

Abstract
We have bred a Pkd1 floxed allele with a nestin-Cre expressing line to generate cystic mice with preserved GFR to address the pathogenesis of complex ADPKD phenotypes. Hypertension affects about 60% of these patients before loss of renal function, leading to significant morbimortality. Cystic mice were hypertensive at 5 and 13 weeks of age, a phenotype not seen in non-cystic controls and Pkd1-haploinsufficient animals, which do not develop renal cysts. Fractional sodium excretion was reduced in cystic mice at these ages. Angiotensinogen gene expression was higher in cystic than non-cystic kidneys at 18 weeks, while ACE and the AT1 receptor were expressed in renal cyst epithelia. Cystic animals displayed increased renal cAMP, cell proliferation and apoptosis. At 24 weeks mean arterial pressure and fractional sodium excretion did not significantly differ between the cystic and non-cystic groups, whereas cardiac mass increased in cystic mice. Renal concentrating deficit is also an early finding in ADPKD. Maximum urine osmolality and urine nitrite excretion were reduced in 10–13 and 24-week-old cystic mice, deficits not found in haploinsufficient and non-cystic controls. A trend of higher plasma vasopressin was observed in cystic mice. Thus, cyst growth most probably plays a central role in early-stage ADPKD-associated hypertension, with activation of the intrarenal renin-angiotensin system as a key mechanism. Cyst expansion is also likely essential for the development of the concentrating deficit in this disease. Our findings are consistent with areas of reduced perfusion in the kidneys of patients with ADPKD.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic life-threatening disease, with an estimated prevalence of 1:400–1000.1 By the age of 60 years, more than half of the patients reach end-stage renal disease.2 While mutations in one of two genes, PKD1 (polycystic kidney disease 1) or PKD2 (polycystic kidney disease 2), cause this disorder, approximately 85% of the cases are linked to the PKD1 locus.3 Mutations in PKD1, in turn, are usually associated with a more severe phenotype than PKD2.4,5 Although most patients seek medical attention due to kidney manifestations, ADPKD is a systemic illness and includes extrarenal phenotypes most often represented by liver cysts, intracranial aneurysms and heart valve abnormalities.6
The pathogenesis of critical and complex phenotypes is still unresolved in ADPKD. Systemic arterial hypertension (SAH) is one of the most common findings in this disorder, occurring in approximately 60% of cases prior to a significant loss of renal function7 and 10 years earlier than in the general population.8 SAH is a major risk factor for cardiovascular disease and accounts for significant morbidity and mortality in this patient population.9,10 The mechanisms involved in ADPKD-associated hypertension, however, are not completely understood, although renal abnormalities influence its genesis and maintenance.11 It is currently thought that activation of the renin-angiotensin system (RAS), in response to cyst expansion and secondary vascular compression, plays a central role in the development of hypertension in this disease.7,11 This model, however, has not been proven yet. Additional data suggest that lower levels of nitric oxide (NO) as a result of ADPKD-associated endothelial dysfunction12 contribute to increased angiotensin II generation through a local increase in oxidative stress. In addition, reduced cardiac relaxation that is typically associated with this disorder may lead to a reduction in renal blood flow and increase in angiotensin II formation.11
The ADPKD renal phenotype is also classically associated with a concentrating deficit, one of the earliest manifestations of the disease.13 This abnormality is usually mild and not associated with polyuria or polydipsia, but can be detected in childhood.14 The pathogenesis of this concentrating defect is not known but disruption of the corticomedullary architecture due to cyst growth, a principal cell defect and/or development of tubulointerstitial alterations have been proposed as possible causes. These mechanisms, however, are not consistent with its early presentation.15 Previous data exclude a central nervous cause, since vasopressin levels have been found to be high in ADPKD patients.15 Increased plasma vasopressin in this illness16,17 has been postulated to contribute to the degree of hypertension. Notably, the observation that aquaporin-2 is upregulated in animal models of polycystic kidneys, as opposed to what is seen in the central and most nephrogenic forms of diabetes insipidus, suggests that the abnormality is positioned distally to the synthesis of this molecule.18,19
In order to test whether cystic disease is the principal determinant of hypertension and the concentrating defects, we have extensively evaluated and characterized these complex phenotypes in two independent PKD1 orthologous mouse models. We found that Pkd1-haploinsufficient animals were normotensive and had normal concentrating capacity whereas cystic animals were hypertensive at the ages of 5 and 13 weeks, showed increased kidney expression of RAS components and exhibited concentrating defects at 10–13 and 24 weeks of age. These findings suggest that cyst formation is critical for the pathogenesis of both these classic manifestations of ADPKD and that RAS activation plays a significant role in the pathogenesis of hypertension in this disorder.
RESULTS
Pkd1cond/cond:Nestincre and Pkd1+/− mouse models
We have used two Pkd1-deficient mouse models in the current work. Using a Pkd1 floxed allele and a nestin-Cre transgene, we have generated homozygous animals for this conditional allele with a mosaic pattern of gene inactivation, via the excision of the exons 2–4 (Pkd1cond/cond:Nestincre; CY).20 This model, used in a previous study by Shillingford et al21, has developed a milder renal phenotype in our animal care facility than the one reported by these investigators. We have also worked with heterozygous mice for a Pkd1-null mutation (Pkd1+/−; HT).20,22 While the CY mice present cystic kidneys without a Pkd1-haploinsufficient cell background, the HT animals display non-cystic kidneys in the setting of Pkd1-haploinsufficiency by 15 weeks (wk) of age.20,22
Serial sections of CY and non-cystic (Pkd1cond/cond; NC) left kidneys confirmed the presence of cysts and microcysts only in CY mice (data not shown). The average number of cysts present in 15-wk CY left kidneys was 22±6; as expected, no cysts were observed in 15-wk NC left kidneys. Total kidney weight (TKW)/body weight (BW) ratios were significantly higher in CY than NC mice at the ages of 5, 15 and 24 wk (Figure 1A, Table 1). Interestingly, heart weight (HW)/BW was also higher in CY animals at 24 wk (Figure 1B, Table 1).
Figure 1.

(A) Comparative analysis of total kidney weight/body weight (TKW/BW) ratios between NC and CY kidneys at 5, 15 and 24 wk, and between WT and HT kidneys at 15 wk. * p<0.05 versus NC; b p<0.05 versus 24 wk. TKW/BW ratios were compared using the nonpaired t test and ANOVA, with the data presented as mean±SD; n/group in Table 1. (B) Comparative analysis of heart weight/body weight (HW/BW) ratios between NC and CY kidneys at 5, 15 and 24 wk, and between WT and HT kidneys at 15 wk. * p<0.05 versus NC. HW/BW ratios were compared using the nonpaired t test and ANOVA, with the data presented as mean±SD; n/group in Table 1. (C) Representative images of NC and CY kidneys and representative histological sections of CY kidneys at 5, 15 and 24 wk of age. (D) Comparative analysis of cAMP levels between NC and CY kidneys at 13 wk of age. * p<0.05 versus NC; comparison performed using the nonpaired t test, with the data presented as mean±SD; n/group in Table 1. (E) Vascular compression determined by a cyst in a CY mouse kidney. Original magnification, ×400. (F) Cystic index analysis in CY kidneys at 5, 15 and 24 wk of age. b p<0.05 versus 24 wk. Comparisons were carried out using the Mann-Whitney and Kruskal-Wallis test, with data expressed as median (lower quartile to upper quartile).
Table 1.
