

Abstract
T/C28a2 immortalized juvenile human chondrocytes were employed to determine the extent to which activation of Signal Transducers and Activators of Transcription-1 (STAT1) occurred in response to recombinant human interleukin-6 (rhIL-6) or rhIL-6 in combination with the soluble IL-6 receptor (sIL-6R). Two forms of STAT1, STAT1A and STAT1B, were identified on SDS-PAGE and western blotting with anti-STAT1 antibody. Western blotting revealed that STAT1 was constitutively phosphorylated (p-STAT1). Although incubation of T/C28a2 chondrocytes with rhIL-6 (50 ng/ml) increased p-STAT1A by Δ=22.3% after 30 min, this percent difference failed to reach significance by Chi-square analysis. Similarly, no effect of rhIL-6 (Δ=+10.7%) on p-STAT1B was seen at 30 min. In contrast, although the combination of rhIL-6 plus sIL-6R had no effect on p-STAT1A, rhIL-6 plus sIL-6R increased p-STAT1B by Δ=73.3% (p<0.0001) after 30 min compared to the control group and by Δ=56.7% (p<0.0001) compared to rhIL-6 alone. Janex-1, a Janus kinase-3-specific inhibitor (100 μM) partially reduced the effect of rhIL-6 on p-STAT1B by Δ=27.7% (p<0.05). The results of this study showed that STAT1A/STAT1B was constitutively activated in T/C28a2 chondrocytes. Although rhIL-6 increased p-STAT1B to a small extent, the combination of rhIL-6 plus sIL-6R was far more effective in stimulating STAT1B phosphorylation compared to controls or rhIL-6 alone. These data support the likelihood that although JAK3-mediated activation of STAT1 in T/C28a2 chondrocytes may involve the IL-6/IL-6R/gp130 pathway, these results indicated that STAT1 activation in response to IL-6 preferentially involved IL-6 trans-signaling via sIL-6R.
Keywords: Chondrocyte, Rheumatoid arthritis, Cytokine
Introduction
Mammalian signal transducers and activators of transcription (STAT) are cytoplasmic proteins comprised of Src homology-2 regions which are found in 7 forms, STAT1, 2, 3 and 4, STAT5a, STAT5b, and STAT6 [1-3]. STAT proteins are phosphorylated by one of several activated Janus kinases, JAK1, 2, 3, 5A, 5B, 6 following cytokine and/or growth factor binding to cytokine-specific receptors [2-5]. Phosphorylated STAT proteins (p-STATs) form STAT-homodimers and/or STAT-heterodimers in the cytoplasm where they undergo nuclear translocation. In the nucleus p-STAT proteins are potent transcription factors through their ability to bind to STAT-responsive DNA sequences in gene promoter regions [6]. Under certain conditions, un-phosphorylated STATs (U-STATs) may also possess transcription factor activity [7-9].
STAT-responsive DNA motifs regulate the expression of many pro-inflammatory and anti-inflammatory cytokine genes, including interleukin-6 (IL-6), oncostatin M, IL-10, interferon-γ (INF-γ), and tumor necrosis factor-α, as well as cytokine receptor genes, such as IL-2Rα and IL-18R1 [6]. These genes have been shown to modulate the transcription of several molecules known to be critical to cell cycle progression (e.g. Fos, Cyclin-D, p21waf1, CDC25A, c-Myc, PIM1) [10], cell survival (e.g. Bcl-2, Bcl-XL, β2-macroglobulin) [11] as well as hematopoietic stem cell [12] and T-cell development [13]. Innate [14] and adaptive immune responses [15] are also regulated by activated STAT proteins.
Recently, the JAK/STAT signaling pathway was identified as a potential target for pharmacologic intervention for rheumatoid arthritis (RA) mainly because several cytokines and growth factors known to activate JAK/STAT were found at significantly elevated levels in the sera and synovial fluid of RA patients [16-19]. Based on the results from these studies, the growth factors and pro-inflammatory cytokines that appear to be most relevant to the pathogenesis and progression of RA which also activate JAK/STAT, include fibroblast growth factor [20], platelet-derived growth factor [21], INF [22], IL-7 [23], IL-17 [24], IL-23 [25], and most prominently, members of the IL-6 cytokine family, including, IL-6, oncostatin M, ciliary neutrotrophic factor, leukemia inhibitory factor, cardiotrophin-1 and adiponectin [26-31].
In general, the IL-6-type cytokines phosphorylate the identical group of JAK-receptor complexes. However, in the case of IL-6, STAT1 or STAT3 were preferentially activated through the classical or “canonical” IL-6/IL-6R/gp130-receptor-mediator signaling [3,6,32,33] (Figure 1). However, there is less evidence for STAT1 activation occurring via the interaction of IL-6 with membrane-bound IL-6R (mIL-6R) or the “non-canonical” trans-signaling pathway involving the interaction between IL-6 and the soluble IL-6R (sIL-6R) (Figure 1).
