

Abstract
A single amino acid mutation near the active site of the CAPN5 protease was linked to the inherited blinding disorder, autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV, OMIM #193235). In homology modeling with other calpains, this R243L CAPN5 mutation was situated in a mobile loop that gates substrate access to the calcium-regulated active site. In in vitro activity assays, the mutation increased calpain protease activity and made it far more active at low concentrations of calcium. To test whether the disease allele could yield an animal model of ADNIV, we created transgenic mice expressing human (h) CAPN5R243L only in the retina. The resulting hCAPN5R243L transgenic mice developed a phenotype consistent with human uveitis and ADNIV, at the clinical, histological and molecular levels. The fundus of hCAPN5R243L mice showed enhanced autofluorescence (AF) and pigment changes indicative of reactive retinal pigment epithelial cells and photoreceptor degeneration. Electroretinography showed mutant mouse eyes had a selective loss of the b-wave indicating an inner-retina signaling defect. Histological analysis of mutant mouse eyes showed protein extravasation from dilated vessels into the anterior chamber and vitreous, vitreous inflammation, vitreous and retinal fibrosis and retinal degeneration. Analysis of gene expression changes in the hCAPN5R243L mouse retina showed upregulation of several markers, including members of the Toll-like receptor pathway, chemokines and cytokines, indicative of both an innate and adaptive immune response. Since many forms of uveitis share phenotypic characteristics of ADNIV, this mouse offers a model with therapeutic testing utility for ADNIV and uveitis patients.
Introduction
The molecular basis of uveitis (intraocular inflammation) is poorly understood (1–7). Until recently, primary (non-syndromic) uveitis was not linked to any causative gene, since families with inherited uveitis are rare. We ascertained two large kindreds with an inherited uveitis; autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV, OMIM #193235). ADNIV patients' eyes develop a severe autoinflammatory uveitis and retinal degeneration with vitreous inflammation, vitreous and retinal fibrosis, cataract, cystoid macular edema and retinal neovascularization. Our studies of human autopsy eyes indicated that ADNIV uveitis is primarily driven by cell-mediated immunity (8,9). Proteomic profiling of vitreous biopsies revealed that interleukin-6 (IL-6) was elevated in ADNIV, suggesting a stimulated inflammatory or autoimmune response (10). Nevertheless, like many other uveitis patients, ADNIV patients respond poorly to conventional immunosuppressive therapies and incompletely to corticosteroids (11).
At different disease stages, ADNIV mimics severe, progressive uveitis, retinitis pigmentosa, proliferative diabetic retinopathy and proliferative vitreoretinopathy (8,9,11–16), a set of eye diseases that account for a large fraction of visual morbidity and blindness (17). Discoveries about the causes of ADNIV might have therapeutic implications for a wide variety of ocular diseases. To find causative genes for ADNIV, two ADNIV kindreds were analyzed via whole exome sequencing, and each harbored a unique missense mutation in the coding region of CAPN5, a calcium-activated cysteine protease expressed in photoreceptor cells (18,19). We recently identified a third ADNIV family (20), and all three mutations lie in the catalytic domain of CAPN5 (21,22). Our homology modeling to calpains generated a three-dimensional structure for calpain-5 (23–26), which showed all mutations were in a calcium-sensitive, flexible loop that gates substrate access to the active site (20,25,27). However, the effect of the mutation on calpain activity was not known until it was tested in this study.
Despite CAPN5 expression in multiple tissues, ADNIV patients only develop disease in the eyes. It is not clear whether disease is driven by the immune system or by the retina. Retina-specific CAPN5 vitreoretinopathy was tested in a mouse model, using lentiviral transduction to express one of the ADNIV human CAPN5 mutant cDNAs, hCAPN5R243L (16). Lentiviral vectors were injected into the sub-retinal space of perinatal mice. Retinal expression of the disease allele was sufficient to recapitulate the major features of the ADNIV phenotype: loss of the electroretinogram (ERG) b-wave, photoreceptor degeneration and inflammatory gene expression (16). Despite these changes, the inflammatory phenotype was limited compared with the human disease phenotype, likely because the lentivirus delivery method, although rapid, has a number of shortcomings. First, effects are usually localized near the site of injection and the number of transduced cells is low (typically 20–30% of cells). Thus it was not surprising that the lentivirus model caused a less severe inflammatory response (16), a limited loss of ERG function, and death in a minor fraction of photoreceptors and cells lining the inner nuclear layer of the eye. In addition, with lentiviral transduction, the mutation was not transmitted through generations, so the system offers a limited utility for testing therapeutics. Given the encouraging results of ERG b-wave reduction, photoreceptor degeneration and inflammatory gene expression with the hCAPN5R243L lentiviral studies, we were optimistic that a transgenic mouse expressing the hCAPN5R243L transgene in the retina would provide a better model of the ADNIV disease.
Results
The ADNIV DII gating-loop mutation increases CAPN5 catalytic activity
All calpains have a proteolytic core with two subdomains (domains IIa and IIb) that contain four flexible loops that undergo significant conformational changes upon binding calcium in CAPN1 (Fig. 1A), shifting the enzyme into an active form (26,28,29). Although areas of domain II (DII) are highly conserved, CONSURF analysis (data not shown) and previous studies indicate that the mobile loops are among the more variable features near calpain active sites (23), which may give the calpain family its wide range of calcium sensitivity and substrate specificity. The four flexible loops are highlighted in the sequence alignment comparing CAPN5 to CAPN1 (Fig. 1B). In both ADNIV cohorts, the second flexible loop in DIIb contains a CAPN5 point mutation. This loop-2 has been implicated in gating substrate access to the catalytic groove (23,26). Calcium binds to other loops within the proteolytic core (green spheres), eliciting a conformational change in the DIIb gating loop-2 that opens the catalytic groove and activates the enzyme (23,26,28,29).
Figure 1.

Generation of a recombinant mini-CAPN1/5 hybrid with loop 2 of proteolytic domain IIb (DIIb) swapped from CAPN5 into the homologous region in mini-CAPN1. The mini-calpain system allows the catalytic activity of domain II to be isolated and assayed, since full-length calpains are generally not stable. (A) Cartoon representation of the rat mini-CAPN5 structure with calcium bound (green spheres), highlighting the active site residues (yellow sticks), the four flexible loops (pink) and the ADNIV mutations (red dotted circle). (B) Sequence alignment of domain II of rat mini-CAPN1 and human mini-CAPN5 highlighting the flexible loops (pink box). (C) Schematic representation showing proteins used in our modified mini-calpain system. Mini-CAPN1, a hybrid mini-CAPN1/5 with loop 2 of mini-CAPN1 swapped out with that of mini-CAPN5 and a hybrid mini-CAPN1/5 harboring the p.R243L ADNIV mutation.
Based on structural modeling (13), our hypothesis was that a CAPN5 gain-of-function might arise if the ADNIV mutations disrupt the DII gating loop-2 and increase substrate access to the catalytic groove. We aimed to recreate the disease scenario in mice, but first needed to confirm the gain-of-function phenotype for a DIIb gating loop-2 mutation. Testing calpain enzymatic activity in vitro, however, is not straightforward, because purified CAPN5 is inherently unstable (unpublished observation), and its substrates are not known (so no in vitro activity assay has been developed). To circumvent these problems, we designed a CAPN1/5 hybrid, in which the DIIb gating loop-2 from human CAPN5 (amino acids KAVTAADMEARLACG) replaced the corresponding sequence in the proteolytic core of a rat mini-CAPN1 (NISDIRDLEAITFKN; Fig. 1C). This hybrid mini-calpain strategy has been used previously to purify and test the enzymatic activity of other calpains (23) and takes advantage of a robust purification method and established activity assay for rat mini-CAPN1.