Body weight (BW), Total kidney weight (TKW)/BW, Heart weight (HW)/BW, renal cAMP and cystic index in NC, CY, WT and HT male mice.
| Parameters | BW (g; Mean ± SD) | TKW/BW (%; Mean ± SD) | HW/BW (%; Mean ± SD) | Renal cAMP (pmol/mL; Mean ± SD) | Cystic Index [%; Median (Lower Quartile to Upper Quartile)] |
|---|---|---|---|---|---|
| NC - 5 weeks (n = 5) | 15.8 ± 2.3b | 1.518 ± 0.203 | 0.641 ± 0.033 | – – | – – |
|
| |||||
| CY - 5 weeks (n = 6) | 17.3 ± 2.7b | 2.198 ± 0.612* | 0.693 ± 0.110 | – – | 8.63 (5.23 – 10.79)b |
|
| |||||
| NC - 13 weeks (n = 6) | 9.92 ± 1.83 | – – | |||
| NC - 15 weeks (n = 9) | 19.3 ± 0.80b | 1.629 ± 0.167 | 0.691 ± 0.088 | – – | |
|
| |||||
| CY - 13 weeks (n = 7) | 14.78 ± 6.33* | – – | |||
| CY - 15 weeks (n = 9) | 19.8 ± 2.1b | 1.869 ± 0.289*, b | 0.718 ± 0.050 | 12.86 (5.67 – 17.83) | |
|
| |||||
| NC - 24 weeks (n = 6) | 28.0 ± 1.8 | 1.636 ± 0.165 | 0.647 ± 0.095 | – – | – – |
|
| |||||
| CY - 24 weeks (n = 6) | 25.9 ± 1.8* | 2.373 ± 0.649* | 0.779 ± 0.120* | – – | 17.31 (13.61 – 31.22) |
|
| |||||
| WT - 15 weeks (n = 8) | 20.1 ± 0.9 | 1.816 ± 0.217 | 0.841 ± 0.135 | – – | – – |
|
| |||||
| HT - 15 weeks (n = 8) | 19.6 ± 0.8 | 1.964 ± 0.196 | 0.852 ± 0.130 | – – | – – |
TKW/BW, HW/BW and renal cAMP were compared using the nonpaired t test.
BW and cystic index were compared using the Mann-Whitney test.
Group-time analyses was performed using ANOVA or Kruskal Wallis.
p < 0.05 versus NC.
p < 0.05 versus 24 weeks.
These models represent, therefore, the two cellular/genetic environments found in the human ADPKD kidneys: the HT mice show almost exclusively Pkd1+/− renal cells but do not display cysts, reproducing the background cell environment found in ADPKD1 patients; CY mice, on the other hand, have cysts presumably formed by Pkd1-/− cells, reproducing the ADPKD1 cystic phenotype and its expected consequences, however in a predominant wild-type cell (nonexpressing Cre) background. Illustrations of CY and NC kidneys are presented in Figure 1C. All experiments were performed in male mice to avoid potential gender-related experimental heterogeneity.
We have also quantified cyclic AMP (cAMP) in kidney tissue at 13 wk of age, having found higher levels in CY (14.78±6.33 pmol/mL) than NC mice (9.92±1.83 pmol/mL; p<0.05, Figure 1D, Table 1). Occasional compression exerted by renal cysts on blood vessels could be seen in CY kidneys, as shown in Figure 1E.
A quantitative analysis of cystic involvement was carried out based on uniformly spaced CY kidney sections. This evaluation revealed significantly higher indexes in CY mice at 24 wk compared with 5 wk (17.31% [13.61–31.22) versus 8.63% (5.23–10.79); p<0.05], a trend of lower indexes at 5 wk compared with 15 wk [8.63% (5.23–10.79) versus 12.86% (5.67–17.83)], and a trend of higher values at 24 wk than 15 wk [17.31% [13.61–31.22) versus 12.86% (5.67–17.83)] (Figure 1F, Table 1).
Blood pressure analysis
Invasive mean arterial pressure (MAP) measured at 13 wk of age revealed higher levels in CY mice compared to NC animals (149.79±4.66 versus 132.86±4.21 mmHg; p<0.001, Figure 2A, Table 2). We did not observe, however, a difference in MAP between HT and wild-type (Pkd1+/+; WT) mice (Figure 2B, Table 2). Following such findings, we extended this analysis to younger and older cystic animals, showing significantly higher MAP in CY compared to NC mice at the age of 5 wk (129.47±6.78 versus 118.81±9.07 mmHg; p<0.05, Figure 2A, Table 2). Cystic animals, however, did not present significantly higher MAP at 24 wk, although a trend for more elevated MAP in CY than NC mice has been observed (Figure 2A, Table 2).
Figure 2.

Comparative analyses of mean arterial pressure: (A) between NC and CY mice at 5, 13 and 24 wk of age; and (B) between WT and HT mice at 13 wk. * p<0.05 versus NC; *** p<0.001 versus NC; aaa p<0.001 versus 13 wk; b p<0.05 versus 24 wk. MAP was compared using the nonpaired t test and ANOVA, with the data presented as mean±SD; n/group in Table 2.
Table 2.
Mean Arterial Pressure (MAP), Serum creatinine (SCr), SUN and Cystatin C in NC, CY, WT and HT male mice.
| Parameters | MAP (mmHg, Mean ± SD) | SCr [mg/dL; Median [Lower Quartile to Upper Quartile]) | SUN (mg/dL, Mean ± SD) | Cystatin C (mg/L, Mean ± SD) |
|---|---|---|---|---|
| NC - 5 weeks | 118.81 ± 9.07 (n = 6) | 0.46 (0.36 to 0.55) (n = 6) | 27.88 ± 1.33 (n = 6) | – – |
|
| ||||
| CY - 5 weeks | 129.47 ± 6.78*, aaa, b (n = 6) | 0.44 (0.30 to 0.56)b (n = 6) | 29.23 ± 3.21 (n = 6) | – – |
|
| ||||
| NC - 10–13 weeks | 0.37 (0.35 to 0.38) (n = 9) | 25.23 ± 1.22 (n = 9) | ||
| NC - 13 weeks | 132.86 ± 4.21 (n = 6) | 3.21 ± 0.50 (n = 6) | ||
| CY - 10–13 weeks | 0.32 (0.30 to 0.35) ** (n = 9) | 26.70 ± 1.43* (n = 9) | ||
| CY - 13 weeks | 149.79 ± 4.66***, b (n = 6) | 3.61 ± 0.53 (n = 7) | ||
| NC - 24 weeks | 131.64 ± 13.08 (n = 6) | 0.23 (0.21 to 0.34) (n = 6) | 27.80 ± 4.62 (n = 6) | – – |
|
| ||||
| CY - 24 weeks | 139.95 ± 7.20 (n = 6) | 0.33 (0.22 to 0.37) (n = 7) | 30.14 ± 5.10 (n = 7) | – – |
|
| ||||
| WT - 10–13 weeks | 0.38 (0.36 to 0.39) (n = 8) | 26.27 ± 1.16 (n = 8) | – – | |
| WT - 13 weeks | 127.54 ± 2.99 (n = 8) | |||
|
| ||||
| HT - 10–13 weeks | 0.36 (0.34 to 0.36) (n = 8) | 25.34 ± 1.18 (n = 8) | – – | |
| HT - 13 weeks | 131.03 ± 4.36 (n = 6) | |||
MAP, SUN and cystatin C were compared using the nonpaired t test.
SCr was compared using the Mann-Whitney test.
Group-time analyses was performed using ANOVA or Kruskal Wallis.
p < 0.05 versus NC.
p < 0.01 versus NC.
p < 0.01 versus NC.
p < 0.001 versus 13 weeks.
p < 0.05 versus 24 weeks.
