

Abstract
Anchoring pharmacologic agents to the vascular lumen has the potential to modulate critical processes at the blood–tissue interface, avoiding many of the off-target effects of systemically circulating agents. We report a novel strategy for endothelial dual targeting of therapeutics, which both enhances drug delivery and enables targeted agents to partner enzymatically to generate enhanced biologic effect. Based on the recent discovery that paired antibodies directed to adjacent epitopes of platelet endothelial cell adhesion molecule (PECAM)-1 stimulate each other’s binding, we fused single-chain fragments (scFv) of paired anti-mouse PECAM-1 antibodies to recombinant murine thrombomodulin (TM) and endothelial protein C receptor (EPCR), endothelial membrane proteins that partner in activation of protein C (PC). scFv/TM and scFv/EPCR bound to mouse endothelial PECAM-1 with high affinity (EC50 1.5 and 3.8 nM, respectively), and codelivery induced a 5-fold increase in PC activation not seen when TM and EPCR are anchored to distinct cell adhesion molecules. In a mouse model of acute lung injury, dual targeting reduces both the expression of lung inflammatory markers and trans-endothelial protein leak by as much as 40%, as compared to either agent alone. These findings provide proof of principle for endothelial dual targeting, an approach with numerous potential biomedical applications.—Greineder, C. F., Brenza, J. B., Carnemolla, R., Zaitsev, S., Hood, E. D., Pan, D. C., Ding, B.-S., Esmon, C. T., Chacko, A. M., Muzykantov, V. R. Dual targeting of therapeutics to endothelial cells: collaborative enhancement of delivery and effect.
Keywords: drug delivery, PECAM-1, thrombomodulin, EPCR
Targeted delivery of therapeutics to endothelial cells (ECs) is a powerful paradigm with the capacity to ameliorate a wide variety of human illnesses (1, 2). The vascular endothelium is a critical organ in cardiovascular and systemic inflammatory diseases, such as sepsis, stroke, and acute lung injury (ALI) (3, 4) and is an increasingly important target for antineoplastic therapies (5). The ability to anchor pharmacologic agents to the vascular lining has long been sought as a means to generate localized biologic effect [e.g., the restoration of blood flow at a site of vascular occlusion, or the cessation of blood flow to a growing tumor (6, 7)]. Potentially more advantageous would be the codelivery of paired therapeutics that could interact and generate a local enzymatic reaction, amplifying the biologic effect at the site of disease. To realize this goal, paired cargoes must be delivered with sufficient proximity to allow their enzymatic partnering, without compromising drug delivery. We recently described a phenomenon with precisely these characteristics, in which antibodies to adjacent, distinct epitopes of platelet endothelial cell adhesion molecule (PECAM)-1 CD31 actually increased each other’s binding, both in vitro and in vivo (8). We hypothesized that this collaborative enhancement effect would provide a platform for dual targeting of therapeutics to ECs.
In this study, we demonstrated the feasibility and utility of endothelial codelivery, using as a case study the enhancement of the protein C (PC) pathway by paired endothelium-targeted biotherapeutics. The PC pathway is an endogenous endothelial system that has long been a target for pharmacologic intervention, in part because of its role in regulating vascular permeability, the innate immune response, and the coagulation cascade (9). At the heart of this pathway is thrombomodulin (TM), a transmembrane endothelial glycoprotein that binds and alters the enzymatic specificity of thrombin (10). Whereas thrombin increases endothelial permeability and induces proinflammatory endothelial activation, it does not exert these effects when bound to TM. Instead, the thrombin/TM complex cleaves the zymogen (PC) to generate activated protein C (APC). APC inhibits coagulation, stabilizes the endothelial barrier, and mitigates activation and apoptosis of ECs (11). In addition to TM, a key partner molecule, the endothelial protein C receptor (EPCR), has a critical role in the protective functions of this system. APC generation by the thrombin/TM complex is accelerated when PC is bound to EPCR (12). In addition, EPCR appears to be required for many of the anti-inflammatory and endothelial protective effects of the PC pathway (13, 14). The TM-EPCR-APC system becomes dysfunctional in the presence of EC injury and inflammatory activation. TM and EPCR are cleaved or shed from the EC surface, and their expression is suppressed by a variety of inflammatory mediators (15, 16). This phenomenon, demonstrated in a variety of animal models and patients with severe forms of sepsis, contributes to disease pathogenesis and end-organ injury (17–19).
To reverse this pathogenic process and test the viability of our dual-targeting approach, we designed biotherapeutic fusion proteins, consisting of either recombinant TM or EPCR, linked to single-chain variable antibody fragments (scFvs) directed at paired PECAM-1 epitopes. Cotreatment of ECs with scFv/TM and scFv/EPCR caused a marked increase in the activation of PC, with evidence of both collaborative enhancement of binding and enzymatic partnering of the paired cargoes. We extended our in vitro observations to studies in mice, in which cotreatment with scFv/EPCR specifically enhanced the pulmonary targeting of scFv/TM and increased the generation of APC in vivo. When tested in a mouse model of ALI, the fusion proteins worked synergistically in the event that both were delivered to PECAM-1, but not when they were anchored to distinct surface determinants. The results provide both proof of principle for the general strategy of dual targeting of therapeutics to ECs and specific data to support this novel and translational therapy for ALI and other diseases involving inflammatory vascular injury.
MATERIALS AND METHODS
Cell lines
Lysate from the Mec13.3 hybridoma (herein referred to as Mec13) was a generous gift of Dr. Annunciata Vecchi (Mario Negri Institute of Pharmacology Research, Milan, Italy). MS1 cells were purchased from American Type Culture Collection (Manassas, VA, USA) and maintained in DMEM with 10% fetal bovine serum and 1% antibiotic-antimycotic (Life Technologies, Grand Island, NY, USA).
Antibodies and other reagents
The anti-TM polyclonal antibody AF3894 was purchased from R&D Systems (Minneapolis, MN, USA). A second anti-TM polyclonal antibody, sc-7097, used in Western blot analyses, was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-EPCR blocking mAb 1560, was supplied by the Esmon laboratory. A horseradish peroxidase (HRP)-conjugated anti-FLAG (M2-HRP) antibody was obtained from Sigma-Aldrich (St. Louis, MO, USA). The HRP-conjugated anti-goat secondary antibody (sc-2056) was from Santa Cruz Biotechnology. Purified recombinant human PC was a generous gift of Dr. Sriram Krishnaswamy (Joseph Stokes Research Institute, Children’s Hospital of Philadelphia, PA, USA). LPS (serotype B4) was purchased from Sigma-Aldrich. The bovine thrombin used in cell experiments was also purchased from Sigma-Aldrich, whereas the thrombin used in in vivo APC generation experiments was from GE Healthcare Life Sciences (Piscataway, NJ, USA). The APC substrate S-2366 was purchased from Diapharma (West Chester, OH, USA).