Figure 1.
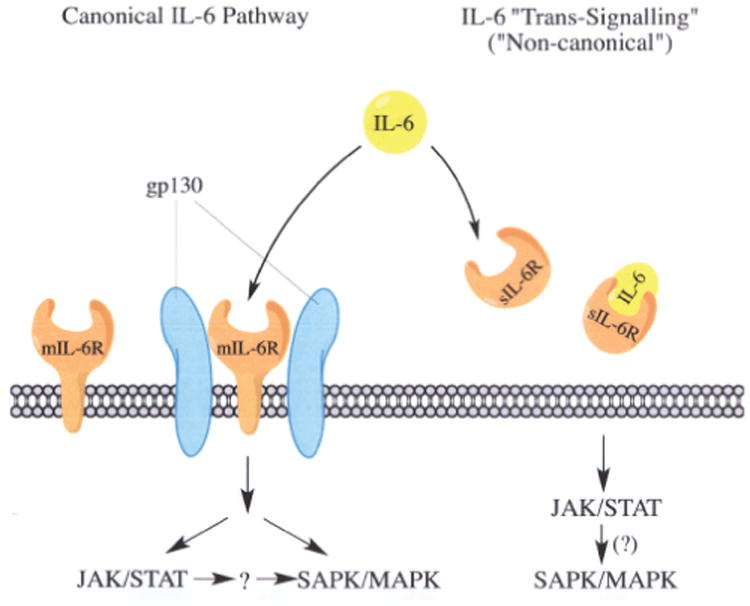
Activation of JAK/STAT by IL-6. The membrane-bound IL-6R (mIL-6R) pathway (far left), canonical IL-6/IL-6R/gp130 pathway (middle), and “non-canonical” IL-6 trans -signaling pathway (far right) is shown where IL-6 interacts with sIL-6R. The canonical IL-6/IL-6R/gp130 pathway is known to activate JAK/STAT and SAPK/MAPK signaling [3]. The JAK/STAT pathway activated by IL-6/IL-6R/gp130 may also activate the SAPK/MAPK pathway via a “cross-talk” mechanism (→?→) [3]. JAK/STAT could also be activated by IL-6 via mIL-6R in cells lacking the gp130 subunit [53]. Under these conditions activated STAT proteins may also initiate SAPK/MAPK signaling (↓?).
The deregulation of the JAK/STAT pathway by IL-6 and/or the IL-6 family of cytokines generally favors aberrant immune-cell and activated synoviocyte survival in RA synovial tissue [16,33-35] as well as “apoptosis-resistance” [34-36] the latter likely resulting from a constellation of cellular and molecular transitions perpetuating the inflammatory response [36]. However, fewer studies have been devoted to elucidating the extent to which activation of JAK/STAT signaling by the IL-6-family of cytokines may also affect articular chondrocyte survival and/or apoptosis in inflammatory arthritis.
The principal objective of this study was to determine the response of the immortalized human chondrocyte cell line, T/C28a2 [37,38] to recombinant human IL-6 (rhIL-6) alone or in combination with soluble IL-6 receptor (sIL-6R) with particular reference to their effects on U-STAT1 and p-STAT1 proteins. The T/C28a2 chondrocyte line was employed because these cells were previously shown to produce several cartilage-specific extracellular matrix proteins and to have additional characteristics of authentic human chondrocytes [37-41]. These include Type II collagen and aggrecan, expression of the adiponectin receptor, activation of MAPK signaling in response to IL-1β in a manner similar to the response of authentic human chondrocytes to IL-1β where activation of p38 MAPK and c-Jun-amino-terminal kinase (JNK) was reported [42,43] as well as PI3K/Akt/mTOR and protein kinase A signaling (Table 1). The T/C28a2 chondrocytes also were shown to express the cartilage-specific transcription factor SOX-9, the latter termed the “master-regulator of transcription” for several cartilage-specific genes, including COL2A1 and AGRN [38].
Table 1.
Some major phenotypic characteristics of t/c28a2 juvenile human chondrocytes.