Glomerular filtration, cystatin C and tubular function analyses
We found no serum creatinine (SCr) difference between CY and NC mice at the ages of 5 and 24 wk, although slightly lower levels have been found in CY animals at 10–13 wk (Figure 3A, Table 2). Serum urea nitrogen (SUN) also did not differ between these groups at 5 and 24 wk, but mildly higher values were detected in the cystic versus noncystic mice at 10–13 wk (26.70±1.43 versus 25.23±1.22 mg/dL; p<0.05, Figure 3B, Table 2). No significant SCr and SUN differences, however, were verified between HT and WT animals at 10–13 wk of age (Figures 3A and B, Table 2). As an additional method to indirectly assess glomerular filtration in CY and NC mice, we evaluated their serum cystatin C levels at 13 wk. No significant difference, however, was observed between the two groups (Figure 3C, Table 2).
Figure 3.

Analyses of serum creatinine (A) and SUN (B) between NC and CY mice at 5, 10–13 and 24 wk of age, and between WT and HT animals at 10–13 wk. * p<0.05 versus NC; ** p<0.01 versus NC; b p<0.05 versus 24 wk. SCr was compared using the Mann-Whitney and Kruskal-Wallis test, with the data expressed as median (lower quartile to upper quartile), while SUN was compared using the nonpaired t test and ANOVA, with data presented as mean±SD; n/group in Table 2. (C) Analysis of serum cystatin C between NC and CY mice at the age of 13 wk. Cystatin C was compared using the nonpaired t test, with the data presented as mean±SD; n/group in Table 2.
Next we studied the renal handling of Na+ and K+. We found that CY mice had a significantly lower fractional excretion of Na+ (FENa) compared with NC animals at 10–13 wk (0.60±0.06% versus 0.74±0.09%; p<0.001), a finding that was reproduced at 5 wk (0.57±0.22% versus 0.88±0.15%; p<0.01) but not at 24 wk (Figure 4A, Table 3). Serum Na+ (SNa) analysis revealed lower values in CY compared with NC mice at 10–13 wk [137 (136 to 139) versus 143 mEq/L (142 to 144); p<0.01], however this difference was not found at 5 and 24 wk (Figure 4B, Table 3). No serum K+ (SK) difference was detected between these groups at the three analyzed ages (Table 3). Transtubular K+ gradient (TTKG), in turn, did not differ between CY and NC mice at all evaluated ages (Table 3, Supplemental Figure S1).
Figure 4.
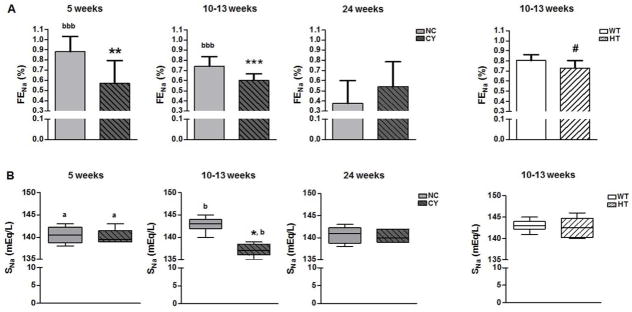
Analyses of fractional excretion of Na+ (A), and serum Na+ (B) between NC and CY male mice at 5, 10–13 and 24 wk of age, and between WT and HT animals at 10–13 wk. *** p<0.001 versus NC; ** p<0.01 versus NC; * p<0.05 versus NC; # p<0.05 versus WT; a p<0.05 versus 10–13 wk; b p<0.05 versus 24 wk; bbb p<0.001 versus 24 wk. FENa was compared using the nonpaired t test and ANOVA, with the data presented as mean±SD, whereas SNa was analyzed by the Mann-Whitney and Kruskal-Wallis test, with the data expressed as median (lower quartile to upper quartile); n/group in Table 3.
Table 3.
Fractional excretion of Na+ (FENa), Transtubular K+ gradient (TTKG), Serum Na+ (SNa), Serum K+ (SK) and Maximum urine osmolality (UosmMax) in NC, CY, WT and HT male mice.
| Parameters | FENa (%,Mean ± SD) | TTKG (Mean ± SD) | SNa [mEq/L; Median (Lower Quartile to Upper Quartile)] | SK [mEq/L; Median (Lower Quartile to Upper Quartile)] | UosmMax (mOsm/kg H2O, Mean ± SD) |
|---|---|---|---|---|---|
| NC - 5 weeks (n = 6) | 0.88 ± 0.15bbb | 3.00 ± 0.85 | 140 (139 to 142)a | 4.0 (3.5 to 4.5)a | 3045 ± 694 |
| CY - 5 weeks (n = 6) | 0.57 ± 0.22** | 3.61 ± 1.09 | 139 (139 to 141)a | 4.0 (3.9 to 4.1)a | 2705 ± 543 |
| NC - 10–13 weeks (n = 9) | 0.74 ± 0.09bbb | 2.51 ± 0.49 | 143 (142 to 144)b | 4.9 (4.8 to 4.9)b | 3199 ± 288 |
| CY - 10–13 weeks (n = 9) | 0.60 ± 0.06*** | 2.71 ± 0.57 | 137 (136 to 139)**, b | 4.8 (4.7 to 5.0)b | 2734 ± 195*** |
| NC - 24 weeks (n = 6) | 0.38 ± 0.22 | 3.45 ± 0.47 | 141 (139 to 142) | 4.6 (4.3 to 4.7) | 3127 ± 303 |
| CY - 24 weeks (n = 7) | 0.54 ± 0.25 | 4.31 ± 1.65 | 140 (139 to 142) | 4.6 (4.2 to 4.9) | 2518 ± 582* |
| WT - 10–13 weeks (n = 8) | 0.81 ± 0.06 | — | 143 (142 to 144) | 4.8 (4.7 to 4.9) | 3067 ± 176 |
| HT - 10–13 weeks (n = 8) | 0.73 ± 0.07# | — | 142 (140 to 144) | 4.8 (4.6 to 4.9) | 3158 ± 123 |
FENa, TTKG and UosmMax were compared using the nonpaired t test.
SNa and SK were compared using the Mann-Whitney test.
Group-time analyses was performed using ANOVA or Kruskal Wallis.
p < 0.05 versus NC.
p < 0.01 versus NC.
p < 0.001 versus NC.
p < 0.05 versus WT.
p < 0.05 versus 10–13 weeks.
p < 0.05 versus 24 weeks.
p < 0.001 versus 24 weeks.
Interestingly, HT mice exhibited a lower FENa compared to WT animals at 10–13 wk of age, although the difference was less striking than that observed between CY and NC mice (Figure 4A, Table 3). There was no significant difference in SNa or SK between the HT and WT groups at this age (Table 3).
Plasma renin, plasma vasopressin and serum aldosterone
Since the renin-angiotensin-aldosterone system has been implicated in ADPKD-associated hypertension, we evaluated this system in our animal models. We did not detect a significant difference in plasma renin (evaluated at 13 wk), plasma vasopressin (14 wk) and serum aldosterone (15 wk) between CY and NC mice (Figure 5A, Table 4). Despite considerable variability, there was a trend toward higher plasma vasopressin in CY animals [515.0 pg/mL (187.6 to 1265.0) versus 71.3 pg/mL (54.3 to 472.8); p=0.13, Figure 5A, Table 4)]. The plasma renin, plasma vasopressin and serum aldosterone levels did not differ between HT and WT mice (Figure 5B, Table 4).
Figure 5.

