

Abstract
The newest technologies for DNA sequencing have led to the determination of the primary structure of the genomes of organisms, mainly prokaryotes, with high efficiency and at lower costs. However, the presence of regions with repetitive sequences, in addition to the short reads produced by the Next-Generation Sequencing (NGS) platforms, created a lot of difficulty in reconstructing the original genome in silico. Thus, even today, genome assembly continues to be one of the major challenges in bioinformatics specifically when repetitive sequences are considered. In this paper, we present an approach to assemble repetitive regions in prokaryotic genomes. Our methodology enables (i) the identification of these regions through visual tools, (ii) the characterization of sequences on the extremities of gaps and (iii) the extraction of consensus sequences based on mapping of raw data to a reference genome. We also present a case study on the assembly of regions that encode ribosomal RNAs (rRNA) in the genome of Corynebacterium ulcerans FRC11, in order to show the efficiency of the strategies presented here. The proposed methods and tools will help in finishing genome assemblies, besides reducing the running time and associated costs.
Availability
All scripts are available at http://github.com/dcbmariano/maprepeat
Keywords: bioinformatics, genome assembly, finishing assemblies, repetitive sequences
Background
Recently, the Next-Generation Sequencing (NGS) platforms have led to the determination of primary structure of DNA with high efficiency and accuracy as well as at a lower cost mainly for prokaryotic genomes such as bacteria. Despite of such great advances, the new sequencing platforms are still unable to read with precision all the genome in a single run. It is necessary to fragment the DNA molecules before the sequencing, after which an in silico strategy has to be employed to reassemble these fragments based on the orientation of the individual reads. This process is known as genome assembly [1].
Several algorithms, models and tools have been used for the reconstruction of genomes after sequencing. However, such genomes may present sequences that repeat several times over a chromosome, e.g., regions codifying the ribosomal RNA (rRNA), transposases, regions of phages and plasmids. The assembly of these regions poses a significant challenge having high complexity for the assembler software [2].
In order to resolve the problem of repetitive sequences and to finish genome assemblies within a reasonable time, it becomes necessary to employ manual curation of gaps or even new sequencing of regions next to the gaps, which increases the cost of the assembly process. Thus, the steps required to finish the genome assembly are the major hurdles both in terms of cost and time [3].
Here, we propose a pipeline for assembly of repetitive regions, mainly in bacterial genomes, using the genome of an organism phylogenetically close to the one under study as reference. The proposed strategy enables the scaffolding of the contigs obtained by de novo assembly, including repetitive regions based on the extraction of the consensus sequence from the reads mapped into the reference genome (Figure 1).