Mini-CAPN1/5 hybrids were created in three maltose-binding protein (MBP)-tagged forms: one containing the wild-type (WT) CAPN5 DIIb gating loop-2, a form expected to be catalytically active; another, carrying a p.C81S mutation at the catalytic triad, which was expected to be inactive; and a third carrying the disease-causing mutation (p.R243L), which we hypothesized would show a gain-of-function. Introduction of the CAPN5 gating loop did not affect cleavage activity. The CAPN1/5 WT hybrid (positive control) robustly cleaved a fluorescent, peptide substrate (AC-LLY-AFC; Fig. 2A and B) recognized by mini-CAPN1. Adding a peptide substrate inhibitor abolished all cleavage activity (Fig. 2A). In addition, the negative-control p.C81S mutant, with its defective catalytic triad, showed no activity (Fig. 2B). These controls demonstrated that the mini-CAPN1/5 hybrid with the DIIb gating loop-2 from WT CAPN5 displays characteristic CAPN1 activity.
Figure 2.
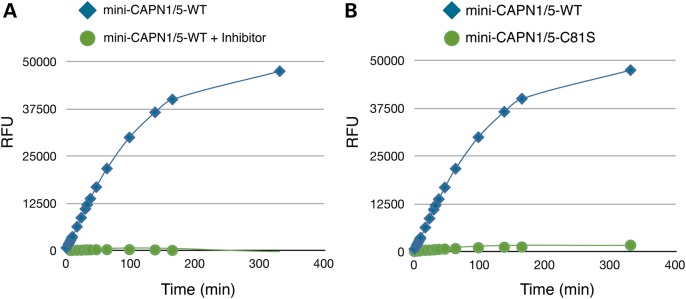
A mini-CAPN1/5 hybrid is active in in vitro assays. A mini-CAPN1/5 is sensitive to a calpain inhibitor as well as mutation of the catalytic cysteine residue (C81S) that renders all calpains enzymatically inactive. (A) Time course of substrate peptide (AC-LLY-AFC) shows catalytic activity of CAPN1/5 hybrid with (green) and without (blue) a calpain peptide inhibitor (Z-LLY-AFC). (B) Catalytic activity of CAPN1/5 hybrid bearing a mutation of the catalytic cysteine residue (C81S; green) renders it inactive. RFU, relative fluorescent units.
Next, the hybrid containing the p.R243L mutant DIIb gating loop-2 was compared with the WT hybrid. The disease mutant had significantly increased calpain catalytic activity at all time points and calcium concentrations tested compared with the WT (Fig. 3). This suggests that the gain-of-function mutation relieves CAPN5 of strict calcium regulation. Indeed, the mutant hybrid cleaved almost as much substrate in the presence of 0.1 mm calcium (Fig. 3B) as did the WT hybrid at 100 mm calcium (Fig. 3D). These results support the assertion that the DIIb gating loop-2 controls calpain catalytic activity, and mutations in that loop can increase enzymatic catalysis. Moreover, these experiments strongly suggest that the effect of the ADNIV mutation is to increase CAPN5 catalytic activity, an idea consistent with the dominant inheritance, high penetrance and disease severity observed in mutant CAPN5 vitreoretinopathy patients. This insight was next supported by physiologic testing of the CAPN5R243L mutation in vivo.
Figure 3.

R243L ADNIV mutation increases catalytic activity of a recombinant mini-CAPN1/5 hybrid. The R243L mutation renders the mini-CAPN1/5 hybrid over 300% more sensitive to calcium (activity is a ratio normalized to mini-CAPN1; control is the reaction without calpain). (A) A time-course comparison of the CAPN1/5 hybrid and a hybrid bearing the R243L mutation, showing catalysis of a standard peptide substrate at 0.01 mm calcium, (B) 0.10 mm calcium, (C) 1.0 mm calcium and (D) 100 mm calcium. RFU, relative fluorescent units.
A transgenic mouse model expresses ADNIV-associated hCAPN5R243L in the retina
To test the idea that a mouse expressing the hyperactive CAPN5 mutation in the retina would develop an ADNIV phenotype and yield a preclinical model for uveitis, we tested the effects of retina-specific expression of hCAPN5R243L. Transgenic lines were created that expressed a full-length cDNA of the hCAPN5R243L mutant, using a 4.2 kb mouse opsin promoter, conferring retinal-specific expression (see targeting construct in Fig. 4A and B and Materials and Methods). Transgenic mice were backcrossed into the C57BL/6J background, yielding B6xCBA-Tg(Rho-hCAPN5R243L)vbm mice. Immunoblotting directed against the MYC tag located in the transgenic construct confirmed the human mutant (hCAPN5R243L) protein was expressed in the retina (Fig. 4C).
Figure 4.
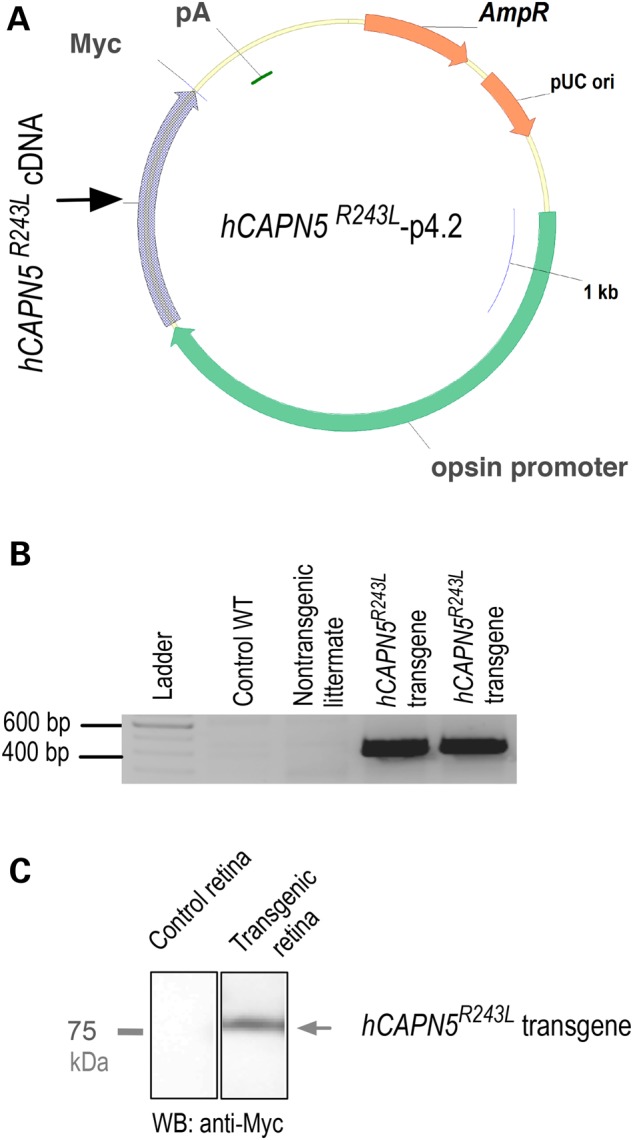
Transgenic ADNIV mutant construct. (A) A p4.2 construct designed for opsin promoter-driven expression of a Myc-tagged human CAPN5R243L transgene (pA, polyadenylation signal; AmpR, ampicillin-resistant selection gene; pUC ori, origin of replication). (B) The hCAPN5R243L transgene expression was detected by reverse transcriptase polymerase chain reaction (RT-PCR) in two transgenic mice as compared with a WT control mouse and a non-transgenic mouse from the same litter. (C) Anti-Myc immunoblot confirms transgene expression in the retinal extract.