Comparative analyses of plasma renin, plasma vasopressin and serum aldosterone between NC and CY (A) and between WT and HT (B) mice. Plasma renin, plasma vasopressin and serum aldosterone were analyzed using the Mann-Whitney and Kruskal-Wallis test, with data expressed as median (lower quartile to upper quartile); n/group in Table 4.
Table 4.
Plasma renin, plasma vasopressin and serum aldosterone in NC, CY, WT and HT male mice.
| Parameters | Plasma renin [pg/mL; Median (Lower Quartile to Upper Quartile)] | Plasma vasopressin [pg/mL; Median (Lower Quartile to Upper Quartile)] | Serum aldosterone [ng/dL; Median (Lower Quartile to Upper Quartile)] |
|---|---|---|---|
| NC (n = 8) | 2.45 (1.42 to 4.19) | 71.3 (54.3 to 472.8) | 26.9 (15.7 to 37.7) |
| CY (n = 9) | 2.71 (0.79 to 2.98) | 515.0 (187.6 to 1265.0) | 30.4 (26.5 to 53.0) |
| WT (n = 8) | 2.31 (1.15 to 12.63) | 57.7 (52.8 to 109.6) | 26.3 (18.4 to 33.7) |
| HT (n = 8) | 2.46 (2.18 to 3.29) | 54.6 (48.0 to 72.5) | 27.6 (22.0 to 32.6) |
Plasma renin, plasma vasopressin and serum aldosterone were compared using the Mann-Whitney test.
Angiotensinogen, renin and angiotensin-converting enzyme gene expression in cystic and non-cystic kidneys
In addition to its primary synthesis and secretion by hepatic cells, angiotensinogen can be also produced locally in the kidneys and constitutes a component of the intrarenal RAS.23 The angiotensinogen mRNA expression level was elevated in CY mouse kidneys at 18 wk of age (1.76±0.65 arbitrary units; AU) compared to NC organs (1.06±0.39 AU; p<0.05, Figure 6A); no significant difference, however, was observed at 5 and 24 wk (Figure 6A). There was no difference in renin and angiotensin-converting enzyme (ACE) gene expression levels between CY and NC kidneys at 18 wk (Figures 6B and 6C).
Figure 6.

(A) Comparative analyses of angiotensinogen mRNA expression between NC and CY mouse kidneys at the ages of 5 wk (n = 6 and n = 5, respectively), 18 wk (n = 7 and n = 8) and 24 wk (n = 5 and n = 6). * p<0.05 versus NC; aa p<0.01 versus 18 wk; b p<0.05 versus 24 wk; bbb p<0.001 versus 24 wk. (B) Comparative analyses of renin and (C) angiotensin-converting enzyme mRNA expression between NC (n = 7) and CY (n = 8) kidneys at 18 wk. Angiotensinogen levels were compared using the nonpaired t test and ANOVA, with the data presented as mean±SD. Renin and ACE mRNA levels were compared using the nonpaired t test, with the data presented as mean±SD. (D) ACE staining in NC (a) and CY mouse kidneys (b, c, d). NC kidney: no signal is detected in tubules, glomeruli and blood vessels. CY kidney: positive signal detected in cystic epithelium and tubules, and absence of staining in blood vessel. Original magnification, ×400. (E) AT1R staining in NC (a, b) and CY mouse kidneys (c, d). NC kidney: no signal is detected in tubules and glomeruli, and positive staining is seen in vascular structures. CY kidney: positive signal detected in cystic epithelium and vascular structures. Original magnification, ×400.
ACE and AT1 receptor immunohistochemical expression in cystic and non-cystic kidneys
Next we studied expression of ACE and AT1 receptor in 15-wk-old mice. At this time point, CY kidneys showed nonuniform immunohistochemical ACE staining in renal cyst epithelia, as well as occasional positive signal in tubular segments (Figure 6D). We did not observe ACE staining in the vasculature of CY kidneys. In contrast, NC mouse kidneys showed no ACE staining in either tissue compartment.
Blood vessels from both CY and NC mouse kidneys exhibited positive AT1 receptor (AT1R) staining (Figure 6E). In addition, cystic epithelia in the CY kidneys stained for AT1R at 15 wk. There was no AT1R signal in glomeruli or in normal-appearing tubules in both CY and NC kidneys (Figure 6E).
Urinary excretion of nitric oxide metabolites
The urinary excretion of nitrite (UENO2) was lower in CY than NC mice at 10–13 wk [8.5 μmol/g of creatinine (3.2 to 10.1) versus 13.9 μmol/g of creatinine (9.9 to 18.1); p<0.01) and at 24 wk of age [4.3 μmol/g of creatinine (3.9 to 5.2) versus 5.5 μmol/g of creatinine (4.8 to 7.8); p<0.05) but no significant difference was detected at 5 wk (Figure 7A, Table 5). In contrast, the urinary excretion of nitrate (UENO3) did not differ between the two sets of animals at the three evaluated ages (Table 5, Supplemental Figure S2A). There was no difference in urinary excretion of these nitric oxide metabolites between HT and WT groups at 10–13 wk of age (Table 5, Figure 1B and Supplemental Figure S2B).
Figure 7.

(A) Comparative analyses of urinary excretion of nitrite between NC and CY mice at the ages of 5, 10–13 and 24 wk, and (B) between WT and HT mice at 10–13 wk. * p<0.05 versus NC; ** p<0.01 versus NC; b p<0.05 versus 24 wk. UENO2 was compared using the Mann-Whitney test for NC and CY comparisons or nonpaired t test for WT and HT comparisons, with the data expressed as median (lower quartile to upper quartile) and mean±SD, respectively; n/group in Table 5.
Table 5.
Urinary excretion of NO2 (UENO2) and Urinary excretion of NO3 (UENO3) in NC, CY, WT and HT male mice.
| Parameters | UENO2 [μmol/g creat; Median (Lower Quartile to Upper Quartile)] | UENO3 (mmol/g creat, Mean ± SD) |
|---|---|---|
| NC - 5 weeks (n = 6) | 7.4 (6.1 to 12.9) | 13.1 ± 3.4bb |
| CY - 5 weeks (n = 6) | 9.0 (5.0 to 16.9) | 12.4 ± 6.3 |
| NC - 10–13 weeks (n = 8) | 13.9 (9.9 to 18.1)b | 12.3 ± 3.1bb |
| CY - 10–13 weeks (n = 9) | 8.5 (3.2 to 10.1)** | 8.2 ± 5.6 |
| NC - 24 weeks (n = 6) | 5.5 (4.8 to 7.8) | 4.6 ± 4.8 |
| CY - 24 weeks (n = 7) | 4.3 (3.9 to 5.2)* | 5.7 ± 3.2 |
| UENO2 (μmol/g creat, Mean ± SD) | UENO3 (mmol/g creat, Mean ± SD) | |
| WT - 10–13 weeks (n = 8) | 14.0 ± 13.0 | 5.7 ± 3.8 |
| HT - 10–13 weeks (n = 8) | 16.0 ± 11.0 | 7.9 ± 3.8 |
UENO2 was compared using the Mann-Whitney or nonpaired t test
UENO3 was compared using the nonpaired t test.
Group-time analyses was performed using ANOVA or Kruskal Wallis.
p < 0.05 versus NC.
p < 0.01 versus NC.
p < 0.05 versus 24 weeks.
p < 0.01 versus 24 weeks.