Cloning of Mec13 VL and VH cDNAs
To target TM and EPCR fusion proteins to distinct paired epitopes on mouse PECAM-1, an scFv derived from the Mec13 mAb was prepared. Mec13.3 cell lysate in TRIzol (Ambion-Life Technologies, Carlsbad, CA, USA) was extracted with chloroform, and the phases were separated by centrifugation. The aqueous phase was removed and mixed with ethanol, and total RNA was purified with the RNeasy kit (Qiagen, Valencia, CA, USA). Combined RT-PCR was performed with the SuperScript One Step RT-PCR kit (Life Technologies). A single full-length VH cDNA was produced by using degenerate 5′ framework region 1 (FR1) primers and a 3′ constant region primer (20). This approach was not possible for the light chain, as PCR with FR1 primers produced only a nonfunctional myeloma-derived cDNA, with an in-frame stop codon in the FR1 region (Supplemental Fig. 1). Purified Mec13 mAb was sent to the University of California Davis Proteomics Core Facility for N-terminal (Edman) sequencing. The first 7 amino acids of the Mec13 light chain were identified and used to design degenerate primers, which allowed cloning of a full-length VL cDNA.
Assembly and expression of Mec13 scFv, soluble EPCR, and Mec13/EPCR constructs
Completed Mec13 VL and VH cDNAs were assembled into an scFv construct, as shown in Supplemental Fig. 1, with VH and VL sequences separated by a (GGGGS)3 linker and a triple FLAG tag appended to the 3′ end (C terminus) for purposes of purification and detection. The extracellular domain of mouse (m)EPCR was cloned by PCR from a previously described full-length cDNA (21), with a C-terminal triple-FLAG tag. Finally, a fusion protein was constructed with mEPCR on the 5′ end (N terminus), separated from the Mec13 scFv by an (SSSSG)2AAA linker. The proteins were arranged in this manner to keep the PC binding site (located on the N-terminal end of EPCR) freely accessible. All proteins were expressed in S2 cells and purified on an anti-FLAG affinity column (Sigma-Aldrich). Purity was assessed with sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE).
Generation of REN-derived stable cell lines
Human mesothelioma (REN)-PECAM cells, which stably express mouse PECAM-1, have been used in our laboratory extensively (8, 22). Similarly, we recently generated REN-intercellular adhesion molecule (ICAM) cells, which stably express mouse ICAM-1 (21). To make REN-TM cells or cells expressing both PECAM/TM and ICAM/TM, we purchased a mouse (m)TM cDNA (containing the entire coding sequence of mTM and a portion of the 5′ and 3′ untranslated regions; nt 87-3482) from OriGene Technologies (Rockville, MD, USA) and cloned into the pcDNA3.1/Zeo(−) vector (Life Technologies). Since REN-PECAM and REN-ICAM cells already stably express the Geneticin resistance gene, this expression vector (which confers resistance to the antibiotic Zeocin; Life Technologies) was used. Each cell type (REN wild-type, REN-PECAM, and REN-ICAM) was transfected with Lipofectamine 2000 (Invitrogen-Life Technologies, Carlsbad, CA, USA) and selected in medium with 250 μg/ml Zeocin and 200 μg/ml Geneticin (Life Technologies).
Live-cell ELISA assays
ELISAs were performed on live cells (21). In most experiments, anti-FLAG (M2)-peroxidase (HRP) conjugate was used as a detection antibody. In ELISA experiments involving more than 1 recombinant protein (e.g., those aimed at measuring collaborative enhancement of binding), anti-FLAG-HRP could not be used, as each fusion protein carries a C-terminal triple-FLAG tag. In these cases, detection was via either a goat anti-mTM or a goat anti-mEPCR polyclonal antibody, with an anti-goat-HRP secondary antibody. ELISA binding data were analyzed with Prism 6.0 software (GraphPad, San Diego, CA, USA).
PC ELISA
ELISAs were performed with the same protocol as just described, except that instead of live cells, fusion proteins were bound to PC immobilized on high-binding plastic wells. Briefly, the wells were incubated overnight at 4°C, with a solution of 4 μg/ml recombinant human PC in Tris-buffered saline. The PC solution was removed, and the plate was blocked with a 3% bovine serum albumin (BSA) solution for 2 h. BSA-coated wells with no PC were used as a control for nonspecific binding.
In vitro PC activation assays
Generation of APC by scFv/TM fusion was assayed (8). Briefly, cell monolayers were incubated with various combinations of TM and EPCR fusion proteins and washed 3 times with media before the addition of 1 nM thrombin and 100 nM PC. In all cases, PC activation occurred at 37°C in assay buffer [20 mM Tris, 100 mM NaCl, 1 mM CaCl2, 0.1% (w/v) BSA, pH 7.5], and the reaction was stopped by addition of an excess of hirudin. In experiments involving MS1 cells, the monolayer was treated with anti-mTM antibody to block endogenous TM and then washed 3 times before incubation with EPCR or TM fusion proteins or both. In experiments involving EPCR blockade, cells were incubated with 300 nM of anti-mEPCR mAb 1560 for 15 min before the addition of PC and thrombin. This antibody has been well characterized and is known to inhibit ∼70% of PC binding, eliminating to a substantial extent the ability of EPCR to accelerate the activation of PC by the thrombin-TM complex (13).
Radiolabeling of 390 scFv/TM
390 scFv/TM was directly radioiodinated with [125I]NaI (Perkin Elmer, Waltham, MA, USA) and purified on Zeba desalting spin columns (ThermoScientific, Rockford, IL, USA). Radiolabeling efficiency was >75%, and free iodine was <5%, after purification.
Animals
Animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals, as adopted by the National Institutes of Health (NIH; Bethesda, MD, USA), under protocols 803320 and 804349, approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Male C57BL/6 mice weighing 20–30 g (Jackson Laboratory, Bar Harbor, ME, USA) were used for all experiments.
Organ biodistribution of 390 scFv/TM
Mice were anesthetized and a tracer dose of [125I]-390 scFv/TM, with or without 2 mg/kg Mec13 scFv/EPCR was injected via the internal jugular vein. The latter was administered in a second injection, immediately after the first, to eliminate any potential effects of mixing the 2 proteins. Each fusion protein was constituted in sterile saline at a total volume of 50 μl. Organs were collected at 30 min after injection for counting on a Wizard2 2470 γ counter (PerkinElmer, Waltham, MA, USA). Data are expressed as a percentage of injected dose per gram of tissue (%ID/g) and are reported as the standard deviation of 4 animals.