| Phenotypic Characteristic | Reference |
|---|---|
| The synthesis of the cartilage proteoglycan biomarker, aggrecan, as well as synthesis of small leucine-rich proteoglycans, decorin and biglycan was confirmed | 37 |
| High-density cultures express the cartilage-specific transcription factor, SOX9 | 38 |
| IL-1β activated ERK1/2, p38 and JNK resulting in increased COX-2 and PGE2 synthesis | 39 |
| Expression of functional adiponectin (ApN) receptors | 40 |
| PGE2 stimulated TLR4 synthesis while activating ERK1/2, PI3K/Akt, PKA/CREB and NF-κB resulting in IL-6 gene expression | 41 |
| PGD2/15d-PGJ2 down-regulated TLR-4, while up-regulating caveolin-1 resulting in inhibition of PGE2-dependent ERK1/2, PI3K and PKA activation | 41 |
In the present study we focused on the extent to which rhIL-6 activated STAT1 primarily because STAT1 has been implicated as a regulator of IL-6 gene expression [6] as well as the finding that activated STAT1 regulates genes pertinent to chondrocyte proliferation, survival and apoptosis [44] including D1/Cdk4 [45], tumor necrosis factor receptor type 1-associated DEATH domain protein, p53 and histone deacetylases [44]. We also treated T/C28a2 chondrocytes with rhIL-6 in the presence and absence of the JAK3-specific inhibitor, WHI-P131 (Janex-1) to determine the extent to which inhibition of JAK3 reduced the phosphorylated form of p-STAT1 without affecting U-STAT1. We now know that JAK3 is abundantly expressed in normal cells of the lymphoid system as well as human lymphoblastic and myeloblastic leukemia cells. Janex-1 is a preferred small molecule inhibitor of JAK3 because it selectively inhibits JAK3 at an IC50 of 78 µM without altering the activity of JAK1 or JAK2, or any other protein tyrosine kinases (IC50 ≥ 350 µM) [46].
Materials and Methods
Materials
Human chondrocytes
The immortalized human juvenile chondrocyte cell line, T/C28a2 [37,38] was obtained from the laboratory of Professor Mary B. Goldring (Hospital for Special Surgery/Weill Medical College of Cornell University (New York, New York.)).
U-STAT1, p-STAT1 and β-actin antibodies
Antibodies which react specifically with U-STAT1 and P-STAT1 or with β-actin were obtained from R and D Systems.
Recombinant human (rh) IL-6 and soluble IL-6 Receptor (sIL-6R)
The cytokine, rhIL-6, and the soluble IL-6 receptor (sIL-6R) were obtained from ProSpec Bio and MyBioSource, respectively.
JAK inhibitor
WHI-P131 (Janex-1) was obtained from Cayman Chemical or EMD4 Biosciences.
Methods
Human T/C28a2 chondrocyte cultures
T/C28a2 chondrocytes were maintained in Dulbecco's Modified Eagle's Medium (DMEM)/F12 medium (1:1 ratio) supplemented with 10% (v/v) fetal bovine serum (FBS). For the measurement of U-STAT1 and p-STAT1 proteins, T/C28a2 chondrocytes were sub-passaged into 60 mm culture dishes at an initial density of 3 × 105 cells. The culture medium was changed every 2-3 days until the cells became confluent, generally within 3-5 days after passage. At that time, the medium was aspirated, chondrocytes washed once with phosphate-buffered saline and DMEM/F12 supplemented with 0.1% FBS (v/v) was added to each culture dish.
Experimental conditions
TC28a2 chondrocytes were incubated under the following conditions: No additions (control group A; see below), dimethyl sulfoxide (DMSO; control group B; see below), rhIL-6 (50 ng/ml), sIL-6R (30 ng/ml); rhIL-6 plus Janex-1 (100 μM) or rhIL-6 plus sIL-6R for zero min at room temperature (baseline) or for 30 min at 37°C. We employed the 30 min time point for analysis of STAT1 protein activation based primarily on our previously published western blot data [47] which showed that activated STAT proteins accumulated in human chondrocyte protein lysate in response to tumor necrosis factor-α for up to 30 min after which time the anti-p-STAT band was undetectable by western blotting.
Furthermore, the rationale for employing the concentrations of rhIL-6 and sIL-6R used in this study was determined from previously published data showing that IL-6 (50 ng/ml) activated chondrocyte JAK/STAT and ERK-MAPK signaling [48]. The concentration of sIL-6R employed in this study (30 ng/ml) was close to the highest concentration of sIL-6R reported in RA synovial fluid (range 10-40 ng/ml) [49]. In that regard, this level of IL-6 in RA synovial fluid is apparently reached as a result of IL-6 secretion from the infrapatellar fat pad into synovial fluid [50].
At the 0 and 30 min time points the culture medium was aspirated, the cell layers washed once with PBS, scraped into PBS and a cell pellet was collected by centrifugation. Protein lysates were prepared from the pelleted cells by sonication.
Western Blots
Western blots were produced as previously described [47]. However, in this study we employed a sequential antibody-probing/stripping technique to measure either U-STAT1 or p-STAT1 after which the immunoblot was stripped and reprobed with the β-actin antibody.
The bands of interest were identified, selected, and the average intensity of both U-STAT and p-STAT proteins and their associated β-actin control blots were measured from scanned images of autoradiographic western blot films using Metamorph® software. The average intensity values from each β-actin western blot were normalized to the highest value measured in control and treatment groups. The values calculated in this manner were then multiplied by the relative intensity values obtained for reactive bands in their affiliated blot (i.e. blots probed with U-STAT or p-STAT antibodies) in order to normalize each band to its corresponding band probed for β-actin. Finally, each of these calculated values were normalized to the experimental treatment (i.e. lane 1 of each blot) to obtain the relative intensity value and bar graphs produced with Excel.