Renal concentrating capacity analysis
We found that the renal concentrating capacity, assessed as the maximum urine osmolality (UosmMax), was significantly lower in CY mice compared with NC animals at 10–13 wk (2734±195 versus 3199±288 mOsm/kg H2O; p<0.001) and at 24 wk of age (2518±582 versus 3127±303 mOsm/kg H2O; p<0.05). Although a similar trend was already detected at 5 wk, the difference between the two groups was not significant (Figure 8A, Table 3). UosmMax did not differ, however, between HT and WT mice at the age of 10–13 wk (Figure 8B, Table 3).
Figure 8.

(A) Comparative analyses of maximum urine osmolality between NC and CY mice at 5, 10–13 and 24 wk of age, and (B) between WT and HT mice at 10–13 wk. *** p<0.001 versus NC; * p<0.05 versus NC. UosmMax was compared using the nonpaired t test and ANOVA, with the data presented as mean±SD; n/group in Table 3.
Cell proliferation and apoptosis in cystic and non-cystic kidneys
Polycystins modulate cell proliferation and are essential to maintain the differentiated phenotype of renal tubule epithelial cells.24,25 A fall in polycystin-1 or -2 below critical levels may also lead to apoptosis, loss of planar cell polarity and extracellular matrix remodeling.24,26,27 We evaluated cell proliferation and apoptosis in our mouse models.
The Ki-67 immunohistochemical analyses revealed an elevated tubular cell proliferation rate in CY [19.4/10,000 cells (10.2 to 36.9)] compared with NC kidneys [5.2/10,000 cells (0.7 to 10.4); p<0.05, Figure 9A and B] at the age of 15 wk. Surprisingly, the cell proliferation rates did not significantly differ between cyst lining cells [18/10,000 cells (0 to 25), n = 8 animals] and tubular cells in CY kidneys. As expected, there were equally low rates of tubular cell proliferation in both HT and WT kidneys (Figure 9A and B).
Figure 9.
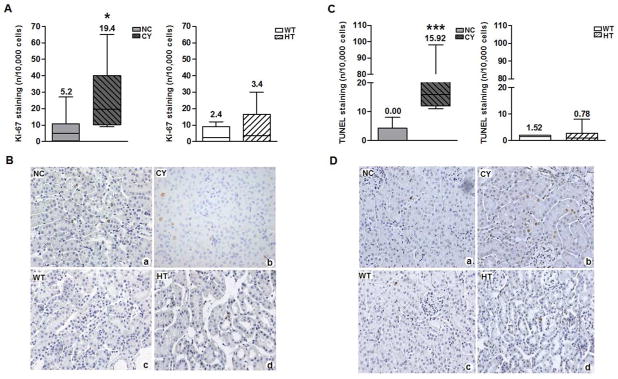
(A) Comparative analyses of tubular cell Ki-67 staining between NC (n = 8; 6025±634 counted cells/animal) and CY (n = 8; 5728±630 counted cells/animal) mouse kidneys and between WT (n = 8; 5860±789 counted cells/animal) and HT (n = 8; 5497±634 counted cells/animal) organs at 15 wk of age; (B) Ki-67 staining representative images. Original magnification, ×400. * p<0.05 versus NC. Tubular Ki-67 staining was compared using the Mann-Whitney test, with the data expressed as median (lower quartile to upper quartile). (C) Comparative analyses of tubular cell TUNEL staining between NC (n = 8; 6122±785 counted cells/animal) and CY (n = 8; 5902±602 counted cells/animal) mouse kidneys and between WT (n = 8; 6218±782 counted cells/animal) and HT (n = 8; 5668±780 counted cells/animal) organs at the age of 15 wk; (D) TUNEL staining representative images. Original magnification, ×400. *** p<0.001 versus NC. Tubular TUNEL staining was compared using the Mann-Whitney test, with the data expressed as median (lower quartile to upper quartile).
We used terminal deoxynucleotidyl transferase-mediated digoxigenin-deoxyuridine nick-end labeling (TUNEL) to study apoptosis in kidneys from 15-wk animals. CY kidney tubules had a significantly higher apoptotic rate [15.9/10,000 cells (12.0 to 24.4)] compared with NC renal tubules [0.0/10,000 cells (0.0 to 3.4), p<0.001; Figure 9C and D]. A trend of higher rate of apoptosis was observed in cyst lining cells [82.2/10,000 cells (3.4 to 102.3), n = 8 animals)] compared with tubular cells in CY kidneys, however the difference between them did not reach significance. There was no difference in tubular apoptotic rates between HT and WT kidneys (Figure 9C and D).
DISCUSSION
The complex mechanisms that underlie pathogenesis of hypertension in ADPKD are incompletely understood.11 It is still debated whether ADPKD-related SAH is primarily caused by vascular/endothelial abnormalities or depends on renal cyst formation and expansion. We addressed this question using two orthologous ADPKD mouse models, with distinct profiles of Pkd1 gene deficiency. HT mice do not develop renal cysts at the analyzed age, representing a pure Pkd1-haploinsufficiency model22 while CY mice are cystic but have preserved glomerular filtration rate (GFR) at the evaluated ages. This latter model reproduces the phenotype observed in humans with early-stage ADPKD. CY mice were hypertensive already at the age of 5 wk and remained so at 10–13 wk, while HT animals had no elevation in MAP when compared to controls.
Cystic mice developed larger kidneys per BW with higher levels of cAMP than noncystic animals, confirming the altered features found in ADPKD. The renal cystic index analysis supports, in addition, the progressive nature of renal disease. We detected no difference in cystatin C levels between CY and NC mice at 13 wk of age. This finding suggests preserved renal function in CY while indicates, in association with no SCr difference between the CY and NC groups at 5 wk, that the slightly lower SCr observed in 10–13-wk CY animals might be secondary to a mildly decreased muscle mass. This hypothesis, however, needs to be investigated. Taken together, the CY and HT results are consistent with the idea that the pathogenesis of ADPKD-associated hypertension is primarily due to cyst formation and enlargement rather than a primary vascular defect or the result of haploinsufficiency. Moreover, at least in our cystic mouse model, renal dysfunction per se does not play a role in the genesis of SAH. Interestingly, a trend for higher MAP was observed in CY compared with NC mice at 24 wk, but not significant, suggesting that a heart damage secondary to SAH is already installed in CY at this age. The finding of heavier hearts per BW in CY animals, in fact, supports this hypothesis.
Five and 10–13-wk CY mice exhibited a lower FENa, accompanied, at 10–13 wk, by a slight increase in SUN compared with their NC controls. These results are consistent with renal vascular compression induced by cystic expansion, sectorial reduced perfusion, RAS activation, and increased urea/Na+ tubular reabsorption. The vascular compression that we observed in CY kidneys is highly supportive of this mechanism. Our plasma renin assay, however, did not reveal higher levels in CY animals, an observation that requires future studies. Notably the FENa difference is no longer detected between CY and NC mice at 24 wk, suggesting that at later stages of the disease the absence of Na+ retention difference may contribute to the loss of significance in MAP difference between the two groups.
Our qPCR assays demonstrated increased angiotensinogen mRNA levels in CY versus NC kidneys at 18 wk of age. These findings support the intrarenal RAS involvement in the pathogenesis of SAH in ADPKD, corroborating previous studies that propose that the intrarenal system plays a significant role in its genesis.1,28 We were unable, however, to detect differences in renin and ACE mRNA expression between CY and NC kidneys. ACE staining was detected in cystic epithelia and some tubular segments of CY kidneys, but was absent in vascular structures. NC kidneys did not show tubular or vascular signal. Positive AT1R staining was also observed in CY renal cystic epithelia, as well as in blood vessels, whereas AT1R signal was restricted to vascular structures in NC kidneys. These findings are similar to previous data that reveal ACE and AT1R expression in cystic epithelia and dilated tubules of human ADPKD kidneys.29 These authors also found angiotensin II staining in cystic epithelia and proximal tubules. Additional studies report increased renin concentration in juxtaglomerular apparatus, arterioles and connective tissue cells surrounding cysts in ADPKD kidneys30, as well as in fibrous tissue distant from vessels.31 Our findings, therefore, provide additional support for the role of intrarenal RAS in the genesis and maintenance of ADPKD-related hypertension.