APC generation in vivo
APC generation in vivo was measured by a published method (23). Briefly, mice were injected via the internal jugular vein with 0.25 mg/kg 390 scFv/TM, with or without 2 mg/kg Mec13 scFv/EPCR. As in biodistribution experiments, the proteins were administered in 2 consecutive injections, to eliminate any chance of direct mixing effects. After 30 min, the mice were injected with 0.85 NIH units of bovine thrombin and 5.2 μg of recombinant human (rh)PC, each freshly constituted in 50 μl of saline. Ten minutes later, blood was collected from the inferior vena cava directly into 3.8% citrate and benzamidine HCl (v/v 2:1). Plasma was isolated, flash frozen, and stored at −80°C until testing for human APC with an antibody-based ELISA (23).
Intratracheal LPS Model
ALI was induced via intratracheal (IT) injection of endotoxin. Injection of LPS, constituted in 100 μl of PBS, was followed immediately by 150 μl of air, to ensure even distribution throughout all distal airspaces. In prophylaxis experiments, fusion proteins or PBS vehicle was injected intravenously 30 min before 2 mg/kg LPS. [125I]-labeled albumin was injected intravenously just before LPS. Six h after induction of lung injury, blood was withdrawn from the inferior vena cava, and the animals were euthanized. A catheter was placed in the pulmonary artery, and the pulmonary circulation was gently flushed with PBS before the harvesting of organs. Radioactivity in the blood and lungs was measured with a Wizard2 2470 γ counter (PerkinElmer). The localization ratio of [125I]-albumin [calculated as (% injected dose present in lung/gram of lung tissue)/blood level] was used as a surrogate for pulmonary edema.
In therapeutic experiments, a lower dose of LPS was used to ensure survival of animals to later time points. LPS (1 mg/kg) was injected IT, followed by intravenous injection of fusion proteins 30 min later. Eighteen hours after induction of lung injury, the animals were euthanized and bronchoalveolar lavage (BAL) was performed via a 19-gauge stainless steel catheter (Harvard Apparatus, Holliston, MA, USA) placed in the trachea and secured with a 5-0 silk suture. Each animal was lavaged twice with 1.0 ml ice-cold PBS. Lavages were pooled and centrifuged at 2000 rpm for 4 min to pellet the cellular content. After hypotonic lysis of RBCs, the leukocytes were resuspended in a known volume of PBS with 1% BSA and counted on a hemocytometer. The total protein concentration in the BAL supernatant was measured with a BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA).
Data analysis and statistics
Results are expressed as means ± sd, unless otherwise noted. Significant differences between means were determined by 1-way ANOVA, followed by the appropriate multiple-comparison (Tukey) test. P < 0.05 was considered statistically significant.
RESULTS
Construction of PECAM-targeted Mec13 scFv/EPCR fusion protein
The previously characterized anti-PECAM scFv/TM (21, 24) contains a single-chain antibody fragment derived from 390, an anti-mouse PECAM-1 mAb. We showed previously that mAb 390 and another anti-PECAM mAb, Mec13, collaboratively enhance each other’s binding to ECs, both in vitro and in vivo (8). As a result, we created a new scFv for fusion to EPCR, this one derived from mAb Mec13. Supplemental Fig. 1 illustrates the molecular design of the Mec13 scFv/EPCR fusion protein and provides a list of all fusion proteins used in the study, their respective targets, and their various functions.
We next tested the function of the individual moieties of the Mec13 scFv/EPCR fusion protein. Specific binding to PECAM-1 was evaluated in the nonendothelial human mesothelioma cell line REN, which has no baseline expression of mouse PECAM-1, ICAM-1, TM, or EPCR (21). Mec13 scFv and the Mec13 scFv/EPCR fusion protein bound to REN cells stably transfected with mouse PECAM-1 (REN-PECAM), but not the control REN cells (Fig. 1A). Likewise, Mec13 scFv/EPCR and soluble (s)EPCR bound to immobilized PC, but not to albumin-coated wells (Fig.1B). We concluded that both the scFv and EPCR moieties of the Mec13 scFv/EPCR fusion are functional, at least in binding to their corresponding ligands.
Figure 1.
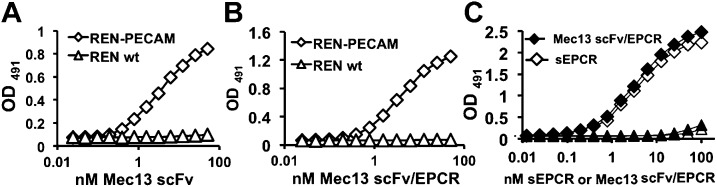
Cell-based ELISAs showing that binding of (A) Mec13 scFv and (B) Mec13 scFv/EPCR fusion protein to PECAM-expressing REN cells was similar. EC50 of 3.5 ± 0.18 and 3.86 ± 0.14 nM, respectively (mean ± sd from 3 independent ELISAs. P > 0.05. Neither protein bound to wild-type REN cells. C) Mec13 scFv/EPCR fusion protein bound to PC in a manner nearly identical to that of soluble (s)EPCR. There was no binding of Mec13 scFv/EPCR or sEPCR to BSA-coated wells. Experiments were performed in triplicate (each data point represents 3 wells). Error bars, sd of technical replicates. Data are representative results of 3 replicate experiments.
Functional activity of Mec13 scFv/EPCR fusion protein
We assessed the ability of Mec13 scFv/EPCR to augment PC activation by the thrombin/TM complex on the cell surface. For this, we generated REN-PECAM-TM cells, which are double-transfected REN cells that demonstrate stable surface expression of mouse PECAM-1 and TM and thrombin-dependent activation of PC (Supplemental Fig. 2A, B). REN-TM and REN-PECAM cells, which express either TM or PECAM-1, were used as controls.
In these stable cell lines, Mec13 scFv/EPCR stimulated activation of PC by the thrombin/TM complex only on cells expressing both TM and PECAM (Supplemental Fig. 2C). Likewise, no effect was seen with Mec13 scFv or sEPCR (Supplemental Fig. 2D), confirming that both moieties of the fusion protein are necessary for this activity.
scFv/EPCR and scFv/TM demonstrate collaborative enhancement of binding to PECAM-1
As discussed above, the scFv/TM and scFv/EPCR fusion proteins were designed with antibody fragments derived from the mAbs 390 and Mec13, based on the finding that these antibodies enhance each other’s binding to PECAM-1. This phenomenon of collaborative enhancement was demonstrated in transfected (i.e., REN-PECAM) cells and MS1 mouse ECs and after intravenous injection in live mice. Moreover, a similar effect was observed in human ECs with distinct paired anti-human PECAM-1 antibodies (8). Given the consistency of this finding, we hypothesized that similar effects would be seen with EPCR and TM fusion proteins directed to paired PECAM-1 epitopes. Indeed, both Mec13 scFv and Mec13 scFv/EPCR increased the binding of 390 scFv/TM to REN-PECAM cells (Fig. 2A). Likewise, 390 scFv/TM increased the binding of Mec13 scFv/EPCR (Fig. 2B).