A “no additions” control group was employed for comparing all of the other incubation conditions except those treatment groups which contained Janex-1. For that group, a control group containing DMSO was employed for comparison purposes because Janex-1 was initially dissolved in DMSO. Then the Janex-1 stock solution was diluted 1:1 with culture medium. Thus, the rhIL-6 plus Janex-1-containing group contained the same concentration of DMSO as did the DMSO control.
Data analysis
The difference in relative band intensities between control and treated groups is expressed as +/-Δ%. The significance of these single point values were analyzed by Chi-square. We set the predicted value difference between control and treated groups as Δ= ± 20% with p<0.05 considered significant.
Results
rhIL-6 increased U-STAT1A but not U-STAT1B
T/C28a2 chondrocytes produced 2 forms of STAT1 (Figure 2). U-STAT1A (band C) and U-STAT1B (band D) migrated to positions on SDS-PAGE previously identified as STAT1A and STAT1B [45,46].
Figure 2.
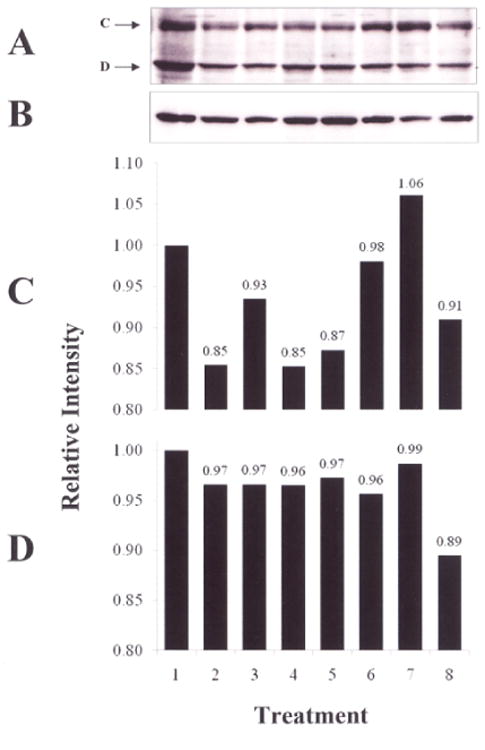
Western blot of U-STAT1. T/C28a2 chondrocytes were incubated for 0 or 30 min with 1) no additions; 2) DMSO; 3) 4) rhIL-6 (50 ng/ml); 5) rhIL-6+Janex-1 (100 μM) or rhIL-6 plus sIL-6R (30 ng/ml). The western blot shows A) U-STAT1A (band C); U-STAT1B (band D); B) β-actin; D) Quantified U-STAT1A and U-STAT1B. Lane 1, No additions, 0 min; Lane 2, DMSO, 0 min; Lane 3, rhIL-6+Janex-1, 0 min; Lane 4, No additions, 30 min; Lane 5, DMSO, 30 min; Lane 6, rhIL-6+Janex-1, 30 min; Lane 7, rhIL-6, 30 min; Lane 8, rhIL-6+sIL-6R, 30 min.
However, only minor changes were detected in the content of U-STAT1A and U-STAT1B among the various groups incubated for 0 min or 30 min with one exception. Incubation with rhIL-6 weakly increased U-STAT1A by Δ%=+24.7 (p>0.5) after 30 min (Figure 2C, lane 7), but did not alter the content of U-STAT1B (Δ%=3.1; p>0.05) (Figure 2D, lane 7). Of note, the combination of rhIL-6 and sIL-6R (Figure 2D, lane 8) did not alter the content of either U-STAT1A (Δ %=7.1; p>0.05) or U-STAT1B (Δ%=7.9; p>0.05) after 30 min, nor did Janex-1 affect these results (Figures 2C and 2D).
STAT1 is constitutively phosphorylated in T/C28a2 chondrocytes
Western blotting detected low levels of p-STAT1A and p-STAT1B at the 0 min time point (Figure 3, lanes 1, 2 and 3) indicative of constitutive phosphorylation of these STAT1 proteins by T/C28a2 chondrocytes.
Figure 3.

Western blot of p-STAT1. T/C28a2 chondrocytes were incubated for 0 or 30 min with 1) no additions; 2) DMSO; 3) 4) rhIL-6 (50 ng/ml); 5) rhIL-6+Janex-1 (100 μM) or rhIL-6 plus sIL-6R (30 ng/ml). The western blot shows A) p-STAT1A (band C); p-STAT1B (band D); B) β-actin; D) Quantified p-STAT1A and p-STAT1B. Lane1, No additions, 0 min; Lane 2, DMSO, 0 min; Lane 3, rhIL-6+Janex-1, 0 min; Lane 4, No additions, 30 min; Lane 5, DMSO, 30 min; Lane 6, rhIL-6+Janex-1, 30 min; Lane 7, rhIL-6, 30 min; Lane 8, rhIL-6+sIL-6R, 30 min.