The clinical and histological phenotype of the hCAPN5R243L mice is consistent with ADNIV
Retinal degeneration in ADNIV patients can be detected non-invasively by measuring autofluorescence (AF) and infrared reflectance (IR) of the fundus (30), a technique we used to compare the phenotypes of our transgenic mouse model to human ADNIV patients (31). AF imaging showed that, compared with the normal human eye (Fig. 5A) and WT mice (Fig. 5D), the fundus of mutant mice displayed enhanced AF from 9 to 18 weeks of age (Fig. 5E and F), similar to the fundus of ADNIV patients (Fig. 5B and C). IR imaging of the retina also showed pigmentary changes indicative of reactive retinal pigment epithelial (RPE) cells and photoreceptor degeneration from 9 to 18 weeks of age (Fig. 5H and I), compared with WT mice (Fig. 5G). At middle stages, CAPN5 vitreoretinopathy in humans is characterized by cataract and retinal neovascularization (11,13,14). At 4 months, hCAPN5R243L mouse eyes exhibited neither obvious clinical signs of cataract nor neovascularization on slit lamp and funduscopic examination.
Figure 5.

AF and IR imaging of human and mouse WT and hCAPN5R243L retinas reveals early retinal degeneration. (A) The normal human AF fundus image. (B) Mid- to late-stage ADNIV eyes show increased AF that may be associated with photoreceptor or RPE cell dysfunction. (C) IR imaging of an ADNIV patient shows scattered pigment accumulation (arrows). Image quality is limited by cataract and vitreous membranes. (D) AF image of a control WT mouse at 18 weeks of age. (E) AF imaging of hCAPN5R243L mice at 9 weeks of age shows increased AF compared with the control. (F) Further autofluorescent increase is observed by 18 weeks of age, as seen in human ADNIV eyes. (G) IR imaging of a control WT mouse fundus. (H) IR imaging of a hCAPN5R243L mouse fundus at 9 weeks of age, and (I) by 18 weeks of age scattered, melanin-like pigment is accumulating in the hCAPN5R243L mouse fundus (arrow).
Patients with CAPN5 vitreoretinopathy display a number of other phenotypic features. Their chronic uveitis is characterized by protein extravasation from dilated vessels into the anterior chamber and vitreous, iris foreshortening, fibrosis in the vitreous and retina and retinal degeneration (Fig. 6) (8,9). To look for similar pathologies in our mouse model, the eyes were analyzed by histology. Pupil-optic nerve sections from mutant eyes appeared normal at 2 months of age, but showed abnormalities at 4 months consistent with intraocular inflammation (Fig. 7B) in comparison to WT mice (Fig. 7A). The hCAPN5R243L eyes showed dense protein extravasation into the anterior chamber, and there were dilated vessels in contracted irises (Fig. 7B and C). At the histological level, subtle evidence of cataracts became detectable that had not been detected clinically. Cataract formation was apparent with posterior migration of lens epithelial cells (Fig. 7D). The vitreous also showed signs of inflammation including cell infiltration, protein extravasation, fibrotic bands and early neovascularization (Fig. 7E), mirroring the effects of mid- to late-stage vitreoretinopathy in patients harboring mutant CAPN5. Epiretinal fibrosis and dilated retinal vessels were found in the histological samples (Fig. 7F and G). There were foci of reactive RPE cells penetrating the outer retina (Fig. 7H), a sign of early photoreceptor degeneration. These histological retinal changes correlated with the clinical AF, IR and ERG changes observed in early-stage CAPN5 vitreoretinopathy human patients. Additionally, a few hCAPN5R243L eyes showed fibrovascular membranes at the optic nerve (Fig. 7I) that had not been detected clinically.
Figure 6.

Human ADNIV pathology. (A) Histology reveals a foreshortened, fibrotic iris with dilated vessels and exudate in the anterior chamber (visualized by Masson's trichrome stain). (B) A cataractous lens with lens epithelial cells (arrow) located along the posterior capsule (visualized by Masson's trichrome stain). (C). RPE cells (arrow) infiltrating the degenerating retina (visualized by H&E stain). (D) Vitreous containing fibrous membranes (arrow) and filled with exudate (visualized by H&E stain). (E) Epiretinal membrane (arrow). (F) Fibrovascular membrane at the optic nerve (arrow). Scale bar = 250 μm.
Figure 7.

The hCAPN5R243L transgenic mouse develops severe uveitis that phenocopies early-stage human ADNIV. (A) Pupil-optic nerve section of a normal mouse eye. (B) Pupil-optic nerve section of a hCAPN5R243L transgenic mouse eye at 4 months of age. The anterior segment shows signs of inflammation with protein exudates in the anterior chamber and a contracted iris with dilated vessels. There are membranes and cells in the vitreous chamber. (C) Magnified view of foreshortened iris and dilated vessels (arrow). (D) Posterior epithelial cells (green arrow) are a sign of posterior subcasular cataract. Vitreous vessels (green arrow) are a sign of early vitreous neovascularization. (E) Vitreous membranes (arrow) correlate with fibrotic bands found in the human vitreous. (F) Control retina. (G) Retinal vessels are dilated (arrow). (H) Reactive retinal pigmented epithelial cells (arrow) in early retinal degeneration. (I) Fibrovascular tissue at optic nerve (arrow).
The hCAPN5R243L mutation reduces visual function in the mouse
During early stages of ADNIV, before patients suffer widespread photoreceptor degeneration, they show loss of the scotopic and photopic b-wave ERG (Fig. 8A and B), indicating inner-retina signaling defects. To test whether this phenotype could be detected in the hCAPN5R243L transgenic mice, ERG testing was performed beginning at 1 month of age. ERG phenotypes found in the transgenic mice showed some variability. However, we found that most phenotypic changes were detectable by 2 months of age in the hCAPN5R243L transgenic mice. Representative traces of visual responses at 8 months of age for the hCAPN5R243L transgenic mice (green) and control mice (blue) show the common ERG phenotype observed in these mice (Fig. 8C and D).
Figure 8.

Electroretinography shows hCAPN5R243L mice have a specific loss of the inner retinal cells and cone cell function. (A) ERG tracing of ADNIV patient shows reduction of b-wave relative to the a-wave (14). The scotopic b-wave was 83% of normal (normal scotopic b-wave was 450 ± 100 μV in this system). (B) The photopic ERG b-wave was ∼32.5% of normal (339 ± 85 μV). The calibration mark before stimulus shows 100 μV. (C) A representative ERG test on an 8 month-old hCAPN5R243L transgenic mouse (green) compared with a control mouse (blue) displays the frequently observed functional phenotype. hCAPN5R243L transgenic mice have a reduced scotopic maximal b-wave amplitude compared with control mice, with the a-wave remaining unchanged. (D) Photopic, cone-specific ERGs showed a reduction in the b-wave maximal amplitude in the hCAPN5R243L transgenic mice compared with controls. Such a pattern correlates to ADNIV patients.
Representative traces for the dark adapted, 3.0 maximal combined ERG showed that, compared with the WT mice (Fig. 8C, blue trace), the hCAPN5R243L transgenic mice generated b-waves with a smaller maximal amplitude (Fig. 8C, green trace). In contrast, the a-wave was unchanged between the control and hCAPN5R243L transgenic mice, similar to human ADNIV patients (Fig. 8A). Under photopic conditions, the maximal b-wave amplitude was lower in the hCAPN5R243L transgenic mice (Fig. 8D, green trace) compared with control mice (Fig. 8D, blue trace), and similar to the b-wave reduction found in human ADNIV patients (Fig. 8B). This suggests hCAPN5R243L transgenic mice lose cone-specific visual function. In a prior study, we reported a cone and inner-retina cell loss-of-function with lentiviral delivery of the single p.R243L mutation in CAPN5 within the mouse eye (16). Here, we see similar effects, which additionally correlate with the ERG phenotype found in ADNIV (32,33), before the appearance of widespread photoreceptor degeneration.
hCAPN5R243L transgenic mice display biomarkers of uveitis
We previously found lentiviral induction of hCAPN5R243L in mouse photoreceptor cells elevated expression of IL-6 mRNA in the mouse retina. IL-6 is a cytokine associated with uveitis and fibrosis (16) and ADNIV patients display high levels of the IL-6 cytokine in their vitreous (10). To determine if the transgenic hCAPN5R243L mice showed elevated IL-6, we performed immunohistochemistry for this profibrotic cytokine. Elevated IL-6 was not seen in retinal cells, but the vitreous contained very high levels of the cytokine in regions clustered near the pars plana and ciliary body (Fig. 9). This anatomic location is a key site where ADNIV patients develop fibrosis despite corticosteroid implantation (15), and the profibrotic properties of IL-6 and its localization may offer some explanation of this disease phenotype.