Figure 2.
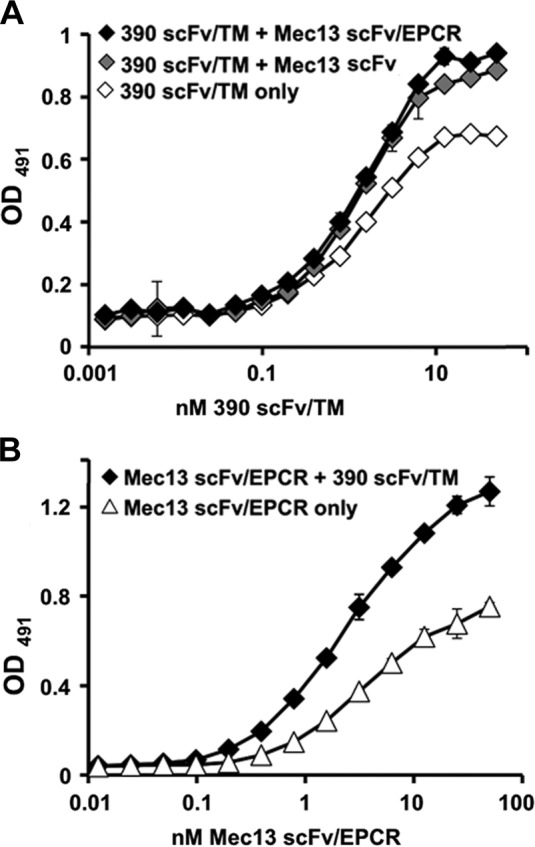
Mec13 scFv/EPCR and 390 scFv/TM demonstrate collaborative enhancement of binding. A) In agreement with experiments performed with intact antibodies, Mec13 scFv and Mec13 scFv/EPCR fusion protein (150 nM) enhanced the binding of 390 scFv/TM to REN-PECAM cells. B) Likewise, 390 scFv/TM (50 nM) enhanced the binding of Mec13 scFv/EPCR. Experiments were performed in triplicate (each data point shown represents 3 wells). Error bars, sd.
Mec13 scFv/EPCR enhances PC activation by 390 scFv/TM on mouse PECAM-1-expressing cells
We next studied APC generation by scFv/TM and scFv/EPCR fusion proteins coanchored to REN-PECAM cells. Mec13 scFv/EPCR demonstrated a dose-dependent enhancement of thrombin-dependent APC generation on cells also treated with 390 scFv/TM, with an ∼5-fold increase at the highest concentration (Fig. 3A).
Figure 3.

Mec13 scFv/EPCR enhances APC generation by 390 scFv/TM via 2 distinct mechanisms. A) Cotreatment of REN-PECAM cells with Mec13 scFv/EPCR produced a dose-dependent increase in APC generation by surface-bound 390 scFv/TM. B) Additional experiments suggest that 2 distinct effects contribute to the action of Mec13 scFv/EPCR fusion protein: an increase in 390 scFv/TM binding and the partnering of cell-bound TM and EPCR. The binding effect is demonstrated by cotreatment of cells with 390 scFv/TM and Mec13 scFv (150 nM). The EPCR-dependent effect is evident in the loss of APC generation when Mec13 scFv/EPCR is blocked with an anti-EPCR monoclonal antibody (300 nM). All experiments were performed in triplicate. Data are means ± sd.
In theory, both enhanced binding (Fig. 2) and functional partnering between the TM and EPCR moieties could have contributed to the 5-fold increase observed. We conducted several experiments to determine the relative contribution of each effect. As shown in Fig. 3B, Mec13 scFv (lacking the EPCR moiety) was used to estimate the binding effect and provided an ∼2-fold stimulation of APC production by 390 scFv/TM. Conversely, we estimated the contribution of enzymatic partnering (the EPCR effect) using mAb 1560, which blocks the interaction of EPCR and murine PC. This antibody inhibits ∼70% of the effect of EPCR on APC production by the thrombin-TM complex (13). In cells treated with Mec13 scFv/EPCR and 390 scFv/TM, blockade of EPCR reduced APC generation by ∼2-fold, to almost exactly the level in cells treated with TM fusion protein and Mec13 scFv (Fig. 3B). Taken together, these data suggest distinct mechanisms that make roughly equivalent contributions to the net effect of Mec13 scFv/EPCR on the generation of APC by 390 scFv/TM.
Mec13 scFv/EPCR, but not anti-ICAM scFv/EPCR, enhances PC activation by 390 scFv/TM on mouse ECs
Although transfected REN cells provide a convenient system for initial testing of fusion proteins, they do not express endogenous endothelial membrane proteins, nor do the extent and distribution of overexpressed molecules necessarily reflect what is present on ECs (21). As a result, we conducted additional experiments in MS1 ECs. These cells express endogenous mouse TM, EPCR, PECAM-1, and ICAM-1, with surface distribution membrane and response to inflammatory activation similar to that seen in primary human ECs [e.g., HUVECs (21)]. To inhibit APC generation by endogenous TM (which complicates the measurement of the fusion-mediated effect), we used a blocking TM antibody (21). This method allowed us to measure PC activation by 390 scFv/TM anchored to the MS1 cells, with and without the addition of Mec13 scFv/EPCR. Similar to what had been seen with REN-PECAM cells, Mec13 scFv/EPCR increased PC activation by 390 scFv/TM. Likewise, the use of Mec13 scFv and the EPCR blocking mAb revealed a similar contribution of 2 distinct mechanisms by which the EPCR fusion protein exerts its effect: collaborative enhancement of binding and enzymatic partnering (Fig. 4).
Figure 4.

Mec13 scFv/EPCR, but not anti-ICAM scFv/EPCR, enhances APC generation by 390/TM on mouse ECs. A) APC generation by 390 scFv/TM was assayed on MS1 ECs, on which endogenous TM had been blocked by anti-TM antibody. Dotted line: residual activity of endogenous TM. Mec13 scFv/EPCR (100 nM) enhanced APC generation by cell-bound 390 scFv/TM. As in the REN-PECAM cell experiments, 2 mechanisms were discerned: a binding effect, determined by cotreatment with Mec13 scFv, and an EPCR-dependent effect, demonstrated by use of an EPCR-block antibody. B) In contrast to Mec13 scFv/EPCR, cotreatment of cells with YN1 scFv/EPCR and 390 scFv/TM did not enhance APC generation.