The importance of circulating RAS in ADPKD-related SAH remains controversial.1,28,32,33 Renin plasma concentration was not different between CY and NC nor between HT and WT mice. Similar results were observed for serum aldosterone. This is consistent with a prior study that showed no difference in plasma renin activity between hypertensive and normotensive ADPKD patients.34
CY animals displayed lower UENO2 than NC mice but no difference in UENO3. Our findings suggest that the decreased NO production associated with human ADPKD35,36 is related to cystic disease and could contribute to SAH. The absence of decreased UENO2 in the hypertensive CY at 5 wk, however, suggests that decrease in NO generation is not essential for the development of SAH. We did not find significant differences in UENO2 and UENO3 between the HT and WT groups which differ from findings reported by other investigators.37 This difference may have occurred due to different ages or genetic backgrounds. At this point, however, we cannot exclude a contributory effect of abnormal vascular reactivity to catecholamines and/or acetylcholine to hypertension in CY mice, determined by the mosaic distribution of Pkd1 deficiency and, potentially, by its effects on polycystin-2 in endothelial and vascular smooth muscle cells.
A hypothesis for ADPKD-associated renal concentrating deficit invokes a primary abnormality in the principal cells. The fact that polyuria develops only after renal cyst detection, however, makes it unlikely.15 The early presentation of concentrating deficits, in turn, argues against a primary role for tubulointerstitial alterations or cyst-dependent disruption of the corticomedullary architecture in determining this phenotype.38 Our data did not reveal a difference in UosmMax between HT and WT mice, suggesting that PKD1 haploinsufficiency does not result in this defect. In contrast, CY mice showed a UosmMax decrease compared to NC at 10–13 and 24 wk, a finding not yet significant at 5 wk. These data support that cyst development and time-dependent structural disruption of renal architecture play a central role in the genesis of concentrating impairment. Moreover, they are consistent with a significant concentration deficit detected in children only with more than 10 cysts39 and a study that shows that this defect correlates with the number of cysts.38
High vasopressin levels could contribute to SAH via effects on V1a or V2 receptors. The former mediates a direct effect on vascular smooth muscle thereby decreasing medullary renal blood flow40 while activation of the latter increases expression of the ENaC β and γ subunits.41 We found a trend of higher vasopressin in CY, though not significant. This lack of significance may be due to the variability in cystic phenotype observed in our model, likely based on the mosaic and nonsynchronous expression of nestin-Cre42, or in the employed assay. The lower SNa in 10–13-wk CY, in addition, suggests an effect of increased vasopressin in these mice.
The higher tubular cell proliferation and apoptotic rates observed in CY kidneys included areas near and distant from cysts. This cell proliferation pattern in tubules differs from a previous report, in which Pkd1 was inactivated at 5 wk and increased cell proliferation was not detected in areas distant from cysts.43 The distinct profiles are likely explained by the fact that in our model Pkd1 inactivation is not synchronous.
Our data, therefore, support the cyst expansion/local hypoperfusion model for hypertension in an orthologous mouse model of ADPKD and implicates intrarenal RAS activation as a critical player in its pathogenesis. Our results, in addition, lead to the conclusion that cyst expansion is determinant for the development of renal concentrating deficit in this cystic mouse. Based on the orthologous nature of this model, these findings bring relevant and applicable mechanistic insights to human ADPKD.
METHODS
Mouse models
The generation of the cystic model was based on a Pkd1 floxed allele (Pkd1cond) that contains lox P sites inserted in introns 1 and 4 and a neomycin cassette flanked by FRT sites inserted in intron 1.20 In the presence of Cre recombinase, exons 2–4 are excised, producing the new allele Pkd1del2–4. In TgN(balancer2)3Cgn mice, Cre recombinase expression is controlled by the nestin promoter and enhancer sequences located in the second intron.44 The Pkd1cond line, generated on a C57BL/6 background, was crossed with a mouse line carrying this transgene, leading to inactivation of the floxed allele following a mosaic pattern. The presence of Pkd1cond alleles and the nestin-Cre transgene was determined using specific PCR reactions. Animals with the Pkd1cond/cond:Nestincre genotype (CY), in turn, develop a renal cystic phenotype that mimics human ADPKD. Pkd1cond/cond (NC) littermates were used as controls.
We have previously reported and characterized a 129Sv mouse line with a Pkd1 null allele.20,21 The construct had part of exon 2 and the entire exon 3 replaced with lacZ cloned in frame to the remainder of exon 2, followed by the neomycin resistance gene (neor). The lacZ-neor 5′ segment contains transcriptional termination sequences that preclude expression of 3′ Pkd1 exons. The mice were genotyped using a three-primer PCR strategy. Heterozygotes for the Pkd1-null mutation (Pkd1+/−, HT) present virtually no renal cysts by 15 wk of age, representing a pure Pkd1-haploinsufficiency model. In this case, wild-type littermates (Pkd1+/+, WT) were employed as controls.
The study was performed in accordance with international standards of animal care and experimentation.
Assessment of renal cyst formation and histology
The mice were anesthetized with intraperitoneal sodium pentobarbital (80 Pg/g BW) and submitted to complete exsanguination and immediate perfusion with Millonig formalin, modified by Carlson. The left kidney was then harvested and fixed in situ with 4% buffered paraformaldehyde, paraffin embedded, and sectioned at 4 μm. Kidney sections were mounted on slides and stained using hematoxylin and eosin for histological analysis. The number of cysts present in each left kidney was calculated by evaluating entire sections obtained at 70-μm intervals.
The cystic index was quantified using kidney sections spaced at 30-μm intervals. The total kidney area and the total cystic area were measured using the Image Pro Plus software (Media Cybernetics Manufacturing, Warrendale, USA) and the cystic index was presented as the percent total cystic area/total kidney area ratio.
Invasive measurement of mean arterial pressure
The mice were submitted to inhalation anesthesia with isoflurane 1.5%. A catheter 4.0 cm in length, 0.4 mm in external diameter and 0.25 mm in internal diameter (Micro-Renathane, Braintree Science Inc, Braintree, USA) was inserted into the right carotid artery to record MAP.45 Prior to implantation, the catheter was filled with saline solution containing heparin 50 IU/mL and occluded with a metal pin. It was exteriorized at the nape with a trocar and the incision sutured. The catheter was then connected to a pressure transducer linked to a data acquisition system (BIOPAC Systems, Santa Barbara, USA) for measurement in mmHg. MAP was assessed 24 h after surgery, allowing animals to recover and return to baseline conditions.
Biochemical determinations
Blood samples were drawn by retro-orbital bleeding. Four hours (h) after this procedure the animals were weighted and housed for 24 h for urine collection, without food and water. The samples were allowed to clot and centrifuged to obtain serum. SUN was determined according to Crocker’s protocol (Celm, Barueri, Brazil) whereas SCr and urine creatinine (UCr) were measured using a colorimetric assay (Labtest, Lagoa Santa, Brazil). Two CY and one NC mice with baseline SUN above 35 mg/dL were removed from 10–13-wk groups to avoid any potential interference of decreased renal function on the evaluated variables. This criterion, however, was not applied to the 24-wk groups due to the stage of the disease. FENa and TTKG were calculated following the equations FENa = (UNa × SCr) / (SNa × UCr) × 100, where UNa is urine Na+, and TTKG = (UK × Sosm) / (SK × Uosm). Cystatin C was measured in serum using an ELISA kit (Enzyme-Linked Immunosorbent Assay, Sigma, St Louis, MO, USA).