Figure 1.
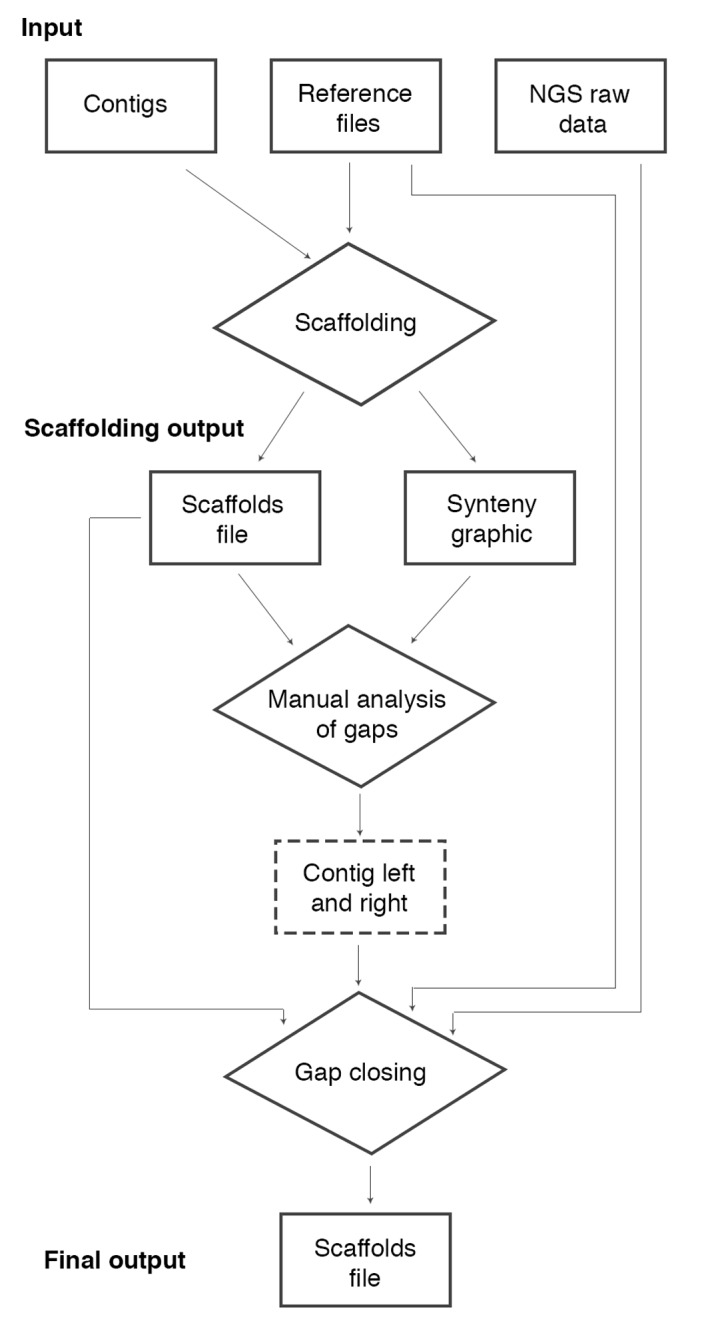
pipeline flowchart. The pipeline receives as input: contigs file, reference files (Fasta and GenBank files) and NGS raw data (reads file). The first step is the scaffolding of the contigs. This step can be realized by a modified version of the CONTIGuator software and it has as output a scaffolds file and a synteny graphic with colored targets indicating repetitive regions in the reference file and the gaps׳ positions in the scaffolds file. Using this file it is possible to conduct a manual analysis to choose two contigs׳ names as neighbors to a gap. Note that in the scaffolds file, we do not have contigs (orientated are called scaffolds), however we preserve this denomination in this flowchart to facilitate the comprehension. After this step, we developed the movednaa.py script to correct the beginning of the scaffold file for circular genomes searching the gene dnaA. We also developed the script cut_left.pl to remove barcodes on raw data, when needed. Thus, one can complete the assembly of repetitive regions based on the extraction of the consensus sequence of the mapping of raw data to the reference genome. To automate this step, we developed a software called MapRepeat. It receives as input the name of the two contigs and the path of the scaffolds file, reference Fasta file and the folder containing the NGS raw data file. MapRepeat has as output a new scaffolds file with a closed gap that was indicated in the step before. To analyze the result we developed the scripts: mcontig.py (to divided scaffold files in Multi-Fasta files breaking Ns regions) and contiginfo.py (to analyze number of gaps, length of the genome, length of larger and smaller contigs, and calculate the N50 value).
Methodology
Inputs:
(1) Multi-FASTA (Multiple FASTA format) file with contigs obtained by an assembler software, like Mira Assembler [4]; (2) Raw data file obtained by sequencing in NGS platforms in FASTQ, FASTQ/XML or FASTA/QUAL format (we recommend a depth coverage of approximately 50-fold for lower run time); (3) Two files of an organism of the same specie or genus to be used as reference: the first must contain a complete genome (nucleotide sequences in Fasta format), and the second has information about genes׳ annotation, in GBK format (GenBank Flat File Format). Both can be obtained from public data banks, like in the FTP utility of NCBI [5].
Determination of repetitive regions next to gaps:
We propose an approach for scaffolding the contigs through the software CONTIGuator v2.7 [6] (Figure 2A). The source code of CONTIGuator was modified to allow inputs as a GBK file. Additionally, these modifications allowed the insertion of colored targets in the synteny graphic generated by CONTIGuator. Targets of blue color indicate regions codifying ribosomal RNA; light blue color indicate regions codifying transposases; green color indicate phages and yellow color indicate plasmids. Through the synteny graphic of CONTIGuator, it is possible to identify a contig׳s neighbors when ordered and oriented properly; consequently, it is also possible to infer the existence of a repetitive region based on a similar region in the reference genome.