We reasoned that the functional synergy observed between scFv/TM and scFv/EPCR is most likely the result of their proximity while anchored to the same surface determinant, PECAM-1. We sought to determine whether anchoring TM and EPCR to different surface molecules on MS1 ECs would have similar effects. Using a recently created anti-ICAM (YN1) scFv (21), we designed and synthesized an anti-ICAM YN1 scFv/EPCR fusion protein (Supplemental Fig. 3A). Before testing the ECs, we first examined the functional activity of the new fusion in a series of experiments similar to those described above for Mec13 scFv/EPCR. The YN1 scFv/EPCR fusion protein bound cells expressing mouse ICAM-1 and TM and enhanced PC activation by the thrombin/TM complex (Supplemental Fig. 3B–E).
Having confirmed ligand binding and the functional activity of YN1 scFv/EPCR, we tested its ability to enhance the function of PECAM-1 anchored 390 scFv/TM on MS1 ECs. In contrast to Mec13 scFv/EPCR, no enhancement of PC activation was seen with the YN1 scFv/EPCR fusion protein, nor was there any effect of the EPCR blockade antibody (Fig. 4B). This result indicates that anchoring TM and EPCR to different membrane molecular determinants does not reproduce the effect seen with dual targeting to adjacent PECAM-1 epitopes.
Mec13 scFv/EPCR enhances pulmonary targeting and APC generation by 390 scFv/TM in vivo
The dual targeting strategy was tested in vivo by sequential injection of scFv/TM and scFv/EPCR fusion proteins in mice. As shown in Fig. 5A, coadministration of Mec13 scFv/EPCR enhanced pulmonary targeting of radiolabeled 390 scFv/TM by ∼4-fold. Next, we assessed the effect of dual targeting on APC generation in vivo. Using a technique recently reported by our laboratory (23), we measured thrombin-dependent PC activation 30 min after intravenous injection of fusion proteins. Fig. 5B demonstrates a significant increase in APC generation in animals given both 390 scFv/TM and Mec13 scFv/EPCR, as opposed to 390 scFv/TM alone. These data match the results from cell culture experiments, indicating that dual targeting of these collaborative fusion proteins in vivo results in both enhanced drug delivery and functional effect (i.e., APC generation).
Figure 5.

Coadministration of Mec13 scFv/EPCR increases pulmonary targeting, in vivo functional activity, and protective effect of 390 scFv/TM in a mouse model of lung inflammation and injury. A) 390 scFv/TM was cleared from the blood rapidly, with only ∼5% ID/g remaining 30 min after intravenous injection. Distribution to the lung was enhanced ∼4-fold by administration of the collaborative fusion Mec13 scFv/EPCR (n = 4 animals per group). B) Dual targeting similarly augmented the functional activity (APC generation) of 390 scFv/TM in vivo (n = 4 animals per group). Dotted line: level of APC generation observed with saline control. When administered prophylactically, 30 min before IT LPS, 390 scFv/TM and Mec13 scFv/EPCR were more effective than either fusion protein alone in reducing (C) pulmonary endothelial activation (VCAM-1 expression). D) Edema, as determined by leakage of [125I]-labeled albumin from the blood into lung interstitium, alveolar space, or both, was also reduced. Coadministration of 390 scFv/TM and YN1 (anti-ICAM) scFv/EPCR did not show the synergistic effect that occurred with the 2 PECAM-1 anchored fusions. E) Dual targeting is also effective as a therapeutic strategy (i.e., 30 min after LPS) demonstrating significant reduction in BAL fluid protein (normalized to total body weight, i.e., milligrams protein/gram animal weight). Neither fusion protein, given individually, had a significant therapeutic effect on BAL protein at this time point (18 h after injury) (n = 4 or 5 animals for each group in all experiments). C, D) Dotted lines: outcomes in control animals that did not receive LPS.
Vascular protective effects of PECAM-1- and ICAM-1-targeted fusion proteins in a mouse model of ALI
We evaluated the dual targeting strategy in a mouse model of lung inflammation/injury. scFv/TM and scFv/EPCR fusion proteins were first tested in a prophylactic setting, with fusion proteins (or PBS vehicle) sequentially injected intravenously before IT LPS challenge. Coadministration of Mec13 scFv/EPCR and 390 scFv/TM—but not of either fusion protein alone—decreased pulmonary endothelial activation, as marked by levels of lung mRNA for VCAM-1 (Fig 5C), ICAM-1, and E-selectin (Supplemental Fig. 4A, B). In some experiments, radiolabeled albumin was injected intravenously just before LPS administration, as a marker of transendothelial plasma protein leakage (experimental timeline shown in Fig. 5D). Again, dual targeting produced a greater reduction in this pathologic outcome than either fusion protein alone. Coadministration of Mec13 and 390 scFvs was tested as a control, to ensure that any effects on barrier function were not simply the result of the 2 antibody fragments binding to PECAM-1. Finally, cotreatment with 390 scFv/TM and YN1 scFv/EPCR resulted in no additional protection, consistent with our observations in MS1 cells. This critical result suggests that it is the proximity of scFv/TM and scFv/EPCR that allows the synergistic effect, such that the same cargoes provide significantly less protection when anchored to distinct endothelial determinants.
Finally, to probe the translational potential of dual targeting as a therapeutic strategy for ALI, we examined the protective effects of the fusion proteins when given after LPS insult. Besides changing the order of the administration of fusion proteins and endotoxin, we selected a later time point, 18 h, for measurement of disease readouts (experimental timeline shown in Fig 5E). As shown, cotreatment with both 390 scFv/TM and Mec13 scFv/EPCR produced a significant reduction in alveolar protein, as measured by BAL, whereas treatment with 390 scFv/TM alone had no distinguishable effect. Neither therapeutic approach had a significant effect on the BAL leukocyte count (Supplemental Fig. 4C)
DISCUSSION
Over the past several decades, hundreds of advanced drug delivery systems have been designed and implemented. Nearly all were intended to control drug pharmacokinetics to prolong effect and limit toxicity (25). More and more, however, drug targeting is focused on the delivery of pharmacologic agents to specific cell types or even subcellular addresses, such that they can exert optimal, localized therapeutic effects (26). These considerations are of particular relevance to biotherapeutics, a rapidly growing class of drugs designed to modify endogenous biologic pathways (27). Although early members of this class, such as erythropoietin and granulocyte colony-stimulating factor, do not need site-specific delivery, the next generation of biologic agents, such as small interfering RNA and lysosomal enzymes, require precise localization to achieve their intended therapeutic effects (28).
One site of particular interest is the luminal membrane of the vascular endothelium, where multiple endogenous systems interact to maintain blood fluidity, regulate vascular permeability, and control inflammation. Although numerous surface determinants have been identified on ECs, relatively few efforts have been made to deliver therapeutics to this key interface between blood and tissue (21, 29). We describe an important and novel technique that significantly advances our capabilities in this regard: the simultaneous anchoring of 2 agents to the luminal endothelial membrane in a manner that allows them to partner enzymatically and generate an enhanced biologic effect. This is, to our knowledge, the first example of colocalized biologic agents generating an effect on ECs in situ and certainly the first instance in which dual targeting enhances the delivery of each therapeutic.
