-Aggregates Vol. 2, ed. Kobayashi T. Ch. 4 119–153World Scientific (2012). [Google Scholar]
- von Berlepsch H., Kirstein S. & Böttcher C. Effect of alcohols on J-aggregation of a carbocyanine dye. Langmuir 18, 7699–7705 (2002). [Google Scholar]
- von Berlepsch H., Kirstein S. & Böttcher C. Controlling the helicity of tubular J-aggregates by chiral alcohols. J. Phys. Chem. B 107, 9646–9654 (2003). [Google Scholar]
- von Berlepsch H. et al. Supramolecular structures of J-aggregates of carbocyanine dyes in solution. J. Phys. Chem. B 104, 5255–5262 (2000). [Google Scholar]
- von Berlepsch H. et al. Stabilization of individual tubular J-aggregates by poly(vinyl alcohol). J. Phys. Chem. B 107, 14176–14184 (2003). [Google Scholar]
- Eisele D. M. et al. Utilizing redox-chemistry to elucidate the nature of exciton transitions in supramolecular dye nanotubes. Nat. Chem. 4, 655–662 (2012). [DOI] [PubMed] [Google Scholar]
- Milota F. et al. Vibronic and vibrational coherences in two-dimensional electronic spectra of supramolecular J-aggregates. J. Phys. Chem. A 117, 6007–6014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin M., Dede Ö. & Akins D. L. Density functional theory and Raman spectroscopy applied to structure and vibrational mode analysis of 1,1',3,3'-tetraethyl-5,5',6,6'-tetrachloro-benzimidazolocarbocyanine iodide and its aggregate. J. Chem. Phys. 134, 064325 (2011). [DOI] [PubMed] [Google Scholar]
- Rich C. C. & McHale J. L. Resonance Raman spectra of individual excitonically coupled chromophore aggregates. J. Phys. Chem. C 117, 10856–10865 (2013). [Google Scholar]
- Butkus V., Zigmantas D., Abramavicius D. & Valkunas L. Distinctive character of electronic and vibrational coherences in disordered molecular aggregates. Chem. Phys. Lett. 587, 93–98 (2013). [Google Scholar]
- Womick J. M. & Moran A. M. Exciton coherence and energy transport in the light-harvesting dimers of allophycocyanin. J. Phys. Chem. B 113, 15747–15759 (2009). [DOI] [PubMed] [Google Scholar]
- Womick J. M. & Moran A. M. Vibronic enhancement of exciton sizes and energy transport in photosynthetic complexes. J. Phys. Chem. B 115, 1347–1356 (2011). [DOI] [PubMed] [Google Scholar]
- Del Rey M., Chin A. W., Huelga S. F. & Plenio M. B. Exploiting structured environments for efficient energy transfer: the phonon antenna mechanism. J. Phys. Chem. Lett. 4, 903–907 (2013). [DOI] [PubMed] [Google Scholar]
- Hochstrasser R. M. Two-dimensional IR-spectroscopy: polarization anisotropy effects. Chem. Phys. 266, 273–284 (2001). [Google Scholar]
- Read E. L. et al. Cross-peak-specific two-dimensional electronic spectroscopy. Proc. Natl Acad. Sci.USA 104, 14203–14208 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel S. Principles of Nonlinear Optical Spectroscopy Oxford University Press (1995). [Google Scholar]
- Perlík V. et al. Vibronic coupling explains the ultrafast carotenoid-to-bacteriochlorophyll energy transfer in natural and artificial light harvesters. J. Chem. Phys. 142, 212434 (2015). [DOI] [PubMed] [Google Scholar]
- Schröter M. et al. Exciton-vibrational coupling in the dynamics and spectroscopy of Frenkel excitons in molecular aggregates. Phys. Rep. 567, 1–78 (2015). [Google Scholar]
- Killoran N., Huelga S. F. & Plenio M. B. Enhancing light-harvesting power with coherent vibrational interactions: a quantum heat engine picture. Preprint at http://arxiv.org/abs/1412.4136 (2015). [DOI] [PubMed]
- Didraga C. et al. Structure, spectroscopy, and microscopic model of tubular carbocyanine dye aggregates. J. Phys. Chem. B 108, 14976–14985 (2004). [Google Scholar]
- Halpin A. et al. Two-dimensional spectroscopy of a molecular dimer unveils the effects of vibronic coupling on exciton coherences. Nat. Chem. 6, 196–201 (2014). [DOI] [PubMed] [Google Scholar]
- Chenu A. & Scholes G. D. Coherence in energy transfer and photosynthesis. Annu. Rev. Phys. Chem. 66, 69–96 (2015). [DOI] [PubMed] [Google Scholar]
- Falke S. M. et al. Coherent ultrafast charge transfer in an organic photovoltaic blend. Science 344, 1001–1005 (2014). [DOI] [PubMed] [Google Scholar]
- Brixner T., Mančal T., Stiopkin I. V. & Fleming G. R. Phase-stabilized two-dimensional electronic spectroscopy. J. Chem. Phys. 121, 4221–4236 (2004). [DOI] [PubMed] [Google Scholar]
- Augulis R. & Zigmantas D. Two-dimensional electronic spectroscopy with double modulation lock-in detection: enhancement of sensitivity and noise resistance. Opt. Express 19, 13126–13133 (2011). [DOI] [PubMed] [Google Scholar]
- Augulis R. & Zigmantas D. Detector and dispersive delay calibration issues in broadband 2D electronic spectroscopy. J. Opt. Soc. Am. B 30, 1770–1774 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-7, Supplementary Notes 1-3 and Supplementary References