Activation of p-STAT1A and p-STAT1B by rhIL-6 and rhIL-6 plus sIL-6R
Incubation of T/C28a2 chondrocytes with rhIL-6 (Figure 3, lane 7), resulted in an increase in p-STAT1A (Δ%=22.3). However, this difference did not reach statistical significance (p>0.05) (Table 2). Janex-1 (Figure 3, Lane 6) partially inhibited the rhIL-6-mediated increase in p-STAT1B (Δ=-27.7%; p<0.05) (Table 2).
Table 2.
The Δ% change (+/-) in p-STAT1A and p-STAT1B after treatment of T/C28a2 chondrocytes with rhIL-6, rhIL-6+Janex-1 or rhIL-6+sIL-6R. The Δ% change (+/-) between the control group and treatment group was calculated from the bar graphs generated by Metamorph® software of the p-STAT1 western blot shown in Figure 3.
| p-STAT | Control Group | Treated Group | +/-Δ% |
|---|---|---|---|
| 1A | No additions, 30′ | rhIL-6+sIL-6R | 0ns |
| 1A | No additions, 30′ | rhIL-6 | +22.3ns |
| 1B | No additions, 30′ | rhIL-6+sIL-6R | +73.3† |
| 1B | No additions, 30′ | rhIL-6 | +10.7ns |
| 1B | rhIL-6, 30′ | rhIL-6+Janex-1 | -27.7†† |
| 1B | DMSO, 30′ | rhIL-6+Janex-1 | -15.7ns |
| 1B | rhIL-6, 30′ | rhIL-6+sIL-6R | +56.6† |
p<0.0001 by Chi-square analysis;
p<0.05 by Chi-square analysis;
not significant by Chi-square analysis
The combination of rhIL-6 plus sIL-6R (Figure 3, lane 8) caused activation of p-STAT1B (Δ=73.3%; p<0.0001) after 30 min with no effect on p-STAT1A (Table 2). Of note, the combination of rhIL-6 plus sIL-6R activated STAT1B to a greater extent when compared to rhIL-6 alone (Δ=56.6%; p<0.0001) (Table 2).
Discussion
These results of this study showed a weak phosphorylation response of STAT1A and STAT1B in T/C28a2 juvenile chondrocytes in response to rhIL-6 with a much stronger STAT1B activation in response to the combination of rhIL-6 and sIL-6R. In addition, this latter incubation condition showed a greater p-STAT1B response compared to rhIL-6 alone. The finding that the JAK3-selective inhibitor, Janex-1, only partially reduced the level of p-STAT1B in response to rhIL-6 suggested the likelihood that other JAK forms, in addition to JAK3, were essential for activation of STAT1A and STAT1B by T/C28a2 chondrocytes.
Although the ultimate significance of these results to cartilage responses in RA remains to be determined, previous studies have shown that phosphorylation of STAT1 was a major regulator of chondrogenesis, but mainly in the context of insulin-like growth factor-3 binding protein [51] or after treatment with interferon-γ [52]. Moreover, the effect of combining IL-6 with sIL-6R on bovine articular chondrocytes correlated with the reduced expression of Type II collagen, aggrecan core protein, and link protein genes. The reduced expression of these cartilage-relevant genes was also associated with the down-regulation of SOX9 as well as with activation of JAK1/JAK2, STAT1/STAT3 and ERK1/2. Of note, activation of STAT proteins, but not ERK1/2 was IL-6-specific [53]. Additional regulatory factors, such as the IL-6-mediated activation of domain containing tyrosine phosphatase-2 (SHP-2), suppressor of cytokine signaling-3 (SOCS-3) and STAT3 nuclear phosphatase was reported to be involved in hepatocyte responses to IL-6 [54]. However, it is not yet known whether these factors also regulate chondrocyte gene responses as well.
Indeed, the focus of the present study represented only a minor component of a larger project designed to determine the effect of rhIL-6 and other IL-6 family members on the activation of 2 other STAT proteins, STAT3 and STAT5, as well for assessing the effect of neutralizing the IL-6 pathway (Figure 1) in immortalized human chondrocyte lines with tocilizumab on matrix metalloproteinase (MMP) synthesis [55] and apoptosis [Manuscripts in preparation]. However, only employing the IL-6R neutralizing monoclonal antibody, tocilizumab, will not likely provide the means for determining the extent to which rhIL-6 activates STAT proteins via the IL-6/IL-6R/gp130 complex, mIL-6R or sIL-6R (Figure 1). This is because previous reports have shown that tocilizumab specifically neutralized both sIL-6R and mIL-6R in addition to the IL-6R/gp130 complex [56,57].
The finding in the present study that the combination of rhIL-6 and sIL-6R had a significant effect on p-STAT1B now raises the likelihood that the IL-6/sIL-6 pathway (Figure 1) is the preferential pathway resulting in the activation of chondrocyte STAT proteins as well as for regulating cytokine and MMP gene expression by these cells, as was previously suggested by Sims and Walsh [58].