Figure 2.
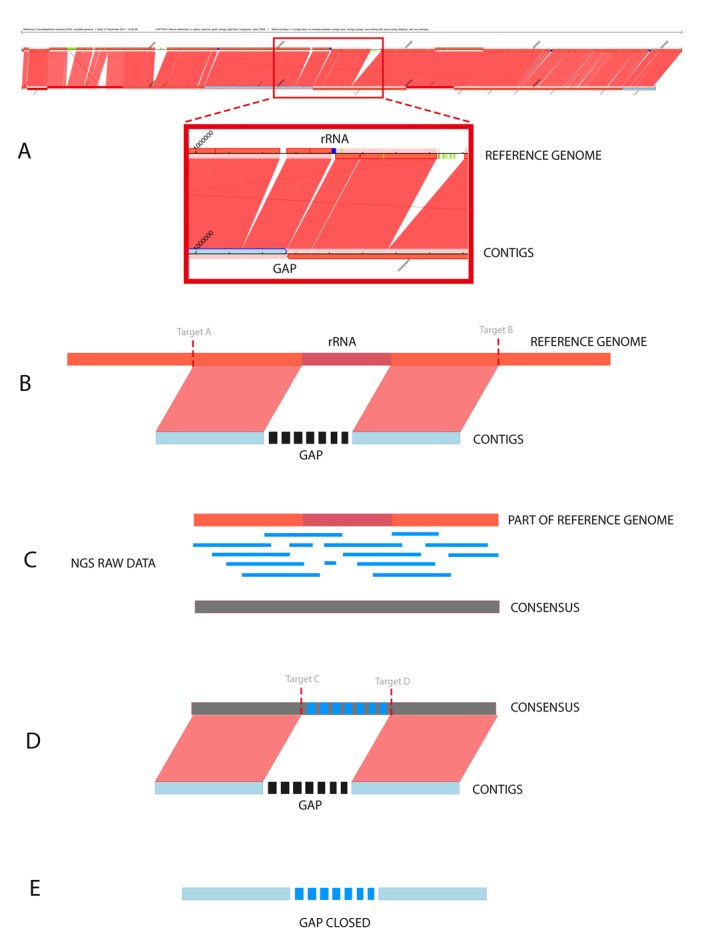
(A) Synteny graphic generated by CONTIGuator. The figure shows the reference genome on top, and below are the contigs aligned with the localization of gaps, which may be used as input parameters for MapRepeat; (B) First step of running MapRepeat. The software uses BLAST to detect whether there are similarities between two neighbors of contigs. If there are similarities, targets will be used to delimit the initial position of similarity with the left contig and the final position of similarity with the right contig. MapRepeat analyzes a region in the left contig until 3,000 pb before the gap and in the right contig until 3,000 pb after the gap; (C) The region between the targets is extracted, and then the raw data of sequencing are mapped against the extracted sequence using Mira assembler. A consensus sequence is generated based on whether there is coverage that proves the existence of this region in the reference genome and also in the genome sequenced; (D) The consensus sequence is aligned against the fragments of the two contigs. New targets (C and D) are used to identify the unknown regions from the contigs file mapped in the consensus; (E) The sequence contained between the targets C and D is extracted and used to close the gap.
Assembly of repetitive regions:
We developed a software, called MapRepeat, that can: (i) infer the position of repetitive regions on the contigs file based on the reference genome; (ii) assemble these regions after the scaffolding process performed by CONTIGuator; and (iii) close gaps. The software was implemented using the high-level programming language Python and the Biopython library. MapRepeat uses as input the following: (i) a FASTA file with the complete genome of the reference organism, (ii) a MultiFASTA file with the contigs, (iii) the directory name with all raw data files, and (iv) the name of the two contigs׳ neighbors of a determinate gap.