Figure 9.
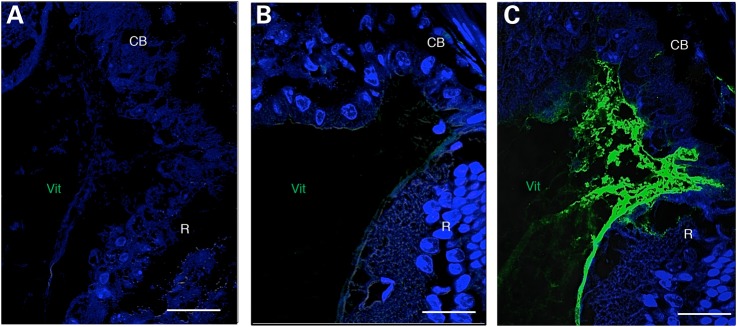
IL-6 is upregulated in the hCAPN5R243L transgenic mouse ciliary body. (A) Control IgG antibody. (B) No IL-6 expression in 4-month-old WT vitreous, as assayed by immunohistochemistry. (C) Vitreous IL-6 expression is upregulated in the hCAPN5R243L mouse anterior vitreous. CB, ciliary body; Vit, vitreous; R, retina. Scale bar = 25 μm.
CAPN5 is expressed in the photoreceptor cell and its nucleus (13), and it can influence transcription of genes that trigger inflammation (18,21,34). We previously found that lentiviral transduction of the mutant CAPN5 alters inflammatory gene expression, but gene expression could only be surveyed after the virus was transduced and was found to correlate to late stages of human ADNIV disease. This current transgenic hCAPN5R243L mouse model allowed us to survey retinal inflammatory gene expression at the earliest stages of disease. Thus, we used quantitative PCR (qPCR) arrays to survey mouse retinal mRNA for 84 inflammatory transcripts, at 1.5 months, before all the signs of inflammatory retinal degeneration were clinically and histologically detectable. We identified 11 genes that were significantly upregulated compared with control mouse retinas (Fig. 10). Six of the 11 genes represented a novel Toll-like receptor (TLR) pathway (Tlr6, Tlr9, Il1a, Itgb2, Ltb and Myd88), components of which are part of the innate immune system that have been implicated in mouse models of autoimmune uveitis (35). Other upregulated genes included chemokine (C-C motif) ligands (Ccl2, Ccl7 and Ccl11), signals shown to be upregulated in the RPE cells that have come into contact with activated T-cells. The T-cell marker, Cd40, and the innate immunity gene, Crp, were also upregulated in diseased eyes. Elevation of these transcripts might suggest stimulated immune cells infiltrating from both the innate and adaptive arms of the immune system. Alternatively, retinal cells themselves might overexpress some of these molecules, since expression of the TLR pathway has been shown in neurons as well as cells of the immune system (36). These early changes in gene expression implicate several potential drug targets. Four genes were downregulated, including Fos, Bcl6, Il1rap and Nr3c1 (Fig. 10). Interestingly, Nr3c1 is a glucocorticoid receptor, and its lower expression might explain why ADNIV patients have a limited clinical response to glucocorticoid therapy (18,21,34).
Figure 10.
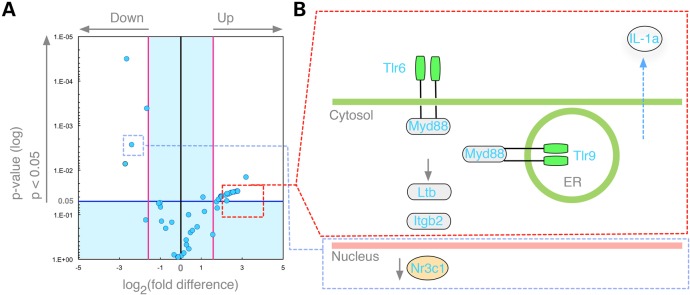
The hCAPN5R243L transgenic retina shows early changes in inflammatory gene expression before any clinical or histological signs of disease. (A) Volcano plot displays genes in the hCAPN5R243L retinas at post-natal day (P) 45 that had statistically significant expression changes that were greater than 2-fold (P < 0.05), when compared with P45 control retinas. Four genes (Fos, Bcl6, IL1rap and Nr3c1) were downregulated (blue dots, upper left). (B) Schematic representation shows that several members of the TLR pathways were among the upregulated inflammatory genes.
Discussion
Similar to human ADNIV patients, mice carrying the transgenic hCAPN5R243L gene develop a robust inflammatory uveitis and vitreoretinopathy. We have used a variety of methods—phenotypic, functional, molecular and cellular—to probe the causes of ADNIV in both humans and our hCAPN5R243L preclinical mouse model. Our evaluation indicates that the human and mouse diseases are similar, even at the molecular level. These results correlate with our previous exploratory studies, in which we introduced hCAPN5R243L into the mouse retina via a lentiviral system, and showed that mice develop an ADNIV phenotype (21). The transgenic mouse more accurately recreates the disease scenario in that the disease allele is expressed in all photoreceptors, rather than a subset of lentiviral-transduced photoreceptor cells, and it is expressed over the lifetime of the mouse. Nevertheless, the mouse does not develop some severe aspects of the human disease. Interestingly, although signs of photoreceptor degeneration were detectable clinically, by AF/IR imaging and ERG analysis, there was not a severe reduction in the outer or inner nuclear layer as anticipated by our preliminary studies (16). However, in contrast to the lentivirus model, the hCAPN5R243L transgenic mouse develops an inflammatory, fibrotic and early vitreous neovascularization phenotype more similar to ADNIV patients.
Overall, the hCAPN5R243L transgenic mouse model of ADNIV recapitulates the clinical phenotype of human ADNIV and provides new insight into the molecular causes of the disease. Human ADNIV retinas are characterized by cells infiltrating the vitreous (8,9); similarly, the hCAPN5R243L mouse eyes showed cells populating the vitreous. In addition, our screen for early biomarkers revealed ADNIV production of specific molecules, such as TLR pathways, and these might be targeted therapeutically. Together, these findings suggest a disease model where molecular factors in the retina are sufficient to trigger uveitis (Fig. 11). Some of the biomarkers detected have been linked to other non-inherited forms of uveitis and retinal degenerations (37–39), and hence we expect this transgenic ADNIV mouse model to have utility for other types of human uveitis. Indeed, the hCAPN5R243L mouse offers distinctions over current animal models of autoimmune uveitis, which are typically generated by ectopically expressing proteins in the retina (e.g. hen egg lysoszyme and beta-galactosidase) or injecting antigens and adjuvants (7,33,40). These models did not develop a constellation of pathologies or naturally develop disease phenotypes without extra manipulation.
Figure 11.

An integrated ADNIV disease model. (A) Histology of the CAPN5R243L uveitis model. (B) The CAPN5R243L disease allele transgene is expressed in retina photoreceptors. Because of the high calcium levels in the retina for phototransduction, CAPN5 mutations in the retina are particularly sensitive. TLR signaling and cyotokine secretion trigger retinal and iris vessel hyperpermeability and a cell-mediated autoinflammatory reaction that includes vitreous inflammation and fibrosis.