As a proof of principle, we have applied this novel approach to enhance the activity of the endothelial PC pathway. Pharmaceutical interest in this system was spurred on by the finding that recombinant human (rh)APC could protect animals from otherwise lethal infusions of bacteria (30). Ultimately, rhAPC became the first drug approved for the treatment of sepsis syndromes in humans (31), although its clinical utility was limited by poor pharmacokinetics, a narrow therapeutic index, and life-threatening systemic toxicity (32). Genetically altered forms of APC have been created that lack the anticoagulant and profibrinolytic effects of wild-type APC, while preserving its anti-inflammatory, antiapoptotic, and cytoprotective activities. Whereas these modifications address some of the limitations of rhAPC (33), significant pharmacokinetic challenges remain, which may limit success in the clinical realm.
Another strategy for augmenting the PC pathway is the infusion of soluble forms of TM. Unlike activated zymogens, soluble TM preserves some of the “on-demand” nature of the endogenous PC pathway, generating a biologic effect primarily at sites of thrombin generation (34). Preliminary studies in animal models and early-stage human clinical trials have demonstrated beneficial effects (35–37). Although each of these approaches has its merits, they all provide inadequate localization to the endothelial membrane. Since circulating APC must compete for EPCR binding sites with a much larger pool of PC zymogen, local generation of APC at the EC surface may have greater capacity to generate protective signaling through endothelial EPCR (14). Indeed, APC signaling through EPCR on other cells, such as neutrophils, may actually worsen outcome in pulmonary or systemic infection by impairing bacterial clearance (38). In contrast, recreating endogenous interactions between TM, PC, and EPCR at the endothelial membrane seems likely to provide greater protection with fewer unintended side effects.
Our laboratory has reported several efforts to achieve precisely this goal: the delivery of recombinant TM to the luminal endothelial membrane—its natural location and, presumably, the site of optimal activity. We created an anti-PECAM scFv/TM fusion protein, which binds to ECs in vitro and after intravenous injection. This protein was found to protect mice from ischemic and inflammatory lung injury, without the bleeding side effects that occur with equipotent doses of recombinant APC (24). Further investigation of the PECAM-targeted scFv/TM fusion protein, however, revealed that its interaction with EPCR differs from that of endogenous TM (21). Although anchoring recombinant TM to other endothelial determinants, such as ICAM-1, improves partnering with endogenous EPCR, PECAM-1 remains a desirable target in many situations, given its panendothelial distribution and high level of expression (39). Furthermore, ECs shed EPCR in response to vascular inflammation (40), and a means of augmenting its presence on the endothelial membrane is therefore likely to be necessary to fully restore the function of the PC pathway.
The application of dual targeting to the TM-EPCR-APC system was informed not only by our previous results, but also by reports from the field of biomaterials, where the PC pathway has gained attention as a means of preventing thrombosis and immune system activation on implantable medical devices (41). TM has been immobilized on polyurethane, polyethylene glycol–modified glass, and even liposomes (42–44). In vitro flow studies have revealed a plateau in the maximum rate of APC generation, despite increasing surface density of recombinant TM (45). This finding has been attributed to a limitation in PC availability caused by the absence of EPCR. Subsequent efforts aimed at coimmobilizing TM and EPCR have achieved substantially higher rates of APC generation and demonstrate that the distance between these molecules is an important factor. In particular, a higher rate of APC generation is observed when the recombinant proteins are immobilized in proximity on polyurethane (<10 nm), as opposed to a random, unordered distribution (46). This report is consistent with our finding that proximity is a critical factor in the dual targeting of enzymatic partners. Codelivery of scFv/TM and scFv/EPCR increases APC generation and provides superior protection in vivo when the fusion proteins are anchored to the same endothelial determinant (PECAM-1), but no synergistic effect occurs when 2 different surface molecules (i.e., ICAM-1 and PECAM-1) are targeted.
In summary, the current report confirms the feasibility of dual targeting of paired therapeutics to the endothelium and demonstrates its utility in the particular case of modulating the PC pathway. Fig. 6 illustrates our key results, including the 2 distinct mechanisms by which Mec13 scFv/EPCR enhances the production of APC by membrane-bound 390 scFv/TM. Our animal data suggest that collaborative TM and EPCR fusion proteins may be worth pursing as translational therapeutics for ALI. Although it is promising that a reduction in BAL protein occurs as much as 18 h after endotoxin injury, after just a single dose of each fusion protein, it is of utmost importance to fully characterize both the pharmacokinetics and therapeutic time window of the scFv/TM and scFv/EPCR fusion proteins. Although the 30-min therapeutic window that we report seems relevant to the emergency medicine setting, the limits of the therapeutic window are unknown and have important implications for future preclinical investigations. The absence of any effect on BAL leukocytes could also be a positive for the translational prospects of these prototherapeutics. Because the recruitment of leukocytes to alveoli is of great importance to pathogen killing, preserving this response is important when both inflammatory injury and pulmonary infection are present, as in the case of ALI associated with sepsis and pneumonia. These intriguing, albeit preliminary, findings will be the subject of ongoing study in our laboratory. Beyond TM and EPCR fusion proteins and the specific clinical setting of ALI, endothelial dual targeting has potential for widespread application. Although the collaborative enhancement effect has so far been observed only with PECAM-1, efforts are under way to determine whether other EC surface molecules (e.g., VE-cadherin and ICAM-1) demonstrate analogous phenomena. Similarly, endogenous endothelial systems other than the PC pathway could be targeted for intervention. Finally, we have recently demonstrated collaborative enhancement of endothelial targeting by nanocarriers, an observation that could enable complementary cargoes or vascular imaging agents to combine and generate signal only at sites of dual delivery. In each case, the critical advantage is the ability to deliver paired agents with nanometer scale proximity while allowing collaborative enhancement—rather than competitive inhibition—of binding.
Figure 6.

Dual targeting of scFv/TM and scFv/EPCR to increase APC production. A) Normal function of the TM/EPCR/PC pathway. Endothelial TM binds thrombin (factor IIa) and activates PC, a process accelerated by EPCR. APC can signal to ECs or bind protein S and exert anticoagulant effects. Va, coagulation factor Va. B) 390 scFv/TM fusion protein anchored to PECAM generates APC, but is unable to partner with endogenous EPCR. APC produced by 390 scFv/TM likely functions in a paracrine fashion. C) Mec13 scFv increases binding of 390 scFv/TM to PECAM-1 via a collaborative enhancement mechanism, resulting in an ∼2-fold increase in APC production. D) The combination of enhanced binding and enzymatic partnering between TM and EPCR further increases APC production to levels roughly equal to endogenous TM/EPCR.