Kidneys from 13-wk-old CY and NC mice were harvested and subsequently homogenized in 0.1 M HCl. Total cAMP was measured in the correponding lysates using an enzymatic immunoassay according to the manufacturer’s protocol (Sigma, St Louis, MO, USA).
Renal concentrating capacity was analyzed by determining UosmMax. The animals were kept in the metabolic cage for additional 8 h following the previous 24-h urine collection time, totalizing 32 h, with food but no water. This period was followed by a subcutaneous injection of desmopressin (DDAVp; 1.0 μg/kg BW) and return to the metabolic cage. After 10 h the mice were weighed and urine was collected to measure Uosm using a Vapro 5500 vapor osmometer (Wescor, Logan, USA).
UENO2 and UENO3 were determined by chemiluminescence using an NO analyzer NOA280; Stevers Instruments, Boulder, USA).
Plasma renin, plasma vasopressin and serum aldosterone
Plasma renin was measured using a commercial immunoradiometric assay kit (Active Renin IRMA, Diagnostic Systems Laboratories, Webster, USA). Plasma vasopressin and serum aldosterone were determined with commercial radioimmunoassay kits (RK-065-07, Phoenix Pharmaceuticals, Phoenix, USA; and TKAL1, Coat-A-Count Aldosterone, Siemens, Los Angeles, USA, respectively).
Real-time RT-PCR
Total RNA was isolated from the right kidneys of 5, 18 and 24 wk-old CY and NC mice using the TRIzol reagent (Invitrogen, Carlsbad, USA). Quantitative real-time RT-PCR was performed using the TaqMan system (Applied Biosystems, Warrington, UK) with cDNA amplification using ImProm II reverse transcriptase (Promega, Madison, USA) and specific assays for renin (Mm02342889_g1), angiotensinogen (Mm00599662_m1), ACE (Mm00802048_m1) and GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) (Mm99999915_g1) as control. Results were obtained with the ΔΔCt methodology and are expressed as arbitrary units (AU).
Immunohistochemistry
Incubation with the primary antibodies was carried out overnight at 4°C in serial sections. A monoclonal IgG1 antibody to ACE (clone 2E2, MAB3502, Chemicon International Inc., Temecula, USA), a polyclonal IgG anti-AT1R antibody (Angio2RNAABR, RDI Division of Fitzgerald Ind., Acton, USA), and a monoclonal IgG2a antibody to Ki-67 (clone TEC-3, M7249, Dako, Carpinteria, USA) were used. Control sections for ACE, AT1R and Ki-67 analyses were incubated with PBS instead of the respective primary antibodies, showing negative or negligible staining in proximal tubules. Ki-67 analyses included the evaluation of eight fields representing renal cortex and two located in the renal medulla for each mouse. The quantification of Ki-67-positive nuclei in renal tubules was performed under light microscopy at ×400 magnification and yielding results expressed as number of positive cells/10,000 cells ratios. ACE and AT1R analyses, on the other hand, comprised the description of the staining pattern, including presence/absence and expression profile in the different kidney structures.
Analyses of apoptosis were performed using the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany). Positive controls were performed in spleen tissue sections, while negative controls were incubated without TdT. Ten different fields were evaluated for each section. The quantification of TUNEL-positive nuclei in renal tubules followed the same protocol applied for Ki-67. Additional analyses compared Ki-67 and TUNEL quantification between cyst lining cells and tubular cells in CY kidneys.
Statistical analyses
The normality test of Komolgorov-Smirnov was applied for all continuous variables. When parametric, the data were analyzed by One-Way ANOVA with Tukey post-test, or nonpaired t test with Welch correction for two groups, and the results were presented as mean and standard deviation. When nonparametric, were used the Kruskal-Wallis test with Müller-Dunn post-test, or Mann-Whitney test for two groups, and the data were presented as median (lower to upper quartiles). We have accepted an α error ≤5% to reject the null hypothesis. The tests were applied using Sigma Plot 9.0 (Jandel ® Corporation) and GraphPad 5.0 (Prism ® Software).
Supplementary Material
Figure S1. Comparative analysis of TTKG between NC and CY mice at the ages of 5, 10–13 and 24 wk. Comparison performed using the nonpaired t test, with data presented as mean±SD; n/group in Table 3.
Figure S2. (A) Comparative analyses of urinary excretion of nitrate between NC and CY mice at the ages of 5, 10–13 and 24 wk, and (B) between WT and HT mice at 10–13 wk. UENO3 was compared using the nonpaired t test and ANOVA, with the data presented as mean±SD; n/group in Table 5.
Acknowledgments
We thank Gregory Germino, M.D., for his suggestions, Isac de Castro, Ph.D., for his valuable help in the statistical analyses, Elia Caldini, Ph.D., for technical suggestions, and Dulce Casarini, Ph.D., for providing essential reagents. This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (grants 2010/17424-0 to LFO and 2009/10748-7 to JMF and LFO), The National Institutes of Health (grant R01DK076017 to TJW) and Laboratórios de Investigação Médica da Faculdade de Medicina da Universidade de São Paulo. These studies utilized resources provided by the NIDDK sponsored Johns Hopkins Polycystic Kidney Disease Research and Clinical Core Center (grant P30DK090868).
Footnotes
DISCLOSURE
None.