Furthermore, we provide evidence of the first example of an overactive calpain causing a dominant, inherited disease. Only one other Mendelian disease has been shown so far to be caused by a mutant calpain: mutations in CAPN3 cause limb-muscle girdle dystrophy type 2A. In this case, however, the disease-associated mutation causes a calpain-3 loss-of-function (41). Excess calpain activity is associated with non-inherited pathologies, such as Alzheimer's disease (calpain 1) and myocardial infarction (calpains 1, 2 and 4) (18–21). Similarly, our biochemical studies demonstrate that the hCAPN5R243L mutation increased calpain activity, such that the mutant protease activity is not only relieved from strict calcium regulation, it is also three orders of magnitude more sensitive to calcium (Fig. 3). Given the major changes in the protein's response to calcium, it is interesting that the hCAPN5R243L mutation is not in a calcium binding loop, but instead, in loop 2, which likely gates access of substrates to the calpain active site (Fig. 1). Interestingly, we have identified a third family with ADNIV (42), and linked their disease to another point mutation in CAPN5, also in the sequence encoding the DIIb mobile gating loop.
Among the calpains, CAPN5 might be particularly susceptible to mutational effects. The calcium residues in the catalytic domain provide one layer of calcium dependence, but most calpains in higher vertebrates have evolved a second regulatory layer by acquiring a calmodulin-like C-terminal domain (34). In contrast, CAPN5 is a ‘non-classical’ calpain that most closely resembles Tra-3, a calpain ortholog conserved in flies and worms that regulates neuronal degeneration and sex determination. In addition, CAPN5 cannot form heterodimers with the regulatory subunit CAPNS1, which might remove yet another layer of potential regulation (18,26). Thus, CAPN5 might rely more heavily on calcium binding in the catalytic domain to control proteolytic activity than other members of the calpain family.
Since CAPN5 is preferentially expressed in the CNS (43), and is genetically linked with retinal disease (13), generalized calpain or CAPN5-specific inhibition is a promising strategy through which to develop future uveitis and retinal disease therapy. Moreover, oral delivery of these inhibitors to the retina may make them particularly effective for treating an eye disorder linked to overactive CAPN5 (44). In a Caenorhabditis elegans genetic model, reducing Tra-3 rescued genetic lesions that cause necrotic (inflammatory) neuronal degeneration (34). Similarly, inhibiting calpain with a small-molecule inhibitor (SNJ-1945) rescued neuronal cell death in models of traumatic brain injury (which elicit necrotic neuronal degeneration) (45). The molecule also blocked experimental autoimmune encephalitis (46), a condition that strongly parallels uveitis. Calpain inhibitors additionally rescued retinal degeneration in several other models, including models of light-induced retinal toxicity (47,48), retinal hypoxia (49), retinitis pigmentosa (50–52), optic neuritis (53,54), diabetic retinopathy (55) and retinal angiogenesis (49,56). Since the inhibitors are not isoform-specific, and RNA sequencing showed expression of at least seven different calpains in the retina (unpublished observation), the main limit of prior studies was that they could not pinpoint the critical calpain isoform. In contrast, our studies clearly link retinal dysfunction to an overactive CAPN5 protease.
CAPN5 expression is highest in the nervous system but our patients do not present with any other diseases, including neurological disease. ADNIV eye disease is the only manifestation of the mutation. Retinal-specific disease might arise for several reasons. First, the extremely high levels of calcium in the retina could make it more susceptible to a CAPN5 that is activated at very low levels of calcium. Alternatively, the retina could be affected by the overactive CAPN5 because some other regulatory mechanism that is redundant in other tissues is lacking in the retina, allowing the ectopic activation of CAPN5 proteolysis.
A third reason for retina-specific disease might lie in an altered CAPN5 target specificity. The natural calpain targets have been difficult to determine, since there is no consensus cleavage site sequence. The calpain family has multiple members and how their precise targets might vary is not currently known. In general, calpains have been shown to cleave a wide variety of exposed peptide surfaces. Nevertheless, the structure and sequence of the calpain proteolytic core revealed that some level of sequence specificity is likely found at the mobile loops of its substrate binding site. Moreover, the loop 2 is among the more variable features among calpains, with each member having a distinctive length and sequence (23). This variability has led others to hypothesize that the mobile loops are good candidates for conferring substrate specificity (23). Since the three ADNIV mutations we uncovered each affect the same DIIb mobile gating loop, they may not only be relieving calcium regulation and boosting catalytic activity but also changing CAPN5 targets, such that a retinal-specific protein is pathologically cleaved, rendering the ADNIV phenotype.
To discriminate between these possibilities it will be advantageous to compare the specificity and sensitivity of proteolytic targets for WT CAPN5 and mutant CAPN5R243L. Understanding how changes in CAPN5 activity cause ADNIV should reveal information about the causes of other diseases linked to aberrant calpain activity. For example, the true targets of calpain and how they are affected by excess proteolysis is currently not clear. With this new hCAPN5R243L transgenic mouse model in hand, however, it might be possible to begin to probe the substrate specificity of calpain proteases.
Materials and Methods
CAPN5 sequence and structure analysis
Using MODELLER 9.14 (57), a homology model of human mini-CAPN5 was generated as previously described (20), using the crystal structure of human mini-CAPN9 as a template, which was the closest match in the Protein Database (PDB). An analysis of the conserved residues in the catalytic domain across various members of the calpain family was performed using the CONSURF server (58–61). The CONSURF scores were mapped to the B-factor column of mini-CAPN1 (PDB ID 2ARY) and visualized using PyMOL (http://www.pymol.org).
CAPN5 cloning, purification and activity assay
Sequences encoding the calpain-1/5 catalytic domain and the inactive calpain-1/5-C81S catalytic domain were cloned into pMal-c5x (New England Biolabs, Ipswich, MA) with a c-terminal His tag. Mini-calpains were expressed as MBP-fusion proteins in BL21(DE3) E. coli and purified on an amylose resin (New England Biolabs). At different calcium concentrations, purified calpain catalytic activity was measured by cleavage of a peptide substrate (AC-LLY-AFC) and quantification on a fluorometric plate reader (Tecan Infinite M200 Pro, Männedorf, Switzerland) using a calpain activity assay and peptide inhibitor (Z-LLY-AFC) according to the manufacturer's instructions (Abcam, ab65308, Cambridge, MA).
Human ADNIV case
The collection of data used in this study was approved by the Institutional Review Board for Human Subjects Research at the University of Iowa, was compliant with the Health Insurance Portability and Accountability Act and adhered to the tenets of the Declaration of Helsinki. Clinical examination and testing were performed as previously described (16). Stereoscopic color fundus images and AF images were obtained using a Topcon TRC 50DX camera (Topcon, Pyramus, NJ). Optical coherence tomography imaging was obtained from the spectral-domain Heidelberg HRA2 Spectralis, version 1.6.1 (Heidelberg Engineering Inc, Vista, CA). A full-field ERG was performed according to international standards. Briefly, the eyes were dilated and dark adapted for 30 min. ERGs were recorded simultaneously from both eyes using Burian-Allen bipolar contact lens electrodes. Evoked waveforms, a 100 μV calibration pulse, and a stimulus artifact were recorded on Polaroid film.
Generation of mutant mouse lines
Transgenic mice were created on a CBA × B6 background (Jackson Laboratory, Bar Harbor, ME). B6CBA-Tg(Rho-hCAPN5R243L)vbm mice were maintained in the Columbia University Pathogen-free Eye Institute Annex Animal Care Services Facility under a 12/12-h light/dark cycle. The line was verified to be negative for mutations in rd1, Gnat2 and rd8 (not shown). The Institutional Animal Care and Use Committee (IACUC) approved all experiments. Mice were used in accordance with the Statement for the Use of Animals in Ophthalmic and Vision Research of the Association for Research in Vision and Ophthalmology, as well as the Policy for the Use of Animals in Neuroscience Research of the Society for Neuroscience.