Supplementary Material
Acknowledgments
The authors thank Dr. Annunciata Vecchi (Mario Negri Institute of Pharmacology Research, Milan, Italy) for her generous gift of Mec13 hybridoma lysate. This work was supported by a Junior Investigator Preliminary/Feasibility Grant Program grant from the Institute of Translational Medicine and Therapeutics at the University of Pennsylvania (to C.F.G.), as well as an American Heart Association Fellow-to-Faculty Grant 10FTF4150053 (to C.F.G.), and a U. S. National Institutes of Health Grant R01-HL091950 from the National Heart, Lung, and Blood Institute (to V.R.M.).
Glossary
- %ID/g
percentage of injected dose per gram of organ weight
- ALI
acute lung injury
- APC
activated protein C
- BAL
bronchoalveolar lavage
- BSA
bovine serum albumin
- EC
endothelial cell
- EPCR
endothelial protein C receptor
- FR1
5′ framework region 1
- HRP
horseradish peroxidase
- ICAM-1
intercellular adhesion molecule-1
- IT
intratracheal
- mAb MS1
mouse EC line
- mEPCR
mouse EPCR
- mTM
mouse TM
- OD491
optical density (absorbance) at 491 nm
- PC
protein C
- PECAM-1
platelet endothelial cell adhesion molecule-1
- REN
human mesothelioma cell line
- rh
recombinant human
- scFv
single-chain variable fragment
- sEPCR
soluble EPCR
- TM
thrombomodulin
- VH
variable domain of antibody heavy chain
- VL
variable domain of antibody light chain
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Muro S., Muzykantov V. R. (2005) Targeting of antioxidant and anti-thrombotic drugs to endothelial cell adhesion molecules. Curr. Pharm. Des. 11, 2383–2401 [DOI] [PubMed] [Google Scholar]
- 2.Greineder C. F., Howard M. D., Carnemolla R., Cines D. B., Muzykantov V. R. (2013) Advanced drug delivery systems for antithrombotic agents. Blood 122, 1565–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aird W. C. (2003) The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101, 3765–3777 [DOI] [PubMed] [Google Scholar]
- 4.Maniatis N. A., Orfanos S. E. (2008) The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 14, 22–30 [DOI] [PubMed] [Google Scholar]
- 5.Thorpe P. E. (2004) Vascular targeting agents as cancer therapeutics. Clin. Cancer Res. 10, 415–427 [DOI] [PubMed] [Google Scholar]
- 6.Muzykantov V. R. (2005) Biomedical aspects of targeted delivery of drugs to pulmonary endothelium. Expert Opin. Drug Deliv. 2, 909–926 [DOI] [PubMed] [Google Scholar]
- 7.Koren E., Torchilin V. P. (2011) Drug carriers for vascular drug delivery. IUBMB Life 63, 586–595 [DOI] [PubMed] [Google Scholar]
- 8.Chacko A.-M., Nayak M., Greineder C. F., Delisser H. M., Muzykantov V. R. (2012) Collaborative enhancement of antibody binding to distinct PECAM-1 epitopes modulates endothelial targeting. PLoS ONE 7, e34958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esmon C. T. (2001) The normal role of Activated Protein C in maintaining homeostasis and its relevance to critical illness. Crit. Care 5, S7–S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esmon C. T. (1995) Thrombomodulin as a model of molecular mechanisms that modulate protease specificity and function at the vessel surface. FASEB J. 9, 946–955 [DOI] [PubMed] [Google Scholar]
- 11.Esmon C. T. (1989) The roles of protein C and thrombomodulin in the regulation of blood coagulation. J. Biol. Chem. 264, 4743–4746 [PubMed] [Google Scholar]
- 12.Stearns-Kurosawa D. J., Kurosawa S., Mollica J. S., Ferrell G. L., Esmon C. T. (1996) The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc. Natl. Acad. Sci. USA 93, 10212–10216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W., Zheng X., Gu J., Hunter J., Ferrell G. L., Lupu F., Esmon N. L., Esmon C. T. (2005) Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J. Thromb. Haemost. 3, 1351–1359 [DOI] [PubMed] [Google Scholar]
- 14.Feistritzer C., Schuepbach R. A., Mosnier L. O., Bush L. A., Di Cera E., Griffin J. H., Riewald M. (2006) Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J. Biol. Chem. 281, 20077–20084 [DOI] [PubMed] [Google Scholar]
- 15.Boehme M. W., Deng Y., Raeth U., Bierhaus A., Ziegler R., Stremmel W., Nawroth P. P. (1996) Release of thrombomodulin from endothelial cells by concerted action of TNF-alpha and neutrophils: in vivo and in vitro studies. Immunology 87, 134–140 [PMC free article] [PubMed] [Google Scholar]
- 16.Xu J., Qu D., Esmon N. L., Esmon C. T. (2000) Metalloproteolytic release of endothelial cell protein C receptor. J. Biol. Chem. 275, 6038–6044 [DOI] [PubMed] [Google Scholar]
- 17.Redl H., Schlag G., Schiesser A., Davies J. (1995) Thrombomodulin release in baboon sepsis: its dependence on the dose of Escherichia coli and the presence of tumor necrosis factor. J. Infect. Dis. 171, 1522–1527 [DOI] [PubMed] [Google Scholar]
- 18.Gu J.-M., Katsuura Y., Ferrell G. L., Grammas P., Esmon C. T. (2000) Endotoxin and thrombin elevate rodent endothelial cell protein C receptor mRNA levels and increase receptor shedding in vivo. Blood 95, 1687–1693 [PubMed] [Google Scholar]
- 19.Faust S. N., Levin M., Harrison O. B., Goldin R. D., Lockhart M. S., Kondaveeti S., Laszik Z., Esmon C. T., Heyderman R. S. (2001) Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N. Engl. J. Med. 345, 408–416 [DOI] [PubMed] [Google Scholar]
- 20.Dübel S., Breitling F., Fuchs P., Zewe M., Gotter S., Welschof M., Moldenhauer G., Little M. (1994) Isolation of IgG antibody Fv-DNA from various mouse and rat hybridoma cell lines using the polymerase chain reaction with a simple set of primers. J. Immunol. Methods 175, 89–95 [DOI] [PubMed] [Google Scholar]
- 21.Greineder C. F., Chacko A.-M., Zaytsev S., Zern B. J., Carnemolla R., Hood E. D., Han J., Ding B.-S., Esmon C. T., Muzykantov V. R. (2013) Vascular immunotargeting to endothelial determinant ICAM-1 enables optimal partnering of recombinant scFv-thrombomodulin fusion with endogenous cofactor. PLoS ONE 8, e80110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun J., Paddock C., Shubert J., Zhang H. B., Amin K., Newman P. J., Albelda S. M. (2000) Contributions of the extracellular and cytoplasmic domains of platelet-endothelial cell adhesion molecule-1 (PECAM-1/CD31) in regulating cell-cell localization. J. Cell Sci. 113, 1459–1469 [DOI] [PubMed] [Google Scholar]