References
- 1.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reed BY, McFann K, Bekheirnia MR, et al. Variation in age at ESRD in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2008;51:173–183. doi: 10.1053/j.ajkd.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 4.Hateboer N, van Dijk MA, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. Lancet. 1999;353:103–107. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 5.Dicks E, Ravani P, Langman D, et al. Incident renal events and risk factors in autosomal dominant polycystic kidney disease: a population and family-based cohort followed for 22 years. Clin J Am Soc Nephrol. 2006;1:710–717. doi: 10.2215/CJN.01581105. [DOI] [PubMed] [Google Scholar]
- 6.Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:173–180. doi: 10.1053/j.ackd.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2009;5:221–228. doi: 10.1038/nrneph.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelleher CL, McFann KK, Johnson AM, et al. Characteristics of hypertension in young adults with autosomal dominant polycystic kidney disease compared with the general U.S. population. Am J Hypertens. 2004;17:1029–1034. doi: 10.1016/j.amjhyper.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 9.Oflaz H, Alisir S, Buyukaydin B, et al. Biventricular diastolic dysfunction in patients with autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:2244–2249. doi: 10.1111/j.1523-1755.2005.00682.x. [DOI] [PubMed] [Google Scholar]
- 10.Johnson AM, Gabow PA. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J Am Soc Nephrol. 1997;8:1560–1567. doi: 10.1681/ASN.V8101560. [DOI] [PubMed] [Google Scholar]
- 11.Chapman AB, Stepniakowski K, Rahbari-Oskoui F. Hypertension in Autossonal Dominant Polycystic Kidney Disease. Adv Chronic Kidney Dis. 2010;17:153–163. doi: 10.1053/j.ackd.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D, Iversen J, Wilcox CS, et al. Endothelial dysfunction and reduced nitric oxide in resistance arteries in autosomal-dominant polycystic kidney disease. Kidney Int. 2003;64:1381–1388. doi: 10.1046/j.1523-1755.2003.00236.x. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Maldonado M, Yium JJ, Eknoyan G, et al. Adult polycystic kidney disease: studies of the defect in urine concentration. Kidney Int. 1972;2:107–113. doi: 10.1038/ki.1972.78. [DOI] [PubMed] [Google Scholar]
- 14.Fick GM, Duley IT, Johnson AM, et al. The spectrum of autosomal dominant polycystic kidney disease in children. J Am Soc Nephrol. 1994;4:1654–1660. doi: 10.1681/ASN.V491654. [DOI] [PubMed] [Google Scholar]
- 15.Torres VE. Vasopressin antagonists in polycystic kidney disease. Kidney Int. 2005;68:2405–2418. doi: 10.1111/j.1523-1755.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- 16.Danielsen H, Nielsen AH, Pedersen EB, et al. Exaggerated natriuresis in adult polycystic kidney disease. Acta Med Scand. 1986;219:59–66. doi: 10.1111/j.0954-6820.1986.tb03276.x. [DOI] [PubMed] [Google Scholar]
- 17.Michalski A, Grzeszczak W. The effect of hypervolemia on electrolyte level and and level of volume regulating hormones in patients with autosomal dominant polycystic kidney disease. Pol Arch Med Wewn. 1996;96:329–343. [PubMed] [Google Scholar]
- 18.Gattone VH, 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9:1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 19.Torres VE, Wang X, Qian Q, et al. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med. 2004;10:363–364. doi: 10.1038/nm1004. [DOI] [PubMed] [Google Scholar]
- 20.Piontek KB, Huso DL, Grinberg A, et al. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J Am Soc Nephrol. 2004;15:3035–3043. doi: 10.1097/01.ASN.0000144204.01352.86. [DOI] [PubMed] [Google Scholar]
- 21.Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21:489–97. doi: 10.1681/ASN.2009040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bastos AP, Piontek K, Silva AM, et al. Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J Am Soc Nephrol. 2009;20:2389–2402. doi: 10.1681/ASN.2008040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobori H, Nangaku M, Navar LG, et al. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 24.Boletta A, Qian F, Onuchic LF, et al. Polycystin-1, the gene product of PKD1, induces resistance to apoptosis and spontaneous tubulogenesis in MDCK cells. Mol Cell. 2000;6:1267–1273. doi: 10.1016/s1097-2765(00)00123-4. [DOI] [PubMed] [Google Scholar]
- 25.Nickel C, Benzing T, Sellin L, et al. The polycystin-1 C-terminal fragment triggers branching morphogenesis and migration of tubular kidney epithelial cells. J Clin Invest. 2002;109:481–489. doi: 10.1172/JCI12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luyten A, Su X, Gondela S, et al. Aberrant regulation of planar cell polarity in polycystic kidney disease. J Am Soc Nephrol. 2010;21:1521–1532. doi: 10.1681/ASN.2010010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres VE, Harris PC. Polycystic kidney disease in 2011: Connecting the dots toward a polycystic kidney disease therapy. Nat Rev Nephrol. 2011;8:66–68. doi: 10.1038/nrneph.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 29.Loghman-Adham M, Soto CE, Inagami T, et al. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol. 2004;287:775–788. doi: 10.1152/ajprenal.00370.2003. [DOI] [PubMed] [Google Scholar]
- 30.Torres VE, Donovan KA, Sicli G, et al. Synthesis of renin by tubulocystic epithelium in autossomal dominant polycystic kidney disease. Kidney Int. 1992;42:364–373. doi: 10.1038/ki.1992.297. [DOI] [PubMed] [Google Scholar]
- 31.Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int. 1988;33:1084–1090. doi: 10.1038/ki.1988.115. [DOI] [PubMed] [Google Scholar]
- 32.Chapman AB, Johnson A, Gabow PA, et al. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med. 1990;323:1091–1096. doi: 10.1056/NEJM199010183231602. [DOI] [PubMed] [Google Scholar]
- 33.Doulton TW, Saggar-Malik AK, He FJ, et al. The effect of sodium and angiotensin-converting enzyme inhibition on the classic circulating renin-angiotensin system in autosomal-dominant polycystic kidney disease patients. J Hypertens. 2006;24:939–945. doi: 10.1097/01.hjh.0000222765.30348.0d. [DOI] [PubMed] [Google Scholar]
- 34.Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol. 1985;5:176–181. doi: 10.1159/000166929. [DOI] [PubMed] [Google Scholar]
- 35.Wang D, Iversen J, Strandgaard S. Endothelium-dependent relaxation of small resistance vessels is impaired in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2000;11:1371–1376. doi: 10.1681/ASN.V1181371. [DOI] [PubMed] [Google Scholar]
- 36.Kocaman O, Oflaz H, Yekeler E, et al. Endothelial dysfunction and increased carotid intima-media thickness in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2004;43:854–860. doi: 10.1053/j.ajkd.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Muto S, Aiba A, Saito Y, et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. 2002;11:1731–1742. doi: 10.1093/hmg/11.15.1731. [DOI] [PubMed] [Google Scholar]
- 38.Seeman T, Dusek J, Vondrák K, et al. Renal concentrating capacity is linked to blood pressure in children with autosomal dominant polycystic kidney disease. Physiol Res. 2004;53:629–634. [PubMed] [Google Scholar]
- 39.Gabow PA, Kaehny WD, Johnson AM, et al. The clinical utility of renal concentrating capacity in polycystic kidney disease. Kidney Int. 1989;35:675–680. doi: 10.1038/ki.1989.38. [DOI] [PubMed] [Google Scholar]
- 40.Cowley AW, Jr, Skelton MM, Kurth TM. Effects of long-term vasopressin receptor stimulation on medullary blood flow and arterial pressure. Am J Physiol. 1998;275(5 Pt 2):R1420–4. doi: 10.1152/ajpregu.1998.275.5.R1420. [DOI] [PubMed] [Google Scholar]
- 41.Nicco C, Wittner M, DiStefano A, et al. Chronic exposure to vasopressin upregulates ENaC and sodium transport in the rat renal collecting duct and lung. Hypertension. 2001;38:1143–1149. doi: 10.1161/hy1001.092641. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Boyle S, Zhao M, et al. Differential expression of the intermediate filament protein nestin during renal development and its localization in adult podocytes. J Am Soc Nephrol. 2006;17:1283–1291. doi: 10.1681/ASN.2005101032. [DOI] [PubMed] [Google Scholar]
- 43.Takakura A, Contrino L, Beck AW, et al. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19:2351–2363. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Betz UA, Vosshenrich CA, Rajewsky K, et al. Bypass of lethality with mosaic mice generated by Cre-loxP-mediated recombination. Curr Biol. 1996;6:1307–1316. doi: 10.1016/s0960-9822(02)70717-3. [DOI] [PubMed] [Google Scholar]
- 45.Ceroni A, Moreira ED, Mostarda CT, et al. Ace gene dosage influences the development of renovascular hypertension. Clin Exp Pharmacol Physiol. 2010;37:490–495. doi: 10.1111/j.1440-1681.2009.05330.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparative analysis of TTKG between NC and CY mice at the ages of 5, 10–13 and 24 wk. Comparison performed using the nonpaired t test, with data presented as mean±SD; n/group in Table 3.
Figure S2. (A) Comparative analyses of urinary excretion of nitrate between NC and CY mice at the ages of 5, 10–13 and 24 wk, and (B) between WT and HT mice at 10–13 wk. UENO3 was compared using the nonpaired t test and ANOVA, with the data presented as mean±SD; n/group in Table 5.