Human CAPN5 cDNA was obtained from Origene (Catalogue # RG202045). DNA constructs for the expression of hCAPN5 contained 4.2 kilobases of the mouse opsin promoter to drive retina-specific expression, the complete open reading frame of the human CAPN5 cDNA and the polyadenylation signal of the mouse protamine gene. The R243L and p.C81S mutations were introduced by a standard PCR-based site-specific mutagenesis strategy (16). The entire CAPN5 cDNA coding region in the transgenic construct was sequenced to confirm introduction of the point mutation and no other inadvertent changes. Vector sequences were excised using SalI and AscI.
Oocytes were obtained from superovulated B6xCBA F1 females mated with B6xCBA F1 males. Control and R243L constructs were injected into the male pronuclei of oocytes under a depression slide chamber. Microinjected oocytes were cultured overnight in M16 (Specialty Media, Phillipsburg, NJ) and transferred into the oviducts of 0.5-day post coitum pseudopregnant B6xCBA F1 females. The resulting transgenic mice were then backcrossed, initially into the C57BL/6J background.
Transgenic mice were identified by analyzing genomic DNA isolated from tail tips. Tissue was homogenized, digested extensively with proteinase K and extracted with phenol. PCR primers targeted sequences originating from exons 12 and 13 of human CAPN5. PCR amplification with the forward primer <5′-CCAGGTGCCGGCCGTTTAAACC-3′ > and the reverse primer <5′-CCAGTGTGAGGGAGACAAAGTC-3′> amplified a 405-bp fragment from the cassette containing the human CAPN5R243L cDNA that had integrated in the mouse genome. A control fragment of 524 bp was amplified using the forward primer <5′- CCTGGGCTAGTCATAGCACGATACCACTCTC-3′> and the reverse primer <5′- TGGTGGTGATGGTGGGGGTTCGATAAGCC-3′>. PCR amplification was performed as follows: 1 cycle at 94°C for 5 min; 35 cycles at 94°C for 30 s and 55°C for 30 s one cycle at 72°C for 7 min.
AF and IR imaging
For the mice, AF fundus imaging was obtained with the Spectralis scanning laser confocal ophthalmoscope (OCT-SLO Spectralis 2; Heidelberg Engineering, Heidelberg, Germany). Pupils were dilated using topical 2.5% phenylephrine hydrochloride and 1% tropicamide (Akorn Inc., Lakeforest, IL, USA). Mice were anesthetized by intraperitoneal injection of 0.1 ml/10 g body weight of anesthesia [1 ml ketamine 100 mg/ml (Ketaset III, Fort Dodge, IA, USA) and 0.1 ml xylazine 20 mg/ml (Lloyd Laboratories, Shenandoah, IA, USA) in 8.9 ml phosphate-buffered saline (PBS)]. Body temperature was maintained at 37°C using a heating pad during the procedure. AF imaging was obtained at 488 nm absorption and 495 nm emission using a 55° lens. IR imaging was performed as previously described (62). Images were taken of the central retina, with the optic nerve located in the center of the image.
ERGs
Mice were dark adapted overnight, manipulations were conducted under dim red light illumination and recordings were made using Espion ERG Diagnosys equipment (Diagnosys LLL, Littleton, MA, USA), as previously described (16,62).
Histochemical analyses
Whole mouse eyes were enucleated at 2-months (12 eyes) and 4-months (14 eyes) of age, fixed, sectioned and stained with hematoxylin and eosin (H&E) as previously described (63). Human eyes were sectioned and stained with H&E or Masson's trichrome as previously described (9).
Immunofluorescence
Sections were deparaffinized and rehydrated with dH2O. Antigen retrieval was performed using 10 mm citrate buffer (pH 6.0) in a 650-watt microwave. Slides were microwaved for 5 min, cooled 5 min, microwaved 5 min, cooled for 30 min and then rinsed in PBS 3 × 3 min. Sections were permeabilized using 0.1% Triton-X-100 in PBS (pH 7.4) for 10 min, followed by 3 × 3 min rinses in PBS. Sections were blocked in 5% normal goat sera (G-9023, Sigma) for 2 h. Primary antibody was incubated using a 1:200 dilution (final 370 µg/ml) rabbit anti-IL6 (ab6672, Abcam) or normal rabbit IgG as a control overnight at 4°C. Sections were rinsed 3 × 3 min in PBS and then incubated with 1.2 µg/ml goat anti-rabbit IgG-biotin conjugated antibody (111-066-047, Jackson ImmunoResearch) for 30 min at RT. Sections were rinsed and incubated with streptavidin-Alexa 647 fluorescent conjugate (S11223, Invitrogen) diluted 1:500 in blocking solution for 1 h and then rinsed again. Slides were mounted with VectaShield containing DAPI (H-1200, Vector Laboratories). Images were collected using a Zeiss 710 confocal microscope with 405 and 633 nm excitation for nuclei and anti-calpain5, respectively.
Real-time PCR array analysis
Mouse retinas were isolated as previously described (64). RNA was extracted from the retina as described above and reverse transcribed using the RT2 First Strand Kit according to manufacturer's instructions (SABiosciences/Qiagen, Valencia, CA). Mouse cDNA was added to SABiosciences RT2 qPCR master mix and then added to the 96 wells of mouse Inflammation and Autoimmunity RT2 Profiler PCR Array System (Product # PAMM-077Z-A; SABiosciences/Qiagen). Quantitative PCR was performed using the Applied Biosystems Model 7000 sequence detection system (Applied Biosystems Inc., Foster City, CA), and Applied Biosystems analysis software SDS 2.3 was used to determine Ct values. Significant gene expression changes were based on fold changes greater than 2 and P-values <0.05.
Authors' Contributions
K.J.W., S.H.T. and V.B.M. designed research; K.J.W., J.Y., W.-H.W., C.-S.L., V.B.M., L.G. and S.H.T. performed research; K.J.W., S.H.T., S.W., A.G.B., D.C., L.G. and V.B.M. analyzed research; K.J.W., A.G.B., S.H.T., D.C., L.G. and V.B.M. wrote the article.
Funding
V.B.M. is supported by the National Institute of Health (K08EY020530, R01EY024665, R01EY025225 and Doris Duke Charitable Foundation, Grant #2013103) and Research to Prevent Blindness New York, NY, USA. S.H.T. is supported by the National Institute of Health Core (5P30EY019007), National Cancer Institute Core (5P30CA013696), the National Institute of Health (R01EY018213) and unrestricted funds from Research to Prevent Blindness, New York, NY, USA. S.H.T. is a member of the RD-CURE Consortium and is supported by Tistou and Charlotte Kerstan Foundation, the Research to Prevent Blindness Physician-Scientist Award, the Schneeweiss Stem Cell Fund, New York State (N09G-302 and N13G-275), and the Foundation Fighting Blindness New York Regional Research Center Grant (C-NY05-0705-0312), the Joel Hoffman Fund, Gale and Richard Siegel Stem Cell Fund, Charles Culpeper Scholarship, Laszlo Bito and Olivia Carino Foundation, Irma T. Hirschl Charitable Trust, Bernard and Anne Spitzer Stem Cell Fund, Professor Gertrude Rothschild Stem Cell Foundation and Gebroe Family Foundation. K.J.W. was supported by the National Institute of Health (5T32EY013933, 5T32DK007647-20) and is supported by the National Cancer Institute (F32CA196065).
Acknowledgements
Melinda Smits and Jessica Skeie provided technical assistance.
Conflict of Interest statement. None declared.