Finally, the finding that STAT1A and STAT1B were constitutively phosphorylated in T/C28a2 chondrocytes is very likely to be another indicator that the phenotype of this immortalized human chondrocyte line includes deregulated activation of JAK/STAT signaling and continuous proliferation. In that regard, we also found that STAT3 and STAT5 were constitutively phosphorylated in T/C28a2 chondrocytes [unpublished data]. Of note, the dichotomy that exists between the activation of STAT3 as a pro-survival STAT protein and STAT1 as a pro-apoptosis STAT protein is a well-recognized component of tumorigenesis [59]. Therefore, in an attempt to address this issue, we are at present employing another juvenile human chondrocyte line, C28/I2 [38], to determine whether or not STAT1, STAT3 and STAT5 proteins are constitutively activated as well.
Acknowledgments
This study was supported, in part, by a contract from Genentech/Roche group awarded to Charles J. Malemud, PhD and Case Western Reserve University (CWRU), and also by the CWRU Visual Sciences Center Core Grant from the National Eye Institute (P30 EY11373).
References
- 1.Ivashkiv LB, Hu X. Signaling by STATs. Arthritis Res Ther. 2004;6:159–168. doi: 10.1186/ar1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 3.Malemud CJ, Pearlman E. Targeting JAK/STAT signaling pathway in inflammatory disorders. Curr Signal Transduct Ther. 2009;4:201–221. [Google Scholar]
- 4.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 5.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- 6.Malemud CJ. Suppression of pro-inflammatory cytokines via targeting of STAT-responsive genes. In: El-Shemy H, editor. Drug Discovery. Rijeka: InTech; 2013. pp. 373–411. [Google Scholar]
- 7.Cheon H, Yang J, Stark GR. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J Interferon Cytokine Res. 2011;31:33–40. doi: 10.1089/jir.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J Biol Chem. 2012;287:14192–14200. doi: 10.1074/jbc.M111.323899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stark GR, Darnell JE., Jr The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barré B, Avril S, Coqueret O. Opposite regulation of myc and p21waf1 transcription by STAT3 proteins. J Biol Chem. 2003;278:2990–2996. doi: 10.1074/jbc.M210422200. [DOI] [PubMed] [Google Scholar]
- 11.Abell K, Watson CJ. The Jak/Stat pathway: a novel way to regulate PI3K activity. Cell Cycle. 2005;4:897–900. doi: 10.4161/cc.4.7.1837. [DOI] [PubMed] [Google Scholar]
- 12.Kaimakis P, Crisan M, Dzierzak E. The biochemistry of hematopoietic stem cell development. Biochim Biophys Acta. 2013;1830:2395–2403. doi: 10.1016/j.bbagen.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011;34:39–49. doi: 10.1016/j.immuni.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemon SM. Induction and evasion of innate antiviral responses by hepatitis C virus. J Biol Chem. 2010;285:22741–22747. doi: 10.1074/jbc.R109.099556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knosp CA, Johnston JA. Regulation of CD4+ T-cell polarization by suppressor of cytokine signalling proteins. Immunology. 2012;135:101–111. doi: 10.1111/j.1365-2567.2011.03520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker JG, Smith MD. The Jak-STAT pathway in rheumatoid arthritis. J Rheumatol. 2005;32:1650–1653. [PubMed] [Google Scholar]
- 17.Malemud CJ, Reddy SK. Targeting cytokines, chemokines and adhesion molecules in rheumatoid arthritis. Curr Rheum Rev. 2008;4:219–234. [Google Scholar]
- 18.Imboden JB. The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol. 2009;4:417–434. doi: 10.1146/annurev.pathol.4.110807.092254. [DOI] [PubMed] [Google Scholar]
- 19.Malemud CJ. Recent advances in neutralizing the IL-6 pathway in arthritis. Open Access Rheumatol Res Rev. 2009;1:133–150. doi: 10.2147/oarrr.s6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malemud CJ. Growth hormone, VEGF and FGF: involvement in rheumatoid arthritis. Clin Chim Acta. 2007;375:10–19. doi: 10.1016/j.cca.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 21.Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D. Angiogenesis in rheumatoid arthritis. Histol Histopathol. 2006;21:557–566. doi: 10.14670/HH-21.557. [DOI] [PubMed] [Google Scholar]
- 22.Conigliaro P, Perricone C, Benson RA, Garside P, Brewer JM, et al. The type I IFN system in rheumatoid arthritis. Autoimmunity. 2010;43:220–225. doi: 10.3109/08916930903510914. [DOI] [PubMed] [Google Scholar]