MapRepeat uses the software BLAST (Basic Local Alignment Search Tool) [7] to determine, in the reference genome, the position of syntenic regions and regions on the extremities of a gap (input contigs׳ neighbors), and it also stores the information on the targets A and B (Figure 2B). Then, the sequence between the targets A and B is extracted to be used in the mapping of raw data through the software Mira version 4.0. Thus it generates a consensus sequence (Figure 2C). BLAST is used one more time to determine the values of the targets C and D, that indicates the position of the beginning and end of a gap in the consensus sequence obtained (Figure 2D). Finally, the sequence contained between the targets C and D is extracted and used to close the gap (Figure 2E).
Case study:
To evaluate the efficacy of the proposed method, the genome of Corynebacterium ulcerans FRC11 (CuFRC11), access number CP009622, was used as a model. CuFRC11 was sequenced using the platform Ion Torrent™ Personal Genome Machine® (PGM) System (Life Technologies, USA) with 200 pb fragment library kit. The de novo assembly was performed with Mira 4.0 and produced a total of 30 contigs, N50 value of 236,335 and depth coverage for reads mapped to ~179× [9]. As reference genome, we used Corynebacterium ulcerans 0102 (Cu0102), access number NC_018101.1. The scaffolding was performed using our modified version of CONTIGuator. The synteny graphic showed the presence of four regions marked with a blue color, i.e., four clusters codifying rRNA. MapRepeat was used to close the gaps among the contigs: (i) frc11_c6 and frc11_c10; (ii) frc11_c7 and frc11_c8; (iii) frc11_c1 and frc11_c3; and (iv) frc11_c4 and frc11_c2. The extraction of a consensus of the mapping in the reference Cu0102 was successful for the four gaps. These were filled with sequence insertions of length 5,402 pb, 6,101 pb, 4,042 pb, and 4,606 pb, respectively. The BLAST online tool was used to prove that the inserted regions contained sequences codifying rRNA.
Discussion
The efficacy of our proposed method and the developed pipeline for resolving gaps in assemblies of bacterial genomes were illustrated by the results of the case study. They represent alternatives for the finishing of assemblies without additional costs, while also allowing for the code to be modified and adapted as per the needs of the pipeline. We point out that the strategies presented here can be performed through other software without great modifications in the final results. For example, the scaffolding process can be performed using the software Mauve [9], in addition with the BLAST web tool to detect the repetitive regions. We can also use the proprietary tool CLC Genomics Workbench (Qiagen, USA) for extraction of the consensus of the raw data mapping (this tool was used for the finalization of CuFRC11 in [8]).
Conclusion
The tools and methods proposed here are good alternatives for improving the process of finishing bacterial genomes, providing a reduction in costs and also accelerating the process. However, we currently have a command line interface for running the pipeline, which may present some difficulties to users with less informatics skills. All the software developed or modified, in addition to the scripts that can help in the genome assembly process have been made available for download at: http://github.com/dcbmariano /maprepeat. The corresponding documentation on the usage and installation of these tools has been included in the supplementary materials.
Prospects for the future
We aim to improve MapRepeat in future by including (i) the automation of the pipeline, (ii) the integration with other steps of the assembly process, and (iii) the construction of a userfriendly web-interface.
Acknowledgments
The authors would like to thank the following funding agencies: Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), Ministério da Pesca e Aquicultura (MPA), and Conselho Nacional de DesenvolvimentoCientífico e Tecnológico (CNPq).
Footnotes
Citation:Mariano et al, Bioinformation 11(6): 276-279 (2015)
References
- 1.Miller JR, et al. Genomics. 2010;95:315. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loman NJ, et al. Nat Rev Microbiol. 2012;10:599. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 3.Ribeiro FJ, et al. Genome res. 2012;22:2270. doi: 10.1101/gr.141515.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. http://mira-assembler.sourceforge.net.
- 5. ftp://ftp.ncbi.nih.gov/genomes/
- 6.Galardini M, et al. Source Code Biol Med. 2011;6:11. doi: 10.1186/1751-0473-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. http://blast.ncbi.nlm.nih.gov/Blast.cgi.
- 8.Benevides L, et al. Genome Announc. 2015;3:1. [Google Scholar]
- 9. http://asap.ahabs.wisc.edu/mauve.