References
- 1.Rose C.D., Doyle T.M., McIlvain-Simpson G., Coffman J.E., Rosenbaum J.T., Davey M.P., Martin T.M. (2005) Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis. J. Rheumatol., 32, 373–375. [PubMed] [Google Scholar]
- 2.Caspi R.R., Silver P.B., Luger D., Tang J., Cortes L.M., Pennesi G., Mattapallil M.J., Chan C.C. (2008) Mouse models of experimental autoimmune uveitis. Ophthalmic Res., 40, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luger D., Caspi R.R. (2008) New perspectives on effector mechanisms in uveitis. Semin. Immunopathol., 30, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caspi R.R. (2010) A look at autoimmunity and inflammation in the eye. J. Clin. Invest., 120, 3073–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zenewicz L.A., Abraham C., Flavell R.A., Cho J.H. (2010) Unraveling the genetics of autoimmunity. Cell, 140, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou R., Horai R., Mattapallil M.J., Caspi R.R. (2011) A new look at immune privilege of the eye: dual role for the vision-related molecule retinoic acid. J. Immunol., 187, 4170–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forrester J.V., Klaska I.P., Yu T., Kuffova L. (2013) Uveitis in mouse and man. Int. Rev. Immunol., 32, 76–96. [DOI] [PubMed] [Google Scholar]
- 8.Mahajan V.B., Vallone J.G., Lin J.H., Mullins R.F., Ko A.C., Folk J.C., Stone E.M. (2010) T-cell infiltration in autosomal dominant neovascular inflammatory vitreoretinopathy. Mol. Vis., 16, 1034–1040. [PMC free article] [PubMed] [Google Scholar]
- 9.Mahajan V.B., Lin J.H. (2013) Lymphocyte infiltration in CAPN5 autosomal dominant neovascular inflammatory vitreoretinopathy. Clin. Ophthalmol., 7, 1339–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahajan V.B., Skeie J.M. (2013) Personalized proteomics for inflammatory retinal disease therapy [abstract]. HUPO 2013 Annual World Congress, Yokohama, Japan.
- 11.Tlucek P.S., Folk J.C., Orien J.A., Stone E.M., Mahajan V.B. (2012) Inhibition of neovascularization but not fibrosis with the fluocinolone acetonide implant in autosomal dominant neovascular inflammatory vitreoretinopathy. Arch. Ophthalmol., 130, 1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azuma M., Shearer T.R. (2008) The role of calcium-activated protease calpain in experimental retinal pathology. Surv. Ophthalmol., 53, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahajan V.B., Skeie J.M., Bassuk A.G., Fingert J.H., Braun T.A., Daggett H.T., Folk J.C., Sheffield V.C., Stone E.M. (2012) Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet., 8, e1003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rowell H.A., Bassuk A.G., Mahajan V.B. (2012) Monozygotic twins with CAPN5 autosomal dominant neovascular inflammatory vitreoretinopathy. Clin. Ophthalmol., 6, 2037–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tlucek P.S., Folk J.C., Sobol W.M., Mahajan V.B. (2013) Surgical management of fibrotic encapsulation of the fluocinolone acetonide implant in CAPN5-associated proliferative vitreoretinopathy. Clin. Ophthalmol., 7, 1093–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wert K.J., Skeie J.M., Bassuk A.G., Olivier A.K., Tsang S.H., Mahajan V.B. (2014) Functional validation of a human CAPN5 exome variant by lentiviral transduction into mouse retina. Hum. Mol. Genet., 23, 2665–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pascolini D., Mariotti S.P. (2012) Global estimates of visual impairment: 2010. Br. J. Ophthalmol., 96, 614–618. [DOI] [PubMed] [Google Scholar]
- 18.Sorimachi H., Ishiura S., Suzuki K. (1997) Structure and physiological function of calpains. Biochem. J., 328(Pt 3), 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croall D.E., Ersfeld K. (2007) The calpains: modular designs and functional diversity. Genome Biol., 8, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassuk A.G., Yeh S., Wu S., Martin D.F., Tsang S.H., Gakhar L., Mahajan V.B. (2015) Structural modeling of a novel CAPN5 mutation that causes uveitis and neovascular retinal detachment. PLoS One, 10, e0122352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goll D.E., Thompson V.F., Li H., Wei W., Cong J. (2003) The calpain system. Physiol. Rev., 83, 731–801. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki K., Hata S., Kawabata Y., Sorimachi H. (2004) Structure, activation, and biology of calpain. Diabetes, 53(Suppl. 1), S12–S18. [DOI] [PubMed] [Google Scholar]
- 23.Moldoveanu T., Campbell R.L., Cuerrier D., Davies P.L. (2004) Crystal structures of calpain-E64 and -leupeptin inhibitor complexes reveal mobile loops gating the active site. J. Mol. Biol., 343, 1313–1326. [DOI] [PubMed] [Google Scholar]
- 24.Davis T.L., Walker J.R., Finerty P.J., Jr, Mackenzie F., Newman E.M., Dhe-Paganon S. (2007) The crystal structures of human calpains 1 and 9 imply diverse mechanisms of action and auto-inhibition. J. Mol. Biol., 366, 216–229. [DOI] [PubMed] [Google Scholar]
- 25.Hanna R.A., Campbell R.L., Davies P.L. (2008) Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature, 456, 409–412. [DOI] [PubMed] [Google Scholar]
- 26.Campbell R.L., Davies P.L. (2012) Structure–function relationships in calpains. Biochem. J., 447, 335–351. [DOI] [PubMed] [Google Scholar]
- 27.Cuerrier D., Moldoveanu T., Inoue J., Davies P.L., Campbell R.L. (2006) Calpain inhibition by alpha-ketoamide and cyclic hemiacetal inhibitors revealed by X-ray crystallography. Biochemistry, 45, 7446–7452. [DOI] [PubMed] [Google Scholar]
- 28.Moldoveanu T., Hosfield C.M., Lim D., Elce J.S., Jia Z., Davies P.L. (2002) A Ca(2+) switch aligns the active site of calpain. Cell, 108, 649–660. [DOI] [PubMed] [Google Scholar]
- 29.Moldoveanu T., Hosfield C.M., Lim D., Jia Z., Davies P.L. (2003) Calpain silencing by a reversible intrinsic mechanism. Nat. Struct. Biol., 10, 371–378. [DOI] [PubMed] [Google Scholar]
- 30.Marsiglia M., Lee W., Mahajan V.B., Zernant J., Delori F.C., Tsang S.H., Sparrow J.R. (2015) Quantitative autofluorescence as a clinical tool for expedited differential diagnosis of retinal degeneration. JAMA Ophthalmol., 133, 219–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang N.K., Fine H.F., Chang S., Chou C.L., Cella W., Tosi J., Lin C.S., Nagasaki T., Tsang S.H. (2009) Cellular origin of fundus autofluorescence in patients and mice with a defective NR2E3 gene. Br. J. Ophthalmol., 93, 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett S.R., Folk J.C., Kimura A.E., Russell S.R., Stone E.M., Raphtis E.M. (1990) Autosomal dominant neovascular inflammatory vitreoretinopathy. Ophthalmology, 97, 1125–1135; discussion 1135–1136. [DOI] [PubMed] [Google Scholar]
- 33.Stone E.M., Kimura A.E., Folk J.C., Bennett S.R., Nichols B.E., Streb L.M., Sheffield V.C. (1992) Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy to chromosome 11q13. Hum. Mol. Genet., 1, 685–689. [DOI] [PubMed] [Google Scholar]
- 34.Syntichaki P., Xu K., Driscoll M., Tavernarakis N. (2002) Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature, 419, 939–944. [DOI] [PubMed] [Google Scholar]