- 23.Carnemolla R., Patel K. R., Zaitsev S., Cines D. B., Esmon C. T., Muzykantov V. R. (2012) Quantitative analysis of thrombomodulin-mediated conversion of protein C to APC: translation from in vitro to in vivo. J. Immunol. Methods 384, 21–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding B.-S., Hong N., Christofidou-Solomidou M., Gottstein C., Albelda S. M., Cines D. B., Fisher A. B., Muzykantov V. R. (2009) Anchoring fusion thrombomodulin to the endothelial lumen protects against injury-induced lung thrombosis and inflammation. Am. J. Respir. Crit. Care Med. 180, 247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan R. (2003) The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2, 347–360 [DOI] [PubMed] [Google Scholar]
- 26.Torchilin V. P. (2006) Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu. Rev. Biomed. Eng. 8, 343–375 [DOI] [PubMed] [Google Scholar]
- 27.Carnemolla R., Shuvaev V. V., Muzykantov V. R. (2010) Targeting antioxidant and antithrombotic biotherapeutics to endothelium. Semin. Thromb. Hemost. 36, 332–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J., Lu Z., Wientjes M. G., Au J. L.-S. (2010) Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 12, 492–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muzykantov V. R. (2013) Targeted drug delivery to endothelial adhesion molecules. ISRN Vasc. Med. 2013, 1–27 [Google Scholar]
- 30.Taylor F. B. Jr., Chang A., Esmon C. T., D’Angelo A., Vigano-D’Angelo S., Blick K. E. (1987) Protein C prevents the coagulopathic and lethal effects of Escherichia coli infusion in the baboon. J. Clin. Invest. 79, 918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernard G. R., Vincent J. L., Laterre P. F., LaRosa S. P., Dhainaut J. F., Lopez-Rodriguez A., Steingrub J. S., Garber G. E., Helterbrand J. D., Ely E. W., Fisher C. J. Jr.; Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. N. Engl. J. Med. 344, 699–709 [DOI] [PubMed] [Google Scholar]
- 32.Opal S. M., LaRosa S. P. (2013) Recombinant human activated protein C as a therapy for severe sepsis: lessons learned? Am. J. Respir. Crit. Care Med. 187, 1041–1043 [DOI] [PubMed] [Google Scholar]
- 33.Kerschen E. J., Fernandez J. A., Cooley B. C., Yang X. V., Sood R., Mosnier L. O., Castellino F. J., Mackman N., Griffin J. H., Weiler H. (2007) Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J. Exp. Med. 204, 2439–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solis M. M., Cook C., Cook J., Glaser C., Light D., Morser J., Yu S. C., Fink L., Eidt J. F. (1991) Intravenous recombinant soluble human thrombomodulin prevents venous thrombosis in a rat model. J. Vasc. Surg. 14, 599–604 [DOI] [PubMed] [Google Scholar]
- 35.Su E. J., Geyer M., Wahl M., Mann K., Ginsburg D., Brohmann H., Petersen K. U., Lawrence D. A. (2011) The thrombomodulin analog solulin promotes reperfusion and reduces infarct volume in a thrombotic stroke model. J. Thromb. Haemost. 9, 1174–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kearon C., Comp P., Douketis J., Royds R., Yamada K., Gent M. (2005) Dose-response study of recombinant human soluble thrombomodulin (ART-123) in the prevention of venous thromboembolism after total hip replacement. J. Thromb. Haemost. 3, 962–968 [DOI] [PubMed] [Google Scholar]
- 37.Saito H., Maruyama I., Shimazaki S., Yamamoto Y., Aikawa N., Ohno R., Hirayama A., Matsuda T., Asakura H., Nakashima M., Aoki N. (2007) Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J. Thromb. Haemost. 5, 31–41 [DOI] [PubMed] [Google Scholar]
- 38.Schouten M., de Boer J. D., Kager L. M., Roelofs J. J. T. H., Meijers J. C. M., Esmon C. T., Levi M., van ’t Veer C., van der Poll T. (2014) The endothelial protein C receptor impairs the antibacterial response in murine pneumococcal pneumonia and sepsis. Thromb. Haemost. 111, 970–980 [DOI] [PubMed] [Google Scholar]
- 39.Newman P. J. (1997) The biology of PECAM-1. J. Clin. Invest. 99, 3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurosawa S., Stearns-Kurosawa D. J., Carson C. W., D’Angelo A., Della Valle P., Esmon C. T. (1998) Plasma levels of endothelial cell protein C receptor are elevated in patients with sepsis and systemic lupus erythematosus: lack of correlation with thrombomodulin suggests involvement of different pathological processes. Blood 91, 725–727 [PubMed] [Google Scholar]
- 41.Sperling C., Salchert K., Streller U., Werner C. (2004) Covalently immobilized thrombomodulin inhibits coagulation and complement activation of artificial surfaces in vitro. Biomaterials 25, 5101–5113 [DOI] [PubMed] [Google Scholar]
- 42.Kishida A., Ueno Y., Fukudome N., Yashima E., Maruyama I., Akashi M. (1994) Immobilization of human thrombomodulin onto poly(ether urethane urea) for developing antithrombogenic blood-contacting materials. Biomaterials 15, 848–852 [DOI] [PubMed] [Google Scholar]
- 43.Han H. S., Yang S. L., Yeh H. Y., Lin J. C., Wu H. L., Shi G. Y. (2001) Studies of a novel human thrombomodulin immobilized substrate: surface characterization and anticoagulation activity evaluation. J. Biomater. Sci. Polym. Ed. 12, 1075–1089 [DOI] [PubMed] [Google Scholar]
- 44.Zhang H., Weingart J., Jiang R., Peng J., Wu Q., Sun X.-L. (2013) Bio-inspired liposomal thrombomodulin conjugate through bio-orthogonal chemistry. Bioconjug. Chem. 24, 550–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tseng P.-Y., Jordan S. W., Sun X.-L., Chaikof E. L. (2006) Catalytic efficiency of a thrombomodulin-functionalized membrane-mimetic film in a flow model. Biomaterials 27, 2768–2775 [DOI] [PubMed] [Google Scholar]
- 46.Kador K. E., Mamedov T. G., Schneider M., Subramanian A. (2011) Sequential co-immobilization of thrombomodulin and endothelial protein C receptor on polyurethane: activation of protein C. Acta Biomater. 7, 2508–2517 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.