- 23.Churchman SM, Ponchel F. Interleukin-7 in rheumatoid arthritis. Rheumatology (Oxford) 2008;47:753–759. doi: 10.1093/rheumatology/ken053. [DOI] [PubMed] [Google Scholar]
- 24.Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. Autoimmune inflammation from the Th17 perspective. Autoimmun Rev. 2007;6:169–175. doi: 10.1016/j.autrev.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Paradowska-Gorycka A, Grzybowska-Kowalczyk A, Wojtecka-Lukasik E, Maslinski S. IL-23 in the pathogenesis of rheumatoid arthritis. Scand J Immunol. 2010;71:134–145. doi: 10.1111/j.1365-3083.2009.02361.x. [DOI] [PubMed] [Google Scholar]
- 26.Malemud CJ. Differential activation of JAK enzymes in rheumatoid arthritis and autoimmune disorders by pro-inflammatory cytokines: potential drug targets. Int J Interferon Cytokine Mediator Res. 2010;2:97–111. [Google Scholar]
- 27.Hams E, Colmont CS, Dioszeghy V, Hammond VJ, Fielding CA, et al. Oncostatin M receptor-beta signaling limits monocytic cell recruitment in acute inflammation. J Immunol. 2008;181:2174–2180. doi: 10.4049/jimmunol.181.3.2174. [DOI] [PubMed] [Google Scholar]
- 28.Tang CH, Chiu YC, Tan TW, Yang RS, Fu WM. Adiponectin enhances IL-6 production in human synovial fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-kappa B pathway. J Immunol. 2007;179:5483–5492. doi: 10.4049/jimmunol.179.8.5483. [DOI] [PubMed] [Google Scholar]
- 29.Silver JS, Hunter CA. gp130 at the nexus of inflammation, autoimmunity, and cancer. J Leukoc Biol. 2010;88:1145–1156. doi: 10.1189/jlb.0410217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan Z, Bau B, Yang H, Aigner T. IL-1beta induction of IL-6 and LIF in normal articular human chondrocytes involves the ERK, p38 and NFkappaB signaling pathways. Cytokine. 2004;28:17–24. doi: 10.1016/j.cyto.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 31.Malemud CJ. Small molecular weight inhibitors of stress-activated and mitogen-activated protein kinases. Mini Rev Med Chem. 2006;6:689–698. doi: 10.2174/138955706777435670. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Badgwell DB, Bevers JJ, 3rd, Schlessinger K, Murray PJ, et al. IL-6 signaling via the STAT3/SOCS3 pathway: functional analysis of the conserved STAT3 N-domain. Mol Cell Biochem. 2006;288:179–189. doi: 10.1007/s11010-006-9137-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malemud CJ. Suppression of autoimmune arthritis by small molecule inhibitors of the JAK/STAT pathway. Pharmaceuticals. 2010;3:1446–1455. doi: 10.3390/ph3051446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tak PP, Firestein GS. Apoptosis in rheumatoid arthritis. In: Winkler JD, editor. Apoptosis and Inflammation-Progress in Inflammation Research. Basel: Birkhauser; 1999. pp. 149–162. [Google Scholar]
- 35.Lewis AC, Malemud CJ. Correction of dysfunctional apoptosis in arthritis by pharmacologic interventions: Focus on altering the activity of inhibitor of apoptosis protein. In: Pandalai SG, editor. Recent Research Developments in Pharmacology. Kerala: Research Signpost; 2011. pp. 69–84. [Google Scholar]
- 36.Malemud CJ. Intracellular signaling pathways in rheumatoid arthritis. J Clin Cell Immunol. 2013;4:160. doi: 10.4172/2155-9899.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kokenyesi R, Tan L, Robbins JR, Goldring MB. Proteoglycan production by immortalized human chondrocyte cell lines cultured under conditions that promote the expression of the differentiated state. Arch Biochem Biophys. 2000;383:79–90. doi: 10.1006/abbi.2000.2044. [DOI] [PubMed] [Google Scholar]
- 38.Finger F, Schörle C, Zien A, Gebhard P, Goldring MB, et al. Molecular phenotyping of human chondrocyte cell lines T/C-28a2, T/C-28a4, and C-28/I2. Arthritis Rheum. 2003;48:3395–3403. doi: 10.1002/art.11341. [DOI] [PubMed] [Google Scholar]
- 39.Lago R, Gomez R, Otero M, Lago F, Gallego R, et al. A new player in cartilage homeostasis: adiponectin induces nitric oxide synthase type II and pro-inflammatory cytokines in chondrocytes. Osteoarthritis Cartilage. 2008;16:1101–1109. doi: 10.1016/j.joca.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Nieminen R, Leinonen S, Lahti A, Vuolteenaho K, Jalonen U, et al. Inhibitors of mitogen-activated protein kinases downregulate COX-2 expression in human chondrocytes. Mediators Inflamm. 2005;2005:249–255. doi: 10.1155/MI.2005.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang P, Zhu F, Konstantopoulos K. Interleukin-6 synthesis in human chondrocytes is regulated via the antagonistic actions of prostaglandin (PG)E2 and 15-deoxy-Δ(12,14)-PGJ2. PLoS One. 2011;6:e27630. doi: 10.1371/journal.pone.0027630. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Singh R, Ahmed S, Malemud CJ, Goldberg VM, Haqqi TM. Epigallocatechin-3-gallate selectively inhibits interleukin-1ß-induced activation of mitogen activated protein kinase subgroup c-Jun N-terminal kinase in human osteoarthritis chondrocytes. J Orthop Res. 2003;21:102–109. doi: 10.1016/S0736-0266(02)00089-X. [DOI] [PubMed] [Google Scholar]