- 35.Fang J., Fang D., Silver P.B., Wen F., Li B., Ren X., Lin Q., Caspi R.R., Su S.B. (2010) The role of TLR2, TRL3, TRL4, and TRL9 signaling in the pathogenesis of autoimmune disease in a retinal autoimmunity model. Invest. Ophthalmol. Vis. Sci., 51, 3092–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lafon M., Megret F., Lafage M., Prehaud C. (2006) The innate immune facet of brain: human neurons express TLR-3 and sense viral dsRNA. J. Mol. Neurosci., 29, 185–194. [DOI] [PubMed] [Google Scholar]
- 37.Horai R., Caspi R.R. (2011) Cytokines in autoimmune uveitis. J. Interferon Cytokine Res., 31, 733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chinnery H.R., McLenachan S., Binz N., Sun Y., Forrester J.V., Degli-Esposti M.A., Pearlman E., McMenamin P.G. (2012) TLR9 ligand CpG-ODN applied to the injured mouse cornea elicits retinal inflammation. Am. J. Pathol., 180, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarallo V., Hirano Y., Gelfand B.D., Dridi S., Kerur N., Kim Y., Cho W.G., Kaneko H., Fowler B.J., Bogdanovich S., et al. (2012) DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell, 149, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasparin F., Takahashi B.S., Scolari M.R., Gasparin F., Pedral L.S., Damico F.M. (2012) Experimental models of autoimmune inflammatory ocular diseases. Arq. Bras. Oftalmol., 75, 143–147. [DOI] [PubMed] [Google Scholar]
- 41.Jia Z., Petrounevitch V., Wong A., Moldoveanu T., Davies P.L., Elce J.S., Beckmann J.S. (2001) Mutations in calpain 3 associated with limb girdle muscular dystrophy: analysis by molecular modeling and by mutation in m-calpain. Biophys. J., 80, 2590–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassuk, A.G., Yeh, S., Wu, S., Martin, D.F., Tsang, S.H., Gakhar, L. and Mahajan, V.B. (2015) Structural modeling of a novel CAPN5 mutation that causes uveitis and neovascular retinal detachment. PLoS One, 10, e0122352. [DOI] [PMC free article] [PubMed]
- 43.Singh R., Brewer M.K., Mashburn C.B., Lou D., Bondada V., Graham B., Geddes J.W. (2014) Calpain 5 is highly expressed in the central nervous system (CNS), carries dual nuclear localization signals, and is associated with nuclear promyelocytic leukemia protein bodies. J. Biol. Chem., 289, 19383–19394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirasaki Y., Yamaguchi M., Miyashita H. (2006) Retinal penetration of calpain inhibitors in rats after oral administration. J. Ocul. Pharmacol. Ther., 22, 417–424. [DOI] [PubMed] [Google Scholar]
- 45.Bains M., Cebak J.E., Gilmer L.K., Barnes C.C., Thompson S.N., Geddes J.W., Hall E.D. (2013) Pharmacological analysis of the cortical neuronal cytoskeletal protective efficacy of the calpain inhibitor SNJ-1945 in a mouse traumatic brain injury model. J. Neurochem., 125, 125–132. [DOI] [PubMed] [Google Scholar]
- 46.Trager N., Smith A., Wallace Iv G., Azuma M., Inoue J., Beeson C., Haque A., Banik N.L. (2014) Effects of a novel orally administered calpain inhibitor SNJ-1945 on immunomodulation and neurodegeneration in a murine model of multiple sclerosis. J. Neurochem., 130, 268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanan Y., Moiseyev G., Agarwal N., Ma J.X., Al-Ubaidi M.R. (2007) Light induces programmed cell death by activating multiple independent proteases in a cone photoreceptor cell line. Invest. Ophthalmol. Vis. Sci., 48, 40–51. [DOI] [PubMed] [Google Scholar]
- 48.Imai S., Shimazawa M., Nakanishi T., Tsuruma K., Hara H. (2010) Calpain inhibitor protects cells against light-induced retinal degeneration. J. Pharmacol. Exp. Ther., 335, 645–652. [DOI] [PubMed] [Google Scholar]
- 49.Hoang M.V., Smith L.E., Senger D.R. (2011) Calpain inhibitors reduce retinal hypoxia in ischemic retinopathy by improving neovascular architecture and functional perfusion. Biochim. Biophys. Acta, 1812, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimazawa M., Suemori S., Inokuchi Y., Matsunaga N., Nakajima Y., Oka T., Yamamoto T., Hara H. (2010) A novel calpain inhibitor, ((1S)-1-((((1S)-1-benzyl-3-cyclopropylamino-2,3-di-oxopropyl)amino)carbonyl)-3-methylbutyl)carbamic acid 5-methoxy-3-oxapentyl ester (SNJ-1945), reduces murine retinal cell death in vitro and in vivo. J. Pharmacol. Exp. Ther., 332, 380–387. [DOI] [PubMed] [Google Scholar]
- 51.Ozaki T., Nakazawa M., Yamashita T., Sorimachi H., Hata S., Tomita H., Isago H., Baba A., Ishiguro S. (2012) Intravitreal injection or topical eye-drop application of a mu-calpain C2L domain peptide protects against photoreceptor cell death in Royal College of Surgeons’ rats, a model of retinitis pigmentosa. Biochim. Biophys. Acta, 1822, 1783–1795. [DOI] [PubMed] [Google Scholar]
- 52.Ozaki T., Ishiguro S., Hirano S., Baba A., Yamashita T., Tomita H., Nakazawa M. (2013) Inhibitory peptide of mitochondrial mu-calpain protects against photoreceptor degeneration in rhodopsin transgenic S334ter and P23H rats. PLoS One, 8, e71650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith A.W., Das A., Guyton M.K., Ray S.K., Rohrer B., Banik N.L. (2011) Calpain inhibition attenuates apoptosis of retinal ganglion cells in acute optic neuritis. Invest. Ophthalmol. Vis. Sci., 52, 4935–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Das A., Guyton M.K., Smith A., Wallace G.t., McDowell M.L., Matzelle D.D., Ray S.K., Banik N.L. (2013) Calpain inhibitor attenuated optic nerve damage in acute optic neuritis in rats. J. Neurochem., 124, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shanab A.Y., Nakazawa T., Ryu M., Tanaka Y., Himori N., Taguchi K., Yasuda M., Watanabe R., Takano J., Saido T., et al. (2012) Metabolic stress response implicated in diabetic retinopathy: the role of calpain, and the therapeutic impact of calpain inhibitor. Neurobiol. Dis., 48, 556–567. [DOI] [PubMed] [Google Scholar]
- 56.Ma H., Tochigi A., Shearer T.R., Azuma M. (2009) Calpain inhibitor SNJ-1945 attenuates events prior to angiogenesis in cultured human retinal endothelial cells. J. Ocul. Pharmacol. Ther., 25, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics, doi:10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glaser F., Pupko T., Paz I., Bell R.E., Bechor-Shental D., Martz E., Ben-Tal N. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics, 19, 163–164. [DOI] [PubMed] [Google Scholar]
- 59.Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res., 33, W299–W302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res., 38, W529–W533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Celniker G., Nimrod G., Ashkenazy H., Glaser F., Martz E., Mayrose I., Pupko T., Ben-Tal N. (2013) ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem., 53, 199–206. [Google Scholar]
- 62.Wert K.J., Davis R.J., Sancho-Pelluz J., Nishina P.M., Tsang S.H. (2013) Gene therapy provides long-term visual function in a pre-clinical model of retinitis pigmentosa. Hum. Mol. Genet., 22, 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mahajan V.B., Skeie J.M., Assefnia A.H., Mahajan M., Tsang S.H. (2011) Mouse eye enucleation for remote high-throughput phenotyping. J. Vis. Exp., 57, doi:10.3791/3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skeie J.M., Tsang S.H., Mahajan V.B. (2011) Evisceration of mouse vitreous and retina for proteomic analyses. J. Vis. Exp., 50, e2795 doi:2710.3791/2795. [DOI] [PMC free article] [PubMed] [Google Scholar]