- 43.Malemud CJ. MAP kinases. In: Buckwalter J, Lotz M, Stoltz JF, editors. OA, Inflammation and Degradation: A Continuum. Vol. 70. Biomedical and Health Research, IOS Press; Amsterdam: 2007. pp. 99–117. [Google Scholar]
- 44.Kim HS, Lee MS. STAT1 as a key modulator of cell death. Cell Signal. 2007;19:454–465. doi: 10.1016/j.cellsig.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Dimco G, Knight RA, Latchman DS, Stephanou A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle. 2010;9:4638–4649. doi: 10.4161/cc.9.23.13955. [DOI] [PubMed] [Google Scholar]
- 46.Sudbeck EA, Liu XP, Narla RK, Mahajan S, Ghosh S, et al. Structure-based design of specific inhibitors of Janus kinase 3 as apoptosis-inducing antileukemic agents. Clin Cancer Res. 1999;5:1569–1582. [PubMed] [Google Scholar]
- 47.Malemud CJ, Sun Y, Pearlman E, Ginley NM, Awadallah A, et al. Monosodium urate and tumor necrosis factor-α increase apoptosis in human chondrocyte cultures. Rheumatology (Sunnyvale) 2012;2:113. doi: 10.4172/2161-1149.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aida Y, Honda K, Tanigawa S, Nakayama G, Matsumura H, et al. IL-6 and soluble IL-6 receptor stimulate the production of MMPs and their inhibitors via JAK-STAT and ERK-MAPK signalling in human chondrocytes. Cell Biol Int. 2012;36:367–376. doi: 10.1042/CBI20110150. [DOI] [PubMed] [Google Scholar]
- 49.Kotake S, Sato K, Kim KJ, Takahashi N, Udagawa N, et al. Interleukin-6 and soluble interleukin-6 receptors in the synovial fluids from rheumatoid arthritis patients are responsible for osteoclast-like cell formation. J Bone Miner Res. 1996;11:88–95. doi: 10.1002/jbmr.5650110113. [DOI] [PubMed] [Google Scholar]
- 50.Ushiyama T, Chano T, Inoue K, Matsusue Y. Cytokine production in the infrapatellar fat pad: another source of cytokines in knee synovial fluids. Ann Rheum Dis. 2003;62:108–112. doi: 10.1136/ard.62.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spagnoli A, Torello M, Nagalla SR, Horton WA, Pattee P, et al. Identification of STAT-1 as a molecular target of IGFBP-3 in the process of chondrogenesis. J Biol Chem. 2002;277:18860–18867. doi: 10.1074/jbc.M200218200. [DOI] [PubMed] [Google Scholar]
- 52.Krejci P, Prochazkova J, Bryja V, Jelinkova P, Pejchalova K, et al. Fibroblast growth factor inhibits interferon gamma-STAT1 and interleukin 6-STAT3 signaling in chondrocytes. Cell Signal. 2009;21:151–160. doi: 10.1016/j.cellsig.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Legendre F, Dudhia J, Pujol JP, Bogdanowicz P. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of SOX9 expression. J Biol Chem. 2003;278:2903–2912. doi: 10.1074/jbc.M110773200. [DOI] [PubMed] [Google Scholar]
- 54.Singh A, Jayaraman A, Hahn J. Modeling regulatory mechanisms in IL-6 signal transduction in hepatocytes. Biotechnol Bioeng. 2006;95:850–862. doi: 10.1002/bit.21026. [DOI] [PubMed] [Google Scholar]
- 55.Lim H, Kim HP. Matrix metalloproteinase-13 expression in IL-1β treated chondrocytes by activation of the p38 MAPK/c-Fos/AP-1 and JAK/STAT pathways. Arch Pharm Res. 2011;34:109–117. doi: 10.1007/s12272-011-0113-4. [DOI] [PubMed] [Google Scholar]
- 56.Mihara M, Kasutani K, Okazaki M, Nakamura A, Kawai S, Sugimoto M, et al. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int Immunopharmacol. 2005;5:1731–1740. doi: 10.1016/j.intimp.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka T, Kishimoto T. Immunotherapy of tocilizumab for rheumatoid arthritis. J Clin Cell Immunol. 2013;S6:001. [Google Scholar]
- 58.Sims NA, Walsh NC. GP130 cytokines and bone remodelling in health and disease. BMB Rep. 2010;43:513–523. doi: 10.5483/bmbrep.2010.43.8.513. [DOI] [PubMed] [Google Scholar]
- 59.Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT. 2012;1:65–72. doi: 10.4161/jkst.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]